

Abstract
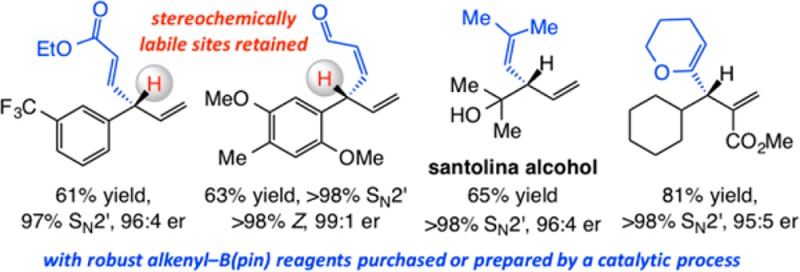
A set of protocols for catalytic enantioselective allylic substitution (EAS) reactions that allow for additions of alkenyl units to readily accessible allylic electrophiles is disclosed. Transformations afford 1,4-dienes that contain a tertiary carbon stereogenic site and are promoted by 1.0–5.0 mol % of a copper complex of an N-heterocyclic carbene (NHC). Aryl- as well as alkyl-substituted electrophiles bearing a di- or trisubstituted alkene may be employed. Reactions can involve a variety of robust alkenyl–(pinacolatoboron) [alkenyl–B(pin)] compounds that can be either purchased or prepared by various efficient, site-, and/or stereoselective catalytic reactions, such as cross-metathesis or proto-boryl additions to terminal alkynes. Vinyl-, E-, or Z-disubstituted alkenyl-, 1,1-disubstituted alkenyl-, acyclic, or heterocyclic trisubstituted alkenyl groups may be added in up to >98% yield, >98:2 SN2′:SN2, and 99:1 enantiomeric ratio (er). NHC–Cu-catalyzed EAS with alkenyl–B(pin) reagents containing a conjugated carboxylic ester or aldehyde group proceed to provide the desired 1,4-diene products in good yield and with high enantioselectivity despite the presence of a sensitive stereogenic tertiary carbon center that could be considered prone to epimerization. In most instances, the alternative approach of utilizing an alkenylmetal reagent (e.g., an Al-based species) represents an incompatible option. The utility of the approach is illustrated through applications to enantioselective synthesis of natural products such as santolina alcohol, semburin, nyasol, heliespirone A, and heliannuol E.
Introduction
Coupling reactions with organoboron compounds are among the most strategically significant processes in chemical synthesis.1 Robust (pinacolato)boron [B(pin)] derivatives are utilized frequently, and alkenylboron species represent one of the more attractive substrate classes;2 the resulting olefin-containing products, particularly if generated stereoselectively, can be functionalized in a number of ways. Site- and enantioselective catalytic protocols involving alkenyl–B(pin) reagents therefore carry substantial value, and their development constitutes a compelling research objective.
One recent line of study in these laboratories has led to the design of enantioselective processes with different types of B-containing starting materials. Cu-catalyzed enantioselective allylic substitution (EAS) processes3,4 involving alkenyl–B(pin) compounds have been of interest, because they form C–C bonds while delivering alkenyl groups of varying substitution patterns that might contain sensitive functionality (e.g., a carbonyl group). The significance of such attributes becomes evident through comparison with the alternative catalytic transformations that entail alkenylaluminum species (Scheme 1A and B). Although the latter nucleophile set can be prepared through reaction of a terminal alkyne (uncatalyzed or promoted by a Ni-based complex) with diisobutylaluminum hydride (dibal–H),5 a number of shortcomings accompany their use.
Scheme 1. State-of-the-Art in Catalytic EAS Reactions Involving Alkenyl Nucleophiles.

One issue is the sensitivity of the metal–hydride reagent as well as the resulting alkenylmetal complex to air and moisture. The relatively high reactivity of the latter species with commonly used functional units, such as a carbonyl unit (unless it is conjugated with the reacting π system4b−4d), also detracts from the approach. Moreover, direct preparation of several types of alkenylaluminum reagents would be challenging (e.g., Z-disubstituted or vinylboron compounds), and it would not be a given that the stereochemical identity of the organometallic reagent would be retained in the course of the ensuing EAS. We have previously reported a sequence involving silyl-substituted alkenylaluminum compounds (Scheme 1B), accessed via a silyl-alkyne and its site-selective hydroalumination.6 The catalytic EAS reactions proved to be high yielding, and the expected 1,4-dienes were formed in up to >98% SN2′ selectivity and >99:1 enantiomeric ratio (er); nonetheless, silylated alkynes had to be prepared initially, and the desired Z alkenes were revealed after protodesilylation. It would be more efficient if Z-alkenylboron reagents were to be employed, especially if accessed through a catalytic stereoselective process; the need for transitory incorporation of a silyl unit would thus be obviated. What is more, reactions of silyl-substituted alkenylaluminum species and sterically demanding electrophiles are inefficient,7 a limitation more likely to be resolved with nonsilylated nucleophilic components. Addition of an unsubstituted alkene can be more easily accomplished by means of a vinyl–B(pin) reagent; as far as we are aware, catalytic EAS reactions involving a vinyl group have not been previously disclosed.
A significant recent advance by Carreira illustrates that a variety of secondary allylic alcohols react with alkenyl(trifluoro)boron potassium salts to generate EAS products in the presence of a chiral phosphoramidite–Ir complex (Scheme 1C).8 Transformations are efficient and proceed in 75:25 to >98:2 branched:linear (SN2′:SN2) ratios and up to >99:1 er. A distinguishing feature of the approach is that unsaturated alcohols serve as starting materials (vs a more activated derivative). Certain deficiencies remain to be addressed however. Substrates are limited to aryl-substituted allylic alcohols with a terminal olefin, affording 1,4-dienes bearing a monosubstituted alkene. The need for 2 equiv of hydrogen fluoride detracts from the practicality of the approach. The protocol requires the use of fluoroboryl compounds, which are typically accessed via alkenyl–B(pin) compounds. The alkenyl–BF3K species employed did not contain a carbonyl-based moiety; EAS with vinylboron entities or alkenylboron reagents that carry a trisubstituted heterocyclic unit were not reported. A notable 2011 disclosure by Hayashi and Shintani focused on NHC–Cu-catalyzed EAS processes with arylboronic acid neopentyl glycol esters; this study included an example of the addition of cyclohexenyl-derived species, which furnished the desired product in 84% yield and complete site selectivity (>99:1 SN2′:SN2) and in 86.5:13.5 er.9
Herein, we put forth a broadly applicable method for catalytic EAS reactions of easily accessible di- or trisubstituted allylic phosphates and alkenyl–B(pin) reagents, resulting in the formation of tertiary C–C bonds (Scheme 2); only the carboxylic ester and acetal-containing alkenylboron reagents have previously been employed in EAS reactions that furnish quaternary carbon centers.10 Products are obtained in 52% to >98% yield, 93:7 to >98:2 SN2′:SN2 selectivity, and 83:17–99:1 er. A key attribute of the approach is that, in many cases, products are obtained in high enantiomeric purity despite containing a sensitive stereogenic center (cf., highlighted acidic protons in Scheme 2); such a challenge was not germane to the previously investigated transformations leading to quaternary carbon stereogenic centers,10 nor did it pose a complication in those performed with allenyl–B(pin) species.11 An assortment of alkenylboron compounds can be used, including those containing a carbonyl, an acetal, or an O- or N-substituted heterocyclic unit. In all cases, except one, organoboron reagents can be used as purchased. Disubstituted (E or Z), trisubstituted (linear or cyclic) variants, as well as vinyl–B(pin) species serve as effective reagents. To the best of our knowledge, there are no existing descriptions of catalytic EAS processes with heterocyclic alkenylboron or vinyl–B(pin) compounds. In some instances, alkenyl–B(pin) reagents are prepared by catalytic site- or stereoselective processes, such as Z-selective cross-metathesis (CM) with commercially available vinyl–B(pin) or α-selective protoboryl additions to terminal alkynes. Catalysts are derived from sulfonate-containing chiral imidazolinium salts synthesized in four steps12 and are converted to N-heterocyclic carbene (NHC) copper complexes through deprotonation and reaction with commercially available CuCl. Utility is illustrated through approaches to enantioselective syntheses of several natural products (cf., Scheme 2).
Scheme 2. NHC–Cu-Catalyzed EAS with Alkenyl–B(pin) Reagents Developed in This Study and Related Applications.

Results and Discussion
1. Catalyst Screening
We began by examining the ability of a selection of chiral NHC–Cu complexes to promote the EAS involving allylic phosphate 1a and commercially available n-alkyl-substituted alkenyl–B(pin) 2(13) to afford diene 3 (Table 1). The transformation carried out in the presence of C2-symmetric 4 and with C1-symmetric species derived from imidazolinium salt 5 proceeded to 38% and 76% conversion, respectively, generating the desired product with low site and enantioselectivity (entries 1,2, Table 1). Similarly low efficiency was observed with phenyl glycinol-derived 6(14) (entry 3); while site selectivity improved substantially, underlining the crucial ability of cuprate complexes (vs monodentate NHC–Cu) in delivering SN2′ mode of addition, inferior er values persisted. 1,4-Diene 3 was formed with reasonable efficiency (91% conv) and complete site selectivity (>98% SN2′) but only in 41:59 er when imidazolinium salt 7 was utilized (entry 4). Particularly noteworthy is that the copper complex derived from NHC–sulfonate 8, which proved to be the most effective for EAS with 2 and the corresponding trisubstituted allylic phosphate leading to a quaternary carbon stereogenic center,10 promoted a minimally enantioselective reaction (entry 5). Finally, we established that bidentate copper complexes generated from 9a,b (entries 6,7), containing a 3,5-disubstituted NAr moiety, deliver 3 efficiently and selectively; the complex derived from 9b afforded the desired product in 97% yield, 98:2 SN2′:SN2 selectivity, and 92:8 er (entry 7, Table 1). Reaction at lower temperatures resulted in reduced efficiency and similar selectivity. At this point, we turned to examining EAS where the more synthetically versatile organoboron reagents are used.
Table 1. Initial Screening of Different NHC–Cu Complexesa.
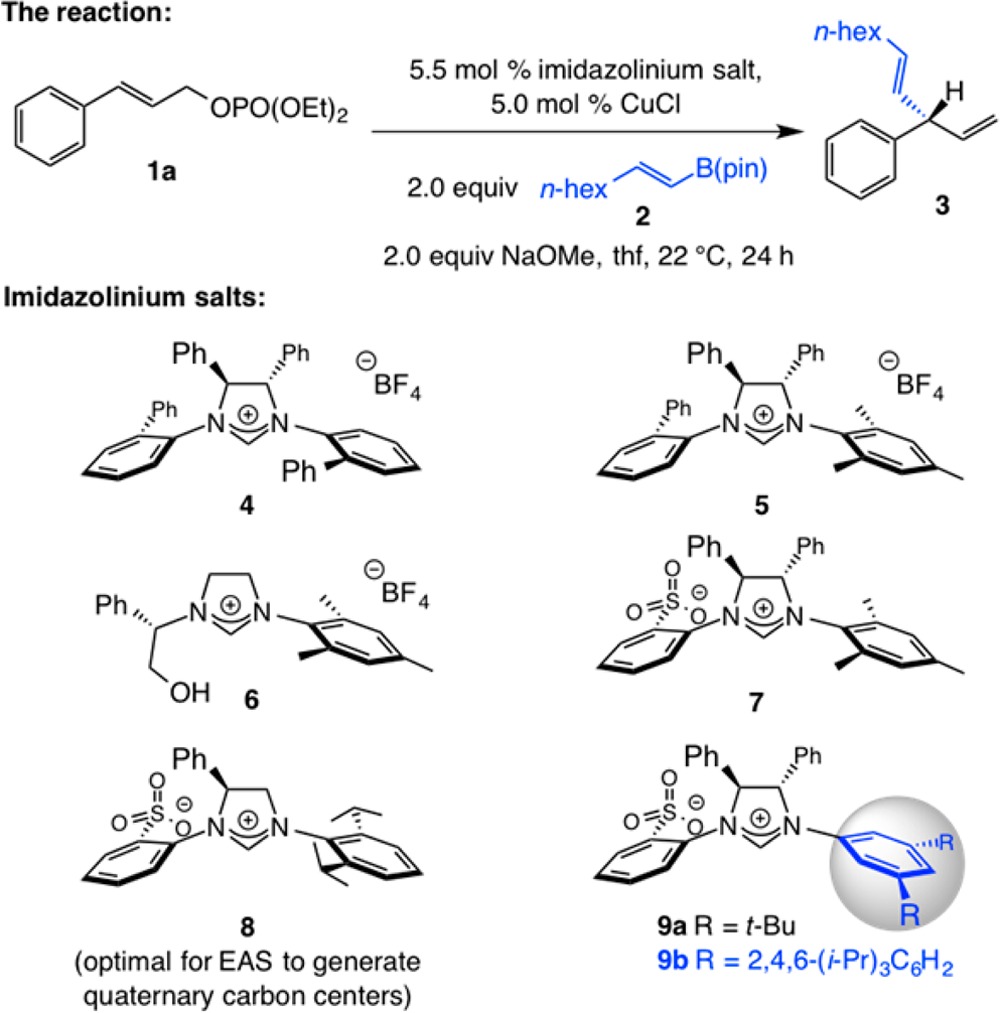
| entry | imidazolinium salt | conv (%)b | yield (%)c | SN2′:SN2b | erd |
|---|---|---|---|---|---|
| 1 | 4 | 38 | 31 | 67:33 | 45:55 |
| 2 | 5 | 76 | 59 | 79:21 | 74:26 |
| 3 | 6 | 38 | 30 | >98:2 | 78:22 |
| 4 | 7 | 91 | 90 | >98:2 | 41:59 |
| 5 | 8 | 72 | 70 | 98:2 | 34:66 |
| 6 | 9a | 98 | 93 | 98:2 | 87:13 |
| 7 | 9b | 97 | 97 | 98:2 | 92:8 |
Reactions performed under N2 atm.
Conversion (allylic phosphate consumption) and site selectivity (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification.
Yield of products (±5%) after purification by silica gel chromatography.
Enantioselectivity (±2%) determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
2. Transformations with Acetal-Substituted Alkenyl–B(pin) Reagent 10
The NHC–Cu-catalyzed EAS involving p-nitrophenyl-substituted allylic phosphate 1b and commercially available alkenylboron species 10 proceeded to >98% conv, affording 11 in 90% yield, 96% site selectivity, and 93:7 er (Scheme 3). Hydrolysis of the acetal group, achieved by subjection of 11 to a suspension of silica gel in Et2O, delivered enal 12b in quantitative yield without detectable loss of enantiomeric purity [93:7 er; >98% enantiospecificity (es)] despite an acidic benzylic proton. Three more examples are provided in Scheme 3. Two involve additions to a disubstituted allylic phosphate, followed by hydrolysis to afford α,β-unsaturated aldehydes 12a and 12c; the latter case entails EAS with a sterically demanding substrate containing an o-bromophenyl moiety. Also shown is a reaction with a trisubstituted allylic phosphate, furnishing unsaturated ester 13, with the acetal unit preserved, in 52% yield, >98:2 SN2′:SN2 selectivity, and 92:8 er. The discrepancy between the conversion level and yield of the desired product in the last case might be due to sensitivity of the acetal group to adventitious hydrolysis upon purification.
Scheme 3. NHC–Cu-Catalyzed EAS with Acetal-Containing Alkenyl–B(pin) Reagent 10.
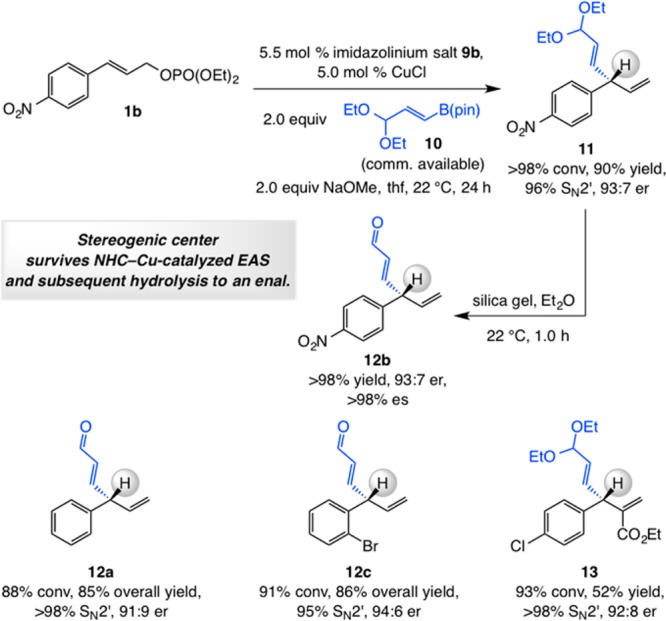
Reactions performed under N2 atm; conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
3. Transformations with Carboxylic Ester-Containing Alkenyl–B(pin) Reagent 14
The catalytic processes involving the commercially available ethyl ester-substituted 14 present a more notable challenge in relation to retention of the kinetically generated stereoisomeric purity (Scheme 4). Here, the resulting stereogenic center must survive the basic conditions needed for EAS reactions for 24 h (vs mild acidic conditions needed for hydrolysis of the acetal-containing products in Scheme 3).15 The most sensitive 1,4-diene generated through the transformations illustrated in Scheme 4 relates to the EAS with m-trifluoromethylphenyl-substituted allylic phosphate 1c; nonetheless, 15c was formed in 61% yield,16 >98% SN2′ selectivity, and 96:4 er; undesired 1,3-diene 16 was obtained in 39% yield. Consistent with the aforementioned complication, synthesis of 15a proceeded in an improved 95% yield with 97% site selectivity and 92.5:7.5 er (Scheme 4). Addition to the sterically demanding o-tolyl substrate leading to 15d (95% yield, 96:4 SN2′:SN2, 92:8 er) illustrates that NHC–Cu-catalyzed EAS can be performed with sterically demanding electrophiles. Alkyl-substituted allylic phosphates are suitable starting materials as well, underscored by the efficient and highly selective formation of 17 (>98% conv, 95% SN2′, 92.5:7.5 er); subsequent treatment with MeLi delivered the tertiary alcohol, and concomitant removal of the acyl group revealed an irregular monoterpene derived from Artemisia annua L., from which the potent antimalarial artemisinin was isolated.17
Scheme 4. NHC-Cu-Catalyzed EAS with Carboxylic Ester-Substituted Alkenyl–B(pin) Reagent 14.

Reactions performed under N2 atm; conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
4. Transformations with Z-Alkenyl–B(pin) Compounds
Our goal in this segment of our investigations was 2-fold. We wished to introduce a catalytic EAS protocol that leads to incorporation of Z alkenes and is of broad scope. Moreover, we aimed to establish a catalytic EAS protocol with the more readily accessible (in many cases through a catalytic stereoselective process) and robust alkenyl–B(pin) systems.8 What renders such a task challenging is the reduced Lewis acidity of the boron center and the larger size of the pinacol moiety, attributes that diminish the rate of transmetalation leading to the NHC–Cu–alkenyl intermediate of the catalytic cycle; such a complication is exacerbated by the increased steric hindrance of a Z alkenyl–B(pin) reagent (vs an E isomer).
4.1. Catalytic EAS with Z Alkenyl–B(pin) 18
We first investigated transformations involving methyl-substituted reagent 18, a compound that can be purchased in the purely Z form (<2% E; Scheme 5). We established that the Cu-catalyzed process involving 1a and 18 proceeds to 90% conversion at ambient temperature within 8 h (Scheme 5); Z-1,4-diene 19a was obtained in 76% yield, with >98% SN2′ selectivity, without any isomerization to the E isomer (>98% Z) and in 95:5 er. An assortment of allylic phosphates can be employed: electrophilic alkenes attached to electron-deficient (cf., 19b), electron-rich (cf., 19c), or sterically demanding (cf., 19d) aryl groups were transformed to the desired products in 76–82% yield, exceptional site selectivity (>98% SN2′), and 97:3–98:2 er. Reactions with electrophilic components containing a trisubstituted alkene, previously determined to be a relatively challenging and more sparsely examined substrate class,18 whether it is a methyl group (20a–c) or a carboxylic ester (21a–c), remained reasonably efficient (74–96% yield), generating little or none of the achiral linear product isomer (≤2% SN2).
Scheme 5. NHC–Cu-Catalyzed EAS with Z-Alkenyl–B(pin) Reagent 18.

Reactions performed under N2 atm; reaction times are 8.0 h for 19a–d, and 24 h for the other cases. Conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
4.2. Catalytic EAS with Z-Alkenyl–B(pin) Compounds Generated by Catalytic Stereoselective Cross-Metathesis (CM)
The feasibility of efficient EAS reactions with Z-alkenyl–B(pin) reagents allows for a two-stage catalytic process that begins with a Z-selective CM. Regarding the first stage, CM transformations promoted by Mo monopyrrolide aryloxide catalysts involving commercially available vinyl–B(pin) (22) were recently shown to proceed efficiently and with high Z selectivity.19
The transformation culminating in the formation of enal 26 demonstrates the effectiveness of the catalytic CM/EAS sequence (Scheme 6). Z-Selective CM of commercially available 22 and a readily accessible silyl ether in the presence of 5.0 mol % Mo complex 23a, generated and used in situ,20 afforded Z-alkenyl–B(pin) 24 in 70% yield and 96:4 Z:E selectivity. The ensuing EAS with allylic phosphate 1d delivered Z-1,4-diene 25 in 81% yield as its pure Z isomer21 with >98% site selectivity and in 99:1 er. Unmasking of the allylic alcohol and subsequent oxidation afforded enantiomerically enriched 26 in 78% overall yield with complete retention of diastereo- and enantiomeric purity (>98% Z and 99:1 er). As demonstrated through syntheses of 27a–c and 28, a variety of 1,4-dienes can be accessed efficiently and with ≥95% SN2′ selectivity and 96:4–99:1 er, and as ≥93% Z isomers.
Scheme 6. Combining Mo-Catalyzed Z-Selective Cross-Metathesis and NHC–Cu-Catalyzed EAS.
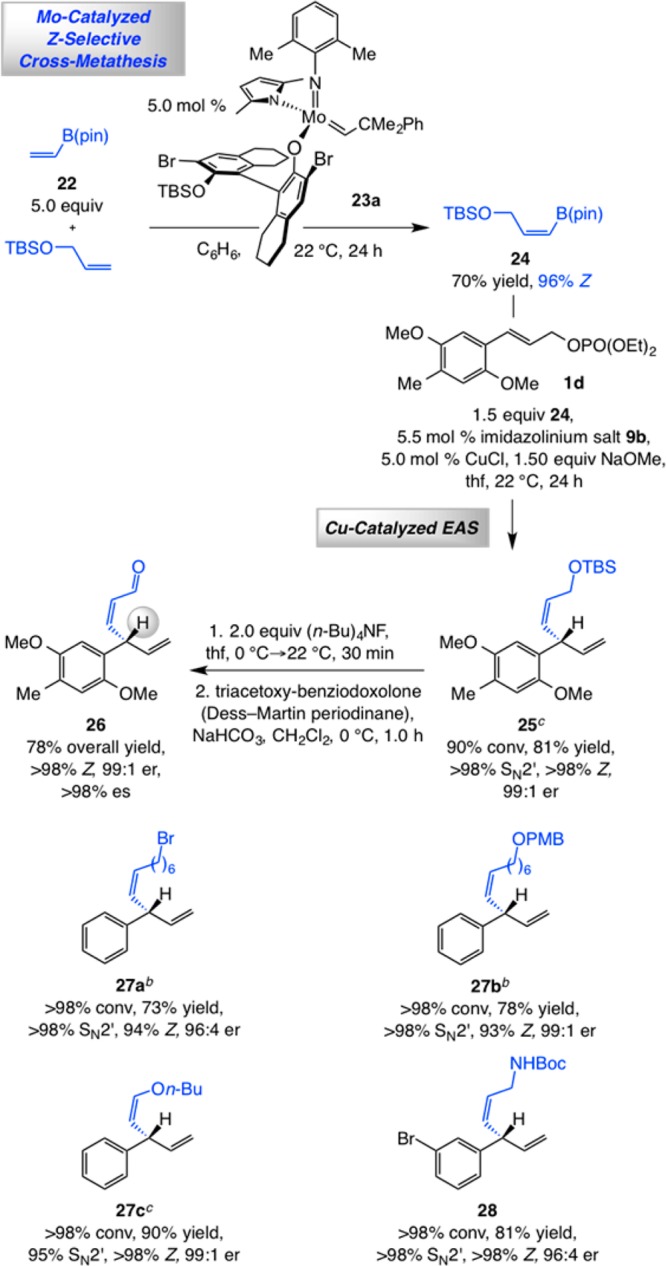
Reactions performed under N2 atm. Z:E ratios for EAS products are the same as the alkenyl–B(pin) used, except noted otherwise. Conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of unpurified product mixtures; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
Conditions were the same as those used for 25, except 1.0 equiv of Z-alkenyl–B(pin), 1.25 equiv of allylic phosphate, and 1.25 equiv of NaOMe were used.
The alkenyl–B(pin) reagent used consisted of a 96:4 Z:E and >98% Z samples for 25 and 27c, respectively (in each case, the pure Z isomer of the EAS product was isolated after silica gel chromatography).
4.3. Catalytic EAS with Z-Alkenyl–B(pin) Compounds with a Large Substituent; Applications to Enantioselective Synthesis of Nyasol, Heliespirone A, and Heliannuol E
When the Z-alkenylboron reagent carries a sizable group, EAS processes proceed with lower site- and enantioselectivity with imidazolinium salt 9b serving as the catalyst precursor. One example (Scheme 7) is the transformation involving the cyclohexyl-containing allylic phosphate 29a and phenyl-substituted alkenylboron species 30 (obtained through Z-selective CM);1931 was obtained in 74:26 SN2′:SN2 and er.
Scheme 7. EAS with Hindered Z-Alkenyl–B(pin) Reagents and Alteration of the Catalyst Structure.
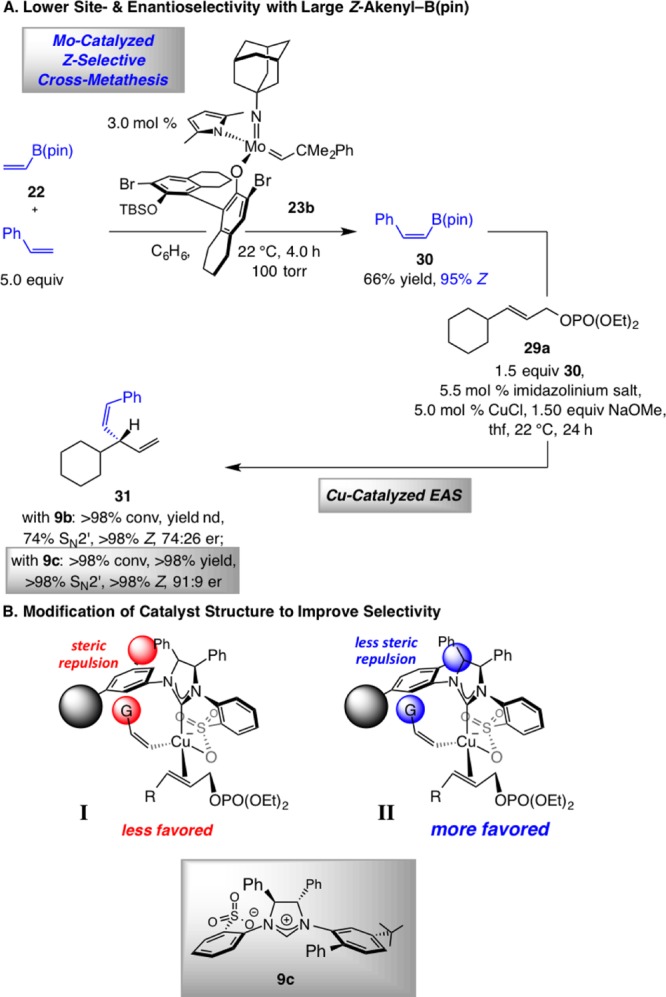
Reactions performed under N2 atm; all alkenyl–B(pin) reagents were obtained through Mo-catalyzed Z-selective cross-metathesis (CM). Conversion (allylic phosphate consumption), site-, and Z/E selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
To address the above issue, we returned to the stereochemical model for catalytic EAS reactions (I, Scheme 7). We considered the possibility that the large group of the Z-alkenylcopper intermediate (G) might elevate the energy of complex I due to steric interactions involving the tri-i-propylphenyl moiety in 9b. Accordingly, modes of transformation involving substrate–catalyst association through the alternative enantiotopic face of the allylic phosphate could become competitive, leading to lowering of er. Our analysis suggested that the latter complex (cf., II), with the protruding group of the NAr group at its ortho position, might be better able to accommodate the sizable alkenyl substituent.22 We therefore prepared chiral imidazolinium salt 9c and probed the ability of the derived NHC–Cu complex in catalyzing the formation of 1,4-diene 31, which was accordingly obtained in quantitative yield and significantly improved >98% SN2′ selectivity and 91:9 er.
To demonstrate utility, we carried out a concise, diastereo- and enantioselective synthesis of nyasol (Scheme 8).23 The requisite p-methoxyphenyl-substituted Z-alkenyl–B(pin) reagent 32 was prepared in 69% yield and 93% stereoselectivity through Mo-catalyzed CM performed with 3.0 mol % 23b and 1.5 equiv of p-methoxystyrene. Subsequent NHC–Cu-catalyzed EAS with 1e afforded 33 in 92% yield, with complete site selectivity, as a single alkene isomer (>98% Z after purification) and in 97:3 er. Nyasol was generated after removal of the methoxy and tosyl groups in 79% overall yield. In contrast to a route reported previously, also involving an NHC–Cu-catalyzed EAS,6 preparation and use of a silyl-substituted alkenylaluminum reagent and subsequent desilylation was not needed. Additionally, unlike the recent route to nyasol involving a phosphine–Ir-catalyzed EAS reaction,8 complete site selectivity was attained (vs 74:26 SN2′:SN2), the presence of hydrogen fluoride was not necessary (see Scheme 1C), and the Z-alkenyl–B(pin) compound could be employed directly without the multistep procedures needed for the formation of the corresponding trifluoroborate salts.24
Scheme 8. Application to Enantioselective Synthesis of Nyasol.

See the Supporting Information for experimental and analytical details.
The possibility of a succinct route for enantioselective synthesis of diol 38 (Scheme 9), used to prepare different members of the heliannuol family of natural products25 as well as the structurally related heliespirones,26 presented us with an opportunity to explore further the scope of the catalytic protocol with a more sterically demanding Z-alkenyl–B(pin) (vs 30 and 32 in Schemes 7 and 8). We established that reaction of 1d with allylic t-butyldimethylsilyl ether 34(27) promoted by 5.0 mol % of the NHC–Cu complex derived from 9c furnishes 1,4-diene 35 in >98:2 SN2′:SN2 selectivity and 98:2 er (>98% Z). That is, the highly encumbered boron center of alkenyl–B(pin) 34 readily participates in metal exchange with the NHC–Cu–OMe intermediate. The resulting sizable NHC–Cu–alkenyl complex undergoes reasonably efficient addition to generate 35; although 18% of the allylic phosphate was recovered, the expected product was isolated in 79% yield (82% conv). When performed on ca. 0.5 g scale, the reaction proceeded to 79% conv, affording 35 in 71% yield (ca. 0.4 g) and with identical levels of selectivity. Desilylation, oxidation, and alkylation of the resulting ketone delivered tertiary alcohol 36 in 60% overall yield (no detectable change in stereoisomeric purity). Attempts at the preparation of the Z-alkenyl–B(pin) containing the tertiary alcohol or the derived silyl ether moiety (obviating the above deprotection/oxidation/alkylation sequence) by hydroboration of the appropriate terminal alkyne27 proved unsuccessful. Site- (chemo-) and diastereoselective epoxidation,28 in the presence of excess Ti(Oi-Pr)4 likely needed due to steric hindrance at the hydroxyl directing group or competitive coordination to Lewis basic OMe groups,29 afforded 37 in 92:8 diastereomeric ratio (dr), which was isolated as a single diastereomer in 76% yield (15% epoxide derived from the terminal alkene was also isolated after chromatography). Reductive cleavage of the epoxide proceeded with >98% site selectivity through a Ti-mediated procedure,30 furnishing 38 in 55% yield. The latter diol has been utilized in the enantioselective synthesis of heliespirone A and C26 (only the former shown in Scheme 9) and heliannuol E.25e
Scheme 9. Application to Enantioselective Synthesis of Heliespirone A and Heliannuol E.

Reactions performed under N2 atm; conversion (allylic phosphate consumption), site selectivities, and diastereoselectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
5. Catalytic EAS with Trisubstituted Alkenyl–B(pin) Reagents
Another class of nucleophiles that have been used less commonly in catalytic EAS reactions involves those that contain a trisubstituted alkene.6,8,9 As far as we are aware, there are no reported examples involving transformations with heterocyclic alkenyl moieties.
5.1. Transformation with an Acyclic Trisubstituted Alkenylboron Reagent To Prepare Santolina Alcohol
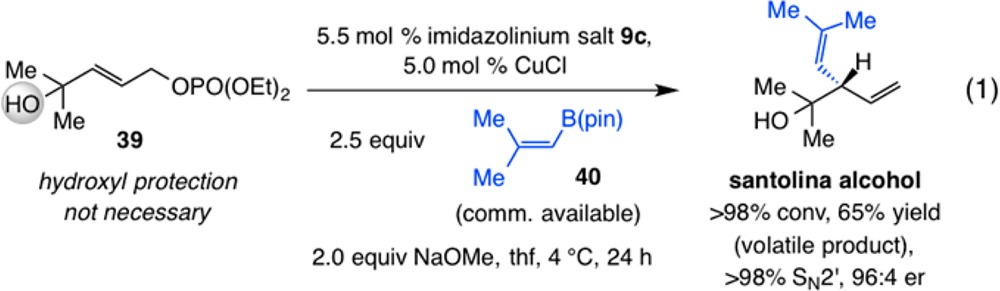
The EAS shown in eq 1 delivers natural product santolina alcohol31 in 65% yield (some loss due to product volatility), complete SN2′ selectivity, and 96:4 er. The catalytic process involves alkyl-substituted substrate 39, which contains a hydroxyl-substituted quaternary carbon center immediately next to the reactive site. Although the transformation involves the sterically demanding reagent 40 (commercially available), enantioselective addition proceeds readily. It is equally noteworthy that protection of the tertiary alcohol, a measure adopted to curtail the competitive and adventitious protonation of the intermediate alkenyl–Cu complex, is not necessary here. Such an advantage is likely the result of the inability of the sterically encumbered hydroxyl unit to approach the NHC–Cu–alkene species; however, use of 2.5 equiv of 40 is needed for achieving complete conversion (vs 1.5–2.0 equiv), perhaps because the above-mentioned complication does remain operative to a limited extent.
 |
1 |
5.2. Transformations with Heterocyclic Trisubstituted Alkenylboron Reagents; Application to Enantioselective Synthesis of Semburin
Various heterocyclic alkenyl–B(pin) compounds can be either purchased or prepared by catalytic protocols,32 including ring-closing metathesis procedures.33 The NHC–Cu-catalyzed EAS reactions in Scheme 10 involve commercially available heterocyclic organoboron reagents, affording products that contain unsaturated NBoc- or O-containing unsaturated rings in 52–98% yield, 93% to >98% site selectivity, and 91:9–98:2 er. A range of allylic phosphates serve as competent reaction partners, and a (pinacolato)boron-bearing trisubstituted cyclic allylic ether (e.g., 44–46, Scheme 10) or enol ether (e.g., 47a,b, 48) readily undergoes addition with high site- and enantioselectivity. Aryl-substituted variants may contain a sterically demanding ortho group (e.g., 42, 45, or 47b, Scheme 10), electron-withdrawing (e.g., 42, 44, 47a), or electron-donating units (e.g., 45). Transformations with alkyl-substituted allylic phosphates (e.g., 43b, 46, 48), those containing a trisubstituted olefin with a methyl (e.g., 43a,b, 46), or a carboxylic ester group (e.g., 44, 47a,b, 48) proceed efficiently and with high site- and enantioselectively. Processes involving unsaturated esters (Scheme 9) were carried out with 2.5 mol % catalyst loading (vs 5.0 mol % for other cases). Except for heterocyclic alkenyl–B(pin) 49 (cf., Scheme 11) where silica gel chromatography is required for optimal results, the commercially available organoboron reagents can be used as received in EAS reactions to generate products with reasonable efficiency and similarly high site- and enantioselectivity as would be observed with the materials that have been purified.34
Scheme 10. NHC–Cu-Catalyzed EAS with Heterocyclic Alkenyl–B(pin) Compounds.
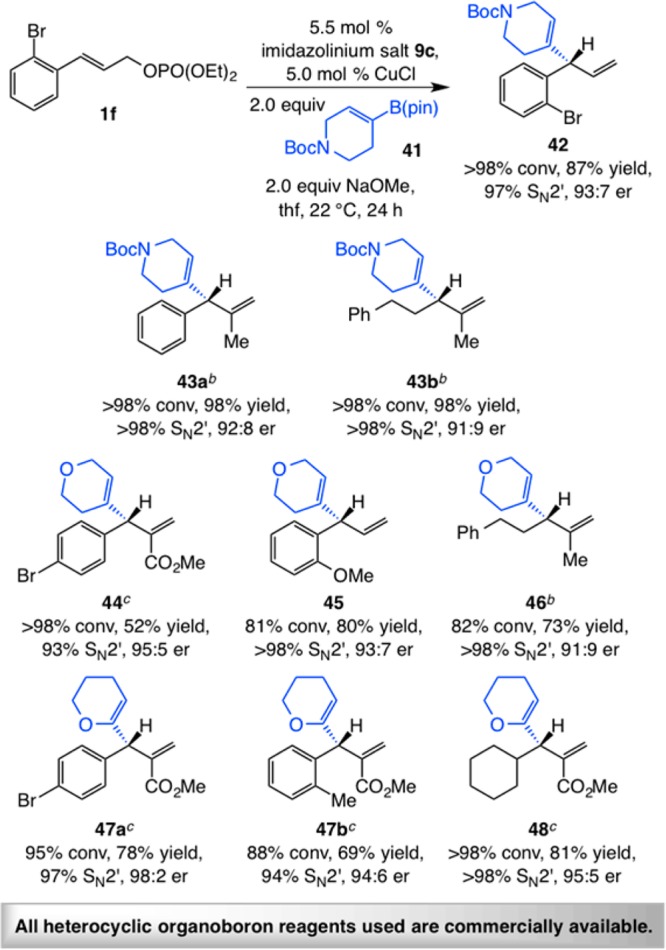
Reactions performed under N2 atm; conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
Reaction performed at 60 °C under otherwise identical conditions as shown for the formation of 42.
Reaction performed at 60 °C with 2.5 mol % 9b (for 47a and 48) or 9c (for 47b) and 25 mol % CuCl under otherwise identical conditions as shown for the formation of 42.
Scheme 11. Application to Diastereo- and Enantioselective Synthesis of Semburin.
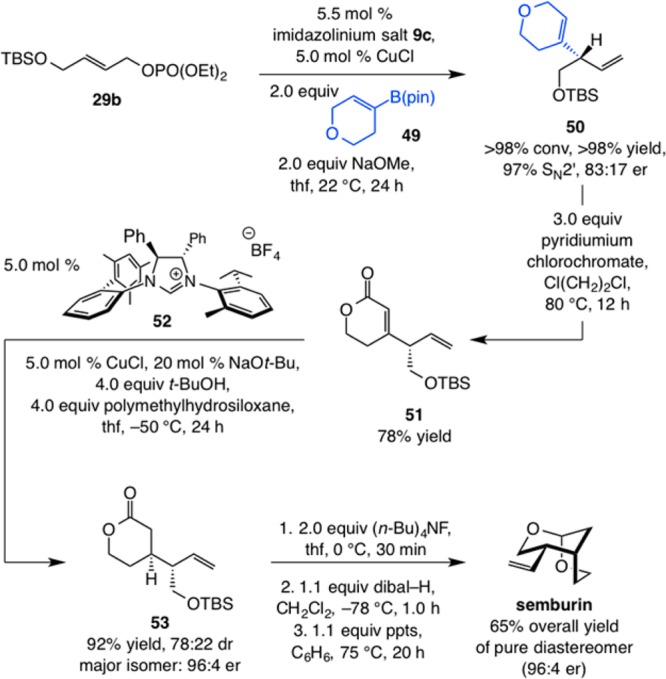
Reactions performed under N2 atm; conversion (allylic phosphate consumption), site-, and diastereoselectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details. ppts = pyridinium p-toluenesulfonate.
The diastereo- and enantioselective synthesis of semburin35 demonstrates utility. The route in Scheme 11 includes a key NHC–Cu-catalyzed stereoselective conjugate reduction developed to complement the EAS process. Formation of unsaturated pyran 50 from reaction of alkyl-substituted allylic phosphate and organoboron reagent 49 is efficient (>98% yield) and site-selective (97% SN2′); nevertheless, the desired product is generated in moderate enantioselectivity (83:17 er); examination of various Cu complexes and silyl ether derivatives did not lead to identification of superior conditions. Oxidation of the heterocyclic moiety to α,β-unsaturated lactone 51 proceeded in 78% yield. At this point, a diastereoselective 1,4-reduction of the cyclic enoate was required. Investigation of different Cu complexes reported previously to be effective for reductions involving polymethylhydrosiloxane as the hydride source38 led to either inefficient and/or minimally selective reactions.39 As an example, when the (achiral) NHC–Cu complex reported to be effective for such transformations was used,38b the desired saturated lactone was obtained in 81% yield and 45:55 dr (in favor of the undesired isomer). We therefore probed a number of chiral imidazolinium salts prepared previously in the context of catalytic enantioselective processes.40 We determined that with the Cu complex derived from C1-symmetric enantiomerically pure heterocyclic salt 52,41 lactone 53 can be obtained in 92% yield and 78:22 dr. Removal of the silyl unit, reduction of the ester group, and cyclization to the desired bicyclic acetal delivered diastereomerically pure semburin in 65% overall yield and 96:4 er after silica gel chromatography.
6. Catalytic EAS with 1,1-Disubstituted Alkenyl–B(pin) Reagents Derived from NHC–Cu-Catalyzed Protoboration of Terminal Alkynes
In addition to catalytic Z-selective CM reactions (cf., Schemes 6–8), another set of processes that can be combined with catalytic EAS processes is NHC–Cu-catalyzed site-selective protyl-boron additions to terminal alkynes. For instance, 1,1-disubstituted alkenyl–B(pin) compounds that can be accessed directly through efficient and α-selective alkyne protoborations in the presence of 1.0 mol % of a Cu complex derived from a commercially available imidazolinium salt.36,37 Alkenyl-B(pin) 56, formed through a transformation with NHC–Cu complex 55 in 91% yield and >98:2 α:β selectivity, was used to prepare 1,4-diene 57a in 89% yield, >98% SN2′ selectivity, and 98:2 er (Scheme 12). Similarly, allylsilane 57b was isolated in 93% yield, >98:2 SN2′:SN2 selectivity, and 97:3 er. Because the B-based reagents are relatively unhindered, the Cu complex derived from 9b was used (cf., Schemes 7 and 8). It should be noted that alkenyl reagents such as 56 (Scheme 12) cannot be synthesized alternatively through hydrometalation reactions due to rapid substrate decomposition.5 Elevated temperatures (60 °C) were required for complete conversion to 57a, perhaps as a result of diminution of reactivity as a result of intramolecular amide chelation to the Cu center, which is absent in the case of 57b, formation of which proceeds to >98% conversion at 22 °C. The catalytic EAS leading to 57a was significantly more sluggish at 22 °C, while delivering identical enantioselectivity (e.g., 37% conv after 24 h formed in 98:2 er).
Scheme 12. Combining Site-Selective NHC–Cu-Catalyzed Alkyne Protoboration and EAS.
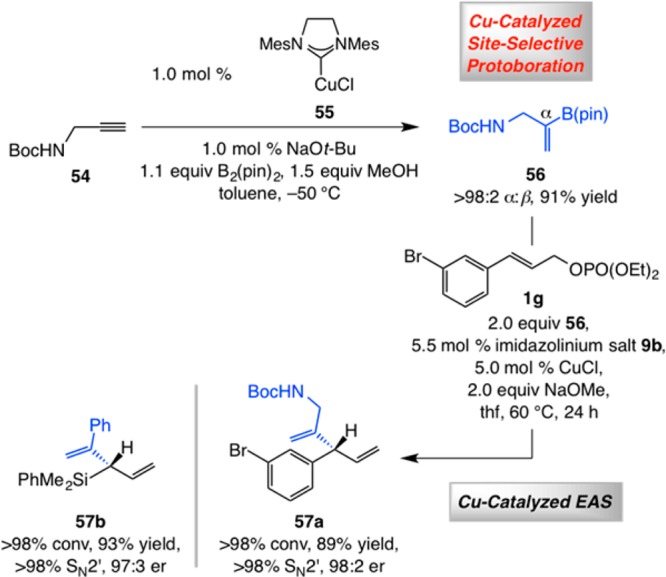
Reactions performed under N2 atm; conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
7. Catalytic EAS with Vinyl–B(pin)
Another noteworthy class of EAS transformations involves the use of robust and commercially available vinyl–B(pin). An assortment of allylic phosphates, including those that contain a sterically demanding, electron-withdrawing, or electron-donating aryl group (59a–d, Scheme 13), undergo EAS in the presence of 2.5 mol % 9b and 25 mol % CuCl to afford the desired products bearing an α,β-unsaturated ester in 68–85% yield, complete SN2′ selectivity, and ≥98:2 er. Addition of a vinyl group can be performed with an alkyl-substituted allylic phosphate, delivering 61 in 66% yield, >98% site selectivity, and 94:6 er. The use of excess CuCl in the reactions in Scheme 13 merits note. Initial screening indicated that in the presence of 10 mol % 9b and 10 mol % CuCl, >98% conversion can be achieved with er values similar to those shown; subsequent investigations revealed that with 10–25 mol % of CuCl present, the amount of the more valuable chiral imidazolinium salt can be decreased. The precise reason for the need for excess CuCl might be attributed to association of the Lewis acidic salt with the carbonyl unit of the ester moiety to facilitate EAS.
Scheme 13. NHC–Cu-Catalyzed EAS with Vinyl–B(pin) as Reagent.
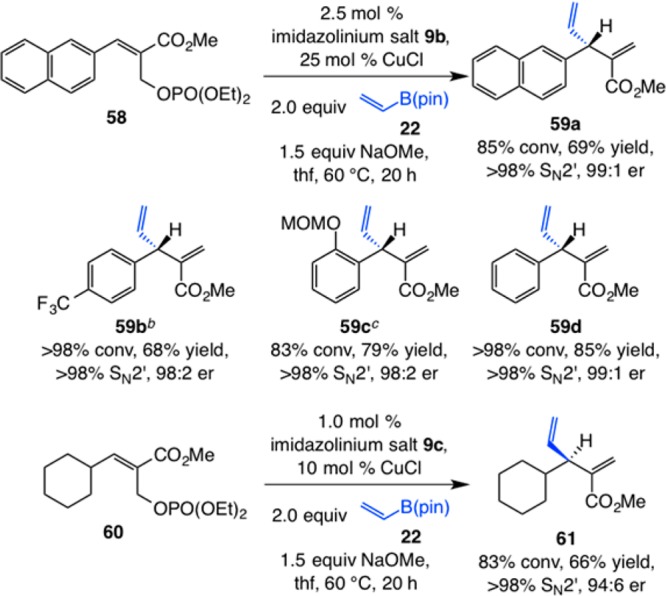
Reactions performed under N2 atm; conversion (allylic phosphate consumption) and site selectivities (±2%) were determined by analysis of 400 MHz 1H NMR spectra of product mixtures prior to purification; yields (±5%) are of products after purification; enantioselectivities (±2%) were determined by HPLC analysis. See the Supporting Information for experimental and analytical details.
Reaction performed with 10 mol % 9b and 10 mol % CuCl.
Reaction performed with 3.0 equiv of 22.
Conclusions
We have developed a generally applicable class of catalytic EAS transformations involving the use of an assortment of readily accessible and/or commercially available alkenyl–B(pin) reagents that possess a number of noteworthy attributes:
(1) The NHC–Cu-catalyzed C–C bond-forming processes have a reasonably general scope. Monosubstituted, E- and Z-1,2-disubstituted, 1,1-disubstituted, as well as acyclic and cyclic trisubstituted alkenyl–B(pin) compounds can be reacted with aryl- or alkyl-containing allylic electrophiles efficiently to afford the desired products in high branched:linear (SN2′:SN2) and enantiomeric ratios. The scope of the approach and the range of ways through which enantiomerically enriched EAS products can be functionalized are underscored through applications to enantioselective synthesis of several small molecule natural products.
(2) Catalytic transformations require an abundant and inexpensive metal salt and proceed readily with alkenyl–B(pin) compounds, robust reagents that can be purchased or prepared through catalytic processes. The strategy of combining Mo-catalyzed Z-selective CM or Cu-catalyzed protoboration of terminal alkynes and NHC–Cu-catalyzed EAS is notable.
(3) Alkenyl moieties bearing conjugated carbonyl units can be added efficiently under conditions that are sufficiently mild to allow the isolation of 1,4-dienes that contain a sensitive stereogenic tertiary carbon center in 61–95% yield and 92:8–96:4 er.
Collectively, the NHC–Cu-catalyzed transformations described above allow access to a considerable range of versatile enantiomerically enriched 1,4-dienes, the synthesis of which would have otherwise been less efficient and concise. Design and development of catalytic methods for efficient EAS processes involving other versatile classes of boron-based reagents and their applications to synthesis of natural products are in progress.
Acknowledgments
This work is dedicated to the fond memory of Professor Harry H. Wasserman. Financial support was provided by the NIH (GM-47480; GM-59426) and the NSF (CHE-1111074). F.G. was a Bristol-Myers Squibb Graduate Fellow. We thank Dr. E. S. Kiesewetter and E. C. Yu for valuable experimental assistance and helpful discussions.
Supporting Information Available
Experimental details for all reactions and analytic details for all enantiomerically enriched products. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hall D. G.Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley–VCH: Weinheim, 2005. [Google Scholar]
- For applications of alkenylboron compounds in catalytic cross-coupling processes, see:; a Reference (1).; b Suzuki A. J. Organomet. Chem. 2002, 653, 83–90. [Google Scholar]; c Negishi E.-i.; Huang Z.; Wang G.; Mohan S.; Wang C.; Hattori H. Acc. Chem. Res. 2008, 41, 1474–1485. [DOI] [PubMed] [Google Scholar]; d Tobisu M.; Chatani N. Angew. Chem., Int. Ed. 2009, 48, 3565–3568. [DOI] [PubMed] [Google Scholar]; For representative syntheses of alkenylboron compounds by Pd-catalyzed cross coupling reactions involving alkenyl bromides and triflates, see:; e Takagi J.; Takahashi K.; Ishiyama T.; Miyaura N. J. Am. Chem. Soc. 2002, 124, 8001–8006. [DOI] [PubMed] [Google Scholar]; For a review on alkenyl trifluoroborate species, accessed via (pinacolato)alkenylboron compounds, see:; f Molander G. A.; Ellis N. Acc. Chem. Res. 2007, 40, 275–286. [DOI] [PubMed] [Google Scholar]
- For reviews on catalytic EAS reactions with “hard” C-based nucleophilic reagents, see:; a Hoveyda A. H.; Hird A. W.; Kacprzynski M. A. Chem. Commun. 2004, 1779–1785. [DOI] [PubMed] [Google Scholar]; b Yorimitsu H.; Oshima K. Angew. Chem., Int. Ed. 2005, 44, 4435–4439. [DOI] [PubMed] [Google Scholar]; c Alexakis A.; Bäckvall J. E.; Krause N.; Pàmies O.; Diéguez M. Chem. Rev. 2008, 108, 2796–2823. [DOI] [PubMed] [Google Scholar]
- For representative studies regarding catalytic EAS reactions performed in these laboratories, see NHC–Mg-catalyzed and with alkylmagnesium halides:; a Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2006, 128, 15604–15605. [DOI] [PubMed] [Google Scholar]; NHC–Cu-catalyzed and with aryl- or heteroarylaluminum reagents:; b Gao F.; Lee Y.; Mandai K.; Hoveyda A. H. Angew. Chem., Int. Ed. 2010, 49, 8370–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]; NHC–Cu-catalyzed and with alkynylaluminium reagents:; c Dabrowski J. A.; Gao F.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 4778–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Dabrowski J. A.; Haeffner F.; Hoveyda A. H. Angew. Chem., Int. Ed. 2013, 52, 7694–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 10961–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama K.; Gao F.; Hoveyda A. H. Angew. Chem., Int. Ed. 2010, 49, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F.; McGrath K. P.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2010, 132, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J. Y.; Sarlah D.; Carreira E. M. J. Am. Chem. Soc. 2013, 135, 994–997. [DOI] [PubMed] [Google Scholar]
- Shintani R.; Takatsu K.; Takeda M.; Hayashi T. Angew. Chem., Int. Ed. 2011, 50, 8656–8659. [DOI] [PubMed] [Google Scholar]
- Gao F.; Carr J. L.; Hoveyda A. H. Angew. Chem., Int. Ed. 2012, 51, 6613–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 1490–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. K.; May T. L.; Baxter C. A.; Hoveyda A. H. Angew. Chem., Int. Ed. 2007, 46, 1097–1100. [DOI] [PubMed] [Google Scholar]
- β-Alkenyl–B(pin) reagents can be prepared by Ni-catalyzed hydroalumination of alkyl- or aryl-substituted terminal alkynes; see ref (5).
- For representative previous applications of Cu complexes derived from 6 and related NHC ligands, see:; a Martin D.; Kehrli S.; d’Augustin M.; Clavier H.; Mauduit M.; Alexakis A. J. Am. Chem. Soc. 2006, 128, 8416–8417. [DOI] [PubMed] [Google Scholar]; b Magrez M.; Le Guen Y.; Baslé O.; Crévisy C.; Mauduit M. Chem.—Eur. J. 2013, 19, 1199–1203. [DOI] [PubMed] [Google Scholar]
- a See ref (10). For the use of NaOt-Bu for activation of arylboron reagents towards additions to CO2 in a Cu-catalyzed process, see:; b Ohishi T.; Nishiura M.; Hou Z. Angew. Chem., Int. Ed. 2008, 47, 5792–5795. [DOI] [PubMed] [Google Scholar]
- Attempts to separate 15c from 16 were unsuccessful; the yield indicated is estimated from analysis of the 1H NMR spectra.
- a Sy L.-K.; Brown G. D. Phytochemistry 2001, 58, 1159–1166. [DOI] [PubMed] [Google Scholar]; b Araki S.; Kambe S.; Kameda K.; Hirashita T. Synthesis 2003, 751–754. [Google Scholar]
- Catalytic EAS reactions with substrates that contain a 1,1,2-trisubstituted olefin have been less frequently investigated as compared to those that contain a disubstituted or the alternative trisubstituted olefin isomer. See:; a Falciola C. A.; Tissot-Croset K.; Alexakis A. Angew. Chem., Int. Ed. 2006, 45, 5995–5998. [DOI] [PubMed] [Google Scholar]; b Gillingham D. G.; Hoveyda A. H. Angew. Chem., Int. Ed. 2007, 46, 3860–3864. [DOI] [PubMed] [Google Scholar]; c Lee Y.; Akiyama K.; Gillingham D. G.; Brown M. K.; Hoveyda A. H. J. Am. Chem. Soc. 2008, 130, 446–447. [DOI] [PubMed] [Google Scholar]; For reactions with activated substrates, see:; d Goldsmith P. J.; Teat S. J.; Woodward S. Angew. Chem., Int. Ed. 2005, 44, 2235–2237. [DOI] [PubMed] [Google Scholar]
- Kiesewetter E. T.; O’Brien R. V.; Yu E. C.; Meek S. J.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2013, 135, 6026–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolmson S. J.; Meek S. J.; Sattely E. S.; Schrock R. R.; Hoveyda A. H. Nature 2008, 456, 933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- In general, the small amount of the E-alkenyl–B(pin) isomer obtained from Mo-catalyzed CM is lost through the NHC–Cu-catalyzed EAS, allowing the enantiomerically enriched 1,4-dienes to be isolated as pure Z isomers. Examination of the 1H NMR spectra of the unpurified reaction mixtures does not indicate the presence of any EAS products that contain an E olefin; it is unlikely that the latter is removed in the course of silica gel chromatography. It is unclear in exactly what way the minor alkenyl isomer is consumed. It is possible that the minor but the less sterically congested and thus more reactive C–B bond of the E-alkenyl–B(pin) is more rapidly converted to the corresponding C–Cu bond (vs the Z isomer) that then undergoes protonation by the small amount (ca. 5 mol %) of MeOH generated due to deprotonation of the imidazolinium salt by NaOMe (to afford the corresponding terminal alkene). The latter possibility thus accounts for some of the differences between the conversion levels and values of yield for isolated and purified compounds.
- Examination of molecular models indicates that binding of the allylic phosphate to the front of the NHC–Cu complex leads to steric repulsion with the protruding phenyl moiety of the dissymmetric NAr unit. In contrast, substrate association from the rear, as shown, allows the bulky phosphate or alkenyl substituent to occupy the quadrant that is rendered relatively unoccupied due to one of the two Ph groups at the backbone of the chiral NHC pointing syn to the neighboring sulfonate bridge. For a detailed discussion on the specific structural attributes of chiral bidentate NHC–metal complexes that contain a sulfonate bridge, see:; a Lee Y.; Li B.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 11625–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Reference (11).
- For isolation and selected previous syntheses, see:; a Iida Y.; Oh K.-B.; Saito M.; Matsuoka H.; Kurata H.; Natsume M.; Abe H. J. Agric. Food Chem. 1999, 47, 584–587. [DOI] [PubMed] [Google Scholar]; b Jeong S. J.; Higuchi R.; Ono M.; Kuwano M.; Kim Y.-C.; Miyamoto T. Biol. Pharm. Bull. 2006, 26, 1721–1724. [DOI] [PubMed] [Google Scholar]; c Reference (6).; d Reference (8).
- a Molander G. A.; Ellis N. M. J. Org. Chem. 2008, 73, 6841–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a Rh- or Ir-catalyzed method for synthesis of Z-alkenyl–B(pin) compounds through trans hydroboration of terminal alkynes, see:; b Ohmura T.; Yamamoto Y.; Miyaura N. J. Am. Chem. Soc. 2000, 122, 4990–4991. [Google Scholar]
- For isolation, see:; a Masías F. A.; Molinillo J. M. G.; Varela R. M.; Torres A. J. Org. Chem. 1994, 59, 8261–8266. [Google Scholar]; For previous synthesis of heliannuol C, see:; b Kamei T.; Shindo M.; Shishido K. Tetrahedron Lett. 2003, 44, 8505–8507. [Google Scholar]; c Vyvyan J. R.; Oaksmith J. M.; Parks B. W.; Peterson E. M. Tetrahedron Lett. 2005, 46, 2457–2460. [Google Scholar]; d Biswas B.; Sen P. K.; Venkateswaran R. V. Tetrahedron Lett. 2006, 47, 4019–4021. [Google Scholar]; For previous synthesis of heliannuol E, see:; e Liu Y.; Huang C.; Liu B. Tetrahedron Lett. 2011, 52, 5802–5804. [Google Scholar]
- Huang C.; Liu B. Chem. Commun. 2010, 46, 5280–5282and references cited therein. [DOI] [PubMed] [Google Scholar]
- Preparation of Z-alkenyl–B(pin) reagent 34 through stereoselective CM proved to be inefficient, probably as a result of a high degree of steric hindrance imposed by the allylic silyl ether. As a result, trans-selective Rh-catalyzed hydroboration of the corresponding terminal alkyne was used; see ref (24b).
- For a comprehensive review of Ti-catalyzed directed epoxidation of allylic alcohols, see:Johnson R. A.; Sharpless K. B. In Catalytic Asymmetric Synthesis; Ojima I., Ed.; VCH: New York, 1993; pp 103–158. [Google Scholar]
- For a related example, see:; a Kitamura M.; Isobe M.; Ichikawa Y.; Goto T. J. Org. Chem. 1984, 49, 3517–3527. [Google Scholar]; For a review on Ti-catalyzed directed epoxidation of allylic alcohols, see:; b Hoveyda A. H.; Evans D. A.; Fu G. C. Chem. Rev. 1993, 93, 1307–1370. [Google Scholar]
- Dai L.-x.; Lou B.-l.; Zhang Y.-z.; Guo G.-z. Tetrahedron Lett. 1986, 27, 4343–4346. [Google Scholar]
- For investigations regarding the isolation and natural occurrence of santolina alcohol, see:; a Segal R.; Breuer A.; Feurstein I. Phytochemistry 1980, 19, 2761–2762. [Google Scholar]; b Näf-Müller R.; Pickenhagen W.; Willhalm B. Helv. Chim. Acta 1981, 64, 1424–1430. [Google Scholar]; c Filippi J.-J.; Lanfranchi D.-A.; Prado S.; Baldovini N.; Meierhenrich U. J. J. Agric. Food Chem. 2006, 54, 6308–6313. [DOI] [PubMed] [Google Scholar]; For a synthesis in the racemic form, see:; d Moiseenkov A. M.; Czeskis B. A.; Semenovsky A. V. Chem. Commun. 1982, 109–110. [Google Scholar]
- For a method for synthesis of cyclic alkenylboron compounds through the use of β-borylallylsilanes, prepared by Pd-catalyzed B–Si additions to allenes, see:; a Suginome M.; Ohmori Y.; Ito Y. J. Am. Chem. Soc. 2001, 123, 4601–4602. [DOI] [PubMed] [Google Scholar]; For methods involving Pd-catalyzed C–H borylation, see:; b Olsson V. J.; Szabó K. J. Angew. Chem., Int. Ed. 2007, 46, 6891–6893. [DOI] [PubMed] [Google Scholar]; c Selander N.; Willy B.; Szabó K. J. Angew. Chem., Int. Ed. 2010, 49, 4051–4053. [DOI] [PubMed] [Google Scholar]; Cyclic alkenylboron species have been prepared through Ni-catalyzed borylation processes that involve C–O activation:; d Huang K.; Yu D.-G.; Zheng S.-F.; Wu Z.-H.; Shi Z.-J. Chem.—Eur. J. 2011, 17, 786–791. [DOI] [PubMed] [Google Scholar]
- a Renaud J.; Ouellet S. G. J. Am. Chem. Soc. 1998, 120, 7995–7996. [Google Scholar]; b Jang H.; Zhugralin A. R.; Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 7859–7871. [DOI] [PubMed] [Google Scholar]
- For example, 1,4-diene 45 was obtained in 50% yield (65% conv; vs 81% conv and 80% yield), >98% SN2′ selectivity, and 93:7 er when an unpurified sample of the heterocyclic alkenyl–B(pin) was used.
- a Kawamura M.; Ogasawara K. Tetrahedron Lett. 1995, 36, 3369–3372. [Google Scholar]; b Sakai T.; Nakagawa Y.; Iwashita T.; Naoki H.; Sakan T. Bull. Chem. Soc. Jpn. 1983, 56, 3477–3482. [Google Scholar]
- For example, with a formerly reported chiral phosphine, 53 was obtained in a lower yield (ca. 60%), partly as a result of the formation of several unidentifiable byproduct; see:; a Hughes G.; Kimura M.; Buchwald S. L. J. Am. Chem. Soc. 2003, 125, 11253–11258. [DOI] [PubMed] [Google Scholar]; For conjugate reductions with achiral NHC–Cu complexes, see:; b Jurkauskas V.; Sadighi J. P.; Buchwald S. L. Org. Lett. 2003, 5, 2417–2420. [DOI] [PubMed] [Google Scholar]; For a review on catalytic conjugate hydride additions, see:; c Deutsch C.; Krause N.; Lipschutz B. H. Chem. Rev. 2008, 108, 2916–2927. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for complete data regarding the screening studies.
- Lee K.-s.; Hoveyda A. H. J. Org. Chem. 2009, 74, 4455–4462. [DOI] [PubMed] [Google Scholar]
- a Vieira E. M.; Snapper M. L; Hoveyda A. H. J. Am. Chem. Soc. 2011, 133, 3332–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vieira E. M.; Haeffner F.; Snapper M. L; Hoveyda A. H. Angew. Chem., Int. Ed. 2012, 51, 6618–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reference (33b). For related investigations, see:; b Kim H. R.; Jung I. G.; Yoo K.; Jang K.; Lee E. S.; Yun J.; Son S. U. Chem. Commun. 2010, 46, 758–760. [DOI] [PubMed] [Google Scholar]; c Moure A. L.; Arrayás R. G.; Cárdenas D. J.; Alonso I.; Carretero J. C. J. Am. Chem. Soc. 2012, 134, 7219–7222. [DOI] [PubMed] [Google Scholar]; d Park J. K.; Ondrusek B. A.; McQuade D. T. Org. Lett. 2012, 14, 4790–4793. [DOI] [PubMed] [Google Scholar]
- Recent studies illustrate that alkenyl–B(pin) compounds can be obtained through site-selective catalytic protoborations of allenes. See:; a Yuan W.; Ma S. Adv. Synth. Catal. 2012, 354, 1867–1872. [Google Scholar]; b Meng F.; Jung B.; Haeffner F.; Hoveyda A. H. Org. Lett. 2013, 15, 1414–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Semba K.; Fujihara T.; Terao J.; Tsuji Y. Chem.—Eur. J. 2013, 18, 4179–4184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.