

Abstract
Although it has been more than 165 years since the first introduction of modern anesthesia to the clinic, there is surprisingly little understanding about the exact mechanisms by which general anesthetics induce unconsciousness. As a result, we do not know how general anesthetics produce anesthesia at different levels. The main handicap to understanding the mechanisms of general anesthesia is the diversity of chemically unrelated compounds including diethyl ether and halogenated hydrocarbons, gases nitrous oxide, ketamine, propofol, benzodiazepines and etomidate, as well as alcohols and barbiturates. Does this imply that general anesthesia is caused by many different mechanisms Until now, many receptors, molecular targets and neuronal transmission pathways have been shown to contribute to mechanisms of general anesthesia. Among these molecular targets, ion channels are the most likely candidates for general anesthesia, in particular γ-aminobutyric acid type A, potassium and sodium channels, as well as ion channels mediated by various neuronal transmitters like acetylcholine, amino acids amino-3-hydroxy-5-methyl-4-isoxazolpropionic acid or N-methyl-D-aspartate. In addition, recent studies have demonstrated the involvement in general anesthesia of other ion channels with distinct gating properties such as hyperpolarization-activated, cyclic- nucleotide-gated channels. The main aim of the present review is to summarize some aspects of current knowledge of the effects of general anesthetics on various ion channels.
Keywords: General anesthesia, Ion channels, γ-amino-butyric acid type A receptors, Hyperpolarization activated cyclic nucleotide, Potassium channels, Glutamatergic ion channels, Sodium channels
INTRODUCTION
The start of modern anesthesia, through the use of inhaled volatile anesthetics 150 years ago, dramatically revolutionized modern medicine. Dentist Dr. Horace Wells used nitrous oxide for a public demonstration of its powers of intoxication. Another dentist, William Morton, took up Wells’ idea of a gaseous anesthetic, together with the suggestion from Charles Jackson to use ether, to perform a widely known public demonstration of ether anesthesia on 16 October, 1846.
The structural diversity of general anesthetics, from simple chemically inert gases to complex barbiturates, has baffled anesthesiologists, and ideas about how these anesthetics might work have been correspondingly confused. In the early stages, the notion that anesthetics worked “nonspecifically” by dissolving in the lipid bi-layer portions dominated. Although this simple idea could explain the structural diversity of general anesthetics, it is now generally accepted that anesthetics act by binding directly to sensitive target proteins.
Until now, many receptors, molecular targets and neuronal transmission pathways have been shown to contribute to general anesthesia. Among these molecular targets, ion channels are the predominant candidates for general anesthetic effect, in particular γ-aminobutyric acid type A (GABAA), potassium and sodium channels and ion channels activated by acetylcholine, amino-3-hydroxy-5-methyl-4-isoxazolpropionic acid or N-methyl-D-aspartate. In addition, some other ion channels such as hyperpolarization activated cyclic nucleotide (HCN) channels are also involved in general anesthesia (Table 1).
Table 1.
The effects of general anesthetics on ion channels
|
Volatile anesthetics |
Intravenous anesthetics |
|||||||||
| Halothane | Isoflurane | Enflurane | Ether | Ethanol | Propofol | Etomidate | Ketamine | Phentobarbital | Benzodiazepine | |
| GABAA | + | + | + | + | + | + | + | -- | + | + |
| hα1β1 | + | + | + | |||||||
| hα1β2 | + | + | + | + | + | + | + | |||
| hα1β1γ2 | + | + | + | + | + | + | ||||
| hα1β2γ2 | + | + | + | |||||||
| NMDA | -- | -- | -- | -- | -- | -- | ||||
| AMPA/Kainate | ||||||||||
| GluR1 | -- | -- | ||||||||
| GluR3 | -- | -- | -- | -- | ||||||
| GluR2 + 3 | -- | -- | -- | -- | ||||||
| GluR6 | + | + | + | -- | + | -- | ||||
| K+ | ||||||||||
| KV | -- | -- | -- | -- | -- | |||||
| TREK | +/-- | +/-- | +/-- | +/-- | +/-- | |||||
| HCN | -- | -- | -- | -- | ||||||
| Na+ | -- | -- | -- | -- | -- | -- | ||||
+: Effect of agonist; --: Effect of antagonist; GABAA: γ-aminobutyric acid type A.
The main aim of the present review is to summarize some aspects of current knowledge about the function of general anesthetics at different ion channels.
GABAA RECEPTORS AND GENERAL ANESTHESIA
Structure and function of the GABAA receptor
The GABAA receptor is composed of five different subunits (α1-6, β1-3, γ1-3, δ, epsilon, φ, π and ρ1-3) which are encoded by at least 19 mammalian genes, with additional diversity arising in certain regions[1]. In most GABAA receptors, the most common combination of subunits is α, β, and γ, with a ratio of 2:2:1 although the γ subunit may be replaced by δ or epsilon subunits, particularly in brain regions, as shown in Figure 1. These GABAA receptor subunits are densely packed in the cortex, and receptors with the γ2 subunit comprise more than 40% of all GABAA receptors in the brain[2].
Figure 1.
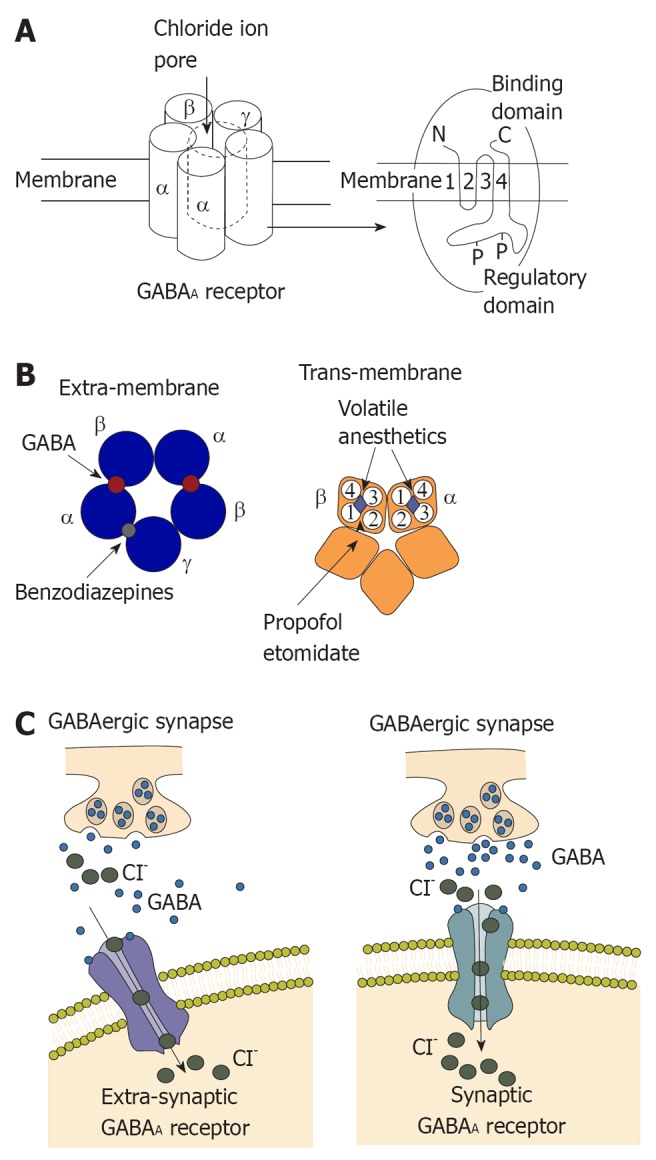
Structure and function of γ- aminobutyric acid type A receptor. A: γ- aminobutyric acid type A (GABAA) receptors commonly contain two α subunits, two β subunits and one γ subunit. Chloride influx through the pore could hyperpolarize the postsynaptic membrane; B: Left: Extra-membrane region of GABAA receptor. The binding sites for GABA are located between α and β subunits and the binding site for benzodiazepines is located between γ and α subunits; Right: Trans-membrane region of GABAA receptor. Four trans-membrane segments form the α subunit. It has been shown that the trans-membrane segment of β subunit is the binding site for propofol and etomidate. This binding site is close to a binding site for volatile anesthetics; C: Activation of the GABAA receptor could increase conductance of the postsynaptic membrane and alter the potential of the membrane because of influx of chloridion. Synaptic receptors could detect GABA at mmol concentration to produce fast inhibitory postsynaptic potentials (IPSPs), and extra-synaptic receptors that detect GABA at μmol concentrations to produce slower IPSPs.
The GABA system is the main inhibitory neurotransmitter pathway in the CNS of mammalian brain, and one-third of all synapses are GABAergic[3]. The GABA system induces inhibition of the central nervous system by generating fast, transient inhibitory postsynaptic currents. Activation of GABAA receptors decreases excitability of the neurons by an influx of chloride, hyperpolarization of the membrane, and shunting of excitatory input. This synaptic inhibition of the GABA system maintains neuronal communication and induces precise timing of action potentials and synchronization of neuronal populations[4,5]. For many years, enhancement of fast inhibition at synapses was widely regarded as the dominate mechanism underlying the effects of many GABAergic drugs.
The α1 and β2 subunit-containing GABAA receptors in the cortex are thought to contribute to the sedative actions of several inhaled anesthetics. Some studies of tool drugs indicate the important role of GABA receptors and its subunits in the anesthetic effect. Tonic current in the thalamic VB neurons may contribute to the sedative action of 4,5,6,7-tetrahydroisoxazolo(5,4-c)pyridin-3-ol (THIP). Although THIP is not commonly used as an anesthetic, it promotes slow wave sleep and produces analgesic, sedative, hypnotic actions and ataxic properties[6]. GABAA receptors that contain the α4 and δ subunit appear to contribute to the sedation effects of THIP. At low concentrations, THIP strongly potentiates the activity of GABAA receptors containing the δ subunit, and enhances a tonic conductance generated by α4δ GABAA receptors[7]. Rotarod performance and spontaneous loco-motor activity were unimpaired by THIP in α4 subunit knock-out mice[8], which suggests that α4δ subunit containing GABAA receptors are necessary for the sedative and ataxic effects of THIP. THIP enhanced the tonic but not the phasic GABAA receptor currents in VB neurons, and had no effect on nRT neurons[9]. Since the sedative actions of THIP were absent in α4 knock-out mice, it is likely that the tonic current mediated by α4β2δ GABAA receptors in VB neurons contributes to anesthetic sedation.
Actions of general anesthetics on GABAA receptors
The enhancement of GABA-activated chloride currents is the main effect of some intravenous general anesthetic such as propofol and etomidate, decreasing neuronal activity by producing hyperpolarization of the neuronal membrane. This is in agreement with the finding that etomidate-mediated sedation also depends on GABAA receptors containing the β2 subunit[9,10], although the specific contribution of thalamic β2 subunits to this effect is uncertain. Propofol and etomidate also enhance function of GABAA receptors to produce immobility[11-13]. In contrast, gaseous general anesthetics such as xenon, nitrous oxide, cyclopropane as well as ketamine have minimal or no effect on GABAA receptor subtypes[14-18].
Compared to other general anesthetics, volatile anesthetics show low potency to a variety of receptors at clinical concentrations[19]. As a result, the determination of the specific sites of effect of volatile anesthetics is a challenge. In addition, behavioral evaluation with volatile anesthetics has some obvious practical difficulties. Even with these handicaps, it has been demonstrated by some carefully designed studies that isoflurane anesthesia is mediated by GABAA receptors. Volatile anesthetics at clinical concentrations could activate GABAA receptors both in vitro and in vivo, using heterologous expression systems and the postsynaptic membrane, respectively[20,21]. The depressive effects of isoflurane, enflurane and halothane on rat neocortical neuron activity were studied using in vivo recordings of spontaneous action-potential firing and in vitro recordings from isolated cortical networks. Sedative concentrations of isoflurane, enflurane and halothane similarly reduced the firing of spontaneous action potentials in vivo and in vitro by approximately 50%. This reduction in neuronal firing strongly correlated with an increase in GABAergic synaptic inhibition. Anesthetics prolonged the time course of GABAA receptor-mediated spontaneous IPSCs from pyramidal neurons in organ cortical cultures with no effect on their frequency or peak amplitude.
At the spinal level the role of inhibitory GABAA receptors on anesthetics actions has been extensively studied. With the evaluation of motor response, MAC of volatile anesthetics was more significantly affected by spinal injections of glycine receptor antagonists than GABAA receptor antagonists[22].
For many years, the binding site of GABAA receptor for volatile anesthetics is still unclear. The binding site for volatile anesthetics on the GABAA receptor was determined to be a binding pocket for volatile anesthetics, by complementary site directed mutagenesis, using general anesthetics of varying molecular size[23]. With the finding of a binding pocket for general anesthetics, the long-held assumption that general anesthetics worked by a nonspecific mechanism was overturned. Dramatic progress has been made in dissecting the behavioral effects of general anesthetics, in particular the subunit combination of GABAA receptors, on anesthetic effect. GABAA receptors containing the α1β2γ2 subunits are enriched at synaptic sites throughout the brain[24]. This suggests that the enhancement of synaptic activity within the cortex could be responsible for anesthetic sedation. The contribution of the cortex to the sedative properties of inhaled anesthetics was studied by Hentschke and colleagues[25]. In recent studies, an anesthetic binding cavity for volatile anesthetics has been identified, critically involving in the α1 subunit[26,27]. Rudolph et al[28], reported that animal behavioral patterns induced by benzodiazepine were moderated by a point mutation on the mouse α1 GABAA subunit. At the same time, barbiturates directly activate and inhibit GABAA receptors by means of positive allosteric modulation depending on their concentration at the receptor. In addition, a mutation in the GABAA α subunit was identified that abolishes the action of barbiturates, although, the potentiating by etomidate on GABAA receptors was not affected. Furthermore, enhancement of GABAA mediated transmissions was also affected by alcohol, indicating an important role of alcohol in mediating its intoxicating effects[29].
The biophysical profile of GABA receptors and their sensitivity to general anesthetics can be dramatically altered by subunit composition[30]. Using chimerical channel construction, Mihic and colleagues discovered a domain, relevant for mediating the effect of volatile anesthetics and etomidate[29], but not propofol[27]. Two key amino acids in GABAA receptor subunits were found to be involved in their interaction with volatile anesthetics. These amino residues may contribute to the molecular binding pockets for general anesthetics[31]. According to important studies, two amino acids in the α1 subunit are the most critical points for general anesthetic effect[27]. Serine 270 is in the trans-membrane segment and while Alanine 291 is near the extracellular regions. For GABAA receptors, replacing Ser 270 with larger amino acid residues in the α1 subunit resulted in a decrease of sensitivity to volatile anesthetics[26,31], while replacement with smaller residues resulted in the opposite effect[26]. Also, replacing the α1 Ser270 residue with histidine resulted in recombinant heteromeric GABAA receptors that were insensitive to isoflurane[26]. However, an additional change to the GABAA receptors, introduced by the α1 (Ser270His) mutation, complicated the interpretation of receptor pharmacology[32]. This problem was addressed by introducing an additional mutation into the α1 subunit, whereby the leucine residue at position 277 was replaced with alanine. This double knock-in mutation, α1 (Ser270His, Leu277Ala), restored normal sensitivity to GABA[29]. These mutations laid the foundation for generating knock-in mice that were partially insensitive to isoflurane. Mice with a double knock-in mutation were used to explain the interaction between GABAA receptors containing α1 subunits to isoflurane anesthesia[33]. Some studies demonstrated that double-mutant mice expressing the α1 (Ser270His, Leu277Ala) subunit was less sensitive to isoflurane, compared to wild-type controls, indicating the important role of α1 subunit in the hypnotic effect of isoflurane. Interestingly, according to the tail clamp test, the immobilizing effect of isoflurane was not affected in these double-mutant mice. Using cued and contextual fear conditioning, the amnesic effect of isoflurane was also unaffected in the α1 (Ser270His, Leu277Ala) mice, comparing to wild-type control, indicating that this subunit is not critical for amnesia induced by isoflurane. This last finding is in contrast to previous work using mouse mutants in which the α1 subunit was knocked out either globally or in the forebrain alone[34]. In other studies with the α1 subunit knock-out mice, the amnesic effect induced by isoflurane was impaired, indicating the role of α1 subunit in isoflurane amnesia. At the same time, the β subunit of GABAA receptors is also important to the binding site of volatile anesthetics, as well as for the behavioral effects of volatile anesthetics[27,35]. In addition, on the β3 subunit when the asparagine residue at position 265 was replaced with methionine or the methionine at position 286 with tryptophan, GABA current potentiated by enflurane was reduced[35]. With β3 (Asn265Met) knock-in mice, isoflurane is slightly less effect at inhibiting the righting reflex in β3 (Asn265Met) mice, suggesting the role of the β3 subunit in isoflurane hypnosis. The immobility induced by isoflurane, however, is significantly impaired in these knock-in mice, as measured by hind limb or tail clamp withdrawal reflex. Additionally, in β3 (Asn265Met) mice, heart rate and core temperature were decreased less by isoflurane[36], indicating the role of the β3 subunit in the effect of isoflurane on circulation. Therefore, neuronal depressive effect and cardiovascular effects induced by volatile anesthetics might be mediated by distinct GABAA receptor subunits[37].
Knock-in mutant mice have been used to determine the GABAA subunits responsible for the sedative and hypnotic actions of etomidate. Some studies indicate that amnesic effect induced by etomidate might contribute to the α5 GABAA receptors in hippocampal region, while the sedative effect of etomidate might be due to other GABAA receptor isoforms. GABAA receptors with some structural modifications (the asparagine at position 265 in the β2 or β3 subunits was replaced with serine or methionine, respectively) were insensitive to etomidate in vitro[9,10]. Etomidate showed low efficacy in reducing spontaneous loco-motor activity in β2 (Asn265Ser) knock-in mice, indicating that GABAA receptors with the β2 subunit were important for the sedative effect of etomidate[9].
In other studies, sedative property of diazepam has been demonstrated to be mediated by the α1 subunit of GABAA receptors. With some different features from general anesthetics, diazepam produces a sedative effect. GABAA receptors which contained a histidine to arginine mutation at position 101 of the α1 subunit were insensitive to diazepam in vitro[28]. Behavioral tests indicated that the sedative effect induced by diazepam were eliminated in knock-in mice that expressed the α1 (His101Arg) mutation[28].
Some other types of GABAA receptors, such as the extra junction GABAA receptors, could be activated by GABA at very low concentrations. Junction GABAA receptors are widely expressed in important brain regions including the hippocampus, thalamus, cortex and cerebellum. Currents mediated by these junctions GABAA receptors are affected by volatile anesthetics at low concentrations[38].
Neuroprotection of anesthetics involve in action of GABA receptors
Recent studies have shown that general anesthetics could produce significant neural protection and/or induce a preconditioning effect against ischemia/reperfusion induced injury. Propofol, a potent antioxidant, has been reported to have neural protective effects, reducing cerebral blood flow and intracranial pressure. Many studies have indicated that propofol pretreatment significantly improves post-resuscitation recovery of neuronal functions. Recent studies have suggested that during the process of resuscitation, the effect of GABAA changes from inhibitory to excitatory, through a mechanism that is closely associated with activation of microglia and down regulation of the K+-Cl- transporter. It has been demonstrated that propofol might protect the neurons by inhibiting the transition of GABAergic inhibition into excitation during resuscitation.
POTASSIUM CHANNELS AND GENERAL ANESTHESIA
Structure and function of potassium channels
Mammalian K+ channel subunits contain two, four or six/seven transmembrane segments, as shown in Figure 2. Members of the two and six/seven transmembrane segments classes are characterized by the presence of a single pore-forming (P) domain, whereas the more recently discovered four transmembrane segment subunits contain two P domains that are arranged in tandem[39-43]. Background K+ channels are transmembrane K+-selective ionic pores that are constitutively open at rest and are central to neural function. Background K+ channels and their regulation by membrane-receptor-coupled second messengers, as well as pharmacological agents, are therefore important in tuning neuronal resting membrane potential, action potential duration, membrane input resistance and, consequently, regulating transmitter release[44,45].
Figure 2.

The trans-membrane structures and subunit formulation of the potassium channels and phylogenetic tree of K2P channels in humans. A: The trans-membrane structures and subunit formulation of the potassium channels. BK channels (background) are made up of four α-subunits and the four β subunits. Structures of Kir or KATP channels are the simplest. Their subunit has two trans-membrane segments connected by a pore loop. Four subunits form a functional channel pore. K2P channels are made of a tetrameric pore made up of two subunits. Subunits of KV channels have six trans-membrane regions and trans-membrane domain S4 acts as the voltage sensor; B: Phylogenetic tree of K2P channels from humans. The chromosomal localization, nomenclature and functional properties of each subunit are indicated. Different colors indicate the functional subgroups. TASK1: TWIK-related acid-sensitive K+; K2P: Two-pore-domain K+.
Background K+ channels are composed of K2P channel subunits, previously called KCNKx subunits, or tandemly arranged P domains in weak inwardly rectifying K+ channel (TWIK) subunits. Two-pore-domain K+ (K2P) channel subunits are made up of four transmembrane segments and two pore-forming domains that are arranged in tandem and function as either homo-or heterodimeric channels. This structural motif is associated with unusual gating properties, including background channel activity and sensitivity to membrane stretch. In one-pore-domain K+ (K1P) channels, four matching P loops are assembled in homo- or heterotetramers (all subunits have a similar P domain sequence, which contains the residues GYG or GFG), whereas in the dimeric K2P channels, the first pore (P1) and P2 domains have different sequences as exemplified by TWIK1 or TWIK-related acid-sensitive K+ 1 (TASK1)[46]. Many K2P channels have a phenylalanine or a leucine in the GXG motif (where X represents any amino acid) of the selectivity filter in the P2 domain instead of a tyrosine[40-41,46]. Therefore, in K2P channels, the pore is predicted to have a two-fold symmetry rather than the classical four-fold arrangement of other K+ channels. Although the selectivity of K2P channels for K+ over Na+ is high [permeability ratio (PNa/PK) < 0.03], these structural differences suggest a more varied permeation and gating compared with K1P channels[39]. K2P channels, including TASK1 and TWIK-related K+ 1 (TREK1), present an instantaneous current component and a second time-dependent component in response to depolarization[47,48]. Furthermore, TREK1 shows a strong outward rectification in a symmetrical K+ gradient instead of the linear current to voltage relationship predicted by the GHK equation[49,50]. The outward rectification of TREK1 is attributed to an external Mg2+ block, which is present at negative membrane potentials, and to an intrinsic voltage- dependent mechanism[49,50]. Transfection of TREK1 (either splice variant) in HEK cells surprisingly produces two populations of channels with different single-channel conductance (about 40 pS and 100 pS in a symmetrical K+ gradient)[51]. Therefore, K2P channels diverge from the constant-field GHK current formulation and are characterized by complex permeation and gating mechanisms[49,52].
Recent in vivo studies have demonstrated that TREK1, the most thoroughly studied K2P channel, has a key role in the cellular mechanisms of neuronal protection, anesthesia, pain and depression[53]. Mechano-gated and acid-activated TREK1 and TREK2 are the hypothetical functional homologues of the Aplysia S-type background K+ channel[53,54]. Recently, genetic inactivation of TREK1 in the mouse has revealed the potential involvement of this K2P channel in a range of neuronal disease states, including pain, ischemia, epilepsy and depression[55-57]. Human TREK1 is highly expressed in the brain, where it is particularly abundant in γ-aminobutyric acid-containing interneurons of the caudate nucleus and putamen[58]. TREK1 is also expressed in the prefrontal cortex, hippocampus, hypothalamus, midbrain serotonergic neurons and sensory neurons of the dorsal root ganglia[55,59-61]. TREK1 is a signal integrator responding to a wide range of physiological and pathological inputs.
Actions of general anesthetics on potassium channels
K2P channels are modulated by a variety of cellular lipids and pharmacological agents, including polyunsaturated fatty acids and volatile general anesthetics. Franks et al[62] identified isoflurane-activated a potassium current in specific neurons of the freshwater snail Lymnaea stagnalis. This current had the characteristics of a leak or background K+ channel because it lacked voltage-dependent activation, was non-inactivating and passed currents closely as predicted by the Goldman-Hodgkin-Katz equation for ion conduction through a passive, K+-selective pore. The Lymnea stagnalis IKAn channel, which has biophysical properties very close to the TREK-1 channel, is activated in a range of volatile anesthetic concentrations corresponding to those needed to produce anesthesia in this mollusc[63]. It was therefore important to establish whether the same close relationship between drug efficacy and anesthetic properties would hold true for humans. Most of the experiments have used mouse TREK-1, for which there is abundant biophysical information[54,60]. However, cloned human TREK-1 channel, which like the mouse TREK-1, was also expressed abundantly in brain and had the same biophysical properties and sensitivity to arachidonic acid and polyunsaturated fatty acids. The effect of anesthetics on this channel was examined with exactly the same techniques that were used for the Lymnea channel[63]. At half-maximal concentrations of volatile anesthetics used in human general anesthesia[63] (chloroform, 0.79 mmol, halothane, 0.21 mmol, isoflurane, 0.31 mmol), the human TREK-1 channel was markedly activated. Subsequently, a unique family of K subunits with two pore-lining sequences (K2P channels) was discovered that had a wide phylogenetic range and was activated by volatile anesthetics at clinically relevant concentrations[64-66]. Activation of these background K+ channels in response to volatile anesthetics results in hyperpolarization and silencing of neuronal activity[62,67]. Members of the family can also be activated by xenon[68] and nitrous oxide[69], and differentially activated by isoflurane stereoisomers[70]. C-terminal regions were critical for anesthetic activation in both TASK and TREK channels. Thus both TREK and TASK are possibly important target sites for these agents[64]. Whole-cell patch-clamp experiments showed that chloroform strongly and reversibly activates TREK-1 expression in transfected cells, and this activation was dose dependent, whereas it depressed TASK only slightly and did not affect TRAAK. Chloroform induced a typical TREK-1 background current, characterized by outward rectification that reversed at the predicted value for EK+. Chloroform reversibly and reproducibly hyperpolarized COS cells expressing TREK-1. Both TREK-1 and TASK, but not TRAAK, were opened by halothane. Halothane-induced TASK current had outward rectification and reversed at the predicted value for EK+. The effects of halothane on TASK were rapid and completely reversible. Isoflurane, like halothane, activated both TREK-1 and TASK channels without altering TRAAK conductance. Like chloroform, diethyl ether opened TREK-1 and did not affect TRAAK, whereas it decreased TASK activity.
In excised outside-out patches, activation by volatile anesthetics was not mediated by second-messenger pathways[64]. A 48-pS TREK-1 channel was opened reversibly and in a dose-dependent manner by halothane. No channel activity was observed in the absence of anesthetic, suggesting that halothane converts inactive channels into active ones. The current-voltage (I-V) curve of the chloroform-sensitive current in an outside-out patch showed the outward rectification previously observed in whole-cell recordings. In the inside-out patch configuration, halothane reversibly opened a 12-pS TASK channel. Without anesthetic, a single TASK channel opened and addition of halothane induced the opening of a second channel, which closed again after washout.
Residues in TREK-1 and TASK proteins that are involved in activation by chloroform and halothane were identified using deletions and chimeras. A deletion of the first 42 amino acids in the amino N-terminal region of TREK-1 affected neither anesthetic-induced nor basal channel opening, suggesting that the amino terminus is not important for anesthetic-induced activation. In contrast, deletion of the last 48 amino acids in the C-terminal region of TREK-1 (TR322) completely suppressed responses to both chloroform and halothane, although it did not affect the basal channel activity. Fusing the C-terminal region of TASK to TR324 did not affect basal activity or restore activation by anesthetics. Further deletion of the C-terminal 72 amino acids in TREK-1 completely abolished both basal and anesthetic-stimulated channel activity. Fusing the C-terminal portion of TASK to TR298 restored basal but not anesthetic-stimulated channel activity. These results demonstrate that anesthetic-mediated TREK-1 opening depends critically on the C-terminal 48 amino acids of the channel[64]. Deletion of the last 147 amino acids in the C-terminal region of TASK did not alter halothane sensitivity, whereas further deletion abolished both basal and halothane induced channel activation. When the C-terminal portion of TREK-1 was fused to TASK, basal but not halothane-induced activity was recovered. These results imply that the region of TASK located between residues 242 and 248 confers sensitivity to halothane[64]. Fusion of the last 48 amino acids of TREK-1 to TASK does not confer sensitivity to chloroform. Moreover, fusion of the last 78 residues of TREK-1 to the anesthetic-resistant channel TRAAK provided no sensitivity to halothane or to chloroform, although the chimera had a prominent basal activity. Introducing the C-terminal portion of TREK-1 has been shown to be essential for both chloroform and halothane. This suggests that the C-terminal region is not the only structural element that confers chloroform sensitivity to TREK-1. Inhalational anesthetics have been proposed to act by binding directly to critical sites on target neuronal proteins[71]. The requirement of segments of the protein sequences situated at the C-terminal of both TREK-1 and TASK for their sensitivity to halothane and chloroform is an indication that anesthetics may bind directly to the channels themselves. However, the possibility remains that these anesthetics bind elsewhere on TASK and TREK-1, and that the identified portions of the C-terminal simply transduce these effects.
Evidence from K2P knock-out mice has further implicated these channels in the mechanism of action of volatile anesthetics. TREK-1 knockout mice were resistant to the effects of volatile anesthetics as determined by the standard MAC assay[57]. Knock-out mice in which other members of K2P channel family have been inactivated (TASK-1 and TASK-3) also show some resistance to the anesthetizing action of volatile anesthetics.
Additional studies with multiple K2P knock-outs will be needed to understand their full importance. Kv channels were first isolated from mutant Drosophila that displayed an abnormal ‘shaking’ reaction upon exposure to ether[72]. The Shaker phenotype arose from inactivation of a voltage-gated K+ channel gene, but in vitro studies of the effects of anesthetics on this channel family found them to be inhibited at supra-clinical anesthetic concentrations[73,74]. Other potential K+ channel targets include voltage-gated K+ channels (Kv) and ATP-activated K+ channels (KATP). However, administration of KATP channel blocking drugs into the neuro-axis did not change isoflurane MAC[75]. Thus, the primary focus of the anesthetic mechanism involving K+ channels remains on background K+ channels.
Some potassium channels are known to play beneficial roles in general anesthesia, cardioprotection and neuro-protection. K2P channels are thought to regulate membrane excitability. In CNS, the TREK channels could be activated by membrane stretch, temperature and H+. It has been shown that TREK channels could be activated by some polyunsaturated fatty acids and volatile general anesthetics which lead to neuro-protective effect. According to a recent study with knockout animals, TREK-1 channels might play an important role in the general anesthetic effect of volatile anesthetics such as halothane, providing an explanation for the neuro-protective effect of general anesthetics.
Potassium channels have been thought to regulate potential in the mitochondrial membrane, respiration rhythmic generation and ion homeostasis. For neuro-protective effects, some potassium channels have been identified in the inner mitochondrial membrane: the KATP channel, the BK (Ca2+) channel (large conductance Ca2+ regulated K+ channels), the voltage-gated K+ channel 1.3 (Kv1.3) channel, as well as the TASK-3 channel. It has been demonstrated that potassium influx to the brain mitochondria by the KATP channel or the BK channel could produce neuro-protective effects on neuron survival under ischemia.
GLUTAMATERGIC ION CHANNELS
Structure and function of glutamatergic ion channels
Glutamate transporters, also called excitatory amino acid transporters, bind and take up extracellular glutamate, a major excitatory neurotransmitter, and regulate glutamatergic neurotransmission in synapses. Glutamatergic neurotransmission can be activated by three distinct families of ligand-gated ion channels: AMPA, kainate and NMDA receptors. Among these ligand-gated ion channels, the NMDA receptor is most important and well-established class.
NMDA is an important chemical molecule (ligand) that selectively acts on the glutamate NMDA receptor (NMDAR). It has been widely demonstrated that NMDARs are important for basic brain function and play a critical role in learning and cognition, memory, and the development of central nervous system hyperactive states. Various chemicals belonging to many drug families have been demonstrated to be NMDAR antagonists.
Actions of general anesthetics on glutamatergic ion channels
Anesthesia, although its exact mechanism is still unclear, is thought to be induced by enhancement of inhibitory neurotransmission or inhibition of excitatory neurotransmission. Block of AMPA receptors can decrease MAC of halothane by 60%[76,77]. Some gaseous anesthetics such as cyclopropane, xenon and nitrous oxide, as well as the intravenous anesthetic ketamine have been shown to reduce excitatory or glutamate mediated synaptic transmission by blocking NMDA receptors on the postsynaptic membrane[14-18]. In addition, both urethane and enflurane have been found to inhibit excitatory flow in NMDA-expressing Xenopus Oocytes by acting with NMDA receptors[78,79]. Hollmann et al[80] demonstrated the reversible dose-dependent inhibition of recombinant NMDA receptors by various volatile anesthetics including isoflurane, sevoflurane and desflurane. These in vitro studies indicate a postsynaptic role of glutamate receptors in general anesthesia. Volatile anesthetics may also reduce the excitatory glutamatergic transmission by presynaptic inhibition[81]. However, some studies with knockout mice have failed to find an obvious role of NMDA receptors on anesthetics actions in vivo[82-84].
Although MAC is believed to predominantly reflect nocuous reaction at the spinal cord level, results suggests that pharmacologic blockade of glutamatergic neurotransmission is sufficient to result in deep anesthesia. Further, the effect of combinations of NMDA and AMPA receptor antagonists on halothane MAC is consistent with an in vivo physiological interaction between the NMDA and AMPA receptors[85]. With the use of the NMDA antagonist magnesium sulfate during general anesthesia for shockwave lithotripsy, a magnesium bolus and infusion can be utilized to reduce analgesic requirements[86].
A theory of anesthesia involving NMDA receptors has been presented, consisting of four hypotheses[87]: (1) The formation of transient higher-order, self-referential mental representations contribute to the states of consciousness. As a result, the brain’s representational activity falls below a critical threshold may lead to loss of consciousness; (2) Higher-order mental representations are initiated by neural cell assemblies; (3) the activation of the NMDA receptor channel complex is involved in the formation of such cell assemblies. The activation of this receptor determines the rate at which such assemblies are generated; and (4) Modification of NMDA-dependent processes is the final common pathway of anesthetic effect. Therefore, the agents which directly inactivate the NMDA synapse obviously have anesthetic potential; while the agents that do not directly affect the NMDA synapse will also exert an anesthetic effect if they inhibit NMDA-dependent processes[87]. For example, halothane anesthesia changes the balance between NMDA mediated cholinergic and GABAergic influences on dopamine release and metabolism. Differential sensitivity to halothane of NMDA receptors expressed by the neurons mediating these modulatory influences, or loss of specific NMDA receptor populations through voltage-dependent Mg2+ block under anesthesia, could underlie these observations[88].
The molecular action of xenon and isoflurane in inhibiting NMDA receptors occurs by binding to the glycine co-agonist site[89]. This finding may lead to the design of new anaesthetics, as some clinically well-tolerated neuronal protective compounds are also known to bind to this site.
HCN CHANNELS
Structure and function of HCN channels
HCN gated channels conduct HCN current (If or Ih), that contributes to multiple membrane properties governing cellular excitability[90-92]. Since its first description in 1979[93], extensive work on the If current has amply demonstrated its role in the generation and neurotransmitter-induced modulation of pacemaker activity in the heart[94] (Figure 3).
Figure 3.
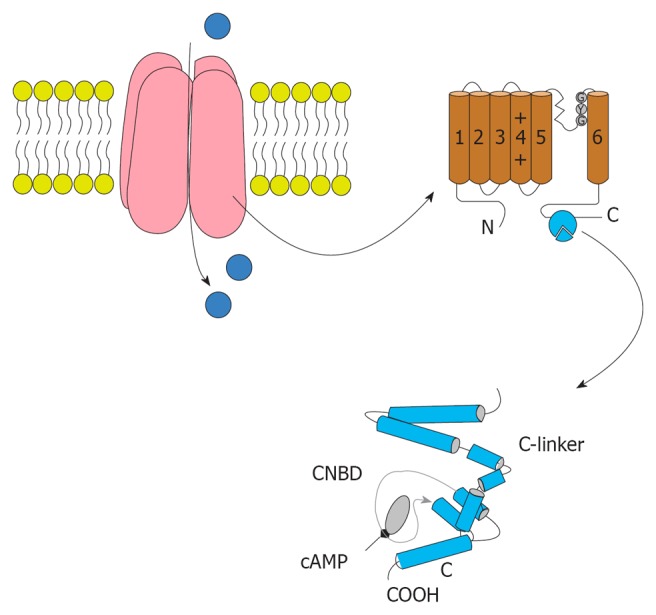
The structure of Hyperpolarization activated cyclic nucleotide channels. Hyperpolarization activated cyclic nucleotide (HCN) channels are made of four subunits. Each subunit contained six trans-membrane segments and S4 acts as the voltage sensor. The pore and filter for ion selection is between S5 and S6. The C-terminal of the HCN channel domain includes the cyclic nucleotide-binding domain (bottom). The domain of the C-linker consists of six a-helices.
HCN currents are encoded by the four member hyperpolarization activated, cyclic nucleotide-regulated gene family (HCN1-4) with a single channel being composed of a homomeric or heteromeric assembly of four HCN subunits[92]. Cloning of four isoforms of HCN channels in the late 1990s showed their correlation to native HCN channels. HCN channels are unevenly distributed on the cell membrane; for example, HCN1 is preferentially expressed on distal dendritic membranes of pyramidal cells in the cortex and hippocampus. Comparison of the properties of native pacemaker channels with those of HCN channels has provided information concerning the composition and molecular features of native channels in different cardiac regions. In addition, HCN channels conduct a cationic current If that contributes to auto-rhythmicity in both the brain and heart. Consistently, dendritic Ih current density and amplitude increases as one moves farther away from the soma[95-98]. Dendritic Ih normalizes temporal summation[95,97,99-100], disconnects somatic and dendritic spike initiation zones[97], and probably limits the development of long-term potential[101]. For example, dendritic expression of HCN1 normalizes somatic voltage responses and spike output in hippocampal and cortical neurons. It was reported previously that HCN2 is predominantly expressed in dendritic spines in reticular thalamic nucleus (RTN) neurons, but the functional impact of HCN2 expression remains unknown.
HCN2 and HCN4 are the two major isoforms present in the thalamic RTN[102], but the relative contribution of the two isoforms to Ih in RTN neurons is unknown. Somatic Ih in RTN neurons is small even at hyperpolarized membrane potentials[103,104]. HCN2 is the major isoform generating Ih in RTN because HCN2 deletion abolishes Ih and reproduces the effects of the HCN channel blocker ZD7288 (4-ethylphenylamino-1, 2-dimethyl-6- methylamino- pyrimidinium chloride). Functional expression of HCN2 in RTN constrains intrinsic excitability and ionotropic glutamate receptor-mediated synaptic integration, thereby reducing spike-dependent GABAergic output. Co-localization of HCN2 channels and the AMPA receptor GluR4 subunit is evident in the spines of RTN neurons, thus providing a structural basis for an interaction between intrinsic and synaptic conductance[105].
The relevance of If to pacemaker generation and modulation makes channels a natural target for drugs aiming to control heart rate pharmacologically. Agents which act by selective inhibition of If have been developed to reduce heart rate, and these drugs have a high potential for treatment of diseases where heart rate reduction is beneficial, such as angina and heart failure. Devices which are able to replace electronic pacemakers and are based on the delivery of a cellular source of pacemaker channels to non-pacing tissue (biological pacemakers) are likely to be developed in the near future for use in therapies for diseases of heart rhythm[106].
Actions of general anesthetics on HCN channels
In the central nervous system, the inhibition of HCN channels by general anesthetics has been suggested to contribute to their anesthesia actions. Inhibition of homomeric HCN1 channels is mediated by anesthetic association with the membrane embedded channel core, a domain that is highly conserved between this isoform and the relatively insensitive HCN2 and 4 subunits. Modeling of the equilibrium and kinetic behavior of HCN1 channels in the absence and presence of anesthetic reveals that gating is best described by models wherein closed and open states communicate by a voltage-independent reaction with no significant equilibrium occupancy of a deactivated open state at non-permissive voltages. Propofol modifies gating by preferentially associating with closed-resting and closed-activated states but a low affinity interaction with the activated open state shapes the effect of the drug under physiological conditions. The mechanism of HCN channel gating provides a framework that will facilitate development of propofol derivates that have altered pharmacological properties and therapeutic potentials.
Activation of native If pacemaker channels and channels formed on heterologous expression of some isoforms of their pore forming HCN subunits, is inhibited by the intravenous general anesthetic propofol (2, 6-diisopropylphenol). Decoupling of HCN channel gating from cAMP and internal protons reveals that changes in these second messengers are neither necessary nor sufficient to account for the actions of propofol. Thus, propofol slows and hyperpolarizes activation of HCN1 channels but it has only weak or no effect on HCN2 and HCN4[107-109] whereas halothane hyperpolarizes HCN1 but suppresses that maximal current carried by HCN2 channels[110,111]. The voltage dependence of If activation is regulated by cAMP[92], internal protons (H+)[112] and several signaling lipids[113-115]. The molecular basis by which lipid messengers alter channel function have not been established. Interestingly, in the case of halothane, HCN isoform selectivity is dependent on the activation status of the cAMP gating ring such that the responses of HCN1 and HCN2 channels are essentially identical when cAMP levels are high or the inhibitory effects of the gating ring are eliminated by deletion[116]. Studies on the effects of propofol on recombinant HCN1, HCN2, and HCN4 channels found that the drug inhibits and slows activation of all three channels at clinically relevant concentrations. In Oocytes expression studies, HCN1 channel activation was most sensitive to slowing by propofol. HCN1 channels also showed a marked hyperpolarizing shift, induced by propofol, in the voltage dependence of activation and accelerated deactivation. Furthermore, propofol reduced heart rate in an isolated guinea pig heart preparation over the same range of concentrations. These data suggest that propofol modulation of HCN channel gating is an important molecular mechanism that can contribute to the depression of central nervous system function and also lead to bradyarrhythmias in patients receiving propofol during surgical anesthesia.
Conventional HCN1 knockout mice were used to test directly the contributions of specific HCN subunits to the effects of isoflurane, an inhalational anesthetic, on membrane and integrative properties of motor and cortical pyramidal neurons in vitro. Compared with wild-type mice, residual If from knockout animals was smaller in amplitude and presented with HCN2-like properties. Isoflurane increased temporal summation of excitatory postsynaptic potentials (EPSPs) in cortical neurons from wild-type mice, an effect predicted by simulation of anesthetic-induced dendritic If inhibition. Accordingly, anesthetic-induced EPSP summation was not observed in cortical cells from HCN1 knockout mice. In wild-type mice, the enhanced synaptic summation observed with low concentrations of isoflurane contributed to a net increase in cortical neuron excitability. HCN channel subunits have been shown to account for distinct anesthetic effects on neuronal membrane properties and synaptic integration. Inhibition of HCN1 by anesthetics in cortical neurons has been shown to contribute to the synaptically-mediated slow-wave cortical synchronization that accompanies anesthetic-induced hypnosis[111].
Na+ CHANNELS AND GENERAL ANESTHESIA
Structure and function of Na+ channels
Voltage-gated Na+ channels have received short shrift as possible anesthetic targets, mainly because early reports failed to demonstrate their significant effects in myelinated axons. However, recently a variety of evidence supports a role for sodium channels in general anesthesia.
The sodium channel family has nine homologous pore-forming α-subunits and these subunits show distinct cellular and sub-cellular distribution, depending on different species and tissues[117]. The pore forming component of sodium channels is a 260 kDa glycoprotein a-subunit, with large intracellular N- and C-terminal. Four internally homologous repeated domains are contained in this subunit (I-IV) and over 50% of the sequence of these domains has been identified. It has been demonstrated that six segments (S1-S6) are contained in each domain and that they form transmembrane α-helices. In addition, four integral membrane glycoprotein subunits have been identified. Generally, the α-subunit is sufficient for the basic functions of sodium channels while expression of β-subunits regulates inactivation and shifts voltage dependence in the direction of more negative potentials. Their modular structures allow interactions between multiple regions of the channel to regulate gating, rapid channel opening and closure.
Functional domains of sodium channels have been identified by many potent toxins[118]. For example, the dinoflagellate toxin (saxitoxin) and the puffer fish poison (tetrodotoxin, TTX) bind to the α-subunit of sodium channel on an extracellular site. For TTX-sensitive sodium channels, Na+ permeability is strongly blocked by these toxins with high potency. In contrast, for TTX-insensitive sodium channels, it is evident that the affinity of TTX to these channels is 200-fold lower. Some lipid soluble steroids, such as veratridine and the frog skin toxin (batrachotoxin) as well as the plant alkaloids (aconitine), bind to the α-subunit of sodium channels on another extracellular site. With a high affinity for the open state of sodium channels, these steroids slow inactivation of sodium channels, resulting in an agonist effect to the ion channels.
Actions of general anesthetics on sodium channels
Voltage-gated sodium channels are regulated by the membrane potential and lead to the passive flux of Na+ into or out of the cell. In most excitable cells and tissues such as nerve, muscle and heart, voltage-gated sodium channels account for the rapid depolarization of action potential[117]. The pharmacological profile as well as ion selectivity of the sodium channel has been explained by a dynamic model of receptor gating. As described by modulated receptor gating, a variety of drugs, such as local anesthetics, class I anti-arrhythmic drugs, and class I anti-epileptic drugs, have shown voltage-dependent and frequency-dependent block of sodium channels. According to this model, these properties are conferred by different drug affinities for the various functional states of the channel (resting, open, inactivated). The evidence that mammalian voltage-gated sodium channels are sensitive to general anesthetics at clinically relevant concentrations comes from careful analysis of anesthetic effects on heterologously expressed sodium channels. It has been demonstrated that one neuronal isoform (Nav1.2) is inhibited by various potent volatile anesthetics by a voltage-independent block of peak current and a hyperpolarizing shift in the steady-state inactivation[119]. In addition, many volatile anesthetics, especially isoflurane, have been demonstrated to inhibit multiple mammalian sodium channel isoforms[120] including Nav1.2[119], Nav1.4 and Nav1.6[121,122], Nav1.5[123], and Nav1.8. Although early studies suggested that the peripheral tetrodotoxin-resistant isoform Nav1.8, expressed in amphibian Oocytes, was resistant to inhaled anesthetics[124], more focused reports in neurons indicate that Nav1.8 is significantly inhibited by isoflurane at concentrations similar to those that inhibit most other isoforms[125]. Potent volatile anesthetics also inhibit native Na+ channels in isolated nerve terminals[122,126] as well as dorsal root ganglion neurons[127]. In contrast, xenon has been found to have no obvious effect on Na+, Ca2+, or K+ channels in isolated cardiomyocytes[128]. However, recent studies suggest that xenon can in fact block neuronal sodium channels at clinically relevant concentrations.
Generally, two principal mechanisms contribute to the inhibition of Na+ channel by volatile anesthetics. These are voltage-independent block of peak currents and enhanced inactivation due to a hyperpolarizing shift in the voltage dependence of steady-state fast inactivation. There are significant differences between isoforms in the contributions of each mechanism to overall inhibition[120,127]. Volatile anesthetics, but not non-immobilizers, also inhibit native neuronal and nerve terminal Na+ channels, supporting the notion that depression of synaptic neurotransmitter release occurs by Na+ channel blocking[120,127]. A recent study demonstrated that NaChBac, a prokaryotic homologue of voltage-gated Na+ channels, is also inhibited by volatile anesthetics[129]. Anesthetic interactions with NaChBac might ultimately allow co-crystallization with anesthetic for three-dimensional structure determinations by X-ray crystallography, as achieved for voltage-gated K+ channels, to determine the site of interaction of anesthetics with a voltage-gated ion channel. It is also intriguing that the binding sites for anesthetics on ion channels exist in prokaryotic homologues, indicating a remarkable evolutionary conservation.
Voltage-gated Na+ channels have been demonstrated to be insensitive to general anesthetics in early studies on myelinated axons. However, smaller diameter unmyelinated fibers and nerve terminals are found to be sensitive to Na+ channel block and do not possess the considerable reserve of conduction seen in myelinated nerves. Many studies summarized earlier demonstrate that inhaled anesthetics partially impair Na+ channel function at MAC (minimum alveolar concentration). Moreover, a variety of evidence supports a role for sodium channels in general anesthesia in vivo, for example the increase in cerebrospinal fluid Na+ concentration increases MAC of halothane (equivalent to ED50) in rats[130]. Intravenous administration of the Na+ channel blocker lidocaine reduces MAC for several volatile anesthetics in rats[131], and intravenous or intrathecal infusions of riluzole, a potent inhibitor of Na+ channels and glutamate release, decrease isoflurane MAC in rats[132]. Finally, intrathecal but not intraventricular administration of veratridine, a toxin that maintains Na+ channels in their open state, increases the MAC for isoflurane in rats. Collectively, these results point to anesthetic inhibition of Na+ channels as a plausible mechanism for the mediation of immobility produced by inhaled anesthetics.
Footnotes
Peer reviewers: Dr. Ayman A Yousef, Department of Anesthesia, Faculty of Medicine, 17 elemam moslem street, Tanta 35217, Egypt; Dr. John F Stover, Professor, Intensive Care Medicine, University Hospital Zürich, 8091 Zürich, Switzerland
S- Editor Gou SX L- Editor Hughes D E- Editor Zheng XM
References
- 1.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 2.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain. Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 3.Bloom FE, Iversen LL. Localizing 3H-GABA in nerve terminals of rat cerebral cortex by electron microscopic autoradiography. Nature. 1971;229:628–630. doi: 10.1038/229628a0. [DOI] [PubMed] [Google Scholar]
- 4.Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P. Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature. 1995;378:75–78. doi: 10.1038/378075a0. [DOI] [PubMed] [Google Scholar]
- 5.Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- 6.Krogsgaard-Larsen P, Frølund B, Liljefors T, Ebert B. GABA(A) agonists and partial agonists: THIP (Gaboxadol) as a non-opioid analgesic and a novel type of hypnotic. Biochem Pharmacol. 2004;68:1573–1580. doi: 10.1016/j.bcp.2004.06.040. [DOI] [PubMed] [Google Scholar]
- 7.Brown N, Kerby J, Bonnert TP, Whiting PJ, Wafford KA. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br J Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra D, Jia F, Liang J, Peng Z, Suryanarayanan A, Werner DF, Spigelman I, Houser CR, Olsen RW, Harrison NL, et al. GABAA receptor alpha 4 subunits mediate extrasynaptic inhibition in thalamus and dentate gyrus and the action of gaboxadol. Proc Natl Acad Sci USA. 2006;103:15230–15235. doi: 10.1073/pnas.0604304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ, Myers J, Sur C, Howell O, Rutter AR, et al. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J Neurosci. 2003;23:8608–8617. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, Zaugg M, Vogt KE, Ledermann B, Antkowiak B, et al. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- 11.Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL. General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J Pharmacol Exp Ther. 2001;297:338–351. [PubMed] [Google Scholar]
- 12.Tomlin SL, Jenkins A, Lieb WR, Franks NP. Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology. 1998;88:708–717. doi: 10.1097/00000542-199803000-00022. [DOI] [PubMed] [Google Scholar]
- 13.Hales TG, Lambert JJ. The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol. 1991;104:619–628. doi: 10.1111/j.1476-5381.1991.tb12479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franks NP, Dickinson R, de Sousa SL, Hall AC, Lieb WR. How does xenon produce anaesthesia. Nature. 1998;396:324. doi: 10.1038/24525. [DOI] [PubMed] [Google Scholar]
- 15.Jevtović-Todorović V, Todorović SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat Med. 1998;4:460–463. doi: 10.1038/nm0498-460. [DOI] [PubMed] [Google Scholar]
- 16.Raines DE, Claycomb RJ, Scheller M, Forman SA. Nonhalogenated alkane anesthetics fail to potentiate agonist actions on two ligand-gated ion channels. Anesthesiology. 2001;95:470–477. doi: 10.1097/00000542-200108000-00032. [DOI] [PubMed] [Google Scholar]
- 17.Zeilhofer HU, Swandulla D, Geisslinger G, Brune K. Differential effects of ketamine enantiomers on NMDA receptor currents in cultured neurons. Eur J Pharmacol. 1992;213:155–158. doi: 10.1016/0014-2999(92)90248-3. [DOI] [PubMed] [Google Scholar]
- 18.Flood P, Krasowski MD. Intravenous anesthetics differentially modulate ligand-gated ion channels. Anesthesiology. 2000;92:1418–1425. doi: 10.1097/00000542-200005000-00033. [DOI] [PubMed] [Google Scholar]
- 19.Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 20.Harrison NL, Kugler JL, Jones MV, Greenblatt EP, Pritchett DB. Positive modulation of human gamma-aminobutyric acid type A and glycine receptors by the inhalation anesthetic isoflurane. Mol Pharmacol. 1993;44:628–632. [PubMed] [Google Scholar]
- 21.Krasowski MD, Harrison NL. General anaesthetic actions on ligand-gated ion channels. Cell Mol Life Sci. 1999;55:1278–1303. doi: 10.1007/s000180050371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonner JM, Antognini JF, Dutton RC, Flood P, Gray AT, Harris RA, Homanics GE, Kendig J, Orser B, Raines DE, et al. Inhaled anesthetics and immobility: mechanisms, mysteries, and minimum alveolar anesthetic concentration. Anesth Analg. 2003;97:718–740. doi: 10.1213/01.ANE.0000081063.76651.33. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- 25.Hentschke H, Schwarz C, Antkowiak B. Neocortex is the major target of sedative concentrations of volatile anaesthetics: strong depression of firing rates and increase of GABAA receptor-mediated inhibition. Eur J Neurosci. 2005;21:93–102. doi: 10.1111/j.1460-9568.2004.03843.x. [DOI] [PubMed] [Google Scholar]
- 26.Koltchine VV, Finn SE, Jenkins A, Nikolaeva N, Lin A, Harrison NL. Agonist gating and isoflurane potentiation in the human gamma-aminobutyric acid type A receptor determined by the volume of a second transmembrane domain residue. Mol Pharmacol. 1999;56:1087–1093. doi: 10.1124/mol.56.5.1087. [DOI] [PubMed] [Google Scholar]
- 27.Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, Mascia MP, Valenzuela CF, Hanson KK, Greenblatt EP, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- 28.Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Möhler H. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 29.Borghese CM, Werner DF, Topf N, Baron NV, Henderson LA, Boehm SL, Blednov YA, Saad A, Dai S, Pearce RA, et al. An isoflurane- and alcohol-insensitive mutant GABA(A) receptor alpha(1) subunit with near-normal apparent affinity for GABA: characterization in heterologous systems and production of knockin mice. J Pharmacol Exp Ther. 2006;319:208–218. doi: 10.1124/jpet.106.104406. [DOI] [PubMed] [Google Scholar]
- 30.Barnard EA, Skolnick P, Olsen RW, Mohler H, Sieghart W, Biggio G, Braestrup C, Bateson AN, Langer SZ. International Union of Pharmacology. XV. Subtypes of gamma-aminobutyric acidA receptors: classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 31.Nishikawa K, Jenkins A, Paraskevakis I, Harrison NL. Volatile anesthetic actions on the GABAA receptors: contrasting effects of alpha 1(S270) and beta 2(N265) point mutations. Neuropharmacology. 2002;42:337–345. doi: 10.1016/s0028-3908(01)00189-7. [DOI] [PubMed] [Google Scholar]
- 32.Hall AC, Rowan KC, Stevens RJ, Kelley JC, Harrison NL. The effects of isoflurane on desensitized wild-type and alpha 1(S270H) gamma-aminobutyric acid type A receptors. Anesth Analg. 2004;98:1297–1304, table of contents. doi: 10.1213/01.ane.0000111108.78745.ad. [DOI] [PubMed] [Google Scholar]
- 33.Sonner JM, Werner DF, Elsen FP, Xing Y, Liao M, Harris RA, Harrison NL, Fanselow MS, Eger EI, Homanics GE. Effect of isoflurane and other potent inhaled anesthetics on minimum alveolar concentration, learning, and the righting reflex in mice engineered to express alpha1 gamma-aminobutyric acid type A receptors unresponsive to isoflurane. Anesthesiology. 2007;106:107–113. doi: 10.1097/00000542-200701000-00019. [DOI] [PubMed] [Google Scholar]
- 34.Sonner JM, Cascio M, Xing Y, Fanselow MS, Kralic JE, Morrow AL, Korpi ER, Hardy S, Sloat B, Eger EI, et al. Alpha 1 subunit-containing GABA type A receptors in forebrain contribute to the effect of inhaled anesthetics on conditioned fear. Mol Pharmacol. 2005;68:61–68. doi: 10.1124/mol.104.009936. [DOI] [PubMed] [Google Scholar]
- 35.Siegwart R, Jurd R, Rudolph U. Molecular determinants for the action of general anesthetics at recombinant alpha(2)beta(3)gamma(2)gamma-aminobutyric acid(A) receptors. J Neurochem. 2002;80:140–148. doi: 10.1046/j.0022-3042.2001.00682.x. [DOI] [PubMed] [Google Scholar]
- 36.Lambert S, Arras M, Vogt KE, Rudolph U. Isoflurane-induced surgical tolerance mediated only in part by beta3-containing GABA(A) receptors. Eur J Pharmacol. 2005;516:23–27. doi: 10.1016/j.ejphar.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 37.Zeller A, Arras M, Jurd R, Rudolph U. Mapping the contribution of beta3-containing GABAA receptors to volatile and intravenous general anesthetic actions. BMC Pharmacol. 2007;7:2. doi: 10.1186/1471-2210-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orser BA. Lifting the fog around anesthesia. Sci Am. 2007;296:54–61. doi: 10.1038/scientificamerican0607-54. [DOI] [PubMed] [Google Scholar]
- 39.Patel AJ, Honoré E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci. 2001;24:339–346. doi: 10.1016/s0166-2236(00)01810-5. [DOI] [PubMed] [Google Scholar]
- 40.Lesage F, Lazdunski M. Molecular and functional properties of two-pore-domain potassium channels. Am J Physiol Renal Physiol. 2000;279:F793–F801. doi: 10.1152/ajprenal.2000.279.5.F793. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci. 2001;2:175–184. doi: 10.1038/35058574. [DOI] [PubMed] [Google Scholar]
- 42.Talley EM, Sirois JE, Lei Q, Bayliss DA. Two-pore-Domain (KCNK) potassium channels: dynamic roles in neuronal function. Neuroscientist. 2003;9:46–56. doi: 10.1177/1073858402239590. [DOI] [PubMed] [Google Scholar]
- 43.Kim D. Fatty acid-sensitive two-pore domain K+ channels. Trends Pharmacol Sci. 2003;24:648–654. doi: 10.1016/j.tips.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Castellucci V, Kandel ER. Presynaptic facilitation as a mechanism for behavioral sensitization in Aplysia. Science. 1976;194:1176–1178. doi: 10.1126/science.11560. [DOI] [PubMed] [Google Scholar]
- 45.Franks NP, Lieb WR. Volatile general anaesthetics activate a novel neuronal K+ current. Nature. 1988;333:662–664. doi: 10.1038/333662a0. [DOI] [PubMed] [Google Scholar]
- 46.O'Connell AD, Morton MJ, Hunter M. Two-pore domain K+ channels-molecular sensors. Biochim Biophys Acta. 2002;1566:152–161. doi: 10.1016/s0005-2736(02)00597-7. [DOI] [PubMed] [Google Scholar]
- 47.Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA. Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem. 2000;275:16969–16978. doi: 10.1074/jbc.M001948200. [DOI] [PubMed] [Google Scholar]
- 48.Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J. 2005;24:44–53. doi: 10.1038/sj.emboj.7600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bockenhauer D, Zilberberg N, Goldstein SA. KCNK2: reversible conversion of a hippocampal potassium leak into a voltage-dependent channel. Nat Neurosci. 2001;4:486–491. doi: 10.1038/87434. [DOI] [PubMed] [Google Scholar]
- 50.Maingret F, Honoré E, Lazdunski M, Patel AJ. Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K(+) channel. Biochem Biophys Res Commun. 2002;292:339–346. doi: 10.1006/bbrc.2002.6674. [DOI] [PubMed] [Google Scholar]
- 51.Xian Tao Li V, Zuzarte M, Putzke C, Preisig-Müller R, Isenberg G, Daut J. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res. 2006;69:86–97. doi: 10.1016/j.cardiores.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 52.Patel AJ, Honoré E. Anesthetic-sensitive 2P domain K+ channels. Anesthesiology. 2001;95:1013–1021. doi: 10.1097/00000542-200110000-00034. [DOI] [PubMed] [Google Scholar]
- 53.Honoré E. The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci. 2007;8:251–261. doi: 10.1038/nrn2117. [DOI] [PubMed] [Google Scholar]
- 54.Patel AJ, Honoré E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283–4290. doi: 10.1093/emboj/17.15.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thümmler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, et al. Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci. 2006;9:1134–1141. doi: 10.1038/nn1749. [DOI] [PubMed] [Google Scholar]
- 56.Alloui A, Zimmermann K, Mamet J, Duprat F, Noël J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, et al. TREK-1, a K+ channel involved in polymodal pain perception. EMBO J. 2006;25:2368–2376. doi: 10.1038/sj.emboj.7601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J. 2004;23:2684–2695. doi: 10.1038/sj.emboj.7600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hervieu GJ, Cluderay JE, Gray CW, Green PJ, Ranson JL, Randall AD, Meadows HJ. Distribution and expression of TREK-1, a two-pore-domain potassium channel, in the adult rat CNS. Neuroscience. 2001;103:899–919. doi: 10.1016/s0306-4522(01)00030-6. [DOI] [PubMed] [Google Scholar]
- 59.Medhurst AD, Rennie G, Chapman CG, Meadows H, Duckworth MD, Kelsell RE, Gloger II, Pangalos MN. Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res. 2001;86:101–114. doi: 10.1016/s0169-328x(00)00263-1. [DOI] [PubMed] [Google Scholar]
- 60.Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C, Lazdunski M. Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J. 1996;15:6854–6862. [PMC free article] [PubMed] [Google Scholar]
- 61.Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. Cns distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci. 2001;21:7491–7505. doi: 10.1523/JNEUROSCI.21-19-07491.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franks NP, Lieb WR. Stereospecific effects of inhalational general anesthetic optical isomers on nerve ion channels. Science. 1991;254:427–430. doi: 10.1126/science.1925602. [DOI] [PubMed] [Google Scholar]
- 63.Lopes CM, Franks NP, Lieb WR. Actions of general anaesthetics and arachidonic pathway inhibitors on K+ currents activated by volatile anaesthetics and FMRFamide in molluscan neurones. Br J Pharmacol. 1998;125:309–318. doi: 10.1038/sj.bjp.0702069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patel AJ, Honoré E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci. 1999;2:422–426. doi: 10.1038/8084. [DOI] [PubMed] [Google Scholar]
- 65.Gray AT, Winegar BD, Leonoudakis DJ, Forsayeth JR, Yost CS. TOK1 is a volatile anesthetic stimulated K+ channel. Anesthesiology. 1998;88:1076–1084. doi: 10.1097/00000542-199804000-00029. [DOI] [PubMed] [Google Scholar]
- 66.Gray AT, Zhao BB, Kindler CH, Winegar BD, Mazurek MJ, Xu J, Chavez RA, Forsayeth JR, Yost CS. Volatile anesthetics activate the human tandem pore domain baseline K+ channel KCNK5. Anesthesiology. 2000;92:1722–1730. doi: 10.1097/00000542-200006000-00032. [DOI] [PubMed] [Google Scholar]
- 67.Winegar BD, Owen DF, Yost CS, Forsayeth JR, Mayeri E. Volatile general anesthetics produce hyperpolarization of Aplysia neurons by activation of a discrete population of baseline potassium channels. Anesthesiology. 1996;85:889–900. doi: 10.1097/00000542-199610000-00026. [DOI] [PubMed] [Google Scholar]
- 68.Gruss M, Bushell TJ, Bright DP, Lieb WR, Mathie A, Franks NP. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol Pharmacol. 2004;65:443–452. doi: 10.1124/mol.65.2.443. [DOI] [PubMed] [Google Scholar]
- 69.Mathie A, Veale EL. Therapeutic potential of neuronal two-pore domain potassium-channel modulators. Curr Opin Investig Drugs. 2007;8:555–562. [PubMed] [Google Scholar]
- 70.Liu C, Au JD, Zou HL, Cotten JF, Yost CS. Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth Analg. 2004;99:1715–122, table of contents. doi: 10.1213/01.ANE.0000136849.07384.44. [DOI] [PubMed] [Google Scholar]
- 71.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 72.Papazian DM, Schwarz TL, Tempel BL, Jan YN, Jan LY. Cloning of genomic and complementary DNA from Shaker, a putative potassium channel gene from Drosophila. Science. 1987;237:749–753. doi: 10.1126/science.2441470. [DOI] [PubMed] [Google Scholar]
- 73.Haydon DA, Urban BW. The actions of some general anaesthetics on the potassium current of the squid giant axon. J Physiol. 1986;373:311–327. doi: 10.1113/jphysiol.1986.sp016049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Friederich P, Urban BW. Interaction of intravenous anesthetics with human neuronal potassium currents in relation to clinical concentrations. Anesthesiology. 1999;91:1853–1860. doi: 10.1097/00000542-199912000-00040. [DOI] [PubMed] [Google Scholar]
- 75.Zucker JR. ATP-sensitive potassium channel agonists do not alter MAC for isoflurane in rats. Anesthesiology. 1992;76:560–563. doi: 10.1097/00000542-199204000-00012. [DOI] [PubMed] [Google Scholar]
- 76.McFarlane C, Warner DS, Todd MM, Nordholm L. AMPA receptor competitive antagonism reduces halothane MAC in rats. Anesthesiology. 1992;77:1165–1170. doi: 10.1097/00000542-199212000-00018. [DOI] [PubMed] [Google Scholar]
- 77.Minami K, Wick MJ, Stern-Bach Y, Dildy-Mayfield JE, Brozowski SJ, Gonzales EL, Trudell JR, Harris RA. Sites of volatile anesthetic action on kainate (Glutamate receptor 6) receptors. J Biol Chem. 1998;273:8248–8255. doi: 10.1074/jbc.273.14.8248. [DOI] [PubMed] [Google Scholar]
- 78.Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–38, table of contents. doi: 10.1097/00000539-200202000-00015. [DOI] [PubMed] [Google Scholar]
- 79.Lin LH, Chen LL, Harris RA. Enflurane inhibits NMDA, AMPA, and kainate-induced currents in Xenopus oocytes expressing mouse and human brain mRNA. FASEB J. 1993;7:479–485. doi: 10.1096/fasebj.7.5.7681790. [DOI] [PubMed] [Google Scholar]
- 80.Hollmann MW, Liu HT, Hoenemann CW, Liu WH, Durieux ME. Modulation of NMDA receptor function by ketamine and magnesium. Part II: interactions with volatile anesthetics. Anesth Analg. 2001;92:1182–1191. doi: 10.1097/00000539-200105000-00020. [DOI] [PubMed] [Google Scholar]
- 81.Maclver MB, Mikulec AA, Amagasu SM, Monroe FA. Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology. 1996;85:823–834. doi: 10.1097/00000542-199610000-00018. [DOI] [PubMed] [Google Scholar]
- 82.Joo DT, Gong D, Sonner JM, Jia Z, MacDonald JF, Eger EI, Orser BA. Blockade of AMPA receptors and volatile anesthetics: reduced anesthetic requirements in GluR2 null mutant mice for loss of the righting reflex and antinociception but not minimum alveolar concentration. Anesthesiology. 2001;94:478–488. doi: 10.1097/00000542-200103000-00020. [DOI] [PubMed] [Google Scholar]
- 83.Sonner JM, Vissel B, Royle G, Maurer A, Gong D, Baron NV, Harrison N, Fanselow M, Eger EI. The effect of three inhaled anesthetics in mice harboring mutations in the GluR6 (kainate) receptor gene. Anesth Analg. 2005;101:143–18, table of contents. doi: 10.1213/01.ANE.0000152615.53435.B4. [DOI] [PubMed] [Google Scholar]
- 84.Sato Y, Kobayashi E, Murayama T, Mishina M, Seo N. Effect of N-methyl-D-aspartate receptor epsilon1 subunit gene disruption of the action of general anesthetic drugs in mice. Anesthesiology. 2005;102:557–561. doi: 10.1097/00000542-200503000-00013. [DOI] [PubMed] [Google Scholar]
- 85.McFarlane C, Warner DS, Dexter F. Interactions between NMDA and AMPA glutamate receptor antagonists during halothane anesthesia in the rat. Neuropharmacology. 1995;34:659–663. doi: 10.1016/0028-3908(95)00029-6. [DOI] [PubMed] [Google Scholar]
- 86.Kaymak C, Yilmaz E, Basar H, Ozcakir S, Apan A, Batislam E. Use of the NMDA antagonist magnesium sulfate during monitored anesthesia care for shockwave lithotripsy. J Endourol. 2007;21:145–150. doi: 10.1089/end.2006.0195. [DOI] [PubMed] [Google Scholar]
- 87.Flohr H, Glade U, Motzko D. The role of the NMDA synapse in general anesthesia. Toxicol Lett. 1998;100-101:23–29. doi: 10.1016/s0378-4274(98)00161-1. [DOI] [PubMed] [Google Scholar]
- 88.Whitehead KJ, Rose S, Jenner P. Halothane anesthesia affects NMDA-stimulated cholinergic and GABAergic modulation of striatal dopamine efflux and metabolism in the rat in vivo. Neurochem Res. 2004;29:835–842. doi: 10.1023/b:nere.0000018858.64265.e9. [DOI] [PubMed] [Google Scholar]
- 89.Dickinson R, Peterson BK, Banks P, Simillis C, Martin JC, Valenzuela CA, Maze M, Franks NP. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor by the anesthetics xenon and isoflurane: evidence from molecular modeling and electrophysiology. Anesthesiology. 2007;107:756–767. doi: 10.1097/01.anes.0000287061.77674.71. [DOI] [PubMed] [Google Scholar]
- 90.Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- 91.Gauss R, Seifert R. Pacemaker oscillations in heart and brain: a key role for hyperpolarization-activated cation channels. Chronobiol Int. 2000;17:453–469. doi: 10.1081/cbi-100101057. [DOI] [PubMed] [Google Scholar]
- 92.Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- 93.Brown HF, DiFrancesco D, Noble SJ. How does adrenaline accelerate the heart. Nature. 1979;280:235–236. doi: 10.1038/280235a0. [DOI] [PubMed] [Google Scholar]
- 94.DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annu Rev Physiol. 1993;55:455–472. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- 95.Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:848. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- 96.Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic I(h) in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- 97.Berger T, Larkum ME, Lüscher HR. High I(h) channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- 98.Kole MH, Hallermann S, Stuart GJ. Single Ih channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. J Neurosci. 2006;26:1677–1687. doi: 10.1523/JNEUROSCI.3664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart G, Spruston N. Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci. 1998;18:3501–3510. doi: 10.1523/JNEUROSCI.18-10-03501.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koch U, Grothe B. Hyperpolarization-activated current (Ih) in the inferior colliculus: distribution and contribution to temporal processing. J Neurophysiol. 2003;90:3679–3687. doi: 10.1152/jn.00375.2003. [DOI] [PubMed] [Google Scholar]
- 101.Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 102.Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- 103.Abbas SY, Ying SW, Goldstein PA. Compartmental distribution of hyperpolarization-activated cyclic-nucleotide-gated channel 2 and hyperpolarization-activated cyclic-nucleotide-gated channel 4 in thalamic reticular and thalamocortical relay neurons. Neuroscience. 2006;141:1811–1825. doi: 10.1016/j.neuroscience.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 104.Rateau Y, Ropert N. Expression of a functional hyperpolarization-activated current (Ih) in the mouse nucleus reticularis thalami. J Neurophysiol. 2006;95:3073–3085. doi: 10.1152/jn.00922.2005. [DOI] [PubMed] [Google Scholar]
- 105.Ying SW, Jia F, Abbas SY, Hofmann F, Ludwig A, Goldstein PA. Dendritic HCN2 channels constrain glutamate-driven excitability in reticular thalamic neurons. J Neurosci. 2007;27:8719–8732. doi: 10.1523/JNEUROSCI.1630-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.DiFrancesco D. Serious workings of the funny current. Prog Biophys Mol Biol. 2006;90:13–25. doi: 10.1016/j.pbiomolbio.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 107.Cacheaux LP, Topf N, Tibbs GR, Schaefer UR, Levi R, Harrison NL, Abbott GW, Goldstein PA. Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J Pharmacol Exp Ther. 2005;315:517–525. doi: 10.1124/jpet.105.091801. [DOI] [PubMed] [Google Scholar]
- 108.Chen X, Shu S, Bayliss DA. Suppression of ih contributes to propofol-induced inhibition of mouse cortical pyramidal neurons. J Neurophysiol. 2005;94:3872–3883. doi: 10.1152/jn.00389.2005. [DOI] [PubMed] [Google Scholar]
- 109.Ying SW, Abbas SY, Harrison NL, Goldstein PA. Propofol block of I(h) contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur J Neurosci. 2006;23:465–480. doi: 10.1111/j.1460-9568.2005.04587.x. [DOI] [PubMed] [Google Scholar]
- 110.Sirois JE, Pancrazio JJ, III CL, Bayliss DA. Multiple ionic mechanisms mediate inhibition of rat motoneurones by inhalation anaesthetics. J Physiol. 1998;512(Pt 3):851–862. doi: 10.1111/j.1469-7793.1998.851bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen X, Shu S, Kennedy DP, Willcox SC, Bayliss DA. Subunit-specific effects of isoflurane on neuronal Ih in HCN1 knockout mice. J Neurophysiol. 2009;101:129–140. doi: 10.1152/jn.01352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Munsch T, Pape HC. Modulation of the hyperpolarization-activated cation current of rat thalamic relay neurones by intracellular pH. J Physiol. 1999;519 Pt 2:493–504. doi: 10.1111/j.1469-7793.1999.0493m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pian P, Bucchi A, Robinson RB, Siegelbaum SA. Regulation of gating and rundown of HCN hyperpolarization-activated channels by exogenous and endogenous PIP2. J Gen Physiol. 2006;128:593–604. doi: 10.1085/jgp.200609648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zolles G, Klöcker N, Wenzel D, Weisser-Thomas J, Fleischmann BK, Roeper J, Fakler B. Pacemaking by HCN channels requires interaction with phosphoinositides. Neuron. 2006;52:1027–1036. doi: 10.1016/j.neuron.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 115.Fogle KJ, Lyashchenko AK, Turbendian HK, Tibbs GR. HCN pacemaker channel activation is controlled by acidic lipids downstream of diacylglycerol kinase and phospholipase A2. J Neurosci. 2007;27:2802–2814. doi: 10.1523/JNEUROSCI.4376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen X, Sirois JE, Lei Q, Talley EM, Lynch C, Bayliss DA. HCN subunit-specific and cAMP-modulated effects of anesthetics on neuronal pacemaker currents. J Neurosci. 2005;25:5803–5814. doi: 10.1523/JNEUROSCI.1153-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- 118.Cestèle S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- 119.Rehberg B, Xiao YH, Duch DS. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84:1223–133; discussion 27A. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- 120.OuYang W, Hemmings HC. Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology. 2007;107:91–98. doi: 10.1097/01.anes.0000268390.28362.4a. [DOI] [PubMed] [Google Scholar]
- 121.Ouyang W, Herold KF, Hemmings HC. Comparative effects of halogenated inhaled anesthetics on voltage-gated Na+ channel function. Anesthesiology. 2009;110:582–590. doi: 10.1097/ALN.0b013e318197941e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ratnakumari L, Hemmings HC. Inhibition of presynaptic sodium channels by halothane. Anesthesiology. 1998;88:1043–1054. doi: 10.1097/00000542-199804000-00025. [DOI] [PubMed] [Google Scholar]
- 123.Stadnicka A, Kwok WM, Hartmann HA, Bosnjak ZJ. Effects of halothane and isoflurane on fast and slow inactivation of human heart hH1a sodium channels. Anesthesiology. 1999;90:1671–1683. doi: 10.1097/00000542-199906000-00024. [DOI] [PubMed] [Google Scholar]
- 124.Shiraishi M, Harris RA. Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J Pharmacol Exp Ther. 2004;309:987–994. doi: 10.1124/jpet.103.064063. [DOI] [PubMed] [Google Scholar]
- 125.Herold KF, Nau C, Ouyang W, Hemmings HC. Isoflurane inhibits the tetrodotoxin-resistant voltage-gated sodium channel Nav1.8. Anesthesiology. 2009;111:591–599. doi: 10.1097/ALN.0b013e3181af64d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ouyang W, Wang G, Hemmings HC. Isoflurane and propofol inhibit voltage-gated sodium channels in isolated rat neurohypophysial nerve terminals. Mol Pharmacol. 2003;64:373–381. doi: 10.1124/mol.64.2.373. [DOI] [PubMed] [Google Scholar]
- 127.Ratnakumari L, Vysotskaya TN, Duch DS, Hemmings HC. Differential effects of anesthetic and nonanesthetic cyclobutanes on neuronal voltage-gated sodium channels. Anesthesiology. 2000;92:529–541. doi: 10.1097/00000542-200002000-00037. [DOI] [PubMed] [Google Scholar]
- 128.Stowe DF, Rehmert GC, Kwok WM, Weigt HU, Georgieff M, Bosnjak ZJ. Xenon does not alter cardiac function or major cation currents in isolated guinea pig hearts or myocytes. Anesthesiology. 2000;92:516–522. doi: 10.1097/00000542-200002000-00035. [DOI] [PubMed] [Google Scholar]
- 129.Ouyang W, Jih TY, Zhang TT, Correa AM, Hemmings HC. Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J Pharmacol Exp Ther. 2007;322:1076–1083. doi: 10.1124/jpet.107.122929. [DOI] [PubMed] [Google Scholar]
- 130.Tanifuji Y, Eger EI. Brain sodium, potassium, and osmolality: effects on anesthetic requirement. Anesth Analg. 1978;57:404–410. doi: 10.1213/00000539-197807000-00007. [DOI] [PubMed] [Google Scholar]
- 131.Zhang Y, Laster MJ, Eger EI, Sharma M, Sonner JM. Lidocaine, MK-801, and MAC. Anesth Analg. 2007;104:1098–102, tables of contents. doi: 10.1213/01.ane.0000260318.60504.a9. [DOI] [PubMed] [Google Scholar]
- 132.Xing Y, Zhang Y, Stabernack CR, Eger EI, Gray AT. The use of the potassium channel activator riluzole to test whether potassium channels mediate the capacity of isoflurane to produce immobility. Anesth Analg. 2003;97:1020–104, table of contents. doi: 10.1213/01.ANE.0000077073.92108.E7. [DOI] [PubMed] [Google Scholar]