

Background: Overexpression of the epigenetic repressor KDM2A promotes lung tumorigenesis.
Results: Transcriptional inhibition of HDAC3 expression by KDM2A releases cell cycle and proinvasive genes from HDAC3-mediated repression and positively regulates cell proliferation and invasiveness.
Conclusion: KDM2A-mediated repression of HDAC3 is linked to KDM2A-promoted tumorigenicity.
Significance: Our findings provide a novel epigenetic insight into how KDM2A promotes lung tumorigenesis and have implications for therapeutic intervention.
Keywords: Chromatin, Gene Regulation, Histone Modification, Lung Cancer, Transcription Repressor
Abstract
Dysregulated expression of histone methyltransferases and demethylases is an emerging epigenetic mechanism underlying cancer development and metastasis. We recently showed that the histone H3 lysine 36 (H3K36) demethylase KDM2A (also called FBXL11 and JHDM1A) is necessary for tumorigenic and metastatic capabilities of KDM2A-overexpressing non-small cell lung cancer (NSCLC) cells. Here, we report that KDM2A transcriptionally represses the histone deacetylase 3 (HDAC3) gene by removing methyl groups from dimethylated H3K36 at the HDAC3 promoter in KDM2A-overexpressing NSCLC cells. KDM2A depletion reduced expression levels of cell cycle-associated genes (e.g. CDK6) and cell invasion-related genes (e.g. NANOS1); these levels were rescued by ectopic expression of KDM2A but not its catalytic mutant. These genes were occupied and down-regulated by HDAC3. HDAC3 knockdown significantly recovered the proliferation and invasiveness of KDM2A-depleted NSCLC cells as well as the levels of CDK6 and NANOS1 expression in these cells. Similar to their previously reported functions in other cell types, CDK6 and NANOS1 were required for the proliferation and invasion, respectively, of KDM2A-overexpressing NSCLC cells. In a mouse xenograft model, HDAC3 depletion substantially restored the tumorigenic ability of KDM2A knockdown cells. These findings reveal a novel cancer-epigenetic pathway in which the antagonistic effect of KDM2A on HDAC3 expression releases cell cycle-associated genes and cell invasion-related genes from HDAC3 repression and indicate the importance of this pathway for tumorigenicity and invasiveness of KDM2A-overexpressing NSCLC cells.
Introduction
Histone lysine methylation is considered a key chromatin mark that mediates epigenetic and transcriptional regulation of gene expression (1, 2). Unlike histone acetylation, which is exclusively associated with gene activation, this modification is linked to activation or silencing of gene expression (3, 4). The methylation effect depends on the lysine residues involved, among which are histone H3 lysine 4 (H3K4),3 H3K9, H3K27, H3K36, H3K79, and H4K20 (5). For example, methylated H3K36 is connected to gene activation, whereas methylated H3K27 is related to gene silencing. Like histone acetylation, which can be removed by histone deacetylases (HDACs), histone lysine methylation is reversibly regulated: it is catalyzed by histone lysine methyltransferases (1, 2) and can be removed by histone lysine demethylases (KDMs) (6, 7). Notably, this modification exists at three different states (i.e. mono-, di-, and trimethylation).
Lung cancer is the most prevalent cause of cancer-related mortality in the United States and worldwide (8). Non-small cell lung cancer (NSCLC) is responsible for up to 85% of lung cancer and includes adenocarcinomas, squamous cell carcinomas, and large cell carcinomas. In NSCLC, genetic mutations and abnormalities in kinase signaling pathway members have been well documented (9). For instance, in lung adenocarcinomas, activating mutations for oncogenes frequently occur in K-RAS and epidermal growth factor receptor gene, whereas mutations in tumor suppressor genes, such as LKB1, are also detected (9, 10). Thus, much research focus for NSCLC has been put on understanding the kinase signaling pathway.
Recently, it has been increasingly evident that dysregulation of histone methylation and its modifiers may be an important factor in lung tumorigenesis and metastasis (11, 12). Alterations in histone lysine methylation are associated with clinical prognosis of lung cancer (13, 14). For example, global levels of trimethylated H4K20, a gene-repressive mark, are decreased in squamous cell carcinomas, and its low levels correlate with poor prognosis (14). In addition to methylation marks, some histone methylation modifiers are known to have altered expression levels in lung cancer. For instance, the H3K27 methyltransferase EZH2 and the H3K36 methyltransferase MMSET (also called WHSC1 and NSD2) are significantly overexpressed in lung tumors (15). Recently, we showed that the H3K36 demethylase KDM2A (also known as FBXL11 and JHDM1A) is frequently up-regulated in NSCLC tumor samples and promotes the tumor growth and invasive abilities of NSCLC cells (16). Consistent with the notion that KDM2A is associated with transcriptional repression by erasing methyl groups from the gene activation mark dimethylated H3K36 (H3K36me2) (17–19), we also reported that KDM2A increases cellular levels of phospho-ERK1/2, a type of oncogenic signal, by down-regulating the expression of dual specificity phosphatase 3 (DUSP3) whose protein dephosphorylates ERK1/2 (16).
To gain greater insights into how KDM2A regulates cell proliferation and invasion, we sought to identify more KDM2A target genes. Here, we provide evidence that HDAC3 is an important target gene of KDM2A. Transcriptional repression of the HDAC3 gene by KDM2A-catalyzed H3K36 demethylation up-regulates HDAC3 target genes, including the cell cycle-associated gene CDK6 and the cell invasion-related gene NANOS1 in two KDM2A-overexpressing NSCLC cell lines. Furthermore, our results suggest that epigenetic repression of HDAC3 expression by KDM2A is needed for the tumorigenic and invasive abilities of KDM2A-overexpressing NSCLC cells.
EXPERIMENTAL PROCEDURES
Samples, Reagents, Antibodies, and Animals
H1975 and H1792 NSCLC cell lines were purchased from ATCC. Cell culture reagents were purchased from Invitrogen; all other chemicals were from Sigma-Aldrich. The KDM2A-specific antibodies (NB100-74602) were purchased from Novus Biologicals. Additional antibodies were purchased as follows: anti-HDAC3 (40968), anti-H3K36me2 (39256), anti-H3K9ac (39138), anti-H3K14ac (39616), and anti-H4ac (39227) from Active Motif; anti-H3K9me3 (07-442) from Millipore; anti-H3 (ab1971) from Abcam; and anti-β-actin (A5441) from Sigma-Aldrich. Anti-CDK6 (14052-1-AP, Proteintech) and anti-NANOS1 (LS-C164739, Life Span Biosciences) were used for immunohistochemical staining. HRP-conjugated anti-mouse IgG and HRP-conjugated anti-rabbit IgG were from Santa Cruz Biotechnology. The nude mice were purchased from MD Anderson Cancer Center, and their care and use were approved by MD Anderson's Institutional Animal Care and Use Committee.
In Vitro Gene Silencing Using siRNA
For knockdown experiments, siRNAs against KDM2A, HDAC3, CDK6, and NANOS1 were purchased from Dharmacon or Integrated DNA Technologies, Inc. (IDT). The siRNA sequences are listed in Table 1. As controls, siRNA against luciferase GL3 RNA (siLuc) and siControl were used. Cells (5 × 104) in a 6-well plate were transfected with siRNAs at a final concentration of 100 nm using Lipofectamine RNAiMAX (Invitrogen). Following 72–96 h of incubation, cells were harvested for mRNA and protein analysis or used for cell proliferation and invasion assays.
TABLE 1.
PCR primers and siRNAs
F, forward; R, reverse.
| Gene | Application | Sequence (5′–3′) |
|---|---|---|
| CDK6 F | RT-qPCR | TCG ATG AAC TAG GCA AAG ACC |
| CDK6 R | RT-qPCR | AGG TGG GAA TCC AGG TTT TC |
| NEK7 F | RT-qPCR | ACT TGG AGA TCT TGG GCT TG |
| NEK7 R | RT-qPCR | GAC AGC CAA GAG ACC AGA TG |
| NANOS1 F | RT-qPCR | GGT CGG CTC GAC ATG GGA CG |
| NANOS1 R | RT-qPCR | CAC ACC CAG CCT TCG CCG TT |
| RAPH1 F | RT-qPCR | GCC AAC TTT TCT TAC CGC TTC |
| RAPH1 R | RT-qPCR | GAT TTG ACG CTT ACT GTT TCC TG |
| β-Actin F | RT-qPCR | GCA CTC TTC CAG CCT TCC |
| β-Actin R | RT-qPCR | TGT CCA CGT CAC ACT TCA TG |
| CDK6 F | ChIP-qPCR | CGG AGA GAG TGC TGG TAA CTC CTT |
| CDK6 R | ChIP-qPCR | TGC GAG TGT CAG TCG GCT CT |
| NEK7 F | ChIP-qPCR | TTC GGC TCC AGT AGG GAA AC |
| NEK7 R | ChIP-qPCR | CTG CCC GAT GGA GGC TT |
| NANOS1 F | ChIP-qPCR | GGA GGA GTG GGC CCG ATA AA |
| NANOS1 R | ChIP-qPCR | AAA GCC TCC ATG GGC GGG |
| RAPH1 F | ChIP-qPCR | CAG TCA GTC AGT CAG TCA GTC AGT |
| RAPH1 R | ChIP-qPCR | AGG GCG AGG CTA ACC ACT CA |
| siLuc | siRNA (Dharmacon) | Sense, 5′-AACTTACGCTGAGTACTTCGA-dTdT-3′ |
| Antisense, 5′-AAUCGAAGUACUCAGCGUAAGUU-3′ | ||
| siKDM2A-3 | siRNA (Dharmacon) | Sense, 5′-AACAAGGAGAGUGUGGUGUUU-dTdT-3′ |
| Antisense, 5′-AAACACCACACUCUCCUUGTT-3′ | ||
| siKDM2A-4 | siRNA (Dharmacon) | Sense, 5′-AAUUACGAAGCCUCACACUAU-dTdT-3′ |
| Antisense, 5′-AUAGUGUGAGGCUUCGUAATT-3′ | ||
| siControl | siRNA (IDT) | Sense, 5′-UCGAAGUACUCAGCGUAAGTT-3′ |
| Antisense, 5′-CUUACGCUGAGUACUUCGATT-3′ | ||
| siHDAC3-3 | siRNA (IDT) | Sense, 5′-CCCGCAUCGAGAAUCAGAACUCACG-3′ |
| Antisense, 5′-CGUGAGUUCUGAUUCUCGAUGCGGGUG-3′ | ||
| siHDAC3-9 | siRNA (IDT) | Sense, 5′-GUCAACGACAUUGUGAUUGGCAUCC-3′ |
| Antisense, 5′-GGAUGCCAAUCACAAUGUCGUUGACAU-3′ | ||
| siCDK6-1 | siRNA (IDT) | Sense, 5′-AGAGUGCAAAUUAGAACUAGAAAGT-3′ |
| Antisense, 5′-ACUUUCUAGUUCUAAUUUGCACUCUAC-3′ | ||
| siCDK6-3 | siRNA (IDT) | Sense, 5′-GGUGUUGUCAGUACUAUAAAUGCTT-3′ |
| Antisense, 5′-AAGCAUUUAUAGUACUGACAACACCUA-3′ | ||
| siNANOS1-1 | siRNA (IDT) | Sense, 5′-GGAGUAGACAGAGUUGUAUGGCCTG-3′ |
| Antisense, 5′-CAGGCCAUACAACUCUGUCUACUCCCA-3′ | ||
| siNANOS1-3 | siRNA (IDT) | Sense, 5′-GGACCACUAUGAACAGAUCAGCGCA-3′ |
| Antisense, 5′-UGCGCUGAUCUGUUCAUAGUGGUCCUU-3′ | ||
| siScramble | siRNA (IDT) | Sense, 5′-GUGUAUGUAGUGCGGUAGUAU-3′ |
| Antisense, 5′-AUACUACCGCACUACAUCAAC-3′ |
Double knockdown of KDM2A and HDAC3 were performed, and its effect was compared with that of individual knockdown. In brief, cells (2–5 × 104) were seeded in a 6-well plate and transfected with one of four different combinations of siRNAs: 1) siControl (70 nm), 2) siKDM2A-3 (35 nm) + siControl (35 nm), 3) siHDAC3-9 (35 nm) + siControl (35 nm), and 4) siKDM2A-3 (35 nm) + siHDAC3-9 (35 nm). It should be noted that siControl was added to obtain a final concentration of siRNAs of 70 nm. For transfection, Lipofectamine RNAiMAX was used. After 24 h of incubation, cells were retransfected with the same amount of siRNAs. An additional 72 h later, cells were harvested for mRNA analysis or used for cell proliferation and invasion assays.
Quantitative RT-PCR
Total RNA was isolated using RNeasy kits (Qiagen) according to the manufacturer's instructions. Then cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. Quantitative PCR was performed in triplicate using SYBR Green and gene-specific primers (Table 1). Messenger RNA levels were quantified using CFX Manager software (Bio-Rad) and were normalized to β-actin mRNA levels. The relative mRNA levels represent the -fold change compared with the levels in control cells.
Cell Proliferation and Invasion Assays
To assess cell proliferation, 1 × 104 cells/well in 24-well plates were seeded in triplicate, and cell number were counted at the indicated time points. For the cell invasion assay, the Boyden chamber assay with a modification was performed. In brief, cells (1 × 105) were seeded on the Matrigel-coated membrane in the inserts. After 18 h of incubation, cells that had invaded the Matrigel and migrated to the other side of the membrane were stained and counted.
Cell Cycle Analysis and BrdU Incorporation Assays
For cell cycle analysis, cells were fixed in cold 75% ethanol for 30 min at 4 °C and washed twice in PBS. Then cells were incubated at 37 °C for 30 min in a buffer containing 50 μg/ml propidium iodine, 5 mm MgCl2, 10 mm Tris-HCl (pH 7.0), and 25 μg/ml RNase A. DNA contents were analyzed using flow cytometry (BD FACSCanto II, BD Biosciences).
For BrdU incorporation assays, 1–2 × 105 cells were seeded in 96-well plates in triplicate. BrdU reagent was added to each well, and the plate was incubated for 24 h. BrdU incorporation was measured at 450 nm absorbance according to the manufacturer's protocol (Millipore).
Rescue Experiments by Ectopic Expression
To test the effects of ectopic expression of KDM2A in H1975 and H1792 cells, the cDNAs encoding KDM2A, its catalytic mutant mKDM2A, and GFP were individually cloned into the mammalian expression vector pFLAG-CMV2 as described previously (16). The resulting expression plasmids (2 μg) were transfected into either siControl- or siKDM2A-treated cells (2–5 × 105 cells) in 60-mm dishes using 10 μl of Lipofectamine 2000 (Invitrogen). These cells were harvested after 72 h of incubation for further analysis, such as quantitative RT-PCR.
Quantitative ChIP Assay
A ChIP assay was performed as described previously (20). After chromatin immunoprecipitation using specific antibodies, DNA was purified using the Qiagen PCR purification kit. Purified DNAs for KDM2A, HDAC3, and the histone marks of interest were amplified by quantitative PCR (qPCR) and normalized to input. Relative occupancy indicates the change over the control value. Enrichment represents PCR values from specific antibody relative to those from IgG (e.g. anti-KDM2A/IgG and anti-HDAC3/IgG).
Mouse Xenograft Study
To determine whether the effect of KMD2A knockdown on tumorigenesis is dependent on HDAC3, three groups of cells (shControl-treated, KDM2A-depleted, and KDM2A/HDAC3-depleted H1792 cells) were compared for their tumorigenicity in a subcutaneous xenograft model. KDM2A-depleted cells were generated using shRNA against KDM2A as described previously (21). KDM2A/HDAC3-depleted cells were generated by treating KDM2A-depleted cells with 50 nm siHDAC3-9 using Lipofectamine RNAiMAX. For comparison, the other two groups of cells were also transfected with 50 nm siScramble (a control siRNA). After a 24-h incubation, all three groups of cells were retransfected with the same amounts of siRNAs at the same concentrations. An additional 72 h later, cells were harvested and suspended in RPMI 1640 medium without serum. Cells (1.5 × 106) were subcutaneously injected into the dorsal flanks of male nude mice (8 weeks old). At least five mice were injected for each group and observed for 10 weeks for tumor formation. The ellipsoid volume formula (1/2 × L × W × H) was used to calculate the tumor volume. Ten weeks after the injection, the mice were killed, and their tumors and lungs were collected. For pathological analysis, hematoxylin and eosin (H&E) and immunochemical staining was performed.
Statistical Analysis
For statistical analysis, each experiment was performed in triplicate and repeated at least three times. Data are presented as the mean ± S.E. (error bars). Statistical significance was tested by the two-tailed Student's t test. * (p < 0.05), ** (p < 0.01), and *** (p < 0.001) indicate statistically significant differences. GraphPad Prism software was used for all statistical analyses.
RESULTS
KDM2A Indirectly Up-regulates Expression of Cell Cycle-associated Genes and Cell Invasion-related Genes in KDM2A-overexpressing NSCLC Cells
In our effort to better understand the mechanisms by which KDM2A may regulate the proliferation and invasion of NSCLC cells, we revisited our recent whole genome mRNA expression data in which a number of genes were commonly modulated by two different siKDM2As in two KDM2A-overexpressing NSCLC cell lines, H1975 and H1792. Because our recent study showed that KDM2A knockdown decreased the S phase percentages and invasive abilities of H1975 and H1792 cells (16), we paid our attention to cell cycle-associated genes and cell invasion-related genes. In particular, the cell cycle-associated genes CDK6 and NEK7 and the invasion-related genes NANOS1 and RAPH1 were of interest because these genes were down-regulated by KDM2A knockdown (16). CDK6 was demonstrated to be often overexpressed in lung tumors (22). CDK6 activity promotes expression of cell cycle regulators and assembly of the prereplication complex (23), and pharmacologic inhibition of CDK6 induces cell cycle arrest (24). NEK7, a centrosomal kinase, regulates proper spindle assembly and mitotic progression (25). NANOS1 is overexpressed in some human invasive lung carcinomas and increases the production of MT1-matrix metalloproteinase to induce matrix metalloproteinase-dependent invasion of carcinoma cells (26). RAPH1 is known to promote cell proliferation and to play an important role in cell motility (27).
To confirm our previous microarray results, we compared expression levels of the CDK6, NEK7, NANOS1, and RAPH1 genes between KDM2A-depleted cells and control siRNA-treated cells using quantitative RT-PCR. Two KDM2A-overexpressing NSCLC cell lines H1975 and H1792 were examined. Our results ensured that these genes were down-regulated by KDM2A knockdown (Fig. 1, A and B). Next, we examined whether ectopic expression of KDM2A restores expression of the KDM2A-regulated genes CDK6, NEK7, NANOS1, and RAPH1 in KDM2A-depleted cells. Wild-type KDM2A and its catalytic mutant mKDM2A were transiently expressed as described previously (16). Exogenous expression of wild-type KDM2A, but not mKDM2A, significantly rescued expression of these genes in H1975 and H1792 cells (Fig. 1, C–F, showing data for CDK6 and NANOS1; similar data are not shown for NEK7 and RAPH1). These results indicate that KDM2A and its demethylase activity may be requisite for expression of the CDK6, NEK7, NANOS1, and RAPH1 genes.
FIGURE 1.

Transcriptional expression of cell cycle-associated genes and invasion-related genes is positively regulated by KDM2A in the KDM2A-overexpressing NSCLC cell lines H1975 and H1792. A and B, analysis of CDK6, NEK7, NANOS1, RAPH1, DOCK4, and ZNF652 mRNA levels in H1975 (A) and H1792 (B) cells after KDM2A knockdown. Expression levels were analyzed by quantitative RT-PCR. C–F, rescue experiments for CDK6 (C and D) and NANOS1 (E and F) expression in KDM2A-depleted H1975 (C and E) and H1792 (D and F) cells. After ectopic expression of GFP, wild-type KDM2A, and its catalytic mutant mKDM2A in KDM2A-depleted H1975 and H1792 cells, expression levels were measured by quantitative RT-PCR. The siControl-treated cells were used as controls. G and H, analysis of KDM2A occupancy at CDK6, NEK7, NANOS1, RAPH1, DOCK4, and ZNF652 genes in H1975 (G) and H1792 (H) cells by quantitative ChIP (qChIP). Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
KDM2A is linked to transcriptional repression of its target genes (17–19). Therefore, KDM2A may indirectly up-regulate the CDK6, NEK7, NANOS1, and RAPH1 genes whose expression levels were decreased by KDM2A knockdown. Thus, we determined whether these genes are indirectly or directly regulated by KDM2A using quantitative ChIP assays. Because it has been demonstrated that KDM2A is located at regions spanning transcription start sites (16, 17), we analyzed KDM2A occupancy near those sites in these genes. ChIP results showed that the regions near the transcription start sites in the CDK6, NEK7, NANOS1, and RAPH1 genes were not occupied by KDM2A. In contrast, DOCK7 and ZNF652 whose expression levels were increased by KDM2A knockdown were KDM2A target genes (Fig. 1, G and H). In line with its role in repressing transcription, these results indicate that KDM2A indirectly up-regulates expression of the CDK6, NEK7, NANOS1, and RAPH1 genes while directly down-regulating expression of the DOCK7 and ZNF652 genes.
Expression of the HDAC3 Gene Is Repressed by KDM2A-catalyzed H3K36 Demethylation
Because CDK6, NEK7, NANOS1, and RAPH1 were not direct KDM2A target genes, we hypothesized that these genes may be down-regulated by a transcriptional co-repressor encoded by a KDM2A-repressed gene. Specifically, we reasoned that HDAC3 might be involved in the repression of these genes as our previous microarray results indicated that HDAC3 is a KDM2A-repressed gene (16). HDAC3 is a well known transcriptional co-repressor (28–30). Similar to the microarray results, our quantitative RT-PCR data showed that HDAC3 expression levels were up-regulated in KDM2A knockdown cells (Fig. 2A). In addition, our ectopic experiments demonstrated that expression of wild-type KDM2A, but not its catalytic mutant mKDM2A, significantly repressed expression levels of HDAC3 in KDM2A-depleted cells (Fig. 2, B and C). These results indicated that KDM2A and its enzymatic activity are critical for KDM2A-mediated repression of the HDAC3 gene.
FIGURE 2.

KDM2A represses HDAC3 expression by demethylating H3K36me2 at the HDAC3 promoter. A, the effect of KDM2A knockdown on HDAC3 mRNA levels in H1975 and H1792 cells. Cells were transfected with two different siKDM2As (siKDM2A-3 and -4). B and C, analysis of HDAC3 mRNA levels in KDM2A-depleted H1975 (B) and H1792 (C) cells after ectopic expression of GFP, wild-type KDM2A, and the catalytic mutant mKDM2A. HDAC3 mRNA levels were measured by quantitative RT-PCR. D, schematic representation of the promoter region of the HDAC3 gene. Arrows indicate the PCR-amplified region. TSS, transcription start site. E, analysis of KDM2A occupancy at HDAC3 gene in H1975 and H1792 cells by quantitative ChIP (qChIP). F and G, analysis of occupied levels of KDM2A, H3K36me2, H3K9me3, and H3 at the HDAC3 promoter region in H1975 (F) and H1792 (G) cells by quantitative ChIP. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether KDM2A is recruited to the HDAC3 gene, we examined enrichment levels of KDM2A over control IgG at a proximal promoter region of the HDAC3 gene using quantitative ChIP assays (Fig. 2D). Our results showed that KDM2A occupied the proximal promoter near the transcription start site in the HDAC3 gene, indicating that HDAC3 is a KDM2A target gene (Fig. 2E). These results are in agreement with our previous study showing that KDM2A is localized near the transcription start site of the DUSP3 gene (16). Because KDM2A removes methyl groups from H3K36me2, we determined the effect of KDM2A knockdown on H3K36me2 levels at the proximal promoter of the HDAC3 gene. Quantitative ChIP results demonstrated that KDM2A knockdown resulted in increased H3K36me2 levels at the proximal promoter of the HDAC3 gene in H1975 and H1792 cells (Fig. 2, F and G). These results indicate that KDM2A-catalyzed demethylation of H3K36me2 at the HDAC3 promoter plays an important role in KDM2A-mediated repression of the HDAC3 gene.
HDAC3 Directly Represses Expression of the CDK6, NEK7, NANOS1, and RAPH1 Genes
To examine the possibility that HDAC3 may down-regulate expression levels of CDK6, NEK7, NANOS1, and RAPH1 genes, we depleted HDAC3 in H1792 and H1975 cells using two different siRNAs against HDAC3 (Fig. 3, A and B). Quantitative RT-PCR results showed that HDAC3 depletion by two independent siHDAC3s increased expression levels of these genes (Fig. 3, C and D). Next, we examined whether HDAC3 is recruited to CDK6, NEK7, NANOS1, and RAPH1 genes using quantitative ChIP. Our results showed that HDAC3 occupied the regions spanning the transcription start sites at these genes (Fig. 4, A–D), consistent with the previous report showing that chromatin peaks of HDAC3 cover the transcription start sites (31). These results indicate that HDAC3 directly represses CDK6, NEK7, NANOS1, and RAPH1 genes and support the idea that KDM2A up-regulates expression of these genes by repressing HDAC3 expression at transcriptional levels.
FIGURE 3.

HDAC3 represses expression of the cell cycle-associated genes CDK6 and NEK7 and the invasion-related genes NANOS1 and RAPH1. A and B, analysis of HDAC3 knockdown efficacy in H1975 and H1792 cells. Cells were treated with two siRNAs against HDAC3 (siHDAC3-3 and siHDAC3-9). HDAC3 mRNA and protein levels were measured by quantitative RT-PCR (A) and Western blot analysis (B), respectively. C and D, the effect of HDAC3 knockdown on CDK6, NEK7, NANOS1, RAPH1, and GPR157 mRNA levels in H1975 (C) and H1792 (D) cells. Expression levels of individual genes were analyzed by quantitative RT-PCR. Data are presented as the mean ± S.E. (error bars). **, p < 0.01; ***, p < 0.001.
FIGURE 4.

HDAC3 occupies the regions spanning the transcription start sites in the CDK6, NEK7, NANOS1, and RAPH1 genes. A–D, analysis of chromatin occupancy of HDAC3 at the CDK6 (A), NEK7 (B), NANOS1 (C), and RAPH1 (D) genes in H1975 and H1792 cells by quantitative ChIP (qChIP). A diagrammatic representation of individual genes is also shown. Arrows indicate the PCR-amplified regions (a and b). TSS, transcription start site. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To assess whether HDAC3 represses its target genes by deacetylation, we examined the effect of HDAC3 knockdown on histone acetylation levels at the HDAC3 target genes, such as CDK6 and NANOS1. Consistent with the deacetylase activity of HDAC3, our ChIP results demonstrated that HDAC3 depletion increased acetylation levels in histones H3 and H4 at these genes in H1792 and H1975 cells (Fig. 5, A–D). Similar to no obvious effect of KDM2A knockdown on cellular levels of H3K36me2, HDAC3 depletion did not affect total cellular levels of H3K9 acetylation and H4 acetylation, suggesting that HDAC3 erases histone acetylation in a gene-specific manner (Fig. 5E).
FIGURE 5.

HDAC3 knockdown increases acetylation levels in histones H3 and H4 at the promoters of the CDK6 and NANOS1 genes. A–D, analysis of chromatin levels of HDAC3, H3K9ac, H3K14ac, H4ac, and H3 at the promoter regions of the CDK6 (A and B) and NANOS1 genes (C and D) in H1975 (A and C) and H1792 (B and D) cells. Chromatin levels of proteins and histone marks were measured by quantitative ChIP. Anti-H3 was used as a ChIP control. E, the effect of KDM2A or HDAC3 knockdown on total cellular levels of H3K36me2, H3K9 acetylation, and H4 acetylation. Histone marks were examined by Western blot analysis. H3 and β-actin were used as internal loading controls. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01.
In KDM2A-overexpressing NSCLC Cells, CDK6 Positively Regulates Cell Proliferation, whereas NANOS1 Is Required for Cellular Invasiveness
As mentioned earlier, it has been shown that CDK6 regulates cell cycle progression in non-lung cancer cell lines (e.g. U2OS), whereas NANOS1 modulates cellular invasion in the breast cancer cell lines Hs578T and BT549 and the transformed lung epithelial cell line BZR. To assess whether CDK6 and NANOS1 play similar roles in KDM2A-overexpressing NSCLC cells, we assessed the effect of CDK6 or NANOS1 knockdown on the proliferation and invasiveness of H1792 cells. CDK6 depletion strongly inhibited cell proliferation and BrdU incorporation into the replicated DNA of S phase cells but only weakly impeded cellular invasiveness (Fig. 6, A–D). On the contrary, NANOS1 knockdown only marginally inhibited cell proliferation and BrdU incorporation but markedly abrogated cellular invasiveness (Fig. 6, E–H). These results suggest that in KDM2A-overexpressing NSCLC cells CDK6 is associated largely with cell proliferation, whereas NANOS1 is related mainly to cellular invasiveness.
FIGURE 6.

CDK6 knockdown inhibits mainly cell proliferation, whereas NANOS1 knockdown impedes largely cell invasiveness. A–D, the effect of CDK6 knockdown on the proliferation and invasiveness of H1792 cells. CDK6 mRNA levels were analyzed in siControl-treated and CDK6-depleted (siCDK6-1 or -3) cells using quantitative RT-PCR (A). Cell proliferation (B), BrdU incorporation (C), and invasion (D) assays were performed. E–H, the effect of NANOS1 knockdown on the proliferation and invasiveness of H1792 cells. NANOS1 mRNA levels were analyzed in siControl-treated and NANOS1-depleted (siNANOS1-1 or -3) cells using quantitative RT-PCR (E). Cell proliferation (F), BrdU incorporation (G), and invasion (H) assays were performed. d, days. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
KDM2A-mediated Repression of HDAC3 Expression Is Important for in Vitro Cell Proliferation and Invasion as Well as Xenograft Tumor Formation
To determine whether KDM2A regulates the CDK6 and NANOS1 genes in an HDAC3-dependent manner, we reduced HDAC3 in KDM2A-depleted H1975 and H1792 cells using siHDAC3 (Fig. 7, A and B). HDAC3 knockdown significantly restored CDK6 and NANOS1 mRNA levels in KDM2A-depleted H1975 and H1792 cells (Fig. 7, C–F). In addition, HDAC3 depletion significantly rescued deficient proliferation of KDM2A-depleted cells by restoring the S phase population (Fig. 8, A–C). Consistent with this, HDAC3 knockdown increased BrdU incorporation in KDM2A-depleted cells (Fig. 8D). Interestingly, HDAC3 depletion did not affect the G2/M phase population that was increased by KDM2A knockdown (Fig. 8C). Similar to its effects on cell proliferation, HDAC3 depletion significantly recovered cellular invasiveness (Fig. 9, A and B). These results indicate that transcriptional repression of HDAC3 by KDM2A may contribute to cell proliferation and invasiveness by releasing cell cycle-associated genes and invasiveness-related genes from HDAC3 repression.
FIGURE 7.

HDAC3 knockdown significantly increases expression of the CDK6 and NANOS1 genes in KDM2A-depleted NSCLC cells. A and B, analysis of HDAC3 and KDM2A mRNA levels in siControl-treated, KDM2A-depleted (siKDM2A-3), and KDM2A/HDAC3-depleted (siKDM2A-3 + siHDAC3-9) cells by quantitative RT-PCR. H1975 (A) and H1792 (B) cells were treated with siControl, siKDM2A-3, or a mixture of siKDM2A-3 and siHDAC3-9. C–F, the effect of HDAC3 knockdown on CDK6 and NANOS1 mRNA levels in KDM2A-depleted cells. Expression levels of the CDK6 (C and D) and NANOS1 (E and F) genes in H1975 (C and E) and H1792 (D and F) cells were measured by quantitative RT-PCR. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 8.

HDAC3 knockdown rescues proliferation defect of KDM2A-depleted NSCLC cells. A and B, the effect of HDAC3 knockdown on the proliferation of KDM2A-depleted H1975 (A) and H1792 (B) cells. C, the effect of single and double knockdown of KDM2A and HDAC3 on S phase cell percentages. The percentages of sub-G1, G1, S, and G2/M phase cells were analyzed. D, BrdU incorporation assay. d, days. Data are presented as the mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
FIGURE 9.
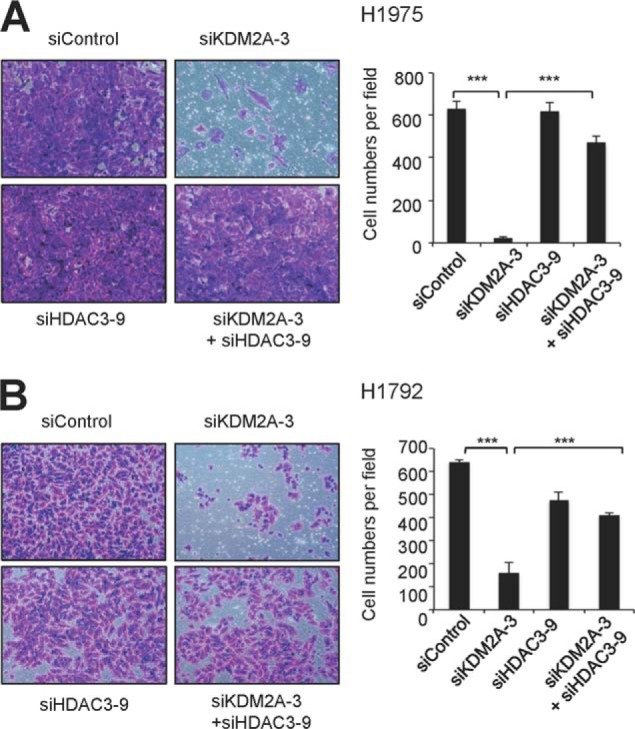
HDAC3 knockdown restores deficient invasiveness of KDM2A-depleted NSCLC cells. A and B, the effect of HDAC3 knockdown on the invasiveness of KDM2A-depleted H1975 (A) and H1792 (B) cells. Representative images of invaded cells are shown (left panels), and cells were counted (right panels). Data are presented as the mean ± S.E. (error bars). ***, p < 0.001.
To determine the effect of KDM2A-mediated repression of HDAC3 on the tumorigenic ability of NSCLC cells in vivo, we depleted HDAC3 in stably KDM2A-depleted H1792 cells using siHDAC3, subcutaneously injected double knockdown cells into mice, and monitored tumor growth. Similar to siRNA-based transient knockdown of KDM2A, stable knockdown of KDM2A increased HDAC3 expression (Fig. 10, A and B). In agreement with our previous report (16), most mice (n = 4 of 5) that were injected with shLuciferase-treated H1792 cells had tumors within 3 months (Fig. 10C and Table 2). In sharp contrast, no mice injected with KDM2A-depleted cells had detectable tumors (n = 0 of 5) (Fig. 10C and Table 2). Interestingly, HDAC3 depletion greatly restored defective tumorigenic abilities of KDM2A-depleted H1792 cells (Fig. 10, C and D, and Table 2). Tumors were confirmed by H&E staining, and CDK6 and NANOS1 levels in a control tumor were comparable with those in a tumor that was originated from double knockdown cells (Fig. 10, E and F). Results obtained from this subcutaneous xenograft experiment indicate that in vivo formation of tumors by KDM2A-overexpressing NSCLC cells may require KDM2A-mediated repression of HDAC3 expression.
FIGURE 10.

HDAC3 knockdown greatly recovers impaired tumorigenicity of KDM2A-depleted NSCLC cells in a mouse xenograft model. A and B, analysis of KDM2A (A) and HDAC3 (B) mRNA levels in control (shLuciferase), stably KDM2A-depleted (shKDM2A-1), and KDM2A/HDAC3-depleted H1792 cells. Stably KDM2A-depleted H1792 cells (shKDM2A-1) were transfected with siHDAC3-9 to generate double (KDM2A/HDAC3) knockdown cells. C and D, the effect of HDAC3 knockdown on tumor development of KDM2A-depleted cells in a subcutaneous xenograft model. Three different groups of cells (shControl, shKDM2A-1, and shKDM2A-1 + siHDAC3-9) were subcutaneously injected into five mice per group. Representative tumors (10 weeks after subcutaneous injection) are shown in dotted circles and indicated by black arrows (C). Tumor volumes were monitored and plotted for 10 weeks (D). E, representative images of H&E staining of xenograft tumor samples (shControl group and shKDM2A-1 + siHDAC3-9 group) and normal skin tissues (shKDM2A-1 group) obtained from the injection sites (10 weeks after subcutaneous injection). Scale bars are indicated (upper panels, 200 μm; lower panels, 100 μm). F, representative images of immunohistochemical staining of NANOS1 and CDK6 in xenograft tumor samples. Immunohistochemical staining was performed only using tumor tissues from the shControl group and shKDM2A-1 + siHDAC3-9 group. Normal mouse skin tissues from the shKDM2A-1 group were not used for staining because they were not a proper control. CDK6 and NANOS1 levels were quantified using the Chromavision Automated Cellular Imaging System (ACIS-III) from Dako and compared between the shControl group and shKDM2A-1 + siHDAC3-9 group: CDK6, 1 versus 0.73; NANOS, 1 versus 0.54. G, in this hypothetical model, KDM2A transcriptionally represses the HDAC3 gene. Subsequently, cell cycle-associated genes (e.g. CDK6) and invasiveness-related genes (e.g. NANOS1) are released from HDAC3-mediated repression. Data are presented as the mean ± S.E. (error bars). **, p < 0.01; ***, p < 0.001.
TABLE 2.
The effect of HDAC3 depletion in stably KDM2A-depleted H1792 cells on tumor formation in a murine subcutaneous xenograft model
| Treatmenta | Mice treated (n) | Mice with lung tumors (n) |
|---|---|---|
| shControl | 5 | 4 |
| shKDM2A-1 | 5 | 0 |
| shKDM2A-1 + siHDAC3 | 5 | 4 |
a Stably KDM2A-depleted (shKDM2A-1) cells, double knockdown (shKDM2A-1 + siHDAC3) cells, and shControl-treated cells were subcutaneously implanted into mice. At the 10th week after injection, numbers of tumor-bearing mice were counted.
DISCUSSION
In this study, our results provided evidence that KDM2A represses expression of the HDAC3 gene by demethylating H3K36me2 at the HDAC3 promoter. Consistent with our previous study showing that KDM2A positively regulates the S phase cell population and cellular invasiveness (16), the results presented here indicate that KDM2A enhances expression of the cell cycle-associated genes CDK6 and NEK7 and the cell invasion-related genes NANOS1 and RAPH1 in KDM2A-overexpressing NSCLC cells. Interestingly, expression of these genes was directly repressed by the transcriptional co-repressor HDAC3. Therefore, our current study uncovered the molecular mechanism in which KDM2A-mediated repression of HDAC3 expression may antagonize the transcriptional repression of cell cycle-associated genes and invasiveness-related genes by HDAC3 in KDM2A-overexpressing lung cancer cells (i.e. KDM2A ⊣ HDAC3 ⊣ cell cycle/invasiveness genes (e.g. CDK6 and NANOS1)) (Fig. 10G). In addition, KDM2A-mediated regulation of HDAC3 expression may be largely dependent on the catalytic activity of KDM2A because the KDM2A catalytic mutant did not repress HDAC3 expression.
It is known that KDM2A acts as a positive regulator for somatic cell reprogramming and cellular antisenescence (32, 33). Our previous data suggest that KDM2A overexpression promotes NSCLC tumorigenesis and metastasis by increasing ERK1/2 signaling (16). Data reported here show that HDAC3 depletion significantly revived the tumorigenic and invasive abilities of KDM2A-depleted cells and restored expression of the cell cycle-associated gene CDK6 and the invasion-related gene NANOS1 in KDM2A-depleted cells. Thus, this work supports the notion that KDM2A-mediated repression of HDAC3 expression contributes to NSCLC tumorigenesis and invasion at least in part by up-regulating certain cell cycle-associated and invasion-related genes. On the basis of our previous and current studies, it is possible that KDM2A promotes tumorigenicity and invasiveness of KDM2A-overexpressing NSCLC cells by controlling two pathways for cell cycle and cell invasion: 1) KDM2A ⊣ DUSP3 ⊣ ERK1/2 → cell cycle/invasiveness and 2) KDM2A ⊣ HDAC3 ⊣ cell cycle/invasiveness genes. Interestingly, treatment of KDM2A-overexpressing cells with the MEK1/2 inhibitor U0126 (10 μm) that consequently impedes ERK1/2 phosphorylation did not show any obvious effect on HDAC3 levels, suggesting that these two pathways may be independent (data not shown).
Certain histone modifiers regulate gene expression in a cooperative mode. The H3K9 methyltransferase G9a and the H3K4 demethylase JARID1a co-repress gene expression (34), and the H3K4 demethylase LSD1 and HDAC1 and -2 cooperatively repress their co-target genes (35). The H3K27 demethylase UTX and H3K4 methyltransferase MLL4 (also known as ALR and KMT2D) may co-activate their co-target genes (36). In contrast, some histone modifiers antagonize the function of others on the same genes. For example, polycomb repressive complex 2 containing the H3K27 methyltransferase EZH2 may enable cancer cells to resist cellular senescence by inhibiting expression of the cellular senescence genes p16INK4A and p14ARF (37), whereas the H3K27 demethylase JMJD3 may neutralize EZH2 function by activating the same genes (38). Interestingly, the results reported here indicate that the transcriptional co-repressor KDM2A counteracts the gene-repressive effects of HDAC3 on cell cycle-regulatory genes and cell invasion-associated genes by transcriptionally down-regulating the HDAC3 gene in a catalytic activity-dependent manner. To our knowledge, the epigenetic repression of HDAC3 function by KDM2A is a distinct mode of antagonistic regulation in which a histone lysine demethylase opposes the function of a histone deacetylase via transcriptional repression.
As many as 70% of eukaryotic genes contain CpG islands, which represent genomic regions that have higher than average genome-wide levels of CpG dinucleotides (39). Blackledge et al. (17) reported that via its CXXC domain KDM2A interacts with many transcription start site-associated CpG islands. Interestingly, the HDAC3 gene has a CpG island spanning the predicted transcription start site (data not shown). Thus, it is possible that KDM2A is recruited to the HDAC3 promoter through its association with the CpG-dense region. However, the transcriptional regulation of HDAC3 expression seems more complex. Although our analysis of the tumor database The Cancer Genome Atlas showed a trend for an inverse correlation between KDM2A and HDAC3 mRNA levels in NSCLC tumors, there was no significant correlation between their levels in a range of NSCLC cell lines (data not shown). These results indicate that KDM2A is a key control factor for HDAC3 expression in certain types of lung cancer cell lines and lung tumors but that other gene-regulatory factors may also be involved in modulating the HDAC3 promoter. Further studies are required for a better understanding of how HDAC3 expression is regulated.
HDAC inhibitors have been recently developed as anticancer agents. Multiple mechanisms underlying the mode of action of HDAC inhibitors have been described, including induced expression of tumor suppressor p21 and the proapoptotic ligand TRAIL (40–42). Recently, Chen et al. (43) reported that the HDAC inhibitor trichostatin A may increase the expression of the potential tumor suppressor cluster Dleu2/miR-15a/16-1 via HDAC3 inhibition in two NSCLC cell lines, A549 and H1299. In contrast, our results suggest that HDAC3 is involved in tumor suppression in the NSCLC cell lines H1975 and H1792. These seemingly contradictory results might result from a cell line-dependent function of HDAC3. Thus, it is possible that HDAC3 may act as an oncogenic factor by repressing tumor suppressor genes in A549 and H1299, whereas it may play a tumor-suppressive role in H1975 and H1792 by down-regulating expression of cell proliferation-related genes and proinvasive genes.
In addition, the pathologic role of HDAC3 in cancer development might be tissue-dependent. In gastric, prostatic, and colorectal cancer samples, HDAC3 overexpression was significantly associated with poor prognosis (44–46). In agreement with these reports, it has been shown that HDAC3 appeared to be up-regulated and to repress the tumor suppressor gene p21 in colorectal cancer cells (47). In contrast, other studies reported that liver-specific HDAC3 knock-out mice develop hepatoma (48) and that HDAC3 maintains genomic stability (48) and potentiates apoptosis by down-regulating the proto-oncogene c-Jun (28), indicating an antitumor function for HDAC3. In support of the tumor-suppressive role of HDAC3, the results reported here showed that HDAC3 knockdown restored defective proliferation and invasiveness of KDM2A-depleted cells. Our additional results indicate that HDAC3 represses cell cycle-associated genes and invasiveness-related genes. Thus, our findings suggest that the inhibition of antitumor function of HDAC3 by KDM2A at transcriptional levels may be requisite for the growth and invasion of KDM2A-overexpressing NSCLC tumors.
Acknowledgement
We are thankful to Luanne Jorewicz for manuscript editing.
This work was supported, in whole or in part, by National Institutes of Health Grants CA157919 and GM095659 (to M. G. L.). This work was also supported by Cancer Prevention and Research Institute of Texas Grant RP110183 (to M. G. L.), a fund from the Center for Cancer Epigenetics at The University of Texas MD Anderson Cancer Center (to M. G. L.), a scholar fellowship from the Center for Cancer Epigenetics at MD Anderson Cancer Center (to S. S. D.), and a fellowship from the Odyssey Program and the Estate of C. G. Johnson, Jr. at MD Anderson Cancer Center (to H. A.).
- H3K4
- histone H3 lysine 4
- HDAC
- histone deacetylase
- NSCLC
- non-small cell lung cancer
- KDM
- histone lysine demethylase
- H3K36me2
- dimethylated H3K36
- DUSP3
- dual specificity phosphatase 3
- ac
- acetylation
- qPCR
- quantitative PCR.
REFERENCES
- 1. Sims R. J., 3rd, Nishioka K., Reinberg D. (2003) Histone lysine methylation: a signature for chromatin function. Trends Genet. 19, 629–639 [DOI] [PubMed] [Google Scholar]
- 2. Martin C., Zhang Y. (2005) The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6, 838–849 [DOI] [PubMed] [Google Scholar]
- 3. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 4. Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T. K., Koche R. P., Lee W., Mendenhall E., O'Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E. S., Bernstein B. E. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greer E. L., Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi Y., Whetstine J. R. (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 25, 1–14 [DOI] [PubMed] [Google Scholar]
- 7. Klose R. J., Zhang Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8, 307–318 [DOI] [PubMed] [Google Scholar]
- 8. Siegel R., Naishadham D., Jemal A. (2013) Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 [DOI] [PubMed] [Google Scholar]
- 9. Herbst R. S., Heymach J. V., Lippman S. M. (2008) Lung cancer. N. Engl. J. Med. 359, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharma S. V., Bell D. W., Settleman J., Haber D. A. (2007) Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 [DOI] [PubMed] [Google Scholar]
- 11. Feinberg A. P., Tycko B. (2004) The history of cancer epigenetics. Nat. Rev. Cancer 4, 143–153 [DOI] [PubMed] [Google Scholar]
- 12. Feinberg A. P., Ohlsson R., Henikoff S. (2006) The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 7, 21–33 [DOI] [PubMed] [Google Scholar]
- 13. Barlési F., Giaccone G., Gallegos-Ruiz M. I., Loundou A., Span S. W., Lefesvre P., Kruyt F. A., Rodriguez J. A. (2007) Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol. 25, 4358–4364 [DOI] [PubMed] [Google Scholar]
- 14. Van Den Broeck A., Brambilla E., Moro-Sibilot D., Lantuejoul S., Brambilla C., Eymin B., Khochbin S., Gazzeri S. (2008) Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin. Cancer Res. 14, 7237–7245 [DOI] [PubMed] [Google Scholar]
- 15. Asangani I. A., Ateeq B., Cao Q., Dodson L., Pandhi M., Kunju L. P., Mehra R., Lonigro R. J., Siddiqui J., Palanisamy N., Wu Y. M., Cao X., Kim J. H., Zhao M., Qin Z. S., Iyer M. K., Maher C. A., Kumar-Sinha C., Varambally S., Chinnaiyan A. M. (2013) Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol. Cell 49, 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wagner K. W., Alam H., Dhar S. S., Giri U., Li N., Wei Y., Giri D., Cascone T., Kim J. H., Ye Y., Multani A. S., Chan C. H., Erez B., Saigal B., Chung J., Lin H. K., Wu X., Hung M. C., Heymach J. V., Lee M. G. (2013) KDM2A promotes lung tumorigenesis by epigenetically enhancing ERK1/2 signaling. J. Clin. Investig. 123, 5231–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blackledge N. P., Zhou J. C., Tolstorukov M. Y., Farcas A. M., Park P. J., Klose R. J. (2010) CpG islands recruit a histone H3 lysine 36 demethylase. Mol. Cell 38, 179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frescas D., Guardavaccaro D., Kuchay S. M., Kato H., Poleshko A., Basrur V., Elenitoba-Johnson K. S., Katz R. A., Pagano M. (2008) KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 7, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsukada Y., Fang J., Erdjument-Bromage H., Warren M. E., Borchers C. H., Tempst P., Zhang Y. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 [DOI] [PubMed] [Google Scholar]
- 20. Dhar S. S., Lee S. H., Kan P. Y., Voigt P., Ma L., Shi X., Reinberg D., Lee M. G. (2012) Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 26, 2749–2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alam H., Kundu S. T., Dalal S. N., Vaidya M. M. (2011) Loss of keratins 8 and 18 leads to alterations in α6β4-integrin-mediated signalling and decreased neoplastic progression in an oral-tumour-derived cell line. J. Cell Sci. 124, 2096–2106 [DOI] [PubMed] [Google Scholar]
- 22. Ma H., Chen J., Pan S., Dai J., Jin G., Hu Z., Shen H., Shu Y. (2011) Potentially functional polymorphisms in cell cycle genes and the survival of non-small cell lung cancer in a Chinese population. Lung Cancer 73, 32–37 [DOI] [PubMed] [Google Scholar]
- 23. Braden W. A., McClendon A. K., Knudsen E. S. (2008) Cyclin-dependent kinase 4/6 activity is a critical determinant of pre-replication complex assembly. Oncogene 27, 7083–7093 [DOI] [PubMed] [Google Scholar]
- 24. Michaud K., Solomon D. A., Oermann E., Kim J. S., Zhong W. Z., Prados M. D., Ozawa T., James C. D., Waldman T. (2010) Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 70, 3228–3238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Regan L., Fry A. M. (2009) The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol. Cell. Biol. 29, 3975–3990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bonnomet A., Polette M., Strumane K., Gilles C., Dalstein V., Kileztky C., Berx G., van Roy F., Birembaut P., Nawrocki-Raby B. (2008) The E-cadherin-repressed hNanos1 gene induces tumor cell invasion by upregulating MT1-MMP expression. Oncogene 27, 3692–3699 [DOI] [PubMed] [Google Scholar]
- 27. Lyulcheva E., Taylor E., Michael M., Vehlow A., Tan S., Fletcher A., Krause M., Bennett D. (2008) Drosophila pico and its mammalian ortholog lamellipodin activate serum response factor and promote cell proliferation. Dev. Cell 15, 680–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xia Y., Wang J., Liu T. J., Yung W. K., Hunter T., Lu Z. (2007) c-Jun downregulation by HDAC3-dependent transcriptional repression promotes osmotic stress-induced cell apoptosis. Mol. Cell 25, 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wen Y. D., Perissi V., Staszewski L. M., Yang W. M., Krones A., Glass C. K., Rosenfeld M. G., Seto E. (2000) The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. U.S.A. 97, 7202–7207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guenther M. G., Lane W. S., Fischle W., Verdin E., Lazar M. A., Shiekhattar R. (2000) A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 14, 1048–1057 [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pfau R., Tzatsos A., Kampranis S. C., Serebrennikova O. B., Bear S. E., Tsichlis P. N. (2008) Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc. Natl. Acad. Sci. U.S.A. 105, 1907–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang T., Chen K., Zeng X., Yang J., Wu Y., Shi X., Qin B., Zeng L., Esteban M. A., Pan G., Pei D. (2011) The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 9, 575–587 [DOI] [PubMed] [Google Scholar]
- 34. Chaturvedi C. P., Somasundaram B., Singh K., Carpenedo R. L., Stanford W. L., Dilworth F. J., Brand M. (2012) Maintenance of gene silencing by the coordinate action of the H3K9 methyltransferase G9a/KMT1C and the H3K4 demethylase Jarid1a/KDM5A. Proc. Natl. Acad. Sci. U.S.A. 109, 18845–18850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee M. G., Wynder C., Bochar D. A., Hakimi M. A., Cooch N., Shiekhattar R. (2006) Functional interplay between histone demethylase and deacetylase enzymes. Mol. Cell. Biol. 26, 6395–6402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee M. G., Villa R., Trojer P., Norman J., Yan K. P., Reinberg D., Di Croce L., Shiekhattar R. (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318, 447–450 [DOI] [PubMed] [Google Scholar]
- 37. Bracken A. P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Mönch K., Minucci S., Porse B. T., Marine J. C., Hansen K. H., Helin K. (2007) The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 21, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Agger K., Cloos P. A., Rudkjaer L., Williams K., Andersen G., Christensen J., Helin K. (2009) The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saxonov S., Berg P., Brutlag D. L. (2006) A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U.S.A. 103, 1412–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chopin V., Toillon R. A., Jouy N., Le Bourhis X. (2004) P21(WAF1/CIP1) is dispensable for G1 arrest, but indispensable for apoptosis induced by sodium butyrate in MCF-7 breast cancer cells. Oncogene 23, 21–29 [DOI] [PubMed] [Google Scholar]
- 41. Nakano K., Mizuno T., Sowa Y., Orita T., Yoshino T., Okuyama Y., Fujita T., Ohtani-Fujita N., Matsukawa Y., Tokino T., Yamagishi H., Oka T., Nomura H., Sakai T. (1997) Butyrate activates the WAF1/Cip1 gene promoter through Sp1 sites in a p53-negative human colon cancer cell line. J. Biol. Chem. 272, 22199–22206 [DOI] [PubMed] [Google Scholar]
- 42. Insinga A., Monestiroli S., Ronzoni S., Gelmetti V., Marchesi F., Viale A., Altucci L., Nervi C., Minucci S., Pelicci P. G. (2005) Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat. Med. 11, 71–76 [DOI] [PubMed] [Google Scholar]
- 43. Chen C. Q., Chen C. S., Chen J. J., Zhou L. P., Xu H. L., Jin W. W., Wu J. B., Gao S. M. (2013) Histone deacetylases inhibitor trichostatin A increases the expression of Dleu2/miR-15a/16–1 via HDAC3 in non-small cell lung cancer. Mol. Cell. Biochem. 383, 137–148 [DOI] [PubMed] [Google Scholar]
- 44. Weichert W., Röske A., Gekeler V., Beckers T., Ebert M. P., Pross M., Dietel M., Denkert C., Röcken C. (2008) Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 9, 139–148 [DOI] [PubMed] [Google Scholar]
- 45. Weichert W., Röske A., Gekeler V., Beckers T., Stephan C., Jung K., Fritzsche F. R., Niesporek S., Denkert C., Dietel M., Kristiansen G. (2008) Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 98, 604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weichert W., Röske A., Niesporek S., Noske A., Buckendahl A. C., Dietel M., Gekeler V., Boehm M., Beckers T., Denkert C. (2008) Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 14, 1669–1677 [DOI] [PubMed] [Google Scholar]
- 47. Wilson A. J., Byun D. S., Popova N., Murray L. B., L'Italien K., Sowa Y., Arango D., Velcich A., Augenlicht L. H., Mariadason J. M. (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281, 13548–13558 [DOI] [PubMed] [Google Scholar]
- 48. Bhaskara S., Knutson S. K., Jiang G., Chandrasekharan M. B., Wilson A. J., Zheng S., Yenamandra A., Locke K., Yuan J. L., Bonine-Summers A. R., Wells C. E., Kaiser J. F., Washington M. K., Zhao Z., Wagner F. F., Sun Z. W., Xia F., Holson E. B., Khabele D., Hiebert S. W. (2010) Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 18, 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]