

Background: Sirtuin-1 improves metabolic disease, but oxidants may inhibit it.
Results: Metabolic stress increased glutathione adducts, inactivated endogenous Sirtuin-1, and promoted apoptosis. A novel Sirtuin-1 oxidation-insensitive mutant or glutaredoxin-1 prevented metabolic dysregulation and apoptosis.
Conclusion: A novel Sirtuin-1 mutant circumvents oxidation and more effectively inhibits metabolic dysregulation and apoptosis.
Significance: Oxidative inactivation of Sirtuin-1 contributes to metabolic disease.
Keywords: Glutathionylation, Metabolic Diseases, Oxidative Stress, Polyphenols, Resveratrol, Glutathiolation, High Fat High Sucrose Diet, Reactive Oxygen and Nitrogen Species
Abstract
Sirtuin-1 (SirT1), a member of the NAD+-dependent class III histone deacetylase family, is inactivated in vitro by oxidation of critical cysteine thiols. In a model of metabolic syndrome, SirT1 activation attenuated apoptosis of hepatocytes and improved liver function including lipid metabolism. We show in SirT1-overexpressing HepG2 cells that oxidants (nitrosocysteine and hydrogen peroxide) or metabolic stress (high palmitate and high glucose) inactivated SirT1 by reversible oxidative post-translational modifications (OPTMs) on three cysteines. Mutating these oxidation-sensitive cysteines to serine preserved SirT1 activity and abolished reversible OPTMs. Overexpressed mutant SirT1 maintained deacetylase activity and attenuated proapoptotic signaling, whereas overexpressed wild type SirT1 was less protective in metabolically or oxidant-stressed cells. To prove that OPTMs of SirT1 are glutathione (GSH) adducts, glutaredoxin-1 was overexpressed to remove this modification. Glutaredoxin-1 overexpression maintained endogenous SirT1 activity and prevented proapoptotic signaling in metabolically stressed HepG2 cells. The in vivo significance of oxidative inactivation of SirT1 was investigated in livers of high fat diet-fed C57/B6J mice. SirT1 deacetylase activity was decreased in the absence of changes in SirT1 expression and associated with a marked increase in OPTMs. These results indicate that glutathione adducts on specific SirT1 thiols may be responsible for dysfunctional SirT1 associated with liver disease in metabolic syndrome.
Introduction
Metabolic syndrome including type 2 diabetes associated with insulin resistance, hyperinsulinemia, and plasma dyslipidemias are on the rise worldwide. Non-alcoholic fatty liver disease (NAFLD)3 is a complication of metabolic syndrome. Hepatosteatosis that leads to NAFLD originates from excessive lipid uptake from nutritional sources exceeding hepatocyte β-oxidation, perturbations in lipoprotein synthesis, and defects in excretion. The disease progresses to more severe forms involving inflammation and hepatocyte apoptosis (1).
Activation of Sirtuin-1 (SirT1), a member of the NAD+-dependent class III histone deacetylase family, alleviates metabolic complications (2–4) and prevents the diet-induced development of NAFLD and non-alcoholic steatohepatitis in mice (5). Conversely, hepatocyte-specific deletion of SirT1 induces weight gain and hepatic steatosis in mice fed a Western diet (6). SirT1, via deacetylation of key transcription factors (7–9), activates beneficial metabolic processes that mimic caloric restriction and increase cellular stress resistance. The first non-histone target described for SirT1 was p53, an important regulator of the cell cycle, senescence, and apoptosis. SirT1 deacetylates p53 directly on the C-terminal Lys382 that represses transcriptional activity and consequently inhibits apoptosis. Hyperacetylation of p53 is accompanied by increased apoptosis and stress sensitivity in SirT1-ablated mice (10). Furthermore, studies in patients with non-alcoholic steatohepatitis show that p53 and hepatocyte apoptosis correlate with the progression of metabolic liver disease (1, 11), rendering SirT1 an attractive therapeutic target.
Post-translational control of SirT1 activity is a complex process that is only beginning to be understood. Depending on the co-substrate NAD+, SirT1 activity may be directly controlled by the cellular redox and nutritional status. In a starving cell the NAD+/NADH ratio is expected to rise, thus increasing SirT1 activity and activating mechanisms similar to caloric restriction.
Various post-translational modifications including SUMOylation (12), phosphorylation (13–18), carbonylation (19), and S-nitrosylation (20) were shown to regulate SirT1 activity, localization, and degradation. In addition, the cellular redox status modulates SirT1 activity, notably in neuronal progenitor (21) and skeletal muscle cells (22). Oxidative post-translational modifications may mediate these effects on SirT1 and directly affect activity and protein-protein interactions. We demonstrated that glutathione-SirT1 adducts formed in vitro by S-nitrosoglutathione inhibit the enzyme with an IC50 of 69 μm (23). Another report identified glutathione-cysteine and carbonyl adducts on SirT1 after incubation of lung epithelial cells with hydrogen peroxide and acrolein (19). Furthermore, Snyder and co-workers (20) reported that GAPDH-mediated S-nitrosylation disrupted the structural zinc tetrathiolate module in SirT1 associated with inhibition of activity.
In our current study we demonstrate that oxidative inactivation of SirT1 occurs in HepG2 cells under nitrosative, oxidative, or metabolic stress. Using an oxidant-resistant mutant generated by replacing three essential cysteines with serines, we show that mutant SirT1 retained higher activity, protecting HepG2 cells from oxidative and metabolic stress-induced apoptosis. In a diet-induced established mouse model of metabolic syndrome, diminished SirT1 activity in the fatty liver correlated with reversible oxidative post-translational modifications of SirT1 compared with normal chow-fed mice. These data suggest that the generation of reactive oxygen and nitrogen species in metabolic disease may directly attenuate SirT1 activity via oxidative post-translational modifications, potentially leading to progressive liver disease and metabolic dysfunction.
EXPERIMENTAL PROCEDURES
Reagents, Materials, and Antibodies
N-(Biotinoyl)-N′-iodoacetyl ethylenediamine (BIAM) (B-1591), glutathione ethyl ester, biotin amide (BioGEE), LipofectamineTM, and cell culture media were obtained from Invitrogen. ZebaTM spin desalting columns (40,000 molecular weight cutoff) (87767) and EZ-Link N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (HPDP-biotin) (21341) were obtained from Thermo Fisher Scientific (Waltham, MA). Anti-SirT1 antibody was from Abcam (Cambridge, MA) (ab70239). Anti-acetylated p53 (Lys382) was from Cell Signaling Technology (Danvers, MA) (2525), and cells were lysed according to the manufacturer's protocol. Anti-total p53 (DO-1) (sc-126) antibody was from Santa Cruz Biotechnology (Dallas, TX). Anti-cleaved caspase 3 antibody was from Cell Signaling Technology (9664). The luciferase assay kit was obtained from Promega (Madison, WI). AdEasy and QuikChange site-directed mutagenesis kits were purchased from Agilent Technologies (Santa Clara, CA). Fluor-de-LysTM SirT1 activity assay was from Enzo Life Sciences (Farmingdale, NY). PVDF membrane, SDS-polyacrylamide gels, and other reagents for immunoblotting were obtained from Bio-Rad. Anti-FLAGTM M2 affinity gel (A2220), anti-FLAG M2 antibody (F1804), trypsin, protein A/G-Sepharose, and all chemicals if not otherwise specified were obtained from Sigma-Aldrich.
Experimental Animals
Male C57BL/6J mice (stock number 000664, The Jackson Laboratory, Bar Harbor, ME) (8 weeks of age) were fed a normal chow diet (4.5% fat, 0% sucrose) or a high fat and high sucrose (HFHS) diet (35.5% fat (lard) representing 60% calories, 16.4% sucrose) ad libitum for 5 months (catalogue numbers D09071702 and D09071703, Research Diets, New Brunswick, NJ). The control diet was custom-formulated to match the micronutrients contained in HFHS except for fat and sucrose. Mice were housed in rooms with a 12-h light/dark cycle and in groups of three to four whenever possible. The protocol was approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine. Mice were euthanized after 5 months on the diet, and livers were perfused, excised, snap frozen, and stored in liquid nitrogen or at −80 °C.
Cell Culture and HPHG Treatment
HEK-293 and HepG2 cells (ATCC, Manassas, VA) were maintained in DMEM containing 10% FBS, 100 units of penicillin, and 100 μg/ml streptomycin (Invitrogen) at 37 °C under a 5% CO2, 95% air atmosphere. Cells were plated at 80% confluence 24 h prior to infection or transfection. Infected cells were treated with control (5 mm glucose, 0.67% bovine serum albumin (BSA; fatty acid-free, Sigma-Aldrich)) or high palmitate and high glucose (HPHG) medium (25 mm glucose, 0.4 mm palmitic acid, 0.67% BSA) for 16 h. This gives a final molar ratio of fatty acid/BSA of 4:1. After treatment cells were washed with PBS before being lysed in Laemmli buffer (Bio-Rad).
PUMA Promoter-Luciferase Reporter
The luciferase reporter vector containing the promoter region of the human PUMA gene was from Addgene (16591). Expression plasmids were co-transfected with a luciferase reporter construct (200–300 ng) and pRL-TK (20–50 ng) into recipient cells as described by the manufacturer. pcDNA3.1 was used for control transfection. Luciferase activity was measured 24–48 h post-transfection using a TD-20e tube luminometer (Turner Biosystems, Sunnyvale, CA).
Construction of SirT1 Cysteine Mutants and Expression in HEK-293 Cells
Full-length mouse SirT1 construct in pcDNA3.1 was obtained from Addgene (plasmid 8438) and was used as the template for site-directed mutagenesis. The Cys to Ser mutants were prepared by introducing a single base exchange (C61S, TGT→TCT; C245, TGT→TCT; C260S, TGT→TCT; C318S, TGT→TCT; and C613S, TGC→TCT) by using the QuikChange site-directed mutagenesis kit. Mutants were confirmed by DNA sequencing (Tufts Medical Center, Sequencing Core, Boston, MA). Constructs were transfected into HEK-293T cells using Lipofectamine 2000.
Preparation of S-Nitrosocysteine (Cys-NO)
S-Nitrosoglutathione was replaced here by Cys-NO because of its better cell permeability (24). Cys-NO stock solutions were prepared freshly by mixing equimolar amounts of l-cysteine and NaNO2 under acidic conditions (0.25 m HCl) in the presence of 0.1 mm diethylenetriamine pentaacetate. The concentration of Cys-NO in solution was determined spectrophotometrically at 334 nm (ϵ344 = 900 m−1 cm−1) and adjusted to 500 mm. Dilutions of Cys-NO were prepared in HEN buffer (250 mm Hepes, pH 7.7, 1 mm EDTA, 0.1 mm neocuproine) immediately before the experiment.
Adenoviral Constructs and Infection
pAd-Track SIRT1-FLAG was linearized and recombined with pAdEasy in BJ5183 cells to generate cosmids. Suitable cosmids were digested with PAC-1 and transfected into HEK-293 cells utilizing calcium phosphate. Adenoviral colonies were isolated and propagated in HEK-293A cells (25). Adenovirus was isolated using double cesium chloride gradient centrifugation. Titers were determined by A260 assay and expressed as optical particle units/ml. Cells were serum-reduced, infected with 1 × 1010 optical particle units/ml for 6 h, and incubated for 48 h before further treatment.
Affinity Capture of SirT1 Proteins
Cells were lysed in FLAG immunoprecipitation buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100) and centrifuged for 15 min at 12,000 × g at 4 °C. The supernatant was incubated with anti-FLAG M2 affinity gel for 2 h at 4 °C to immunoprecipitate SirT1. The gel was washed three times with buffer (150 mm NaCl in PBS, pH 7.4) and boiled in 50 μl of loading buffer, and proteins were separated by SDS-PAGE.
SirT1 Activity Measurement
SirT1 activity was tested by Fluor-de-Lys assay. 90 μl of purified SirT1 from anti-FLAG M2 affinity gel was incubated with 1 μl of 10 mm acetylated p53 peptide (Arg-His-Lys-Lys(Ac)-AMC where AMC is 7-amino-4-methylcoumarin) for 30 min at 37 °C with 1 μ1 of 10 mm NAD+ in activity assay buffer (50 mm Tris-HCl, pH 8.0, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2). Then 100 μl of 1 mg/ml concentrated trypsin solution was added to release the 7-amino-4-methylcoumarin fluorophore, which allows quantification of the amount of substrate deacetylated by SirT1. The fluorescence intensity was recorded over 60 min using a Fluoroscan Ascent microplate reader (Thermo Fisher) with excitation set to 375 nm and emission set to 460 nm.
Biotin Switch Assay for Labeling of Reversible Oxidized Cysteines
Labeling with EZ-Link HPDP-biotin or BIAM was used in a biotin switch assay to detect reversibly oxidized cysteines. To purify SirT1, a 10-cm dish of HEK-293 or HepG2 cells was treated with Cys-NO, hydrogen peroxide (H2O2), or HPHG. Cells were lysed in immune precipitation assay buffer containing 100 mm maleimide to block all free thiols and prevent further oxidation. Before affinity isolation of SirT1, excess maleimide was removed by passing the lysates over Zeba spin columns. SirT1 was incubated with anti-FLAG M2 affinity gel for 2 h at 4 °C. Beads were collected by centrifugation at 2,000 × g for 2 min and washed three times with modified PBS (150 mm NaCl in PBS, pH 7.4). Beads were incubated with 2 mm dithiothreitol (DTT) in 100 mm Tris-HCl, pH 7.4 for 1 h at 4 °C and washed three times. Reduced cysteines of SirT1 were labeled with 0.5 mm biotin-HPDP or 0.1 mm BIAM in labeling solution (50 mm Tris-HCl, 0.5 mm biotin-HPDP, 0.5% SDS, pH 7.4) for 1 h at room temperature. Beads were boiled in 30 μl of 2× non-reducing Laemmli buffer for 10 min and loaded on an SDS Tris-glycine gel. To control for specific labeling of cysteines with biotin, aliquots of each sample were incubated with reducing Laemmli buffer.
Measurements of Reactive Oxygen and Nitrogen Species in HepG2 Cells
Intracellular reactive oxygen and nitrogen species generation was assessed by using 2′,7′-dichlorofluorescin diacetate. Reactive oxygen species in the cells oxidize 2′,7′-dichlorofluorescin diacetate, yielding the fluorescent compound 2′,7′-dichlorofluorescein. HepG2 cells were washed with warm PBS and treated for 16 h in the presence or absence of HPHG medium. Cells were washed, and 2′,7′-dichlorofluorescin diacetate (10 μm) was added for 30 min. The medium was removed; cells were washed with warm PBS and lysed in 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100; and 2′,7′-dichlorofluorescein was measured using a Fluoroscan Ascent microplate reader (Thermo Fisher) with excitation set to 498 nm and emission set to 522 nm.
Liver Histology and Analysis
For hematoxylin and eosin (H&E) staining, liver tissue was fixed in 10% phosphate-buffered formalin, embedded in paraffin, and cut into 5-μm sections. Lipids were visualized with oil red O. For this technique livers were embedded in optimal cutting temperature compound, cut into 5-μm cryosections, and fixed with formalin. Sections were stained with oil red O, and the nuclei were stained with alum hematoxylin. Slides were mounted with aqueous mountant. For immunohistochemistry of protein-bound GSH adducts, 5-μm cryosections were fixed in ice-cold methanol followed by incubation with 100 μm N-ethylmaleimide for 10 min at room temperature. The slides were incubated with anti-GSH antibody (Virogen, 101-A) for immunohistological examination. Oil red O-stained lipids and GSH-protein adducts were quantified by ImageJ with the color deconvolution plug-in (26).
Molecular Homology Modeling
The human SirT1 sequence obtained from UniProt database accession number Q96EB6 was submitted to the I-TASSER server and DISOPRED2 server (27, 28). Modeling was performed with PyMOL v0.99.
Detection of SirT1-Glutathione Adducts
HepG2 cells were incubated with biotinylated GSH monoethyl ester (40 μm) for 1 h. The cells were changed to control or HPHG medium for 16 h. The medium was removed, and cells were incubated in HBSS supplemented with 100 mm N-ethylmaleimide for 10 min at 37 °C. The cells were lysed in non-reducing buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm MgCl2, 0.5% sodium dodecyl sulfate, 100 mm maleimide, 1 mm PMSF), and lysates were passed through Zeba columns to remove excess biotinylated GSH. 500 μg of total protein was incubated for 1 h at 4 °C with streptavidin-Sepharose beads. The beads were rinsed three times with buffer (25 mm Tris-HCl, pH 7.4, 500 mm NaCl, 5 mm MgCl2, and 0.1% SDS), and proteins with GSH adducts were released from the beads by incubating with 40 μl of lysis buffer supplemented with 10 mm DTT for 15 min with gentle shaking. Each sample was diluted in Laemmli buffer containing 5% β-mercaptoethanol and separated by SDS-PAGE, and SirT1 was detected by immunoblotting with a total SirT1 antibody (Santa Cruz Biotechnology).
RESULTS
SirT1 Is Sensitive to Nitrosative and Oxidative Stress in the HepG2 Hepatocyte Cell Line
Recently, we identified five redox-sensitive cysteines on SirT1 by mass spectrometry after in vitro oxidation with S-nitrosoglutathione (23). SirT1 activity was reversibly inhibited by oxidative post-translational modifications. Here, to test the biological relevance of reversible SirT1 oxidation, we measured SirT1 activity in transfected HepG2 cells treated with Cys-NO or H2O2. Cys-NO mimics the previous in vitro conditions (23) but, in contrast to S-nitrosoglutathione, permeates cells (24).
Because SirT1 inhibits the promoter activity of p53 by deacetylation of Lys382 (8, 29–31), suppressing p53-mediated apoptosis and senescence, p53 acetylation was measured as a surrogate for biological SirT1 activity. Overexpression of SirT1 wild type (WT) decreased basal acetylated p53 and the proapoptotic cleaved caspase 3 compared with empty vector control in HepG2 cells. Treatment with Cys-NO (0–750 μm; Fig. 1A) or H2O2 (500 μm; Fig. 1C) abrogated the SirT1 effect, suggesting oxidative inhibition of SirT1. To determine whether the previously identified cysteine thiols of SirT1 contribute to oxidative inactivation, various serine mutants were created. The single mutants had little effect in preventing SirT1 inactivation by oxidants measured by p53 transcriptional activity in cells using a PUMA promoter-luciferase reporter. The SirT1 triple mutant (MUT; C61S,C318S,C613S), however, maintained full activity (Fig. 1B). A quintuple SirT1 mutant in which all previously identified cysteines were mutated did not exhibit any superiority over the triple mutant. To further test whether MUT SirT1 is not reversibly oxidized in H2O2-treated cells a biotin switch assay was performed. In this assay reversibly oxidized cysteines are selectively labeled with the thiol-reactive probe HPDP-biotin (Fig. 1D). WT but not MUT SirT1 showed strong reversible oxidation. In summary these findings suggest that the three mutated cysteines of SirT1 are the most sensitive to oxidation and regulate protein function in HepG2 cells.
FIGURE 1.

SirT1 WT inactivation by oxidative and nitrosative stress is abrogated by a triple cysteine mutant in HepG2 cells. HepG2 cells were transiently transfected for 48 h with either FLAG-SirT1 WT, FLAG-SirT1 MUT (mutated residues C61S, C318S, and C613S), or empty vector (pcDNA3.1) as a control. Equal SirT1 overexpression was confirmed by Western blot analysis with an antibody against the FLAG tag. Representative Western blots of three independent experiments are shown. A, cells were treated with Cys-NO (bolus addition) for 8 h at the indicated concentrations. Overexpression of SirT1 suppressed caspase 3 (Casp-3) cleavage and p53 acetylation (Ac-p53) at the SirT1-specific substrate site Lys382. Increased concentrations of Cys-NO inhibited the SirT1-dependent effect, increasing p53 acetylation and proapoptotic cleaved caspase 3. B, p53 transcriptional activity was measured by transfecting a PUMA-specific luciferase reporter into cells. Cys-NO (bolus addition of 500 μm) treatment inhibited the SirT1-dependent (WT) suppression of PUMA promoter activity, but the double, triple, and quintuple mutants (2MUT, 3MUT, and 5MUT) maintained PUMA suppression when treated with Cys-NO. The triple mutant (3MUT) exhibited the greatest effect and was used for the remainder of the experiments (referred to as MUT SirT1). ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, WT SirT1- versus SirT1 mutant-transfected HepG2 cells either under control conditions or treated with Cys-NO. C, cells were treated with H2O2 (500 μm bolus addition) for 8 h. Overexpression of SirT1 WT reduced cleaved caspase 3 in H2O2-treated cells, whereas MUT almost restored control levels. D, reversible oxidative modifications of cysteines were detected in SirT1 WT and MUT with a biotin switch assay and IR-based (LI-COR Biosciences) Western analysis. Transfected cells were treated for 8 h with hydrogen peroxide (bolus addition) (n = 3). Cysteines of WT SirT1 were reversibly modified by H2O2 (500 μm) treatment as demonstrated by HPDP-biotin labeling of the reduced cysteine modification (green, biotin-labeled protein; red, FLAG antibody for detection of SirT1). DTT treatment preceding the labeling procedure (right panel) shows no staining, confirming the specificity of the method for reversible oxidation. For details please refer to the “Experimental Procedures.” ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, WT versus MUT SirT1-transfected HepG2 cells treated with H2O2.
SirT1 MUT Maintains Full Activity in Metabolically Stressed HepG2
Diminished SirT1 activity has been reported in various disease models including diabetes and metabolic syndrome (32, 33). To mimic the effects of metabolic overload HepG2 cells were incubated in standard medium supplemented with HPHG. These cells respond to metabolic stress with apoptosis, stress-induced senescence, and induction of proinflammatory genes (34, 35).
p53 acetylation and cleaved caspase 3 were detected with Western blot analysis at the same time point to ensure equal expression levels of WT and MUT SirT1. HPHG significantly increased p53 acetylation and proapoptotic cleaved caspase 3 in HepG2 cells transfected with empty vector (pcDNA3.1) (Fig. 2, A and B). The HPHG effect was diminished by overexpression of SirT1 WT and completely suppressed by equal expression of SirT1 MUT. Even when SirT1 MUT was less expressed than WT, the superior protective effect of the mutant was clearly evident. Measuring deacetylase activity of FLAG-immunoprecipitated SirT1 WT and MUT further substantiated that SirT1 WT activity was diminished by about 50% by HPHG, but the MUT was fully functional (Fig. 2C).
FIGURE 2.

SirT1 MUT maintained activity and prevented proapoptotic signaling in HPHG-treated HepG2 cells. HepG2 cells were transiently transfected for 48 h with either FLAG-SirT1 WT, FLAG-SirT1 MUT (mutated residues C61S, C318S, and C613S), or empty vector (pcDNA3.1) as a control. SirT1 overexpression was confirmed by Western blot analysis with an antibody against the FLAG tag. A, overexpression of SirT1 WT suppressed p53 acetylation (Ac-p53) at the SirT1-specific substrate site Lys382 and caspase 3 (Casp-3) cleavage in normal medium. In contrast, treating cells with HPHG for 16 h inhibited the SirT1-dependent effect, increasing p53 acetylation and proapoptotic cleaved caspase 3. The SirT1 MUT, however, maintained control levels. B, densitometric analysis of the Western blot results normalized to GAPDH for cleaved caspase 3 or total p53 for acetylated p53. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, WT SirT1 versus empty vector control; *, p < 0.05, WT SirT1- versus MUT SirT1-transfected HepG2 cells treated with HPHG. C, overexpressed FLAG-SirT1 WT or MUT was either isolated from transfected control or cells treated with HPHG. Activity of the isolated enzyme was measured by the Fluor-de-Lys assay. HPHG treatment decreased SirT1 WT, but MUT activity was fully maintained. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, WT SirT1- versus SirT1 MUT-transfected HepG2 cells treated with HPHG. D, p53 transcriptional activity was measured by transfecting a PUMA-specific luciferase reporter together with FLAG-SirT1 WT, FLAG-SirT1 MUT, or empty vector (pcDNA3.1) into cells for 48 h. HPHG treatment for 16 h inhibited the SirT1-dependent suppression of PUMA promoter activity, but the MUT maintained PUMA suppression when cells were exposed to HPHG. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, empty vector versus WT SirT1 and empty vector- versus SirT1 MUT-transfected HepG2 cells in the control group and WT SirT1 versus MUT SirT1 in the HPHG group. E and F, mRNA was isolated, and endogenous PUMA and p21 mRNA levels were measured by quantitative RT-PCR. SirT1 MUT in contrast to SirT1 WT suppressed induction of both PUMA and p21 when cells were treated with HPHG. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.05, WT SirT1 versus MUT SirT1 and vector- versus WT SirT1-transfected HepG2 cells in the HPHG group and for F in the control group; n.s., not significant, empty vector- versus WT SirT1-transfected HepG2 in the HPHG group. Ctrl, control; AFU, arbitrary fluorescent units.
To demonstrate that modulation of SirT1 activity by metabolic stress alters downstream signaling, p53 transcriptional activity and induction of PUMA and p21 mRNA were measured. p21 (WAF1) is important for execution of cell cycle arrest and is associated with cell senescence (30, 36). PUMA or Bcl-2-binding component 3 modulates apoptosis via interaction with the antiapoptotic protein Bcl-2 (37). PUMA binds to Bcl-2 and Bcl-XL and induces mitochondrial membrane permeability transition and caspase activation (37). Using a PUMA promoter-luciferase assay, HPHG significantly increased p53 transcriptional activity in pcDNA3.1- and SirT1 WT-transfected cells (Fig. 2D). Concomitantly, endogenous PUMA and p21 mRNA levels, measured by quantitative RT-PCR, increased (Fig. 2, E and F). Overexpression of SirT1 WT in HPHG-treated cells only marginally affected PUMA promoter activation and mRNA expression of PUMA and p21. In contrast, the SirT1 MUT exhibited promoter activation comparable with untreated cells (Fig. 2D), and endogenous PUMA and p21 mRNA (Fig. 2, E and F) levels were normalized almost to control levels.
Reversible Oxidative Post-translational Modifications Regulate SirT1 Activity in HepG2 Cells
The SirT1 MUT clearly suggests the possibility that reversible oxidative cysteine modifications on SirT1 WT regulate enzyme activity in the metabolic disease milieu. These modifications are generally induced by an increase in reactive nitrogen and oxygen species (RNOS). Indeed, metabolically stressed HepG2 cells (Fig. 3A) demonstrated augmented RNOS formation as measured by 2′,7′-dichlorofluorescein fluorescence (38). RNOS are known to alter the cellular thiol redox status, and as a consequence GSH-protein adducts are expected to be elevated. Both H2O2 as a positive control and HPHG markedly increased GSH-protein adducts in cells as detected using a GSH adduct-specific antibody (Fig. 3B). The specificity of the antibody was confirmed by removal of GSH-protein adducts by DTT reduction prior to Western blot analysis. To directly detect reversible oxidative modifications on SirT1, a biotin switch assay was performed (Fig. 3, C and D). Whereas SirT1 WT exhibited marked biotin labeling indicating reversible oxidative protein modification, the SirT1 MUT was redox-resistant in HPHG-treated cells as there was no detectable increase in reversible cysteine adducts. In contrast, phosphorylation of SirT1 at Ser47 and proteolytic degradation (Fig. 3E) were unchanged and are apparently less important in this model.
FIGURE 3.

Reversible cysteine modifications on SirT1 are increased in HPHG-treated HepG2 cells. A, reactive oxygen and nitrogen species were measured by incubating cells for 30 min with 10 μm 2′,7′-dichlorofluorescin diacetate. The fluorescence of the oxidized dye was measured as arbitrary fluorescent units (AFU) and normalized to cellular protein content. HPHG treatment of cells increased RNOS formation by about 62% compared with control. Unpaired Student's t test was used (n = 3). Error bars, S.D. *, p < 0.005, control versus HPHG-treated HepG2 cells. B, parallel to the increase in RNOS formation, protein S-glutathionylation is increased in HPHG-treated cells measured with Western blot analysis using an anti-GSH antibody. As a positive control cells were treated with 500 μm H2O2 for 15 min. Lysates were treated with 10 mm DTT to remove S-glutathionylation protein adducts before Western blot analysis. C, FLAG-tagged SirT1 WT or MUT was overexpressed in control or HPHG-treated cells to detect reversible cysteine oxidation by a switch assay. HPHG treatment of cells caused reversible oxidation of SirT1 WT but not of MUT compared with control. D, densitometric analysis of the biotin switch results normalized to total SirT1 (Input). ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.01, control versus HPHG-treated HepG2 cells transfected with WT SirT1. E, proteolytic fragments of SirT1 were detected by Western blot analysis with LI-COR Biosciences infrared imaging (IR) or enhanced chemiluminescence (ECL). No significant additional bands of endogenous SirT1 were detected after either HPHG treatment for 16 h or exposure to H2O2 (bolus addition of 500 μm for 15 min). Phosphorylation of endogenous SirT1 (P-SirT1) at Ser47 remained unchanged by either treatment. Ctrl, control; ROS, reactive oxygen species.
Glutaredoxin-1 Maintains Endogenous SirT1 Activity by GSH-Protein Adduct Removal
To further investigate the nature of the reversible oxidative modification on endogenous SirT1, glutaredoxin-1 (Glrx) was overexpressed in HepG2 cells. Glrx catalyzes the reduction of GSH-protein adducts (39) (Fig. 4A). In cells overexpressing SirT1 WT, additional overexpression of Glrx increased SirT1 activity under control conditions, suggesting that GSH adducts on SirT1 are present even in control cells and may have a regulatory function (Fig. 4B). Furthermore, Glrx overexpression preserved SirT1 WT activity after HPHG exposure, which inhibited SirT1 activity in control cells (Fig. 4B), consistent with the concept that GSH-protein adducts inhibit SirT1 activity. To show the effect of HPHG and Glrx on GSH adducts on endogenous SirT1, cell lysates were labeled in a biotin switch assay followed by a pulldown on streptavidin beads for isolation and enrichment of reversibly oxidized proteins. These proteins were analyzed by Western blot and probed for endogenous SirT1. HPHG increased reversibly oxidized SirT1, which was attenuated by overexpression of Glrx (Fig. 4, C and D). In addition, cell lysates (input) probed for endogenous SirT1, GAPDH, or p53 expression showed these to be unaltered by either HPHG or Glrx overexpression. Moreover, levels of acetylated p53 paralleled those of reversibly oxidized thiols on SirT1 and reflected the changes in SirT1 activity modulated by GSH-protein adducts.
FIGURE 4.

Glrx overexpression reduces reversible oxidative modifications and maintains endogenous SirT1 activity in HPHG-treated HepG2 cells. A, protein S-glutathionylation is a dynamic reversible process that is regulated by Glrx. Glutathione-protein adducts in the cytosol and nucleus are specifically reduced by Glrx, which in a thiol exchange reaction transfers the glutathione from the target protein to itself. Glrx regenerates by reacting with free GSH to form oxidized glutathione (GSSG). Finally, oxidized glutathione is reduced to GSH by glutathione reductase, utilizing NADPH as an electron donor and reduction equivalent. B, cells were transiently transfected for 48 h with FLAG-SirT1 WT and Glrx. SirT1 was immunoprecipitated via its FLAG tag, and activity was measured by the Fluor-de-Lys assay. HPHG inhibited SirT1 activity. Overexpression of Glrx increased SirT1 activity even under control conditions and maintained its full activity in HPHG treatment. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.05, control versus HPHG-treated HepG2 cells; *, p < 0.01 Glrx-transfected HepG2 versus empty vector in the control or HPHG-exposed group. C, reversible oxidative cysteine modifications of endogenous SirT1 were detected by a biotin switch assay (BIAM-labeled SirT1). p53 acetylation was measured by Western blot analysis as a surrogate marker for SirT1 activity. Cells treated with HPHG for 16 h showed increased reversible oxidation of endogenous SirT1. Overexpression of Glrx by transfecting cells for 48 h decreased reversible oxidation of endogenous SirT1 and p53 acetylation. D, densitometric analysis of reversibly oxidized (BIAM-labeled SirT1) to total SirT1 and acetylated p53 (Ac-p53) to total p53 in Glrx-overexpressing cells. ANOVA and Bonferroni's post-test were used (n = 3). Error bars, S.D. *, p < 0.001, control versus HPHG-treated HepG2 cells and empty vector versus Glrx-transfected in control or HPHG-exposed HepG2 cells. E, HepG2 cells were incubated with the cell-permeable and biotin-labeled glutathione monoethyl ester (40 μm). GSH adducts on SirT1 were detected by Western blot with streptavidin. HPHG exposure of cells increased SirT1-GSH adducts, which were prevented or reversed by overexpression of Glrx. Ctrl, control; AFU, arbitrary fluorescent units.
Because these results with Glrx support the importance of GSH adducts on SirT1, to show that SirT1 is directly adducted with GSH in cells HepG2 cells were incubated with cell-permeable, biotin-labeled GSH (Fig. 4E). HPHG greatly increased GSH adducts on SirT1 that were removed or prevented from being formed by overexpression of Glrx. These data provide direct evidence that GSH adducts on SirT1 are formed and contribute to the modulation of its activity by Glrx as observed in Fig. 4B.
Oxidation Attenuates Endogenous SirT1 Activity in Livers of Mice Fed a Diet High in Fat and Sucrose
Reversible oxidative modifications of SirT1 were investigated in livers from C57BL/6J mice fed a HFHS diet for 8 months. This mouse model manifests the typical pathophysiology of metabolic syndrome (obesity, diabetes, and hypertension) as reported earlier (40). Histological examination demonstrated a uniformly fatty liver and hepatomegaly as determined by H&E and oil red O staining (Fig. 5, A and B) in the HFHS-fed mice. Immunostaining of liver slices with an antibody against GSH-protein adducts showed that as in HepG2 cells GSH-protein adducts were increased in the altered metabolic milieu in the liver (Fig. 5, A and B). SirT1 activity was measured in liver nuclear extracts (Fig. 5C). HFHS diet significantly attenuated endogenous SirT1 activity without affecting protein expression. An increase in acetylated p53 reflected diminished SirT1 activity in the fatty liver. Using the biotin switch assay, increased reversible protein oxidation of SirT1 correlated with its decreased enzyme activity (Fig. 5, D and E), consistent with oxidative inactivation of SirT1 in livers of HFHS diet-fed mice.
FIGURE 5.

Oxidation of endogenous SirT1 in livers of mice fed a diet high in fat and sucrose correlates with reduced SirT1 activity. Mice were fed with chow or HFHS diet for 5 months as described under “Experimental Procedures.” A, H&E, oil red O, and immunohistochemical GSH protein-adduct staining of liver sections. HFHS diet markedly increased lipids and DTT-reversible GSH-protein adducts in mouse liver. B, oil red O-stained lipids and GSH-protein adducts were quantified by ImageJ (n = 5/group) with the color deconvolution plug-in. Unpaired Student's t test was used (n = 3). Error bars, S.D. *, p < 0.005, chow- versus HFHS-fed mice. C, nuclear extracts isolated from mouse liver homogenates were used to determine NAD+-dependent SirT1 activity with the Fluor-de-Lys assay. SirT1 protein levels in nuclear extracts were analyzed by Western blot to ensure equal protein amounts. Unpaired Student's t test was used (n = 3). Error bars, S.D. *, p < 0.05, chow- versus HFHS-fed mice. D, reversible oxidative cysteine modifications of endogenous SirT1 were detected by a biotin switch assay (BIAM-labeled SirT1) in mouse liver lysates. p53 acetylation (Ac-p53) was measured by Western blot analysis as a surrogate marker for SirT1 activity. Mice fed a HFHS diet showed increased reversible oxidation of endogenous SirT1 that correlated with an increase in p53 acetylation. E, densitometric analysis of the biotin switch results normalized to total SirT1 (Input) and of acetylated p53 normalized to total p53. Unpaired Student's t test was used (n = 3). Error bars, S.D. *, p < 0.005, chow- versus HFHS-fed mice. Ctrl, control; AFU, arbitrary fluorescent units; AU, arbitrary units.
DISCUSSION
Here we provide evidence that SirT1 activity is inhibited by reversible thiol oxidation of three specific cysteines during metabolic stress and that mutation of these cysteines circumvents the oxidation and sustains SirT1 biological activity despite the metabolic stress. These reactive cysteines were reversibly oxidized in RNOS-challenged or metabolically stressed HepG2 cells as well as in livers of high fat and high sucrose -fed mice. Overexpression of the thioltransferase Glrx restored SirT1 activity and prevented proapoptotic signaling induced by metabolic stress. Exchanging the three critical cysteines 61, 318, and 613 to serine resulted in a SirT1 mutant that maintains full biological activity under oxidative and metabolic stress. The SirT1 mutant confirmed that these cysteines are targets of cellular redox control by reversible oxidative modifications. Our evidence supports GSH-protein adducts as the likely mechanism, but intramolecular disulfide formation cannot be ruled out.
Role of SirT1 in Metabolic Liver Disease
NAFLD is a major complication of metabolic syndrome. SirT1 gain of function by pharmacological agonists or SirT1 overexpression alleviates symptoms of metabolic syndrome and prevents the development of both non-alcoholic and alcohol-induced liver steatosis in mice (5, 41, 42). Beneficial metabolic effects and life span extension for SirT1 activation are universal and have been shown from lower organisms to mammals. Our mutant, however, preserved SirT1 function under metabolic stress and exhibited protective effects superior to SirT1 WT overexpression alone. Metabolic stress is known to increase RNOS formation and lowers the thiol redox status via metabolic maladaptation. Altered cellular redox balance promotes oxidative post-translational modifications of SirT1, attenuating its protective biological function. Increased oxidative stress and enhanced formation of lipid peroxidation are associated with NAFLD and non-alcoholic steatohepatitis in patients (43, 44). By maintaining SirT1 activity, perturbations in energy metabolism are corrected for efficiency, similar to effects of caloric restriction, resulting in lower RNOS formation and preserved physiological function. Furthermore, stimulation of SirT1 activity increases the cellular stress tolerance by increasing mitochondrial biogenesis and antioxidant enzymes such as superoxide dismutase. The importance of SirT1 in diet-induced diabetes is further underlined by two classes of oral hypoglycemic drugs that are activated as downstream targets of SirT1: metformin, which stimulates AMP kinase (45, 46), and thiazolidinediones, which are peroxisome proliferator-activated receptor γ agonists (47). Both mediators improve liver steatosis, insulin resistance, and lipid metabolism (47, 48). Because SirT1 is a central regulator upstream of these cascades, preserved SirT1 function against metabolic stress would be expected to stimulate them more effectively to prevent the progression of NAFLD and non-alcoholic steatohepatitis.
Molecular Regulation of SirT1 Activity
The molecular mechanisms leading to diminished SirT1 activity have been extensively studied because of its potential importance in metabolic syndrome and NAFLD. Reports correlating SirT1 expression with disease are inconclusive, often showing that SirT1 is both up-regulated and down-regulated in similar pathologies (49). We (46) and others (33, 50), however, found SirT1 expression unchanged in metabolically stressed HepG2 cells (Fig. 4C) and livers of mice fed a high fat diet (Fig. 5, C and D). Diminished SirT1 activity was apparent in both of our models as measured by p53 acetylation levels and enzyme activity of immunoprecipitated SirT1. Because SirT1 expression does not adequately explain changes in enzyme activity, various post-translational protein modifications (12–18, 23) have been identified and reported to regulate activity, localization (51), and degradation (13). Resveratrol, for example, was recently shown to inhibit cAMP-degrading phosphodiesterases (52), leading to protein kinase A-dependent phosphorylation and activation of SirT1. Metabolic stress has also been demonstrated to trigger phosphorylation of SirT1 by JNK1, initiating its degradation in mouse liver or cleavage in adipose tissue (13). SirT1 phosphorylation remained unchanged in our oxidant-mediated model of disease. Furthermore, no additional cleavage fragments of SirT1 were detected. Thus, in our model, regulation of SirT1 expression by microRNA (53–55) or degradation as reported previously (13) appears not to contribute.
NAD+ is an essential co-substrate of SirT1 that couples the enzyme to cellular redox processes. Some reports have shown diet-induced changes in NAD+ levels, but a recent publication using a similar model reported no significant changes in NAD+ metabolism or associated enzymes (14, 33). Measuring diminished activity of immunoprecipitated SirT1 in the presence of excess NAD+ as we did in nuclear extracts of mice on a high fat diet also excludes changes in NAD+ (Fig. 5C) (23).
Creating a triple cysteine to serine mutant (C61S,C318S,C613S) demonstrated that these residues (23) are the most reactive to oxidant modification and critical to maintenance of SirT1 activity under metabolic and oxidative stress. Overexpression of Glrx in metabolically stressed HepG2 cells significantly decreased reversible cysteine modifications on SirT1 and maintained its biological function. In addition, using biotin-labeled and cell-permeable GSH, increased adduction of SirT1 with GSH during HPHG exposure that was reversed or prevented by Glrx was demonstrated.
Irreversible thiol oxidation of these cysteines as a long term consequence of disease, however, cannot be excluded. Furthermore, polyphenols such as resveratrol that can act as putative activators of SirT1 exhibit potent antioxidant properties that may prevent GSH-SirT1 adduct formation and inactivation.
Structural Properties of SirT1 and Protein-Protein Interactions
Currently, the crystal structure of mammalian SirT1 is unsolved, and homology-based modeling is too imprecise to predict the intrinsically disordered N- and C-terminal regions that exhibit minimal homology to other Sirtuin isoforms. Sinclair and co-workers (56) confirmed the solvent accessibility of these regions by deuterium exchange combined with mass spectroscopy. The N-terminal Cys61 and C-terminal Cys613 (Fig. 6, A, C, E, and F) are within areas of high deuterium exchange, implicating these residues as solvent-exposed and susceptible to oxidation. Various regulatory post-translational modifications of SirT1 locate to these regions (Fig. 6B), which could affect the structural conformation of the protein, thus promoting protein-protein interactions or modulating activity (57). Large, polar glutathione adducts may function as such and affect protein-protein interactions.
FIGURE 6.

Homology modeling of human SirT1. A, the three critical cysteines required for redox regulation of SirT1 show high sequence homology among mammalian species. B, map of the most important regulatory phosphorylation and oxidation sites of SirT1. All annotations are for the mouse sequence of SirT1. The color code is as follows: green, C terminus; blue, N terminus; magenta, catalytic domain; turquoise, NAD+-binding domain; orange, allosteric site required for activation by small molecular weight activators. CamK2, calcium-calmodulin-dependent protein kinase 2; ESA, essential for SirT1 activity; MAPK8, mitogen-activated protein kinase; NES, nuclear export signal; NLS, nuclear localization signal; Ox, oxidized residue. C, disordered profile blot for human SirT1 UniProt accession number Q96EB6 obtained by submission to the DISOPRED2 server. The blot clearly indicates that the N- and C-terminal domains of SirT1 contain highly disordered sections. These areas vastly differ from other Sirtuin isoforms and may be important for regulation of activity and interaction with other proteins mediated by post-translational protein modifications. D–F, three-dimensional sequence prediction of human SirT1 UniProt accession number Q96EB6 with I-TASSER. The color code is identical to that in B. The three cysteines, CysMus61-CysHomo67 (E), CysMus318-CysHomo326 (G), and CysMus613-CysHomo623 (F), are modeled. Cys61 and Cys613 are clearly solvent-exposed and may therefore be prone to oxidation. Cys318 is less exposed and may form a disulfide with Cys482.
The core domain of SirT1, which is homologous to other SirT1 members, comprises the deacetylase, NAD+-binding domains, a putative allosteric region required for SirT1 activators, and a highly conserved structural zinc tetrathiolate module. Two reports following our initial observation (20, 58) have shown that zinc-coordinating cysteines can be oxidized, which causes structural perturbations and inhibition of enzyme activity. Homology modeling, however, indicates that these cysteines are oriented inward, tending to protect the thiolates from oxidation. In our experiments only millimolar concentrations of H2O2 could disrupt the structural zinc tetrathiolate module of SirT1. These cysteine thiolates, however, might get oxidized co-translationally before SirT1 is folded and loaded with zinc.
Cys318 is in proximity to the NAD+-binding domain and may either form a GSH adduct or a disulfide with the nearby Cys482 (Fig. 6G). This may alter the binding properties for NAD+ because formation of a disulfide could stiffen and stabilize SirT1 in a defined structure, which is otherwise needed for allosteric changes during cycles of activity. Thioredoxin-1, which targets and reduces protein disulfides, may modulate SirT1 activity (59). However, glutaredoxin-1 may exhibit two activities: a thioltransferase (monothiol mechanism) activity that is specific and efficient for reduction of GSH-protein adducts and a less efficient, nonspecific disulfide reductase activity analogous to the thioredoxin system (39). The latter is based on a second cysteine, which is closely located near the active site and would enable specific reduction of protein disulfide bridges. Therefore, although the data in Fig. 4, B and E, support regulation of Glrx of GSH adducts on SirT1 as being responsible for inhibiting SirT1 activity, some caution is warranted in interpreting our results because Glrx may not only convert GSH-SirT1 adducts. The effects on this protein-protein disulfide reductase activity of Glrx and the potential formation of a disulfide bridge, however, require future investigation.
Contributions of Other Sirtuin Isoforms
In humans seven Sirtuin isoforms, SirT1–SirT7, exist (60). Depending on the isoform and substrate, Sirtuins can exhibit deacetylase and/or ADP-ribosyltransferase activity. The best characterized members of the Sirtuin family are the predominantly nuclear localized SirT1 and mitochondrial SirT3 (61). Both are important regulators of energy homeostasis (9), mitochondrial function, and RNOS formation. Similar to SirT1, SirT3 can be modulated by caloric restriction (62). Both high fat diet-fed (63, 64) mice and patients with type 2 diabetes (65) show decreased SirT3 expression and deacetylase activity. Mechanistic studies showed that SirT3 modulates palmitate-fueled mitochondrial respiration, ATP synthesis, and formation of reactive oxygen species (62).
The remaining isoforms are less well characterized, and specific effects on metabolism and liver function remain unknown. Cytosolic SirT2 preferentially deacetylates tubulin, modulating mitosis in HEK293 cells (66). Mitochondrial ADP-ribosyltransferase SirT4 was reported to oppose effects of caloric restriction in pancreatic beta cells (67). SirT5 exhibits additional activities in the mitochondria including desuccinylase and demalonylase activities that affect enzymes involved in mitochondrial metabolism, e.g. amino acid degradation, the tricarboxylic acid cycle, fatty acid metabolism, and particularly the urate cycle. Surprisingly, a high fat diet revealed no metabolic changes in SirT5 deletion mice other than elevated plasma ammonia levels (68). SirT6 is a nuclear protein, and knock-out mice developed an aging phenotype due to severe genomic instability and adverse metabolic effects on glucose and fat metabolism (69). Mice deficient in SirT7, which is localized in the nucleolus, develop hepatosteatosis by increased Myc activity and endoplasmic reticulum stress (70). In summary, Sirtuins directly or indirectly regulate metabolism, cellular stress responses, RNOS formation, and gene expression. The cysteine sites that we currently describe for SirT1 are not conserved in the various isotypes; therefore, although a similar mechanism might exist for the other isoforms, the oxidation of the cysteines studied here is specific for SirT1. Redox regulation involving both the co-substrate NAD+ and reversible cysteine oxidation, however, may lead to concerted but compartmentalized effects on the Sirtuin family that are capable of modulating metabolism. In addition, some redundant functions were reported for the various Sirtuin isoforms such as proapoptotic signaling via p53. This may in part be mediated through different acetylation sites (71) or subcellular compartments (72).
Summary
SirT1 is a cellular target of redox regulation, and perturbations in the thiol redox status modulate SirT1 activity (Fig. 7). Reversible GSH-SirT1 adducts are either directly introduced by reaction of RNOS with sensitive cysteine thiols of SirT1 or through a thiol exchange reaction once the cellular GSH/oxidized glutathione ratio drops. Diminishing cellular radical formation or activation of reductive pathways including glutaredoxin-1 can restore SirT1 function and improve cellular stress resistance. Our SirT1 mutant lacks this regulation and maintains full SirT1 activity, protecting the cell from oxidative or metabolic stress.
FIGURE 7.
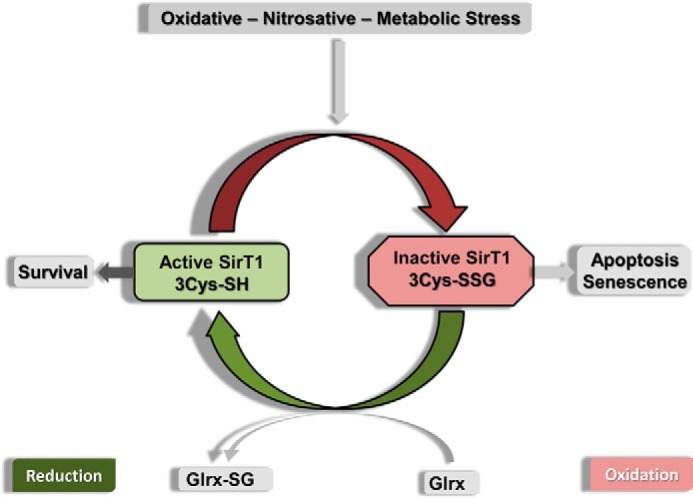
Scheme of SirT1 regulation by GSH-protein adducts. Oxidative stress introduces reversible oxidative modifications of SirT1 including GSH adducts. The enzymatic activity of SirT1 is diminished by GSH adducts, leading to cell apoptosis and senescence. GSH-SirT1 adducts (also referred to as S-glutathionylation) are reduced by Glrx in a thiol exchange reaction, restoring SirT1 activity.
Acknowledgment
We thank Dr. G. Rymarczyk for helpful discussions and assistance with homology modeling.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1 HL 068758 and R37 HL104017 and the NHLBI, National Institutes of Health, Department of Health and Human Services under Contract HHSN268201000031C.
- NAFLD
- non-alcoholic fatty liver disease
- BIAM
- N-(biotinoyl)-N′-iodoacetyl ethylenediamine, thiol-reactive labeling compound
- Cys-NO
- S-nitrosocysteine
- HepG2
- human hepatocellular carcinoma cell line
- HPDP-biotin
- N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide, thiol-reactive labeling compound
- HPHG
- high palmitate and high glucose
- SirT
- Sirtuin
- HFHS
- high fat and high sucrose
- Glrx
- glutaredoxin-1
- MUT
- triple mutant
- RNOS
- reactive nitrogen and oxygen species
- GSH
- glutathione
- ANOVA
- analysis of variance
- PUMA
- p53 up-regulated modulator of apoptosis.
REFERENCES
- 1. Ribeiro P. S., Cortez-Pinto H., Solá S., Castro R. E., Ramalho R. M., Baptista A., Moura M. C., Camilo M. E., Rodrigues C. M. (2004) Hepatocyte apoptosis, expression of death receptors, and activation of NF-κB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 99, 1708–1717 [DOI] [PubMed] [Google Scholar]
- 2. Elliott P. J., Jirousek M. (2008) Sirtuins: novel targets for metabolic disease. Curr. Opin. Investig. Drugs 9, 371–378 [PubMed] [Google Scholar]
- 3. Westphal C. H., Dipp M. A., Guarente L. (2007) A therapeutic role for sirtuins in diseases of aging? Trends Biochem. Sci. 32, 555–560 [DOI] [PubMed] [Google Scholar]
- 4. Potente M., Dimmeler S. (2008) Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle 7, 2117–2122 [DOI] [PubMed] [Google Scholar]
- 5. Pfluger P. T., Herranz D., Velasco-Miguel S., Serrano M., Tschöp M. H. (2008) Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. U.S.A. 105, 9793–9798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Purushotham A., Schug T. T., Xu Q., Surapureddi S., Guo X., Li X. (2009) Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., Mayo M. W. (2004) Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 9. Nemoto S., Fergusson M. M., Finkel T. (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 280, 16456–16460 [DOI] [PubMed] [Google Scholar]
- 10. Cheng H.-L., Mostoslavsky R., Saito S., Manis J. P., Gu Y., Patel P., Bronson R., Appella E., Alt F. W., Chua K. F. (2003) Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 100, 10794–10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Feldstein A. E., Canbay A., Angulo P., Taniai M., Burgart L. J., Lindor K. D., Gores G. J. (2003) Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125, 437–443 [DOI] [PubMed] [Google Scholar]
- 12. Yang Y., Fu W., Chen J., Olashaw N., Zhang X., Nicosia S. V., Bhalla K., Bai W. (2007) SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat. Cell Biol. 9, 1253–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao Z., Zhang J., Kheterpal I., Kennedy N., Davis R. J., Ye J. (2011) Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 286, 22227–22234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gerhart-Hines Z., Dominy J. E., Jr., Blättler S. M., Jedrychowski M. P., Banks A. S., Lim J. H., Chim H., Gygi S. P., Puigserver P. (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD+. Mol. Cell 44, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo X., Williams J. G., Schug T. T., Li X. (2010) DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 285, 13223–13232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nasrin N., Kaushik V. K., Fortier E., Wall D., Pearson K. J., de Cabo R., Bordone L. (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 4, e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasaki T., Maier B., Koclega K. D., Chruszcz M., Gluba W., Stukenberg P. T., Minor W., Scrable H. (2008) Phosphorylation regulates SIRT1 function. PLoS One 3, e4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zschoernig B., Mahlknecht U. (2009) Carboxy-terminal phosphorylation of SIRT1 by protein kinase CK2. Biochem. Biophys. Res. Commun. 381, 372–377 [DOI] [PubMed] [Google Scholar]
- 19. Caito S., Rajendrasozhan S., Cook S., Chung S., Yao H., Friedman A. E., Brookes P. S., Rahman I. (2010) SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 24, 3145–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kornberg M. D., Sen N., Hara M. R., Juluri K. R., Nguyen J. V., Snowman A. M., Law L., Hester L. D., Snyder S. H. (2010) GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 12, 1094–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prozorovski T., Schulze-Topphoff U., Glumm R., Baumgart J., Schröter F., Ninnemann O., Siegert E., Bendix I., Brüstle O., Nitsch R., Zipp F., Aktas O. (2008) Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 10, 385–394 [DOI] [PubMed] [Google Scholar]
- 22. Fulco M., Schiltz R. L., Iezzi S., King M. T., Zhao P., Kashiwaya Y., Hoffman E., Veech R. L., Sartorelli V. (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 12, 51–62 [DOI] [PubMed] [Google Scholar]
- 23. Zee R. S., Yoo C. B., Pimentel D. R., Perlman D. H., Burgoyne J. R., Hou X., McComb M. E., Costello C. E., Cohen R. A., Bachschmid M. M. (2010) Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid. Redox Signal. 13, 1023–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y., Hogg N. (2004) The mechanism of transmembrane S-nitrosothiol transport. Proc. Natl. Acad. Sci. U.S.A. 101, 7891–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haeussler D. J., Evangelista A. M., Burgoyne J. R., Cohen R. A., Bachschmid M. M., Pimental D. R. (2011) Checkpoints in adenoviral production: cross-contamination and E1A. PLoS One 6, e23160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ruifrok A. C., Johnston D. A. (2001) Quantification of histochemical staining by color deconvolution. Anal. Quant. Cytol. Histol. 23, 291–299 [PubMed] [Google Scholar]
- 27. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 [DOI] [PubMed] [Google Scholar]
- 29. Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 30. Langley E., Pearson M., Faretta M., Bauer U.-M., Frye R. A., Minucci S., Pelicci P. G., Kouzarides T. (2002) Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 21, 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen W. Y., Wang D. H., Yen R. C., Luo J., Gu W., Baylin S. B. (2005) Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123, 437–448 [DOI] [PubMed] [Google Scholar]
- 32. Xu S., Jiang B., Hou X., Shi C., Bachschmid M. M., Zang M., Verbeuren T. J., Cohen R. A. (2011) High-fat diet increases and the polyphenol, S17834, decreases acetylation of the sirtuin-1-dependent lysine-382 on p53 and apoptotic signaling in atherosclerotic lesion-prone aortic endothelium of normal mice. J. Cardiovasc. Pharmacol. 58, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Escande C., Chini C. C., Nin V., Dykhouse K. M., Novak C. M., Levine J., van Deursen J., Gores G. J., Chen J., Lou Z., Chini E. N. (2010) Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J. Clin. Investig. 120, 545–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joshi-Barve S., Barve S. S., Amancherla K., Gobejishvili L., Hill D., Cave M., Hote P., McClain C. J. (2007) Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 46, 823–830 [DOI] [PubMed] [Google Scholar]
- 35. Cao J., Dai D.-L., Yao L., Yu H.-H., Ning B., Zhang Q., Chen J., Cheng W.-H., Shen W., Yang Z.-X. (2012) Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol. Cell. Biochem. 364, 115–129 [DOI] [PubMed] [Google Scholar]
- 36. Furukawa A., Tada-Oikawa S., Kawanishi S., Oikawa S. (2007) H2O2 accelerates cellular senescence by accumulation of acetylated p53 via decrease in the function of SIRT1 by NAD+ depletion. Cell. Physiol. Biochem. 20, 45–54 [DOI] [PubMed] [Google Scholar]
- 37. Yu J., Wang Z., Kinzler K. W., Vogelstein B., Zhang L. (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 1931–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ruiz-Ramírez A., Chávez-Salgado M., Peñeda-Flores J. A., Zapata E., Masso F., El-Hafidi M. (2011) High-sucrose diet increases ROS generation, FFA accumulation, UCP2 level, and proton leak in liver mitochondria. Am. J. Physiol. Endocrinol. Metab. 301, E1198–E1207 [DOI] [PubMed] [Google Scholar]
- 39. Mieyal J. J., Gallogly M. M., Qanungo S., Sabens E. A., Shelton M. D. (2008) Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 10, 1941–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin F., Siwik D. A., Luptak I., Hou X., Wang L., Higuchi A., Weisbrod R. M., Ouchi N., Tu V. H., Calamaras T. D., Miller E. J., Verbeuren T. J., Walsh K., Cohen R. A., Colucci W. S. (2012) The polyphenols resveratrol and S17834 prevent the structural and functional sequelae of diet-induced metabolic heart disease in mice. Circulation 125, 1757–1764, S1–S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Deng X.-Q., Chen L.-L., Li N.-X. (2007) The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 27, 708–715 [DOI] [PubMed] [Google Scholar]
- 42. You M., Cao Q., Liang X., Ajmo J. M., Ness G. C. (2008) Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J. Nutr. 138, 497–501 [DOI] [PubMed] [Google Scholar]
- 43. Madan K., Bhardwaj P., Thareja S., Gupta S. D., Saraya A. (2006) Oxidant stress and antioxidant status among patients with nonalcoholic fatty liver disease (NAFLD). J. Clin. Gastroenterol. 40, 930–935 [DOI] [PubMed] [Google Scholar]
- 44. Koruk M., Taysi S., Savas M. C., Yilmaz O., Akcay F., Karakok M. (2004) Oxidative stress and enzymatic antioxidant status in patients with nonalcoholic steatohepatitis. Ann. Clin. Lab. Sci. 34, 57–62 [PubMed] [Google Scholar]
- 45. Zang M., Zuccollo A., Hou X., Nagata D., Walsh K., Herscovitz H., Brecher P., Ruderman N. B., Cohen R. A. (2004) AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J. Biol. Chem. 279, 47898–47905 [DOI] [PubMed] [Google Scholar]
- 46. Hou X., Xu S., Maitland-Toolan K. A., Sato K., Jiang B., Ido Y., Lan F., Walsh K., Wierzbicki M., Verbeuren T. J., Cohen R. A., Zang M. (2008) SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 283, 20015–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahmadian M., Suh J. M., Hah N., Liddle C., Atkins A. R., Downes M., Evans R. M. (2013) PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., Cohen R. A., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kwon H.-S., Ott M. (2008) The ups and downs of SIRT1. Trends Biochem. Sci. 33, 517–525 [DOI] [PubMed] [Google Scholar]
- 50. Nin V., Escande C., Chini C. C., Giri S., Camacho-Pereira J., Matalonga J., Lou Z., Chini E. N. (2012) Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J. Biol. Chem. 287, 23489–23501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tanno M., Sakamoto J., Miura T., Shimamoto K., Horio Y. (2007) Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 282, 6823–6832 [DOI] [PubMed] [Google Scholar]
- 52. Park S.-J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A. L., Kim M. K., Beaven M. A., Burgin A. B., Manganiello V., Chung J. H. (2012) Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 148, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abdelmohsen K., Pullmann R., Jr., Lal A., Kim H. H., Galban S., Yang X., Blethrow J. D., Walker M., Shubert J., Gillespie D. A., Furneaux H., Gorospe M. (2007) Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25, 543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yamazaki Y., Usui I., Kanatani Y., Matsuya Y., Tsuneyama K., Fujisaka S., Bukhari A., Suzuki H., Senda S., Imanishi S., Hirata K., Ishiki M., Hayashi R., Urakaze M., Nemoto H., Kobayashi M., Tobe K. (2009) Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 297, E1179–E1186 [DOI] [PubMed] [Google Scholar]
- 55. Strum J. C., Johnson J. H., Ward J., Xie H., Feild J., Hester A., Alford A., Waters K. M. (2009) MicroRNA 132 regulates nutritional stress-induced chemokine production through repression of SirT1. Mol. Endocrinol. 23, 1876–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hubbard B. P., Gomes A. P., Dai H., Li J., Case A. W., Considine T., Riera T. V., Lee J. E., E S. Y., Lamming D. W., Pentelute B. L., Schuman E. R., Stevens L. A., Ling A. J., Armour S. M., Michan S., Zhao H., Jiang Y., Sweitzer S. M., Blum C. A., Disch J. S., Ng P. Y., Howitz K. T., Rolo A. P., Hamuro Y., Moss J., Perni R. B., Ellis J. L., Vlasuk G. P., Sinclair D. A. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339, 1216–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Costantini S., Sharma A., Raucci R., Costantini M., Autiero I., Colonna G. (2013) Genealogy of an ancient protein family: the Sirtuins, a family of disordered members. BMC Evol. Biol. 13, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jung S.-B., Kim C.-S., Kim Y.-R., Naqvi A., Yamamori T., Kumar S., Kumar A., Irani K. (2013) Redox factor-1 activates endothelial SIRTUIN1 through reduction of conserved cysteine sulfhydryls in its deacetylase domain. PLoS One 8, e65415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Olmos Y., Sánchez-Gómez F. J., Wild B., García-Quintans N., Cabezudo S., Lamas S., Monsalve M. (2013) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid. Redox Signal. 19, 1507–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Haigis M. C., Sinclair D. A. (2010) Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nogueiras R., Habegger K. M., Chaudhary N., Finan B., Banks A. S., Dietrich M. O., Horvath T. L., Sinclair D. A., Pfluger P. T., Tschöp M. H. (2012) Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol. Rev. 92, 1479–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ahn B.-H., Kim H.-S., Song S., Lee I. H., Liu J., Vassilopoulos A., Deng C.-X., Finkel T. (2008) A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kendrick A. A., Choudhury M., Rahman S. M., McCurdy C. E., Friederich M., Van Hove J. L., Watson P. A., Birdsey N., Bao J., Gius D., Sack M. N., Jing E., Kahn C. R., Friedman J. E., Jonscher K. R. (2011) Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 433, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ishikawa S., Li G., Takemitsu H., Fujiwara M., Mori N., Yamamoto I., Arai T. (2013) Change in mRNA expression of sirtuin 1 and sirtuin 3 in cats fed on high fat diet. BMC Vet. Res. 9, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Caton P. W., Richardson S. J., Kieswich J., Bugliani M., Holland M. L., Marchetti P., Morgan N. G., Yaqoob M. M., Holness M. J., Sugden M. C. (2013) Sirtuin 3 regulates mouse pancreatic β cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 56, 1068–1077 [DOI] [PubMed] [Google Scholar]
- 66. North B. J., Marshall B. L., Borra M. T., Denu J. M., Verdin E. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 11, 437–444 [DOI] [PubMed] [Google Scholar]
- 67. Haigis M. C., Mostoslavsky R., Haigis K. M., Fahie K., Christodoulou D. C., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Karow M., Blander G., Wolberger C., Prolla T. A., Weindruch R., Alt F. W., Guarente L. (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic β cells. Cell 126, 941–954 [DOI] [PubMed] [Google Scholar]
- 68. Yu J., Sadhukhan S., Noriega L. G., Moullan N., He B., Weiss R. S., Lin H., Schoonjans K., Auwerx J. (2013) Metabolic characterization of a Sirt5 deficient mouse model. Sci. Rep. 3, 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mostoslavsky R., Chua K. F., Lombard D. B., Pang W. W., Fischer M. R., Gellon L., Liu P., Mostoslavsky G., Franco S., Murphy M. M., Mills K. D., Patel P., Hsu J. T., Hong A. L., Ford E., Cheng H.-L., Kennedy C., Nunez N., Bronson R., Frendewey D., Auerbach W., Valenzuela D., Karow M., Hottiger M. O., Hursting S., Barrett J. C., Guarente L., Mulligan R., Demple B., Yancopoulos G. D., Alt F. W. (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 [DOI] [PubMed] [Google Scholar]
- 70. Shin J., He M., Liu Y., Paredes S., Villanova L., Brown K., Qiu X., Nabavi N., Mohrin M., Wojnoonski K., Li P., Cheng H.-L., Murphy A. J., Valenzuela D. M., Luo H., Kapahi P., Krauss R., Mostoslavsky R., Yancopoulos G. D., Alt F. W., Chua K. F., Chen D. (2013) SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep. 5, 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peck B., Chen C.-Y., Ho K.-K., Di Fruscia P., Myatt S. S., Coombes R. C., Fuchter M. J., Hsiao C.-D., Lam E. W. (2010) SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol. Cancer Ther. 9, 844–855 [DOI] [PubMed] [Google Scholar]
- 72. Li S., Banck M., Mujtaba S., Zhou M.-M., Sugrue M. M., Walsh M. J. (2010) p53-induced growth arrest is regulated by the mitochondrial SirT3 deacetylase. PLoS One 5, e10486. [DOI] [PMC free article] [PubMed] [Google Scholar]