

Abstract
There is emerging evidence for reduced muscle function in children with neurofibromatosis type 1 (NF1). We have examined three murine models featuring NF1 deficiency in muscle to study the effect on muscle function as well as any underlying pathophysiology. The Nf1+/– mouse exhibited no differences in overall weight, lean tissue mass, fiber size, muscle weakness as measured by grip strength or muscle atrophy-recovery with limb disuse, although this model lacks many other characteristic features of the human disease. Next, muscle-specific knockout mice (Nf1muscle−/−) were generated and they exhibited a failure to thrive leading to neonatal lethality. Intramyocellular lipid accumulations were observed by electron microscopy and Oil Red O staining. More mature muscle specimens lacking Nf1 expression taken from the limb-specific Nf1Prx1−/− conditional knockout line showed a 10-fold increase in muscle triglyceride content. Enzyme assays revealed a significant increase in the activities of oxidative metabolism enzymes in the Nf1Prx1−/− mice. Western analyses showed increases in the expression of fatty acid synthase and the hormone leptin, as well as decreased expression of a number of fatty acid transporters in this mouse line. These data support the hypothesis that NF1 is essential for normal muscle function and survival and are the first to suggest a direct link between NF1 and mitochondrial fatty acid metabolism.
INTRODUCTION
Neurofibromatosis type 1 (NF1) is a genetic disorder that is associated with a range of features including superficial and deep neurofibromas, developmental delay affecting both cognitive and motor performance and musculoskeletal complications (1). In recent years, the complexity of the musculoskeletal manifestations has been elaborated on by a number of basic, translational and clinical studies (2). Whereas there has been substantive progress in exploring the bony abnormalities, only recently has muscle emerged as a tissue of interest in individuals with NF1.
While scoliosis and congenital dysplasias (particularly tibial dysplasia) have long been part of the standard diagnostic criteria for NF1, individuals with this condition are also prone to low bone mineral density (BMD) (3,4). While low BMD can correlate with orthopedic complications such as scoliosis, population studies have yet to demonstrate a significant correlation with fracture risk (5,6). These bony manifestations have now been mechanistically linked to several underlying causes. Focal bone defects such as congenital tibial dysplasia have been associated with local double inactivation of NF1, and this may be the basis for some cases of dystrophic scoliosis (7). In contrast, low BMD is more likely to result from haploinsufficiency for NF1 leading to increased osteoclast activity (8). Many additional bone dysplasias associated with NF1 (sphenoid wing dysplasia, pectus excavatum, rib penciling) still have an unknown etiology, but may result from deficiencies in developmental patterning.
In 1994, the NF1 gene product neurofibromin was found to be upregulated during myoblast differentiation, suggesting a potential role in muscle (9). Nevertheless, the deficits in motor skills and co-ordination associated with NF1 have been historically attributed to central nervous system dysfunction (10). Lately, however, a number of clinical reports have identified decreases in muscle size (11,12), strength (13,14) and motor proficiency (15), leading to renewed speculation regarding primary muscle involvement. Muscle weakness could also contribute to systemic osteopenia due to decreased loading, and load-carrying regions of the skeleton are reported as the most affected (3,11). Still, the interactions between muscle and bone, particularly as a modifying factor for BMD and fracture risk, remain unclear.
Recent work with the Nf1Prx1−/− mouse (Prx1-cre driven Nf1 deletion in the limbs) has reinforced the importance of NF1 in muscle development (12). In this study, Nf1Prx1−/− mice were studied from the earliest stages of limb bud morphogenesis. These mice display a congenital myopathy characterized by reduced muscle fibers, reduced muscle force and fibrosis. Neurofibromin is a key negative regulator of Ras signaling, and can activate a variety of downstream pathways including c-Jun N-terminal kinase (JNK), phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways. Muscle extracts from adult Nf1Prx1−/− mice showed increased MAPK signaling, but signaling intermediate cAMP was also dysregulated. Both cAMP and mitochondrial enzyme dysfunction have also been reported in nf1-deficient fruitflies (16). Interestingly, in the fly model, cAMP analogues were able to ameliorate the nf1 phenotype.
While the Nf1Prx1−/− mouse demonstrates a critical role for Nf1 in limb development, Prx1-cre expression affects all mesenchymal lineages of the developing limbs, including adipocytes and vasculature endothelial cells (12). It also does not specifically address whether NF1 heterozygous mice manifest a muscle phenotype. Consequently, we sought to examine muscle function in a range of different NF1 mouse models.
First, we investigated muscle structure and function in the heterozygous Nf1+/− mouse; Nf1−/− knockout is embryonic lethal (17). Grip strength testing was performed and muscle tissue was examined by histology. Next, the capacity of Nf1+/− muscle to respond to stress was tested in a botox-induced disuse atrophy/recovery model. To specifically examine the biological importance of the Nf1 gene in muscle, we generated a muscle-specific double knockout of NF1 using the MyoD-cre and Nf1flox/flox lines (18,19). This Nf1MyoD−/− line showed a severe muscle phenotype resulting in neonatal lethality. Lastly, some of the histological and metabolic features revealed by the Nf1MyoD−/− neonates were examined in Nf1-null muscle tissue from mature Nf1Prx1−/− mice.
RESULTS
Heterozygosity for Nf1 deficiency does not affect muscle size, fiber size or force
Prior studies have indicated that the Nf1+/− mice can manifest some but not all of the clinical features NF1. To examine concordance between clinical reports of human grip weakness (14), and the heterozygous mouse, grip strength of Nf1+/− mice was measured using a mouse grip strength meter. No difference was seen compared to wild-type (WT) littermates (Fig. 1A). No significant difference was seen in body weight with Nf1+/− mice, as has been previously shown (P = 0.16), thus specific force was also unchanged. Examination of muscle fiber size showed initially no difference, although fiber size is known to vary by fiber type. Consequently, sections from the tibialis anterior of Nf1+/− and WT mice were stained for fiber type. No significant differences in fiber size were observed (Fig. 1B).
Figure 1.
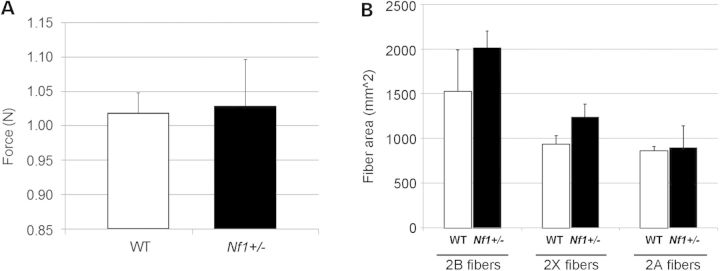
The Nf1+/− mouse line showed (A) no significant difference in grip strength compared with WT controls (P = 0.83), and (B) no significance in fiber size for type 2B, 2X, 2A fibers, respectively.
Nf1+/− mice show no significant difference in recovery following muscle injury
To examine the response of Nf1+/− mice to limb disuse, the hind limb musculature was locally injected with Botox. Mice were analyzed for gait and weight until they were ambulating normally. Botox treatment caused a decrease in ambulation that corresponded with a decrease in body weight in both the WT and Nf1+/− mice (Supplementary Material, Fig. S1). There was no significant difference in the total body weight lost or the time taken to regain this weight in either group. Functionally, there was no difference in the time to restore normal gait.
Lean tissue mass in the affected limb was measured by DEXA (Fig. 2). While this demonstrated rapid loss of lean tissue in the Botox-treated animals over the first 3 weeks followed by progressive recovery, no difference was seen between WT and Nf1+/− mice. Mice were harvested at 1, 2, 3 and 12 weeks for more detailed muscle quantification by microCT (Supplementary Material, Fig. S2A) or muscle wet weight (Supplementary Material, Fig. S2B). Botox induced muscle loss was observed, but Nf1 deficiency did not affect muscle loss or recovery in the heterozygous mouse line.
Figure 2.

Analysis of loss of lean tissue mass in the Botox-treated limb in WT and Nf1+/− mice. Following Botox-induced limb disuse, WT and Nf1+/− mice underwent weekly DEXA analysis to quantify lean tissue mass. In WT and Nf1+/− mouse strains, Botox lead to a significant reduction in lean tissue mass (broken line) compared with untreated control (solid line). There was no difference in loss or recover of lean tissue mass in Nf1+/− mice compared with WT mice with Botox treatment (P > 0.05).
Nf1+/− mice show greater acute cortical bone loss with unloading
Prior analyses had shown no significant difference in bone parameters between Nf1+/− and WT mice, although fracture repair was delayed (20). To examine bone loss and recovery with disuse, bone mineral content and bone volume (BV) were analyzed by X-ray, DXA and microCT.
Analysis of the tibia by microCT showed rapid bone loss following botox treatment in all mice. The Nf1+/− mice exhibited a more rapid loss of cortical bone with unloading (Fig. 3). Loss of trabecular bone and the ability to recover both cortical and trabecular bone at 12 weeks after the resumption of load were statistically no different between Nf1+/− and WT mice.
Figure 3.
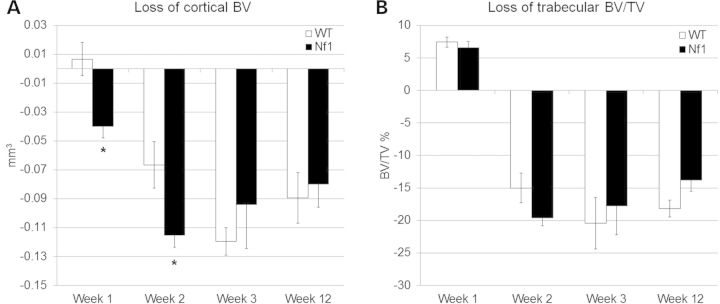
MicroCT analysis of the loss of diaphyseal cortical bone (BV, mm3) and trabecular bone at the metaphysis (BV/TV, %). Bone loss was compared between WT and Nf1+/− genotypes. At the early time points, there was a trend towards a greater initial loss of (A) cortical bone in the Nf1+/− mice immediately following unloading (*P < 0.05). No differences were seen with (B) trabecular bone compartment.
Muscle-specific KO of Nf1 causes decreased body weight and neonatal lethality
To explore the functional role of neurofibromin in muscle tissue, we generated a muscle-specific Nf1 knockout mouse driven by MyoD-cre. Nf1 gene recombination has been previously reported in muscle isolated from this line (21). Conditional knockout mice were analyzed at days 2, 4 and 6, and weighed significantly less than control litter mates (Fig. 4). While smaller, the mutant mice did not show shortened or bowed long bones or impaired mobility. Autopsy of dead Nf1muscle−/− pups showed the presence of milk in their stomachs, evident of some capacity for suckling. No organ failure was apparent in the post-mortem. The primary cause of death was attributed to maternal infanticide, likely due to their inability to thrive compared with littermates.
Figure 4.
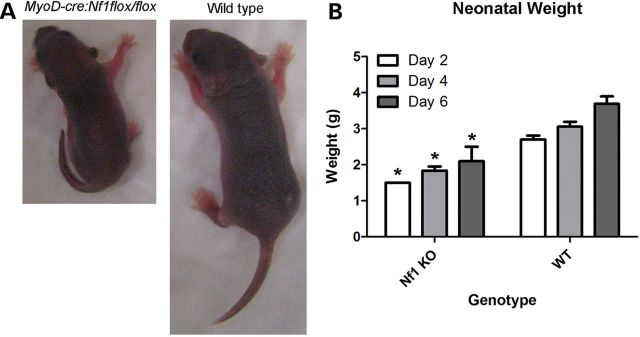
Reduced weight in Nf1muscle−/− mice. In the Nf1muscle−/− mice (MyoD-cre:Nf1flox/flox genotype), neonatal body weight was significantly reduced at day 4 in muscle knockouts compared with WT and heterozygous littermate controls. (A) Representative images of knockout and WT littermate pups. (B) Mean weights of Nf1muscle−/− pups and WT littermates at days 2, 4, and 6 are shown. Multi-group analysis by ANOVA showed significance based on age (P < 0.05) and genotype (P < 0.05).
Ultrastructural analysis of Nf1 knockout muscle tissue
Ultrathin sections of muscle were analyzed from Nf1muscle−/− mice and littermate controls by transmission electron microscopy (EM). While it was hypothesized that force generation may be compromised due to deficiencies in cytoarchitecture, no abnormalities were seen in the muscle sarcomere (Fig. 5A and B). While some abnormal (large or elongated) mitochondria were noted in the Nf1muscle−/− samples, the majority of mitochondria appeared normal and no mitochondrial breakdown was seen. Notably, intramyocellular inclusions that were speculated to contain lipid were observed in the Nf1muscle−/− mice with a greater frequency than controls.
Figure 5.

EM of Nf1muscle−/− muscle. No difference in sarcomeric structure was seen between WT muscle (A) and Nf1muscle−/− muscle (B) at day 4. Globules of what appeared to be lipid were seen in the Nf1muscle−/− samples (C, inset D). Scale bar = 1 µm (A–C), 500 nm (D).
Altered muscle metabolism in Nf1muscle−/− and Nf1Prx1−/− mice
Nf1muscle−/− mice failed to thrive and by day 6 showed significant morbidity and mortality. Tissue from day 4 pups was examined to determine any initial changes in muscle histology. No significant difference was seen in muscle fiber size at this time point (P> 0.05). Even at a later time point of day 6, no differences were seen by Gomori Trichrome staining, which is used to detect mitochondrial myopathy (Fig. 6A–D). Based on the lipid noted by EM, an Oil Red O stain was performed and this revealed marked accumulation of lipid droplets in the Nf1muscle−/− mouse muscle (Fig. 6E–H, Supplementary Material, Fig. S3). These droplets varied considerably in size but were discrete droplets in the muscle tissue and not adipocytes/intermuscular fat. Quantitation showed a significant increase in the amount of intramyocellular lipid in Nf1muscle−/− muscle (P < 0.01, Fig. 6I).
Figure 6.

Muscle histology of Nf1muscle−/− mice. No differences in muscle histology were seen by Haemotoxylin and Eosin staining (A and B) or Gomori Trichrome staining (C and D) between WT and Nf1muscle−/− samples. Oil Red O staining revealed a dramatic increase in the frequency of lipid droplets in the muscle (E, F; zoom in G, H), which was confirmed by quantitation (I).
Non-quantitative NADH and succinate dehydrogenase (SDH) staining suggested that metabolic activity may be affected in the muscle-specific knockout mice (data not shown); therefore quantitative metabolic activity assays were performed on muscle extracts from Nf1muscle−/− and Nf1Prx1−/− mice. Due to the neonatal lethality of the Nf1muscle−/−, whole hind limbs were collected at day 2. In addition, we obtained mature muscle tissue from Nf1Prx1−/− mice. This latter strain has the Nf1 gene knocked out in the entire limb, including the myofibers, adipocytes and vascular cells, yet the mice survive as previously reported (12). Muscle tissue was dissected from the hind limbs of 2 months Nf1Prx1−/− mice. Mitochondrial enzymes SDH, citrate synthase (CS), β-hydroxyacyldehydrogenase (BHAD) and medium-chain acyl-CoA dehydrogenase (MCAD) were tested and compared with WT littermate controls. No differences were seen in the Nf1muscle−/− samples, but there were 2-fold increases in SDH, BHAD and MCAD activity for the Nf1Prx1−/− samples relative to littermate controls (P < 0.05) (Fig. 7).
Figure 7.

Mitochondrial enzyme assays for in Nf1 knockout muscle. Enzyme assays were performed on muscle extracts from 2-day-old Nf1muscle−/− mice and 2-month-old Nf1Prx1−/− mice with littermate controls. SDH (A), CS (B), BHAD (C) and MCAD (D) were tested for all samples. No significant difference in metabolic enzyme activity was noted for Nf1muscle−/− neonatal muscle for any genotype or enzyme. In contrast, mature NF1-null muscle from Nf1Prx1−/− mice showed significant increases in SDH, BHAD and MCAD activity relative to WT controls (P < 0.01).
Further analysis of the Nf1Prx1−/− mice showed considerable increases in triglyceride content of almost 10-fold (Fig. 8A). This was consistent with observation of increased intramyocellular fat deposits in the neonatal Nf1muscle−/− line. Muscle extracts from the quadriceps of Nf1Prx1−/− mice and littermate controls were analyzed by western panels (Fig. 8B). No differences were seen in the expression of OXPHOS complexes, PPARγ1,2 or UCP3. Notably, however, significant increases were seen in the expression of fatty acid synthase (FAS) and leptin. We also observed decreased levels of fatty acid transport proteins carnitine palmitoyl transferase-1, CD36 and FATP4.
Figure 8.
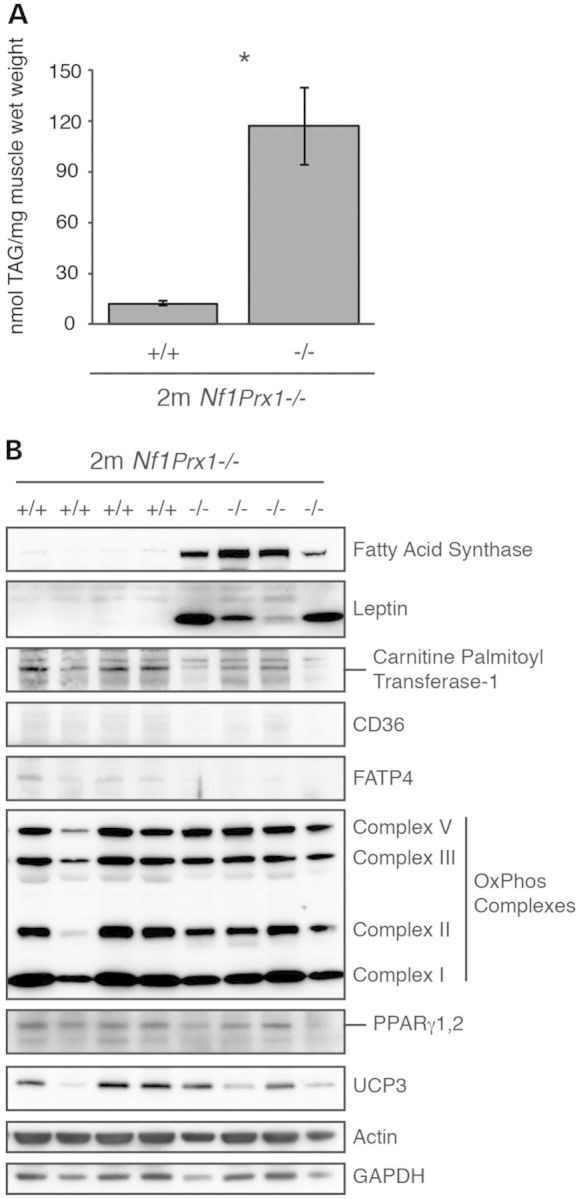
Nf1Prx1−/− mice have increased muscle fat content and altered expression of fat metabolism proteins. Triglyceride content was significantly higher in Nf1Prx1−/− mice compared with WT littermates (n = 5, mean ± 95% CI, P < 0.01) (A). Western blots of quadriceps muscle extracts from Nf1Prx1−/− mice and WT littermates (n = 4) equally loaded for total protein are shown (B). Nf1Prx1−/− mice have higher expression of FAS and leptin and lower expression of carnitine palmitoyl transferase-1, CD36 and FATP4 compared with WT littermates, while there were no differences in expression of OxPhos complexes, PPARγ1,2 or UCP3. Actin and GAPDH are shown as loading controls.
DISCUSSION
NF1 belongs to the RASopathy class of diseases, and the NF1 gene product is a negative regulator of Ras GTPase activity. The RASopathies are a family of diseases that include Noonan syndrome, Costello syndrome and cardiofaciocutaneous syndrome. Since initiating our study in multiple mouse lines, the paradigm of NF1 muscle has advanced to suggest that bone and muscle involvement could be a feature of all RASopathies (22,23). To further study this concept, Stevenson and coworkers undertook studies of muscle function in jumping and grip strength tests (24,25). Their 2011 study used force plate and hand-held dynamometer testing to show that hip extension strength was decreased in NF1 patients. Nevertheless, overall jumping and hopping force production was not significantly different, potentially reflecting compensation by other muscles groups and/or insufficient statistical power. A subsequent study in 2012 measured grip strength in a range of RASopathies and found peripheral muscle weakness in all patient cohorts, including NF1 (22).
In this study, we have gone on to examine the effects of NF1 deficiency on muscle in a number of different genetically modified mouse models. Previous work with the Nf1Prx1−/− mouse that lacks Nf1 in the limb mesenchyme revealed that this can lead to muscle weakness and a dystrophy-like phenotype (12). Additional fat has also been observed in this line (26). However, this is the first study to also examine muscle in the Nf1+/− and Nf1muscle−/− mouse lines, which show less and more severe phenotypes, respectively.
The Nf1+/− mouse model was generated in 1994 and has been extensively explored as a model of human disease (17). In some instances, this line adequately reflects the clinical features of NF1, such as showing impaired fracture healing that parallels orthopedic NF1 patients (20). In this study, we also show that Nf1+/− mice lose cortical bone more rapidly after Botox-induced immobilization, although their subsequent bone recovery is normal. This is consistent with the Nf1+/− line demonstrating a mild bone phenotype, although heterozygous mice fail to recapitulate many of the disease features of NF1, such as the systemic issues with bone density and bone metabolism (4,5,11). Consequentially, numerous murine models featuring double inactivation of Nf1 have been established in order to phenocopy human manifestations not specifically associated with NF1 double inactivation, including systemic osteopenia (27).
In this study, we found no detectable muscle phenotype in the Nf1+/− mice, based on histology and grip strength. While there was a trend towards a larger fiber area in 2B and 2X fibers, this did not reach statistical significance. In contrast, clinical studies have shown individuals with NF1 present with a mean decrease of at least 50% in raw handgrip strength (24). Fiber size and fiber type proportions have not been reported in individuals with NF1. In addition, immobilization-induced muscle/lean tissue loss was not different in the Nf1+/− mice versus controls, even though cortical bone loss was greater. Together, these data suggest that the Nf1+/− strain represents a poor system for modeling weakness or muscle dysfunction reported in individuals with NF1.
Subsequently, we analyzed two additional models where Nf1 was double-inactivated either in cells of the muscle lineage using a MyoD-cre transgene (Nf1muscle−/−), or in the mesenchyme of the developing limb using a Prx1-cre transgene (Nf1Prx1−/− mouse) (18,28). Unlike the Nf1+/− mice, significant phenotypic effects were seen in the tissue-specific knockouts, indicating important development functions for Nf1 in mouse muscle. This has previously been reported in the Nf1Prx1−/− strain with a focus on the early embryonic specification and patterning of muscle, and this report demonstrated increases in MAPK signaling (12). In this study, we have identified a novel mechanism that suggests a regulatory role for Nf1 in mitochondrial function in mammalian muscle.
Several lines of evidence point towards the existence of a previously unrecognized mitochondrial deregulation component of NF1 pathogenesis. First, Nf1 has been implicated in the regulation of mitochondrial function in the Drosophila model (16). In this model, it has been shown that loss of nf1 causes decreased ATP synthesis and increased reactive oxygen species (ROS) production, yielding shorter life span of the fruit fly. Conversely, nf1 overexpression increased complex I activity and decreased ROS, but CS activity remained unchanged. In the Nf1Prx1−/− mouse, we noted an increase in SDH activity consistent with increased mitochondrial function, but CS activity was unchanged. While the same pathways were affected as in the fly models, the direction of change was inverted (loss of Nf1 increased rather than decreased mitochondrial activity). This difference may be attributed to differences between the species, and in muscle it has been previously reported that there is increased cAMP activation in the Nf1Prx1−/− mouse (12), whereas the fly model showed decreased cAMP (16).
Furthermore, in Drosophila, the effects of nf1 on mitochondrial function were found to be independent of Ras signaling (16). However, this may not be true for mammalian cells where Ras may have a more profound influence on mitochondrial respiration. Other RASopathies can affect cellular metabolism, for example in vitro expression of a mutant D61G SHP2 (which results in Noonan syndrome) decreased ATP levels and mitochondrial membrane potential and increased ROS (29). Clinically, mitochondrial dysfunction has been reported in a range of RASopathies (30,31). Intriguingly, in mouse fibroblasts expressing activated V12 H-Ras, a transient increase in oxidative phosphorylation (OXPHOS) was reported to precede increased glycolysis and decreased OXPHOS (32). Thus, the molecular regulation of mitochondrial metabolism in Nf1-deficient muscle is likely to be complex and involve crosstalk between multiple pathways.
Notably, in neonatal muscle from the Nf1muscle−/− line, no significant changes were seen in oxidative enzyme activity; however, the noted increase in intramyocellular lipid indicated fundamental changes in fat metabolism. Overall, the increases in SDH, BHAD and MCAD activity seen in the Nf1Prx1−/− strain but not the Nf1muscle−/− are likely attributable to the differences in mouse ages used and relative maturity of the tissues. However, the presence of increased metabolic activity in non-myogenic cells in the Nf1Prx1−/− strain cannot be completely discounted and increased adipocytes have been reported in this line (26).
Further experiments will need to be undertaken to address key mechanistic and translational questions still remaining. While conditions affecting mitochondrial lipid oxidation (including deficiencies in the acyl CoA dehydrogenase family of enzymes) can lead to lipid accumulations (33), this is normally associated with impaired enzyme function, rather than increased activity as was noted in the Nf1Prx1−/− mice. Interestingly, the Nf1muscle−/− muscle showed lipid accumulation without mitochondrial enzyme dysfunction, potentially suggesting that the latter may develop with time as a consequence of increased lipid being present. Also consistent with this model, increases were detected in FAS, as an enzyme involved with regulating lipid biosynthesis, and leptin, a circulating hormone involved with regulating appetite and metabolism. Intriguingly, regulatory feedback may exist between neurofibromin and cellular lipid metabolism; NF1 protein stability is regulated by the ETEA/Ubxd8 ubiquitase 1, which itself is a cellular sensor of the unsaturated fatty acids (34). In addition, the observed decreases in fatty acid transport proteins could impair fat trafficking and contribute to the accumulation of intramyocellular lipids. Finally, robust experimental evidence will need to confirm whether the metabolic alterations reported in these mouse models are present in human NF1 muscle.
In conclusion, data from these mouse models support the concept that hypotonia, decreased strength and motor function in individuals with NF1 may result from abnormal Ras or cAMP signaling in the muscle, rather than being attributable purely to central nervous system dysfunction. The Nf1+/− model has previously been shown to not reproduce many of the clinical features of NF1 and we show that it does not replicate the muscle phenotype seen in humans. This may imply that human muscle is more dependent on Ras regulation than mouse muscle. Nevertheless, the Nf1muscle−/− and Nf1Prx1−/− strains show significant muscle involvement and NF1 appears to be an important regulator of muscle metabolism.
MATERIALS AND METHODS
Mouse colonies
Animal experiments were approved by the Westmead Hospital Animal Ethics Committee or the Children's Hospital at Westmead/Children's Medical Research Institute Animal Ethics Committee. Nf1+/− mice were a gift from Prof Luis Parada (UT Southwestern, TX, USA) (17). Nf1flox mice originally generated by Prof Parada were sourced from the National Cancer Institute (NCI) mouse repository (Bethesda, MD, USA) (35). The MyoD-Cre mouse line expresses the Cre-recombinase gene under the control of the MyoD promoter (18). This mouse line was a gift from A/Prof David Goldhamer (University of Connecticut, CN, USA). All colonies were maintained on a C57/B6 background. A PCR-based method was used to differentiate between Nf1flox/flox and Nf1flox/+ genotypes and the test for the presence of the MyoD-Cre transgene. Nf1Prx1−/− mice were bred in the Max Planck Institute for Molecular Genetics (Berlin, Germany) by crossing Nf1flox/flox and Prx1-Cre strains (12).
Grip strength test
A grip strength meter (Columbus Instruments) was used to test mouse forearm grip strength as recorded in Newtons (N). Mice were held by the base of the tail and allowed to grip the trapeze with their front paws and then pulled with their body parallel to the floor. Each mouse was trialed 10 times with the highest and lowest readings excluded; the remaining readings were averaged. A single experienced tester was used blinded to genotype.
Botox induced unloading
Botox injection has previously been used to injure muscle and cause unloading of the bone (36). Following this principle, WT and Nf1+/− mice, 48 total, aged 8 weeks were treated with Botox (1 µg) via four intramuscular injections to induce unloading of the right hind limb. Mice were analyzed by video analysis of gait until gait was considered normal, and weighed and X-rayed weekly. Weekly DEXA scans (dual X-ray absorptiometry) were also performed using a Lunar PIXImus DEXA Scanner (GE Lunar PIXImus). Time points were at 1, 2, 3 and 12 weeks. Endpoint analysis included microCT of the Botox and contra-lateral limb using a Skyscan 1174 scanner (Skyscan Corp) and isolated muscle weight measurements.
Electron microscopy
Muscle tissue was harvested from d4 post-natal pups of a MyoD-Cre; Nf1flox/+ × Nf1flox/flox cross. This generated Nf1muscle−/−, Nf1muscle+/− and control samples. Specimens were treated with Karnovsky's fixative (R. Boadle, Institute of Clinical Pathology and Medical Research, Westmead Hospital, NSW, Australia) for 3 h at room temperature and stored in 1 × MOPS buffer. Samples were then prepared for semi-thin and ultra-thin section and examined under transmission EM.
Histological staining
Hind limb muscles were collected immediately after cull covered in cryo-preservation medium (Tissue-Tek) and frozen in partly thawed isopentane pre-chilled in liquid nitrogen. Cryosected muscle underwent trichrome stain to determine abnormalities in connective tissue or large protein accumulations. Sections were incubated in Harris Haematoxylin pH 2.3 for 5 min. Slides were then rinsed five times in distilled water and incubated in trichrome solution for 60 min [0.6% chromotype 2R (C-3143, Sigma), 0.3% Fast Green (F 7258, Sigma), 0.6% phosphotungstic acid (P 6395, Sigma), and 1% glacial acetic acid, pH 3.4]. Slides were then rinsed in 0.2% acetic acid followed by dehydration through ethanol gradients (70, 95, 100%) and mounting. Oil Red O stain was used to determine the presence of lipid accumulation in the muscle tissue. Cryosections were rinsed in 60% isopropanol before staining with Oil Red O stain solution. Stain solution is prepared from a stock solution (0.5 g Oil Red O in 100 ml isopropanol) dissolved 3:2 in dH2O. Slides were incubated for 15 min in stain solution, before rinsing with 60% isopropanol. Slides were counterstained with hematoxylin and rinsed in water before mounting in aqueous media. Quantification was performed using BioQuant Analysis System to select red oil droplets within the section (Nashville, TN, USA).
Fiber size measurements were performed on transverse 8 μm sections from the tibialis anterior. Sections were stained by immunohistochemistry for fiber type using antibodies against myosin isoforms, and membrane was staining was performed using an anti-dystrophin antibody. Fiber size measurements were performed with users blinded to genotype. Cross-sectional fiber diameter was defined as the length of the longest chord perpendicular to the longest distance that stretches through the center of the fiber. Fiber diameters were measured using calibrated Image-Pro Plus 2.0 software (Media Cybernetics, Silver Spring, MD, USA). Methods for immunostaining and quantitation were performed as previously published with no modifications to the protocol (37).
Enzyme assays
Extracts were prepared from the snap frozen quadriceps muscles of 8-week-old Nf1Prx1−/− and Nf1Prx1+/+ mice and from snap frozen skinned hind-limbs of 2-day-old Nf1 Nf1muscle−/−, Nf1muscle+/− and Nf1muscle+/+ mice. The activities of the enzymes CS (EC 4.1.3.7) 3-hydroxyacyl-CoA dehydrogenase (BHAD, EC 1.1.1.35), medium chain acyl-CoA dehydrogenase (MCAD, EC 1.3.99.3) and succinate dehydrogenase (SDH, 1.3.5.1) were determined by spectrophotometry in muscle homogenates using methods described previously (38). Assays were performed at 30°C in duplicate and values were averaged. Activities were corrected for total protein content as measured using the BioRad Protein Assay (500-0006), according to the manufacturer's instructions, for BSA standards and extracts in duplicate. The mg protein in each extract was used to convert the activities of each enzyme (mU) to specific activities (mU/mg or nmol/min/mg protein).
Muscle triglyceride assay
Muscle triglycerides were determined in quadriceps muscles from 8-week-old Nf1Prx1−/− and control (Nf1Prx1+/+) mice using a colorimetric assay kit (Triglycerides GPO-PAP; Roche Diagnostics, Indianapolis, IN, USA).
Western blots
Western blots were performed on quadriceps muscles from 8-week-old Nf1Prx1−/− and Nf1Prx1+/+ mice. Samples were prepared in 4% SDS lysis buffer and total protein content was determined using a BCA assay kit (Pierce 23225) with a BSA standard curve. Eight microgram of total protein/sample was run on 4–12% Bis-Tris precast gels (Invitrogen) in MOPS buffer. Proteins were transferred to PVDF membrane. Antibodies used for western blotting were Fatty Acid Synthase (Cell Signalling C20G5 1:1000), Carnitine Palmitoyl Transferase-1 Muscle (Alpha Diagnostics CPT1-M 1:1000), Western Blotting Detection Kit for OxPhos Complexes (Mito Science MS601 1:10000), CD36 (Santa Cruz sc-9154 1:1000), PPARγ1,2 (Biomol SA-206 1:1000), Leptin (Sigma L3410 1:1000), FATP4 (Abcam ab72721 1:1000), UCP3 (Thermo PA1-055 1:1000), Actin (Sigma 5C5 1:10000) and GAPDH (Millipore MAB374 1:10000).
Statistical analyses
Cell culture assays were conducted in triplicate at least. Values are presented as mean ± standard error (SE), and statistical comparisons were made using parametric two-tailed Student's t-tests. P-values of P < 0.05 were considered statistically significant.
For mouse studies, it was unclear whether data fit a normal distribution so more stringent non-parametric statistical tests were performed. Data were tested by Mann–Whitney U (two-independent groups) or a Kruskal–Wallis test (K independent groups) using SPSS Legacy Non-Parametric testing software (SBSS Statistics v19). Again, P values of P < 0.05 were considered statistically significant. Error bars represent the standard error of the mean.
SUPPLEMENTARY MATERIAL
FUNDING
A.S. has received funding supported for his NF1 research program from the Children's Tumor Foundation (New York, USA) and the National Health and Medical Research Council of Australia (NHMRC) Project Grant Scheme. K.Q. and J.S. are supported by a Research Fellowships from the NHMRC. N.T. is supported by a Future Fellowship from the Australian Research Council. G.C. is supported by a Research Fellowship from the NHMRC. Lauren Peacock and Kathy Mikulec (The Children's Hospital at Westmead) assisted with animal surgery. Ross Boadle (Institute of Clinical Pathology and Medical Research, Westmead Hospital) and Dongwei Wang (Kids Research Institute) aided with EM microscopy sample preparation and analysis.
Supplementary Material
Acknowledgments
Conflict of Interest statement. K.S. currently works for PRA International, a private company in the medical field, but is not involved with NF1 research. A.S. and D.G.L. have received research support from Amgen, Novartis, N8 Medical and Celgene Inc. for commissioned research unrelated to this publication. All other authors state they have nothing to declare.
REFERENCES
- 1.Friedman J.M. Neurofibromatosis 1, clinical manifestations and diagnostic criteria. J. Child. Neurol. 2002;17:548–554. doi: 10.1177/088307380201700802. [DOI] [PubMed] [Google Scholar]
- 2.Schindeler A., Little D.G. Recent insights into bone development, homeostasis, and repair in type 1 neurofibromatosis (NF1) Bone. 2008;42:616–622. doi: 10.1016/j.bone.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Gutmann D.H., Aylsworth A., Carey J.C., Korf B., Marks J., Pyeritz R.E., Rubenstein A., Viskochil D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- 4.Kuorilehto T., Pöyhönen M., Bloigu R., Heikkinen J., Väänänen K., Peltonen J. Decreased bone mineral density and content in neurofibromatosis type 1, lowest local values are located in the load-carrying parts of the body. Osteoporos. Int. 2005;16:928–936. doi: 10.1007/s00198-004-1801-4. [DOI] [PubMed] [Google Scholar]
- 5.Lammert M., Kappler M., Mautner V.F., Lammert K., Störkel S., Friedman J.M., Atkins D. Decreased bone mineral density in patients with neurofibromatosis 1. Osteoporos. Int. 2005;16:1161–1166. doi: 10.1007/s00198-005-1940-2. [DOI] [PubMed] [Google Scholar]
- 6.Tucker T., Schnabel C., Hartmann M., Friedrich R.E., Frieling I., Kruse H.P., Mautner V.F., Friedman J.M. Bone health and fracture rate in individuals with neurofibromatosis 1 (NF1) J. Med. Genet. 2009;46:259–265. doi: 10.1136/jmg.2008.061895. [DOI] [PubMed] [Google Scholar]
- 7.Stevenson D.A., Zhou H., Ashrafi S., Messiaen L.M., Carey J.C., D'Astous J.L., Santora S.D., Viskochil D.H. Double inactivation of NF1 in tibial pseudarthrosis. Am. J. Hum. Genet. 2006;79:143–148. doi: 10.1086/504441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X., Chen S., Potter O.L., Murthy S.M., Li J., Pulcini J.M., Ohashi N., Winata T., Everett E.T., Ingram D., et al. Neurofibromin and its inactivation of Ras are prerequisites for osteoblast functioning. Bone. 2005;36:793–802. doi: 10.1016/j.bone.2005.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Gutmann D.H., Cole J.L., Collins F.S. Modulation of neurofibromatosis type 1 gene expression during in vitro myoblast differentiation. J. Neurosci. Res. 1994;37:398–405. doi: 10.1002/jnr.490370312. [DOI] [PubMed] [Google Scholar]
- 10.Feldmann R., Denecke J., Grenzebach M., Schuierer G., Weglage J. Neurofibromatosis type 1, motor and cognitive function and T2-weighted MRI hyperintensities. Neurology. 2003;61:1725–1728. doi: 10.1212/01.wnl.0000098881.95854.5f. [DOI] [PubMed] [Google Scholar]
- 11.Dulai S., Briody J., Schindeler A., North K.N., Cowell C.T., Little D.G. Decreased bone mineral density in neurofibromatosis type 1, results from a pediatric cohort. J. Pediatr. Orthop. 2007;27:472–475. doi: 10.1097/01.bpb.0000271310.87997.ae. [DOI] [PubMed] [Google Scholar]
- 12.Kossler N., Stricker S., Rödelsperger C., Robinson P.N., Kim J., Dietrich C., Osswald M., Kühnisch J., Stevenson D.A., Braun T., et al. Neurofibromin (Nf1) is required for skeletal muscle development. Hum. Mol. Genet. 2011;20:2697–2709. doi: 10.1093/hmg/ddr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson D.A., Moyer-Mileur L.J., Carey J.C., Quick J.L., Hoff C.J., Viskochil D.H. Case-control study of the muscular compartments and osseous strength in neurofibromatosis type 1 using peripheral quantitative computed tomography. J. Musculoskelet. Neuronal. Interact. 2005;5:145–149. [PubMed] [Google Scholar]
- 14.Souza J.F., Passos R.L., Guedes A.C., Rezende N.A., Rodrigues L.O. Muscular force is reduced in neurofibromatosis type 1. J. Musculoskelet. Neuronal. Interact. 2009;9:15–17. [PubMed] [Google Scholar]
- 15.Johnson B.A., MacWilliams B.A., Carey J.C., Viskochil D.H., D'Astous J.L., Stevenson D.A. Motor proficiency in children with neurofibromatosis type 1. Pediatr. Phys. Ther. 2010;22:344–348. doi: 10.1097/PEP.0b013e3181f9dbc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tong J.J., Schriner S.E., McCleary D., Day B.J., Wallace D.C. Life extension through neurofibromin mitochondrial regulation and antioxidant therapy for neurofibromatosis-1 in Drosophila melanogaster. Nat. Genet. 2007;39:476–485. doi: 10.1038/ng2004. [DOI] [PubMed] [Google Scholar]
- 17.Brannan C.I., Perkins A.S., Vogel K.S., Ratner N., Nordlund M.L., Reid S.W., Buchberg A.M., Jenkins N.A., Parada L.F., Copeland N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- 18.Chen J.C., Mortimer J., Marley J., Goldhamer D.J. MyoD-cre transgenic mice, a model for conditional mutagenesis and lineage tracing of skeletal muscle. Genesis. 2005;41:116–121. doi: 10.1002/gene.20104. [DOI] [PubMed] [Google Scholar]
- 19.Bajenaru M.L., Zhu Y., Hedrick N.M., Donahoe J., Parada L.F., Gutmann D.H. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol. Cell. Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindeler A., Morse A., Harry L., Godfrey C., Mikulec K., McDonald M., Gasser J.A., Little D.G. Models of tibial fracture healing in normal and Nf1-deficient mice. J. Orthop. Res. 2008;26:1053–1060. doi: 10.1002/jor.20628. [DOI] [PubMed] [Google Scholar]
- 21.El-Hoss J., Sullivan K., Cheng T., Yu N.Y., Bobyn J.D., Peacock L., Mikulec K., Baldock P., Alexander I.E., Schindeler A., et al. A murine model of neurofibromatosis type 1 tibial pseudarthrosis featuring proliferative fibrous tissue and osteoclast-like cells. J. Bone. Miner. Res. 2012;27:68–78. doi: 10.1002/jbmr.528. [DOI] [PubMed] [Google Scholar]
- 22.Stevenson D.A., Yang F.C. The musculoskeletal phenotype of the RASopathies. Am. J. Med. Genet. C Semin. Med. Genet. 2011;157:90–103. doi: 10.1002/ajmg.c.30296. [DOI] [PubMed] [Google Scholar]
- 23.Tidyman W.E., Lee H.S., Rauen K.A. Skeletal muscle pathology in Costello and cardio-facio-cutaneous syndromes, developmental consequences of germline Ras/MAPK activation on myogenesis. Am. J. Med. Genet. C Semin. Med. Genet. 2011;157:104–114. doi: 10.1002/ajmg.c.30298. [DOI] [PubMed] [Google Scholar]
- 24.Johnson B.A., Macwilliams B., Carey J.C., Viskochil D.H., D'Astous J.L., Stevenson D.A. Lower extremity strength and hopping and jumping ground reaction forces in children with neurofibromatosis type 1. Hum. Mov. Sci. 2012;31:247–254. doi: 10.1016/j.humov.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevenson D.A., Allen S., Tidyman W.E., Carey J.C., Viskochil D.H., Stevens A., Hanson H., Sheng X., Thompson B.A., Okumura M.J., et al. Peripheral muscle weakness in RASopathies. Muscle Nerve. 2012;46:394–399. doi: 10.1002/mus.23324. [DOI] [PubMed] [Google Scholar]
- 26.El Khassawna T., Toben D., Kolanczyk M., Schmidt-Bleek K., Koennecke I., Schell H., Mundlos S., Duda G.N. Deterioration of fracture healing in the mouse model of NF1 long bone dysplasia. Bone. 2012;51:651–660. doi: 10.1016/j.bone.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 27.Rhodes S.D., Wu X., He Y., Chen S., Yang H., Staser K.W., Wang J., Zhang P., Jiang C., Yokota H., et al. Hyperactive transforming growth factor-β1 signaling potentiates skeletal defects in a neurofibromatosis type 1 mouse model. J. Bone. Miner. Res. 2013 doi: 10.1002/jbmr.1992. [23 May, Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Logan M., Martin J.F., Nagy A., Lobe C., Olson E.N., Tabin C.J. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 29.Lee I., Pecinova A., Pecina P., Neel B.G., Araki T., Kucherlapati R., Roberts A.E., Hüttemann M. A suggested role for mitochondria in Noonan syndrome. Biochim. Biophys. Acta. 2010;1802:275–283. doi: 10.1016/j.bbadis.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleefstra T., Wortmann S.B., Rodenburg R.J., Bongers E.M., Hadzsiev K., Noordam C., van den Heuvel L.P., Nillesen W.M., Hollody K., Gillessen-Kaesbach G., et al. Mitochondrial dysfunction and organic aciduria in five patients carrying mutations in the Ras-MAPK pathway. Eur. J. Hum. Genet. 2011;19:138–144. doi: 10.1038/ejhg.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aeby A., Sznajer Y., Cavé H., Rebuffat E., Van Coster R., Rigal O., Van Bogaert P. Cardiofaciocutaneous (CFC) syndrome associated with muscular coenzyme Q10 deficiency. J. Inherit. Metab. Dis. 2007;30:827. doi: 10.1007/s10545-007-0612-0. [DOI] [PubMed] [Google Scholar]
- 32.de Groof A.J., te Lindert M.M., van Dommelen M.M., Wu M., Willemse M., Smift A.L., Winer M., Oerlemans F., Pluk H., Fransen J.A., et al. Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol. Cancer. 2009;8:54. doi: 10.1186/1476-4598-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laforêt P., Vianey-Saban C. Disorders of muscle lipid metabolism, diagnostic and therapeutic challenges. Neuromuscul. Disord. 2010;20:693–700. doi: 10.1016/j.nmd.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 34.Phan V.T., Ding V.W., Li F., Chalkley R.J., Burlingame A., McCormick F. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol. Cell. Biol. 2010;30:2264–2279. doi: 10.1128/MCB.01450-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y., Ghosh P., Charnay P., Burns D.K., Parada L.F. Neurofibromas in NF1, Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warner S.E., Sanford D.A., Becker B.A., Bain S.D., Srinivasan S., Gross T.S. Botox induced muscle paralysis rapidly degrades bone. Bone. 2006;38:257–264. doi: 10.1016/j.bone.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garton F., Seto J.T., North K.N., Yang N. Validation of an automated computational method for skeletal muscle fibre morphometry. Neuromusc. Disord. 2010;20:540–547. doi: 10.1016/j.nmd.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 38.MacArthur D.G., Seto J.T., Chan S., Quinlan K.G., Raftery J.M., Turner N., Nicholson M.D., Kee A.J., Hardeman E.C., Gunning P.W., et al. An Actn3 knockout mouse provides mechanistic insights into the association between α actinin-3 deficiency and human athletic performance. Hum. Mol. Genet. 2008;17:1076–1086. doi: 10.1093/hmg/ddm380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.