

Abstract
Aldehyde dehydrogenases (ALDH) participate in multiple metabolic pathways and have been indicated to play a role in several cancerous disease states. Our laboratory is interested in developing novel and selective ALDH inhibitors. We looked to further work recently published by developing a class of isoenzyme-selective inhibitors using similar indole-2,3-diones that exhibit differential inhibition of ALDH1A1, ALDH2, and ALDH3A1. Kinetic and X-ray crystallography data suggest that these inhibitors are competitive against aldehyde binding, forming direct interactions with active-site cysteine residues. The selectivity is precise in that these compounds appear to interact directly with the catalytic nucleophile, Cys243, in ALDH3A1 but not in ALDH2. In ALDH2, the 3-keto group is surrounded by the adjacent Cys301/303. Surprisingly, the orientation of the interaction changes depending on the nature of the substitutions on the basic indole ring structure and correlates well with the observed structure–activity relationships for each ALDH isoenzyme.
Introduction
Aldehyde dehydrogenases (ALDH) comprise a superfamily of enzymes that catalyze the NAD(P)+-dependent oxidation of aldehydes to their corresponding carboxylic acids.1 Enzymes in this superfamily exhibit diversity in their specificity for substrates. Detrimental changes in their contributions to specific metabolic pathways lead to several disease states, including Sjögren–Larsson syndrome, type II hyperprolinemia, hyperammonemia, and alcohol flushing disease as well as cancer.2−6 Using known structural and catalytic attributes for several of these family members has led to the discovery and characterization of some selective chemical modulators for ALDH27−9 and ALDH1/310,11 as well as broad-spectrum modulators.12,13
Our prior work with a broad-spectrum inhibitor demonstrated that the enzyme catalyzed production of a vinyl-ketone intermediate that inhibited ALDH1A1, ALDH2, and ALDH3A1 through the formation of a covalent adduct with their catalytic cysteine residue.12 However, to achieve selective inhibition of particular isoenzymes, molecules that do not rely solely on common mechanistic features may be more desirable. Therefore, this study looks to further that work by characterizing a class of inhibitors that utilize a common mechanistic feature but that can achieve selectivity through elaboration of the common functional group, indole-2,3-dione. We report here the kinetic and structural characterization of a diverse group of substituted indole-2,3-diones, from which selective inhibitors for ALDH1A1, ALDH2, and ALDH3A1 may be derived.
Results
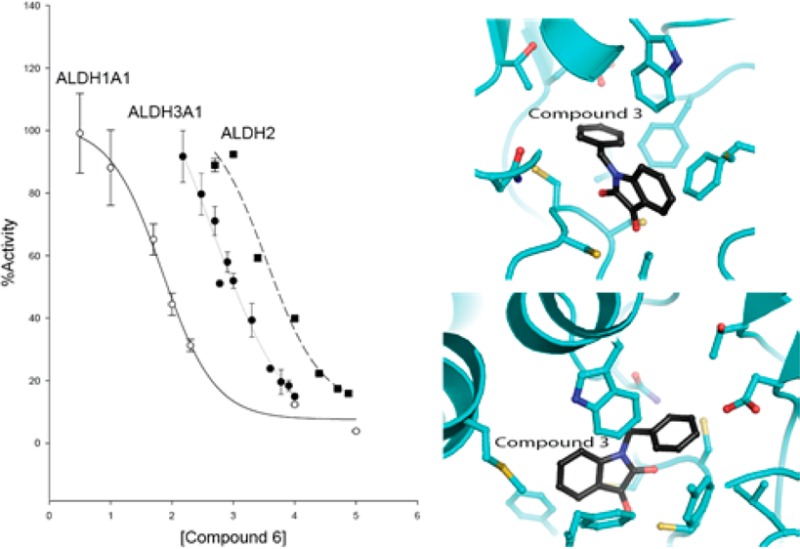
Recently, we reported a class of compounds identified during a high-throughput screen for modulators of ALDH2 that showed nonselective covalent inhibition of ALDH isoenzymes.12 To achieve a more selective inhibition of ALDH isoenzymes, we reasoned that reliance on mechanistic features common to ALDH family members was not desirable. Consequently, we re-evaluated the original high-throughput screening results12,13 for compounds that might demonstrate better isoenzyme selectivity. Re-examination of these screens led to the identification of four ALDH2 inhibitors with structural similarity to five ALDH3A1 inhibitors, some of which showed excellent selectivity toward ALDH3A1.13 To characterize this group of compounds further, we obtained an additional 33 structurally similar analogues from ChemDiv and ChemBridge and evaluated their ability to inhibit ALDH1A1, ALDH2, and ALDH3A1 using NAD(P)+-dependent aldehyde oxidation to measure activity.
The compounds in this study are all derived from the indole-2,3-dione parent compound, but three distinct structural groupings can be created on the basis of the nature of the substitutions to the indole-2,3-dione ring system and their ability to inhibit selected ALDH isoenzymes. Group 1 is represented by substitutions that lack additional ring systems. These were the least selective between ALDH isoenzymes and exhibited low micromolar IC50 values for ALDH2 and middle-to-high nanomolar IC50 values for ALDH1A1 and ALDH3A1 (Table 1).
Table 1.
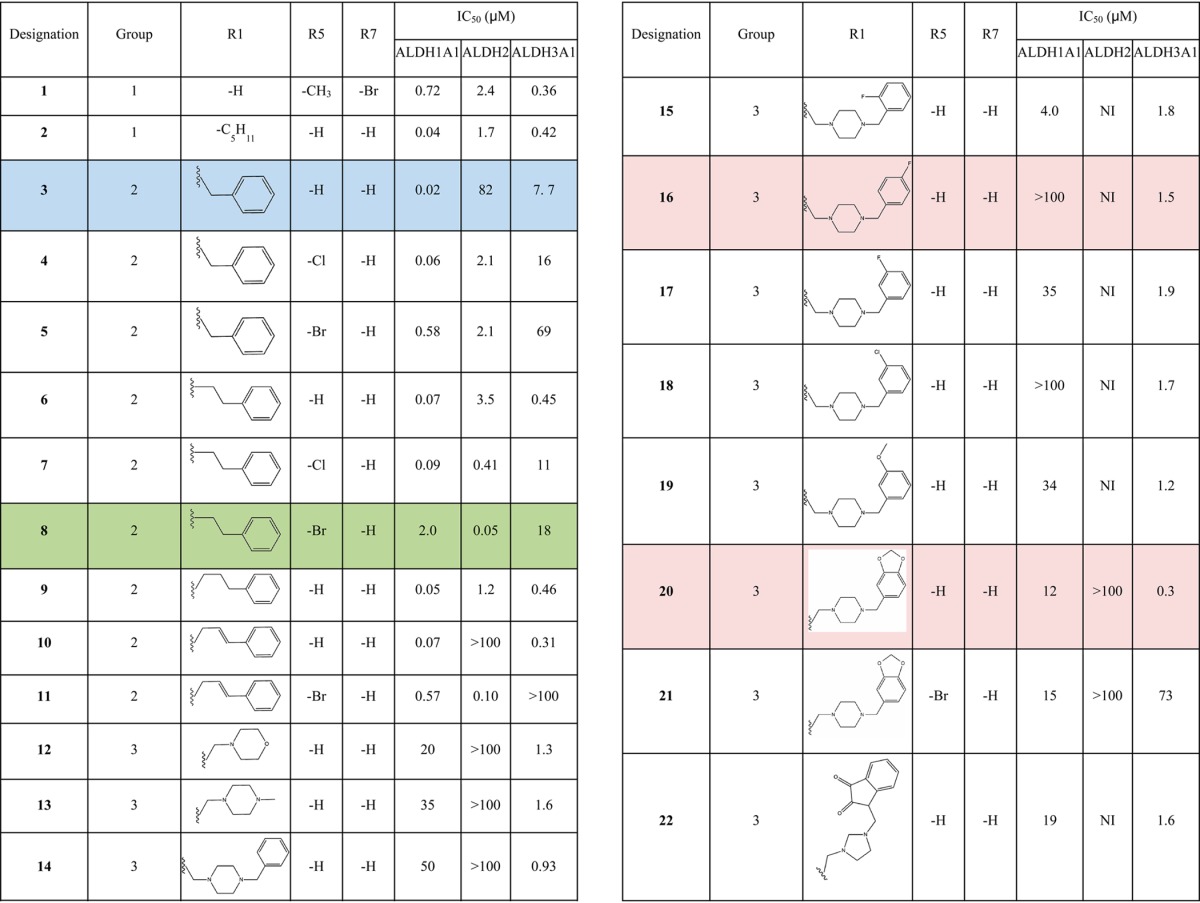
Compounds in group 2 are characterized by the addition of a benzyl moiety via an alkyl chain linker attached to the indole ring nitrogen atom with and without halogen substitutions at the 5-position of the indole ring. This group comprises the most potent inhibitors of ALDH1A1 and ALDH2. However, the nature of the substitutions can shift the potency 380-fold in favor of ALDH1A1 or 40-fold in favor of ALDH2 (1-pentyl-2,3-dihydro-1H-indole-2,3-dione (compound 3) vs 5-bromo-1-(2-phenylethyl)-1H-indole-2,3-dione (compound 8), Table 1). In general, longer alkyl-chain linkers favor ALDH1A1 and ALDH3A1 inhibition. Halogens at the 5-position improve potency toward ALDH2, but 5-bromo-substitutions on the indole ring reduce the potency toward ALDH1A1. Substitution of either a 5-chlorine or 5-bromine on the indole ring severely reduces potency toward ALDH3A1 (1-(2-phenylethyl)-1H-indole-2,3-dione (compound 6) vs 8, Table 1). The addition of a double bond to the linker between the indole and benzyl rings almost eliminates potency toward ALDH2 (1-(3-phenyl-2-propen-1-yl)-1H-indole-2,3-dione (compound 10)), but introduction of the 5-chloro group to the same molecule restores potency (5-chloro-1-[(2E)-3-phenylprop-2-en-1-yl]-2,3-dihydro-1H-indole-2,3-dione (compound 11)).
Group 3 compounds possess either a piperazine, morpholine, or imidazolidine nonaromatic ring linked to the indole nitrogen (Table 1). These compounds tend to be the most selective for hALDH3A1 and show little if any inhibition of ALDH2. Only the compound with a 5-bromo substitution on the indole ring (1-{[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]methyl}-5-bromo-1H-indole-2,3-dione (compound 21)) was a poor inhibitor of hALDH3A1 (Table 1).
To understand the mechanism of inhibition for these compounds better, compounds 1 and 3 were chosen as representative compounds for substrate competition experiments. These inhibitors exhibited noncompetitive mixed-type inhibition with respect to varied coenzyme, and they exhibited competitive inhibition with respect to varied aldehyde substrate for all three ALDH isoenzymes (Tables 2 and 3).
Table 2. Kinetic Inhibition Data versus Varied Coenzyme.
| compound | enzyme | Ki (μM) | α | mode of inhibition |
|---|---|---|---|---|
| 1 | ALDH1 | 0.29 ± 0.05 | 1. 8 | noncompetitive, mixed |
| ALDH2 | 2.2 ± 0.1 | 3.3 | ||
| ALDH3 | 0.50 ± 0.2 | 1.1 | ||
| 3 | ALDH2 | 34 ± 10 | 4.4 | noncompetitive, mixed |
| ALDH3 | 4.0 ± 1 | 2.4 |
Table 3. Kinetic Inhibition Data versus Varied Aldehyde.
| compound | enzyme | Ki (μM) | mode of inhibition |
|---|---|---|---|
| 1 | ALDH1 | 0.90 ± 0.1 | competitive |
| ALDH2 | 1.0 ± 0.1 | ||
| ALDH3 | 0.38 ± 0.05 | ||
| 3 | ALDH2 | 15 ± 8 | competitive |
| ALDH3 | 1.2 ± 0.1 |
Consistent with the kinetic data, the crystal structure of ALDH2 with 3 and 1 bound shows that the compounds bound within the aldehyde substrate binding site with the 3-keto group sandwiched between the active-site cysteine residues 301 and 303 (Figures 1 and 2). The distance of interaction in these models suggest that both cysteine residues are interacting equivalently on either face of the carbonyl carbon but do not appear to have formed formal adducts. There is no evidence of any interaction with the side chain of Cys302. Interestingly, the orientation of their indole-2,3-dione rings are flipped such that the opposing faces are interacting with Cys301 and Cys303 in the 1 versus 3 structures. The 5-methyl substituent of 1 forms hydrophobic interactions with the side chains of Trp177, Leu173, and Met174, whereas the 7-bromo substituent is oriented toward the solvent-exposed exit of the substrate-binding site. In contrast, the 5-position of 3 is oriented toward the solvent, and the 1-benzyl substituent is tucked tightly into the substrate binding side adjacent to the side chain of Phe465, which is displaced from the position found in all other ALDH2 crystal structures. There is an inverse correlation between ordered positioning of the 1-N-benzyl group and that of Phe465, suggesting that one or the other is mobile in this complex. The close contacts between these aromatic rings is likely responsible for the relatively high IC50 exhibited by ALDH2 for 3. Despite the flipped orientations of their common ring system, the indole-2,3-diones of 1 and 3 maintain the same aromatic π-stacking interactions with the side chains of Phe170 and Phe459.
Figure 1.
Interactions of 1 with ALDH2. (A) Side view of the protein surface containing compound 1 (left) and (B) top view with the original unbiased figure of merit, σA-weighted, 2Fo – Fc (blue; contoured at one standard deviation of the map) and Fo – Fc electron density map (green; contoured at 2.5 standard deviations of the map) for 1 prior to its inclusion in the model superimposed on the final refined model.
Figure 2.
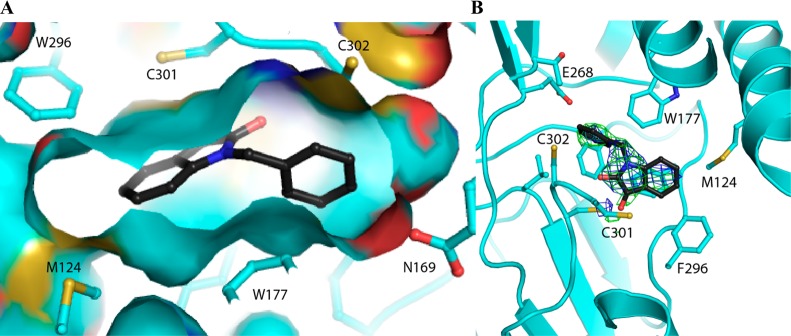
Interactions of 3 with ALDH2. (A) Side view of the protein surface containing compound 3 (left) and (B) top view with the original unbiased figure of merit, σA-weighted, 2Fo – Fc (blue; contoured at one standard deviation of the map) and Fo – Fc electron density map (green; contoured at 2.5 standard deviations of the map) for 3 prior to its inclusion in the model superimposed on the final refined model.
In contrast, the crystal structure of ALDH3A1 in a complex with 1-{[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 20) (Figure 3) shows the 3-keto group of the indole-2,3-dione ring bound within the substrate-binding site and forming an adduct with the active-site nucleophile (Cys243 in ALDH3A1). The distance between Cys243 and the carbonyl-carbon as well as the out-of-plane displacement of the carbonyl oxygen is consistent with the formation of an adduct between these two reactive groups. That this covalent bond is reversible is supported by the fact that addition of dithiothreitol to the reaction solution after preincubation restores the enzymatic activity. There is sufficient electron density to model the indole-2,3-dione and the N-piperazine moiety, but insufficient electron density is present to model the terminal benzyl-dioxol moiety. Computational placement of the benzyl-dioxol moiety onto the crystallographically observed partial structure suggests that the benzyl-dioxol lies at the interface between the exit of the substrate-binding site and bulk solvent, where it can apparently adopt multiple conformations.
Figure 3.
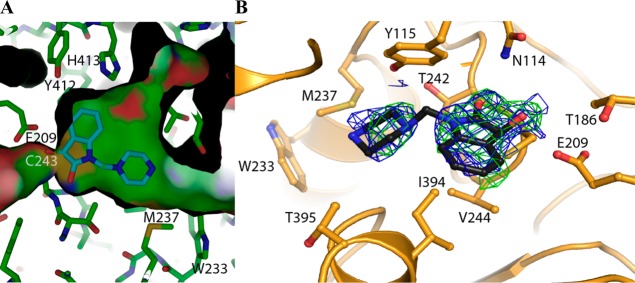
Interactions of 20 with ALDH3A1. (A) Side view of the protein surface containing the crystallgraphically observed portion of compound 20. (B) Top view with the original unbiased figure of merit, σA-weighted, 2Fo – Fc (blue; contoured at one standard deviation of the map) and Fo – Fc electron density map (green; contoured at 2.5 standard deviations of the map) prior to its inclusion in the model superimposed on the final refined model. There is insufficient electron density to fit and refine properly the terminal benzyldioxol group in the structure. Consequently, we have placed and refined only that portion that can be accounted for by the available electron density.
Discussion and Conclusions
There are 19 members of the ALDH superfamily in the human genome, with many of the family members participating in defined metabolic pathways, such as proline, valine, retinal, folate, and GABA metabolism.1 In contrast, several members have less well-defined substrate preferences and constitute a cellular defense system against endogenously and exogenously generated aldehydes. In particular, ALDH1A1, ALDH2, and ALDH3A1 demonstrate broad and overlapping substrate specificities and are frequently expressed in the same tissues or cell types.14 For instance, ALDH1A1 and ALDH3A1 have both been shown to be biomarkers for cancer as well as cancer stem cells.6,15 ALDH1A1 overexpression is indicative of high-grade ductal carcinoma, multiple myeloma, and acute myeloid leukemia.6,16−23 ALDH3A1 is generally found in stratified squamous epithelium,24−26 and both ALDH3A1 and ALDH1A1 confer resistance to the cell-killing effects of cyclophosphamide.27,28 However, ALDH2 has primary contributions to the elimination of acetaldehyde and to the bioactivation of nitroglycerin.5,7,9,29−33 All three isoenzymes are known to metabolize 4-hydroxynonenal.34,35 Thus, selective inhibitors of these general aldehyde-oxidizing enzymes could provide important research tools for the assessment of their individual contributions to their common substrates or cellular functions. Toward this end, we have examined derivatives of indole-2,3-diones for their ability to inhibit differentially these three forms of ALDH.
Members of this class of compounds were identified in separate inhibitory screens on ALDH2 and ALDH3A1, although the diversity of the substitutions on the indole rings of these initial hits suggested that selectivity could be achieved through optimization of those substitutions. That this level of selectivity could be achieved is further supported by the differences in the chemical and surface topologies of their respective substrate-binding sites. ALDH2 and ALDH3A1 share only ∼30% sequence identity, and despite sharing the common catalytic residues they have distinct substrate-binding-site characteristics (Figure 4). Similarly, ALDH2 and ALDH1A1 share 68% sequence identity, and, although their substrate-binding sites are both largely hydrophobic in nature, the middle and exterior regions of the site in ALDH1A1 are considerably wider because of smaller amino acid side chains at positions 124 and 459.36,37 It is in this context that the structure–activity relationships for these substituted indole-2,3-diones should be interpreted.
Figure 4.
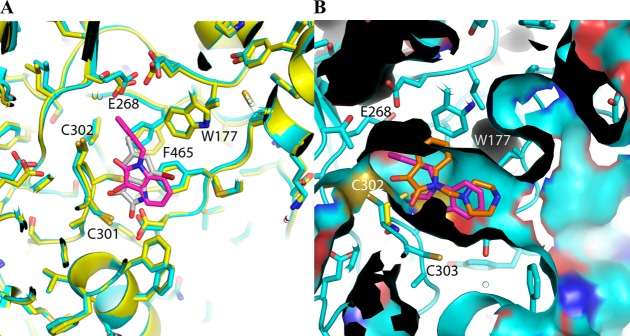
Comparison of ALDiB interactions with active-site cysteines in ALDH2 and ALDH3A1. (A) Crystal structure of ALDH2 with 1 (gray) has been overlaid with that of 3 (magenta) to show how the steric hindrance of the surrounding hydrophobic residues determines the mode of binding. (B) Surface map of 3 (magenta) bound to ALDH2 (cyan) aligned to the crystallographically observed partial structure of 20 (orange) bound to ALDH3A1 (yellow).
The kinetic data is consistent with these inhibitors binding in a manner that is competitive with respect to aldehyde binding. Although the details of the interactions between the ALDiB inhibitors and the enzymes differ in the precise details, all compounds occupy the substrate-binding site in a manner that requires their displacement prior to productive binding of substrate aldehydes. Their mixed-type noncompetitive inhibition patterns with respect to varied coenzyme is consistent with the largely ordered Bi–Bi mechanism followed by ALDH family members that have been characterized in this manner.38,39
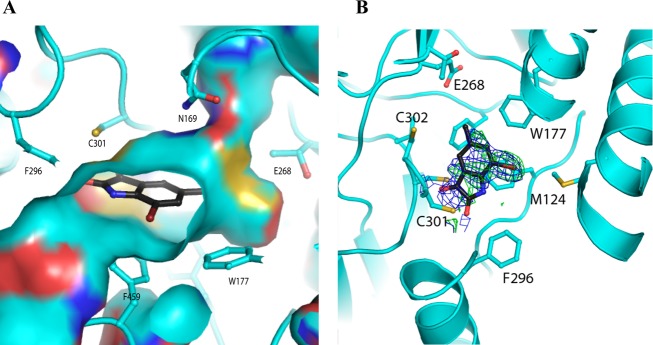
The distinct differences in the nature of the interactions between the indole-2,3-dione rings and ALDH3A1 and ALDH2 highlight the structural and functional differences of these distantly related isoenzymes. Consistent with this level of sequence identity, a structural alignment yields a rmsd of 2.1 Å for 395 similarly positioned Cα atoms. The amino acids lining their respective substrate-binding sites are both largely hydrophobic, but the differences in their positioning and identity create unique topographical features (Figures 3 and 4).
The manner in which 20 is bound within the ALDH3A1 substrate site also provides key information with respect to the manner in which ALDH3A1 selectivity is achieved when compared to the substrate-binding sites of ALDH1A1 and ALDH2 (Figure 3). It is these structural and functional differences that underlie the distinct way these substituted indole-2,3-dione molecules bind to these ALDH active sites. The 3-keto group in these inhibitory molecules is a well-known electrophile, so it was not surprising that this group would seek a reactive nucleophile within the ALDH active site. However, it was surprising that only in ALDH3A1 did these compounds form an adduct with the catalytic nucleophile, as the active site of ALDH2 is generally thought to contain the stronger cysteine nucleophile. ALDH2 generally exhibits low to submicromolar Km values for aldehydes, whereas ALDH3A1 exhibits Km values at least 2 orders of magnitude higher. Why then does 20 bind to the catalytic nucleophile in ALDH3A1 but neither 1 nor 3 does so in ALDH2? An alignment of the respective active sites demonstrates that in order for either 1 or 3 to bind to Cys302 the side chain of Trp177 has to move, but its position is restricted by the side chains of Leu173, Met174, and Leu477 (Figure 4). This explains why the indole-2,3-dione ring cannot bind productively to Cys302 in ALDH2 and why the SAR for ALDH2 was so difficult to understand before the structures of 1 and 3 were available.
The most difficult features of the ALDH2 SAR to understand were the favorability of larger halogens at the 5-position and how the changes in inhibition strength varied as the alkyl linker between the indole ring and a benzene substituent on the N1 position was lengthened. If the indole ring bound similarly to that in ALDH3A1, then we would expect the SAR on halogens at the 5-position to follow that of ALDH3A1; namely, that larger halogens are detrimental to inhibitory potency (Table 1, 8 vs 5-chloro-1-(2-phenylethyl)-1H-indole-2,3-dione (compound 7) vs 6, 20 vs 21). However, this is not the case for ALDH2, as increasing the size of the halogen at the 5-position either has little effect (1-benzyl-5-bromo-1H-indole-2,3-dione (compound 5), 1-benzyl-5-chloro-2,3-dihydro-1H-indole-2,3-dione (compound 4), and 3, Table 1) or increases potency (8 vs 7 vs 6, 10 vs 11, Table 1). In addition, lengthening the alkyl linker to the benzene ring had no real impact on potency, which is opposite that seen with aldehyde substrates where the longer and more hydrophobic the alkyl chain, the better the substrate. However, the presence of a double bond in this linker virtually abolishes inhibitory potency (10 vs 1-(3-phenylpropyl)-1H-indole-2,3-dione (compound 9), Table 1), whereas addition of a 5-chloro group to 10 (11) restores potency. This data suggests that the binding modes for the halogen versus non-halogen-substituted compounds differ. We suggest that the binding mode observed for 3 in ALDH2 is maintained for group 2 compounds lacking halogens at the 5-position (Table 1), as the ability of the longer alkyl chains to adopt new conformations necessary to exit the active site past the position of Cys302 requires a flexible linker. However, this binding mode is inconsistent with the increased potency of halogen substitution at the 5-position because the 5-position of 3 is within van der Waals contact to Val120, Met124, and Phe296. Consequently, the SAR would suggest that 5-halogen-substituted group 2 compounds adopt the position observed for 1, where the 5-methyl substituent will approximate the position of the 5-chloro or 5-bromo substituents and the indole nitrogen is now pointing toward the solvent-exposed exit of the substrate-binding site.
In addition, the ALDH2 crystal structures presented here show that Cys302 is pointing in the direction of the cofactor pocket and away from the substrate pocket, which has been referred to as the resting conformation.40 Lang et al. suggested that this resting conformation contributes to Cys301/303 having the primary role in stimulation of the Cys302 thio-carbonyl adduct formation, lowering the acidity of Cys302.30 Furthermore, comparison of ALDH2 with other enzyme family members suggests that Cys303 interacts with substrates via hydrogen bonding or by an ion–dipole interaction.14 The crystal structures of 1 and 3 presented here suggest that Cys301/303 may contribute to substrate binding through a trapping mechanism and possibly by serving to dehydrate the hydrated aldehydes that form in solution. Weiner and colleagues proposed that elements of the enzyme’s proton-relay system performed this function, but this proposal preceded structure determination by 12 years.47,48 The proton-relay system is identical in ALDH1A1 and ALDH3A1, but the two residues immediately surrounding the catalytic nucleophile in the active-site loop are not. In this regard, ALDH1A1 has Ile at 301 and a single additional Cys residue at 303, whereas ALDH3A1 lacks Cys residues at either equivalent position (Thr and Val, respectively), which correlates with their decreased catalytic efficiency for small aliphatic aldehydes with the greatest level of hydration.49
In contrast to the complex SAR for ALDH2, selectivity for ALDH3A1 is more simply achieved through the addition of a nonaromatic ring system linked by a single carbon atom to the indole nitrogen (Table 1). This addition to the indole ring alone abrogates any potency toward ALDH2 and severely diminishes potency toward ALDH1A1. Addition of hydrophobic substituents at the 4-position of the piperazine ring further improves potency up to 4-fold (1-(4-morpholinylmethyl)-1H-indole-2,3-dione (compound 12) versus 20) while still maintaining a 40-fold selectivity over ALDH1A1. Although the benzyl-dioxol substituent is disordered in our crystal structure, presumably because of the multiple binding modes near the exit of the substrate-binding site, these additional hydrophobic substituents likely interact with the side chains of Trp233 and Met237 (Figure 3). Substitution of the central nonaromatic ring with an aromatic ring reduces potency by about 6-fold (3 versus 12). This is likely due to the generation of an unfavorable steric contact between the aromatic ring and Met237 (Figure 3). Addition of one or more additional linking carbon atoms between the indole and aromatic rings improves potency by more than 15-fold, presumably by bypassing the restrictive space occupied by Met237 (3 vs 6 or 9, Table 1). However, these changes come at the cost of selectivity for ALDH3A1 as neither 6 nor 9 are selective inhibitors for ALDH3A1. Another unique characteristic of ALDH3A1 is the negative impact that substitution of large halogen atoms at the 5-position of the indole ring has on potency. This effect can be explained by the close proximity of the side chains of Phe401, Tyr412, and His413 (Figure 3). Substitution of anything larger than a hydrogen atom would crowd this location.
With the exception of the compounds in group 3, most aliphatic or aromatic N-substituted indole-2,3-diones are potent inhibitors of ALDH1A1 (Table 1). Only the substitution of a nonaromatic ring linked to the indole nitrogen or the presence of a 5-bromo group is detrimental to ALDH1A1 potency. Consistent with its ability to oxidize retinaldehyde with high efficiency, ALDH1A1 has the largest and most hydrophobic substrate-binding site,36 and substituents on the indole nitrogen that enhance hydrophobic interactions improve the potency of these compounds toward this isoenzyme. The structure–activity data for ALDH1A1 is consistent with a binding mode for the substituted indole-2,3-diones that is similar to that for 20 in ALDH3A1. This data is supported by presence of Gly124 in ALDH1A1, rather than Met124 in ALDH2; this substitution provides some additional room for a 5-chloro group but is still insufficient for a 5-bromo substitution, which would still clash with Trp177. 3 is the most selective ALDH1A1 inhibitor (Table 1).
The aim of this work was to develop further a new class of small molecules that can be adapted to selectively inhibit ALDH enzymes. As such, several compounds were identified that show reasonable selectivity (>40-fold) toward ALDH1A1 (3), ALDH2 (8), or ALDH3A1 (20 or 1-{[4-(4-fluorobenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 16)) based on the size and chemical characteristics of each isoenzyme’s substrate-binding site. The competition assays show that these inhibitors are noncompetitive with respect to the coenzyme-binding site and are competitive toward the substrate-binding site. X-ray crystallography data confirms inhibition occurs through direct interactions between the 3-keto group of the indole ring and conserved cysteine residues within the active site. Selective inhibition can be achieved through substitutions at the C5 and N1 positions of the indole ring system. These compounds may act as useful tools in elucidating the contributions of individual ALDH isoenzymes toward specific, as well as overlapping, metabolic pathways.
Experimental Procedures
Materials
Compounds used for the inhibitor screen were found using a 80% structure similarity search against 5-methyl-1H-indole-2,3-dione using the PubChem project.41 The following compounds were purchased from ChemBridge for further in vitro assay screening: 5301889, 1-benzyl-5-bromo-1H-indole-2,3-dione (5); 5192630, 1-(2-phenylethyl)-1H-indole-2,3-dione (compound 6); 7196590, 5-chloro-1-(2-phenylethyl)-1H-indole-2,3-dione (7); 6047303, 5-bromo-1-(2-phenylethyl)-1H-indole-2,3-dione (8); 6378722, 7-bromo-5-methyl-1H-indole-2,3-dione (1); 6433626, 1-(3-phenylpropyl)-1H-indole-2,3-dione (9); 6997087, 1-(3-phenyl-2-propen-1-yl)-1H-indole-2,3-dione (compound 10); 6505720, 1-{[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (20); 8918814, 1-{[4-(4-fluorobenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (16); 8918815, 1-{[4-(3-chlorobenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 18); 8918818, 1-{[4-(2-fluorobenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 15); 8918819, 1-{[4-(3-fluorobenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 17); 8919812, 1-{[4-(3-methoxybenzyl)-1-piperazinyl]methyl}-1H-indole-2,3-dione (compound 19); 6989645, 1-{[4-(1,3-benzodioxol-5-ylmethyl)-1-piperazinyl]methyl}-5-bromo-1H-indole-2,3-dione (21); 5250099, 1,1′-[1,3-imidazolidinediylbis(methylene)]bis(1H-indole-2,3-dione) (compound 22); 5115436, 1-(4-morpholinylmethyl)-1H-indole-2,3-dione (12); 5260280, 1-[(4-methyl-1-piperazinyl)methyl]-1H-indole-2,3-dione (compound 13); and 5115499, 1-[(4-benzyl-1-piperazinyl)methyl]-1H-indole-2,3-dione (compound 14). The following compounds were purchased from ChemDiv for further screening: 0764-0625, 1-pentyl-2,3-dihydro-1H-indole-2,3-dione (compound 2); 0764-0628, 1-benzyl-2,3-dihydro-1H-indole-2,3-dione (3); and 5353-0854, 1-benzyl-5-chloro-2,3-dihydro-1H-indole-2,3-dione (4). On the basis of the SAR data collected, compound BBV-138984, 5-chloro-1-[(2E)-3-phenylprop-2-en-1-yl]-2,3-dihydro-1H-indole-2,3-dione (11), was purchased from MolPort as proof of the selective-inhibition scheme. The NMR spectra confirming >95% compound purity provided by the vendors is included in the Supporting Information. All other chemicals were purchased from Sigma-Aldrich unless otherwise stated.
Protein Purification
Human ALDH1A1 (hALDH1A1), ALDH2 (hALDH2), and ALDH3A1 (hALDH3A1) were expressed and purified as described elsewhere.13,33,42,43
Inhibitor Screens
The ALDH2 and ALDH3A1 screening assays have been reported previously.13 ALDH1A1 was screened using the same assay as that of ALDH2. Inhibitors that emerged from the screens were purchased from ChemBridge or ChemDiv.
Crystallization of ALDH3A1 and ALDH2 Complexes with Inhibitors
Crystals of ALDH3A1 at 3 mg/mL in 10 mM HEPES, pH 7.5, were grown from solutions containing 0.2 M potassium acetate and 18% PEG3350 at 25 °C. Enzyme complexes with inhibitors were generated through direct-soaking experiments by first equilibrating the crystals overnight in a solution containing 2% DMSO, which was then supplemented with 100 μM compound 20. The crystals were directly frozen without any further addition of cryoprotectant.
Crystals of ALDH2 were grown from protein solutions containing 8 mg/mL of ALDH2 in 100 mM ACES, pH 6.2–6.8, 100 mM guanidine-HCl, 10 mM MgCl2, and 14–19% PEG 6000. The ALDH2 complexes with inhibitors were generated through direct-soaking experiments by first equilibrating the crystals overnight in solutions containing 2% DMSO, which was then supplemented with 100 μM of either compound 1 or 3.
All diffraction data were collected at a wavelength of 0.9869 Å and at 100 K. All diffraction data were indexed, integrated, and scaled using the HKL3000 program suite.44 Refinement was performed using Refmac5 within the CCP4 program suite.44,45 The structure of the ALDH3A1 complex with 20 was solved by molecular replacement using the ALDH3A1 apo structure (PDB code 3SZA). The presence of inhibitor was determined after inspection of the initial σ-A weighted Fo – Fc electron density maps. The structures of ALDH2 complexed with 1 and 3 were solved using the coordinates of the refined ALDH2 in the P21 space group after removal of solvent and ligands (1CW3). Data collection and refinement statistics are given in Table 4. The ALDH2 complex with 1 demonstrates high-occupancy binding in subunits A and H and insufficient electron density for unequivocal assignment in the remaining subunits. The ALDH2 complex with 3 shows high-occupancy binding in subunits A, B, E, and H and insufficient electron density for unequivocal assignment in the remaining subunits.
Table 4. Data Collection and Refinement Statistics.
| crystal | ALDH2 + 1 | ALDH2 + 3 | ALDH3 + 20 |
|---|---|---|---|
| PDB code | 4KWG | 4KWF | 4L1O |
| Data Collection | |||
| beamline | APS, GM/CA-CAT | APS, GM/CA-CAT | RAXIS IV+2 |
| wavelength (Å) | 0.98 | 0.98 | 1.54 |
| space group | P21 | P21 | P212121 |
| cell dimensions (Å) | a = 101.5 | a = 102.3 | a = 61.3 |
| b = 176.2 | b = 177.1 | b = 86.4 | |
| c = 101.5 | c = 102.6 | c = 170.2 | |
| cell dimensions (deg) | α = 90.00 | α = 90.00 | α = 90.0 |
| β = 94.92 | β = 94.39 | β = 90.0 | |
| γ = 90.00 | γ = 90.00 | γ = 90.0 | |
| no. of reflections | 193 565 | 150 293 | 39 454 |
| resolution limit (Å) | 2.00 (2.03 – 2.00) | 2.3 (2.34–2.30) | 2.3 (2.34–2.30) |
| completeness (%) | 94.5 (85.9) | 98.9 (97.0) | 96.0 (88.0) |
| redundancy | 3.5 (3.3) | 3.1 (2.9) | 3.4 (2.7) |
| mean I/σI | 7.66 (19.3) | 2.40 (12.88) | 16.3 (6.7) |
| Rmerge (%) | 5.8 (15.8) | 7.2 (44.5) | 6.0 (14.6) |
| Refinement | |||
| resolution range (Å) | 40.5–2.19 | 49.12–2.31 | 48 – 2.30 |
| Rwork/Rfree (%) | 19.3/22.9 | 23.9/29.6 | 17.5/22.2 |
| no. of atoms | |||
| total | 31 640 | 30 636 | 7610 |
| protein | 30 384 | 30 384 | 6913 |
| ligand/ion | 68 | 68 | 12 |
| water | 1162 | 188 | 649 |
| inhibitor | 26 | 72 | 36 |
| average B-factors (Å2) | |||
| total | 21.78 | 53.12 | 18.47 |
| protein | 21.37 | 53.10 | 18.4 |
| ligand/ion | 39.62 | 75.74 | 27.40/36.93 |
| water | 30.74 | 50.23 | 21.03 |
| inhibitor | 53.71 | 51.46 | 32.19 |
| root-mean-square deviations | |||
| bond lengths (Å) | 0.005 | 0.007 | 0.006 |
| bond angles (deg) | 0.969 | 1.188 | 1.143 |
IC50 Determination
IC50 inhibition curves for the inhibitors were measured using the activity of hALDH2, hALDH1A1, and hALDH3A1 as described elsewhere.13 In short, the enzyme was incubated with the inhibitor and coenzyme for 2 min prior to initiation of the reaction with aldehyde substrate. The inhibition curves were fit to the four-parameter EC50 equation using SigmaPlot (version 11, StatSys). All data represent the average of three independent experiments.
Determination of Mode of Inhibition
Inhibition of ALDH activity was measured using the activity of hALDH2, hALDH1A1, and hALDH3A1 to determine the kinetic mode of inhibition versus varied coenzyme or aldehyde substrate. Coenzyme competition was determined by measuring ALDH activity for various concentrations of compound while varying the concentration of NAD+ for hALDH2 and hALDH1A1 and NADP+ for hALDH3A1. Likewise, substrate competition was determined for different concentrations of each compound while varying propionaldehyde concentration for hALDH2 and hALDH1A1 and benzaldehyde concentration for hALDH3A1. In each experiment, the concentration of the nonvaried substrate was set to saturating levels, and the varied substrate concentration ranged at least 10-fold spanning the calculated Km value. Similarly, the concentration range for the inhibitors was varied a minimum of 5-fold, exclusive of the control (no inhibitor) reactions, which spanned the calculated Ki values. The kinetic mode of inhibition was determined by fitting data to the competitive, noncompetitive, and uncompetitive inhibition equations and evaluating the goodness of fit to each equation.46 Data fitting and analysis was performed using SigmaPlot (version 11.0) with the enzyme kinetics module (version 1.3).
Acknowledgments
This work was supported in whole or in part by National Institutes of Health grants R01-AA018123 and R21-AA019746 to T.D.H. and R01-AA11147 to D.M.-R. A.K.-H was supported by R01-AA018123-S1. We thank Lan Chen and the Chemical Genomics facility for access to their instrumentation. Results shown in this article are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We thank Stephan Ginell and Marianne Cuff for their assistance at SBC-CAT 19ID.
Glossary
Abbreviations Used
- ALDH
aldehyde dehydrogenase
- DMSO
dimethyl sulfoxide
- EC50
half maximal effective concentration
- NAD
Nicotinamide adenine dinucleotide
- PEG
poly(ethylene glycol)
- SAR
structure–activity relationship
Supporting Information Available
Comparisons of the kinetic inhibition analysis of the ALDH isoforms and compound structure verification by various analytical techniques. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, New Jersey, with entry codes 4KWG, 4KWF, and 4L1O.
Author Contributions
A.K.-H. performed experiments, data analysis, and manuscript preparation; B.P. performed experiments and data analysis; C.-H.C. performed experiments; D.M.-R. initiated the study and manuscript preparation; and T.D.H. designed the study, data analysis, and manuscript preparation.
The authors declare the following competing financial interest(s): Daria Mochly-Rosen and Che-Hong Chen are the founders and shareholders of ALDEA Pharmaceuticals. However, none of the work described in this study is based on or supported by the company. Che-Hong Chen and Daria Mochly-Rosen are listed as inventors in pending patents filed by Stanford University that are related to this work. Thomas D. Hurley holds significant financial equity in SAJE Pharma, LLC. However, none of the work described in this study is related to, based on, or supported by the company.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Black W.; Vasiliou V. The aldehyde dehydrogenase gene superfamily resource center. Hum. Genomics 2009, 4, 136–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou V.; Pappa A.; Petersen D. R. Role of aldehyde dehydrogenases in endogenous and xenobiotic metabolism. Chem.-Biol. Interact. 2000, 129, 1–19. [DOI] [PubMed] [Google Scholar]
- Rizzo W. B.; Carney G. Sjögren-Larsson syndrome: Diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2). Hum. Mutat. 2005, 26, 1–10. [DOI] [PubMed] [Google Scholar]
- Akaboshi S.; Hogema B. M.; Novelletto A.; Malaspina P.; Salomons G. S.; Maropoulos G. D.; Jakobs C.; Grompe M.; Gibson K. M. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum. Mutat. 2003, 22, 442–450. [DOI] [PubMed] [Google Scholar]
- Enomoto N.; Takase S.; Takada N.; Takada A. Alcoholic liver disease in heterozygotes of mutant and normal aldehyde dehydrogenase-2 genes. Hepatology 1991, 13, 1071–1075. [PubMed] [Google Scholar]
- Ginestier C.; Hur M. H.; Charafe-Jauffret E.; Monville F.; Dutcher J.; Brown M.; Jacquemier J.; Viens P.; Kleer C. G.; Liu S.; Schott A.; Hayes D.; Birnbaum D.; Wicha M. S.; Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-H.; Budas G. R.; Churchill E. N.; Disatnik M.-H.; Hurley T. D.; Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Miller S.; Younus H.; Vanam R.; Chen C.-H.; Mochly-Rosen D.; Hurley T. D. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat. Struct. Mol. Biol. 2010, 17, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arolfo M. P.; Overstreet D. H.; Yao L.; Fan P.; Lawrence A. J.; Tao G.; Keung W.-M.; Vallee B. L.; Olive M. F.; Gass J. T.; Rubin E.; Anni H.; Hodge C. W.; Besheer J.; Zablocki J.; Leung K.; Blackburn B. K.; Lange L. G.; Diamond I. Suppression of heavy drinking and alcohol seeking by a selective ALDH-2 inhibitor. Alcohol.: Clin. Exp. Res. 2009, 33, 1935–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton J. Role of aldehyde dehydrogenase in cyclophosphamide-resistant L1210 leukemia. Cancer Res. 1984, 44, 5156–5160. [PubMed] [Google Scholar]
- von Eitzen U.; Meier-Tackmann D.; Agarwal D. P.; Goedde H. W. Detoxification of cyclophosphamide by human aldehyde dehydrogenase isozymes. Cancer Lett. 1994, 76, 45–49. [DOI] [PubMed] [Google Scholar]
- Khanna M.; Chen C.-H.; Kimble-Hill A.; Parajuli B.; Perez-Miller S.; Baskaran S.; Kim J.; Dria K.; Vasiliou V.; Mochly-Rosen D.; Hurley T. D. Discovery of a novel class of covalent inhibitor for aldehyde dehydrogenases. J. Biol. Chem. 2011, 286, 43486–43494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli B.; Kimble-Hill A. C.; Khanna M.; Ivanova Y.; Meroueh S.; Hurley T. D. Discovery of novel regulators of aldehyde dehydrogenase isoenzymes. Chem.-Biol. Interact. 2011, 191, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveros-Rosas H.; González-Segura L.; Julián-Sánchez A.; Díaz-Sánchez Á. G.; Muñoz-Clares R. A. Structural determinants of substrate specificity in aldehyde dehydrogenases. Chem.-Biol. Interact. 2013, 202, 51–61. [DOI] [PubMed] [Google Scholar]
- Nalwoga H.; Arnes J. B.; Wabinga H.; Akslen L. A. Expression of aldehyde dehydrogenase 1 (ALDH1) is associated with basal-like markers and features of aggressive tumours in African breast cancer. Br. J. Cancer 2009, 102, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui W.; Wang Q.; Barber J. P.; Brennan S.; Smith B. D.; Borrello I.; McNiece I.; Lin L.; Ambinder R. F.; Peacock C.; Watkins D. N.; Huff C. A.; Jones R. J. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008, 68, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A. M. S.; Wan T. S. K.; Leung J. C. K.; Chan L. Y. Y.; Huang H.; Kwong Y. L.; Liang R.; Leung A. Y. H. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia 2007, 21, 1423–1430. [DOI] [PubMed] [Google Scholar]
- Gerber J. M.; Qin L.; Kowalski J.; Smith B. D.; Griffin C. A.; Vala M. S.; Collector M. I.; Perkins B.; Zahurak M.; Matsui W.; Gocke C. D.; Sharkis S. J.; Levitsky H. I.; Jones R. J. Characterization of chronic myeloid leukemia stem cells. Am. J. Hematol. 2011, 86, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope K. J.; Jin L.; Dick J. E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [DOI] [PubMed] [Google Scholar]
- Kantarjian H. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N. Engl. J. Med. 2002, 346, 645–652. [DOI] [PubMed] [Google Scholar]
- Pearce D. J.; Taussig D.; Simpson C.; Allen K.; Rohatiner A. Z.; Lister T. A. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. 2005, 23, 752–760. [DOI] [PubMed] [Google Scholar]
- Stirewalt D. L.; Meshinchi S.; Kopecky K. J.; Fan W.; Pogosova-Agadjanyan E. L.; Engel J. H.; Cronk M. R.; Dorcy K. S.; McQuary A. R.; Hockenbery D.; Wood B.; Heimfeld S.; Radich J. P. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes, Chromosomes Cancer 2008, 47, 8–20. [DOI] [PubMed] [Google Scholar]
- Sutherland H. J.; Blair A.; Zapf R. W. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood 1996, 87, 4754–4761. [PubMed] [Google Scholar]
- Boelens M. C.; van den Berg A.; Fehrmann R. S. N.; Geerlings M.; de Jong W. K.; te Meerman G. J.; Sietsma H.; Timens W.; Postma D. S.; Groen H. J. M. Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. J. Pathol. 2009, 218, 182–191. [DOI] [PubMed] [Google Scholar]
- Patel M.; Lu L.; Zander D. S.; Sreerama L.; Coco D.; Moreb J. S. ALDH1A1 and ALDH3A1 expression in lung cancers: Correlation with histologic type and potential precursors. Lung Cancer 2008, 59, 340–349. [DOI] [PubMed] [Google Scholar]
- Spira A.; Beane J.; Shah V.; Liu G.; Schembri F.; Yang X.; Palma J.; Brody J. S. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 10143–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreb J.; Muhoczy D.; Ostmark B.; Zucali J. RNAi-mediated knockdown of aldehyde dehydrogenase class-1A1 and class-3A1 is specific and reveals that each contributes equally to the resistance against 4-hydroperoxycyclophosphamide. Cancer Chemother. Pharmacol. 2007, 59, 127–136. [DOI] [PubMed] [Google Scholar]
- Sládek N. E. Aldehyde dehydrogenase-mediated cellular relative insensitivity to the oxazaphosphorines. Curr. Pharm. Des. 1999, 5, 607–625. [PubMed] [Google Scholar]
- Beretta M.; Gruber K.; Kollau A.; Russwurm M.; Koesling D.; Goessler W.; Keung W. M.; Schmidt K.; Mayer B. Bioactivation of nitroglycerin by purified mitochondrial and cytosolic aldehyde dehydrogenases. J. Biol. Chem. 2008, 283, 17873–17880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang B. S.; Gorren A. C. F.; Oberdorfer G.; Wenzl M. V.; Furdui C. M.; Poole L. B.; Mayer B.; Gruber K. Vascular bioactivation of nitroglycerin by aldehyde dehydrogenase-2: Reaction intermediates revealed by crystallography and mass spectrometry. J. Biol. Chem. 2012, 287, 38124–38134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Taniyama M.; Muramatsu T.; Higuchi S.; Ohta S.; Atsumi Y.; Matsuoka K. ALDH2/ADH2 Polymorphism associated with vasculopathy and neuropathy in type 2 diabetes. Alcohol.: Clin. Exp. Res. 2004, 28, 111S–116S. [DOI] [PubMed] [Google Scholar]
- Wenzl M. V.; Beretta M.; Griesberger M.; Russwurm M.; Koesling D.; Schmidt K.; Mayer B.; Gorren A. C. F. Site-directed mutagenesis of aldehyde dehydrogenase-2 suggests three distinct pathways of nitroglycerin biotransformation. Mol. Pharmacol. 2011, 80, 258–266. [DOI] [PubMed] [Google Scholar]
- Ni L.; Sheikh S.; Weiner H. Involvement of glutamate 399 and lysine 192 in the mechanism of human liver mitochondrial aldehyde dehydrogenase. J. Biol. Chem. 1997, 272, 18823–18826. [DOI] [PubMed] [Google Scholar]
- Hartley D. P.; Ruth J. A.; Petersen D. R. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch. Biochem. Biophys. 1995, 316, 197–205. [DOI] [PubMed] [Google Scholar]
- Schaur R. J. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 2003, 24, 149–159. [DOI] [PubMed] [Google Scholar]
- Moore S. A.; Baker H. M.; Blythe T. J.; Kitson K. E.; Kitson T. M.; Baker E. N. Sheep liver cytosolic aldehyde dehydrogenase: The structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases. Structure 1998, 6, 1541–1551. [DOI] [PubMed] [Google Scholar]
- Hurley T. D.; Perez-Miller S.; Breen H. Order and disorder in mitochondrial aldehyde dehydrogenase. Chem.-Biol. Interact. 2001, 130–132, 3–14. [DOI] [PubMed] [Google Scholar]
- Blackwell L.; Motion R.; MacGibbon A.; Hardman M.; Buckley P. Evidence that the slow conformation change controlling NADH release from the enzyme is rate-limiting during the oxidation of propionaldehyde by aldehyde dehydrogenase. Biochem. J. 1987, 242, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho K. K.; Allali-Hassani A.; Hurley T. D.; Weiner H. Differential effects of Mg2+ ions on the individual kinetic steps of human cytosolic and mitochondrial aldehyde dehydrogenases. Biochemistry 2005, 44, 8022–8029. [DOI] [PubMed] [Google Scholar]
- González-Segura L.; Rudiño-Piñera E.; Muñoz-Clares R. A.; Horjales E. The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa provides new insight into the reaction mechanism and shows a novel binding mode of the 2′-phosphate of NADP+ and a novel cation binding site. J. Mol. Biol. 2009, 385, 542–557. [DOI] [PubMed] [Google Scholar]
- Weiner H.; Freytag S.; Fox J. M.; Hu J. H. Reversible inhibitors of aldehyde dehydrogenase. Prog. Clin. Biol. Res. 1982, 114, 91–102. [PubMed] [Google Scholar]
- Weiner H.; Lin F. P.; Sanny C. G. Chemical probes for the active site of aldehyde dehydrogenase. Prog. Clin. Biol. Res. 1985, 174, 57–70. [PubMed] [Google Scholar]
- Bell R. P. The reversible hydration of carbonyl compounds. Adv. Phys. Org. Chem. 1966, 4, 1–29. [Google Scholar]
- Bolton E. E.; Wang Y.; Thiessen P. A.; Bryant S. H.. PubChem: Integrated platform of small molecules and biological activities. In Annual Reports in Computational Chemistry; Ralph A. W., David C. S., Eds.; Elsevier: Boston, MA, 2008; Chapter 12, pp 217–241. [Google Scholar]
- Jeng J.; Weiner H. Purification and characterization of catalytically active precursor of rat liver mitochondrial aldehyde dehydrogenase expressed in Escherichia coli. Arch. Biochem. Biophys. 1991, 289, 214–222. [DOI] [PubMed] [Google Scholar]
- Perez-Miller S. J.; Hurley T. D. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 2003, 42, 7100–7109. [DOI] [PubMed] [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. HKL-3000: The integration of data reduction and structure solution – from diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Segel I. H.Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems; Wiley: New York, 1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.