

Abstract
Nucleic acids are the molecular targets of many clinical anticancer drugs. However, compared with proteins, nucleic acids have traditionally attracted much less attention as drug targets in structure-based drug design, partially because limited structural information of nucleic acids complexed with potential drugs is available. Over the past several years, enormous progresses in nucleic acid crystallization, heavy-atom derivatization, phasing, and structural biology have been made. Many complicated nucleic acid structures have been determined, providing new insights into the molecular functions and interactions of nucleic acids, especially DNAs complexed with small molecule ligands. Thus, opportunities have been created to further discover nucleic acid-targeting drugs for disease treatments. This review focuses on the structure studies of DNAs complexed with small molecule ligands for discovering lead compounds, drug candidates, and/or therapeutics.
Keywords: nucleic acids, structure-based drug discovery, small molecule ligands, modification and derivatization, structures of DNA–ligand complexes
1. INTRODUCTION
Structure-based drug design (SBDD) has been used in the pharmaceutical industry for over 25 years as a guiding approach to identify lead compounds and develop new therapeutics. The success of SBDD mainly depends on the rapid advances in structural biology, which provides the detailed three-dimensional (3D) information of the drug targets and, more importantly, sheds light on the interactions between the targets and small molecule ligands. Based on the structures, novel small molecules can be designed by molecular modeling for additional screening. Combined with the co-crystallization studies, optimized compounds with better specificity and affinity can be further designed.1 The biological macromolecules, including protein and nucleic acid, have naturally become the drug targets. Nucleic acids play important roles in living systems via participation in genetic information storage, replication, transcription, and translation directing protein synthesis. Catalytic RNAs also play critical roles in biological reactions as catalysts.2–5 Thus, targeting nucleic acids can selectively disrupt gene expression for treating virus-caused diseases and cancers at the genetic level, that is, the nucleic acid level. Actually, it has already been established that nucleic acids are the molecular targets of many chemotherapeutic anticancer drugs used clinically. Compared to proteins, however, nucleic acids have traditionally gained much less attention in the SBDD field, probably because it is challenging to specifically recognize nucleic acids by small molecules and it lacks structural information of nucleic acids in complex with lead compounds and drug candidates. Fortunately, over the past several years, the fields of nucleic acid chemistry and molecular and structural biology have made tremendous progresses. Many nucleic acid structures of various complexities have been determined, including more than 300 structures of ligand–DNA or ligand–RNA complexes deposited in the structural databases since 2004. The structural data provide the molecular mechanisms and fundamental functions of many nucleic acids and their complexes with ligands. Therefore, opportunities have been created for further design and development of nucleic acid-targeting drugs to treat genetic and acquired diseases. Herein we mainly highlight the recent advancements in structural studies of complexes of DNAs and small molecules (Table I) for potential drug discovery and investigation of DNA–ligand interactions.
Table I.
Summary of the DNA-Targeting Small Molecules as Drugs or Therapeutic Candidates
| Entry | Name | Chemical structure | DNA sequences | PDB | Ref. |
|---|---|---|---|---|---|
| (A) Covalent binders | |||||
| 1 | Cisplatin | 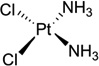 |
5′-CCTCTGGTCTCC-3′ 3′-GGAGACCAGAGG-5′ |
1AIO 3LPV |
15, 16 |
| 2 | Cisplatin | 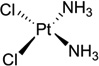 |
5′-CCUCTCTGGACCTTCC-3′ 3′-GGAGAGACCTGGAAGG-5′ |
1CKT | 18 |
| 3 | Cisplatin | 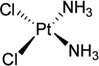 |
5′-TCTTCTGTGCTCACCAC-3′ 5′-GTGGTGAGC-3′ |
2WTF | 20 |
| 4 | Nitrogen mustard | 5′-(CGCGAATTCGCG)2-3′ | 2QEF | 25 | |
| 5 | Mitomycin C |  |
5′-ICACGTCIT-3′ 3′-CGTGCAGCA-5′ |
199D | 27 |
| 6 | 2,7-diaminomitosene | 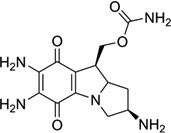 |
5′-(GTGGTATACCAC)2-3′ | 1JOI | 28 |
| 7 | 4′-(hydroxymethyl)-4,5′, 8-trimethylpsoralen |
 |
5′-(CCGGTACCGG)2-3′ | 204D | 29 |
| 8 | AFB-FAPY | 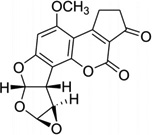 |
5′-CTATGATTCA-3′ 3′-GATACTAAGT-5′ |
2KH3 | 33 |
| (B) Minor and major groove binders | |||||
| 9 | DAPI | 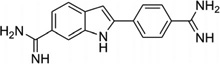 |
5′-(CGCGAATTCGCG)2-3′ | 1D30 | 52 |
| 10 | Berenil |  |
5′-(CGCGAATTCGCG)2-3′ | 2DBE | 54 |
| 11 | Pentamidine |  |
5′-(CGCGAATTCGCG)2-3′ | 1D64 | 55 |
| 12 | Hoechst 33258 |  |
5′-(CGCGAATTCGCG)2-3′ | 8BNA | 53 |
| 13 | Netropsin |  |
5′-(C5-BrCCCCIIIII)2-3′ | 358D | 51 |
| 14 | Distamycin | 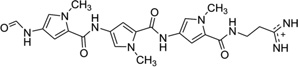 |
5′-d(GTATATAC)2-3′ | 378D | 55 |
| 15 | 8-ring cyclic Py-Im polyamide-α |
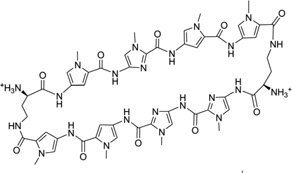 |
5′-(CCAGGCCTGG)2-3′ | 315L | 66 |
| 16 | 8-ring cyclic Py-Im polyamide-β |
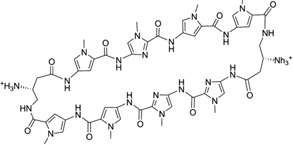 |
5′-(CCAGTACTGG)2-3′ | 3OMJ | 70 |
| 17 | KWKK | 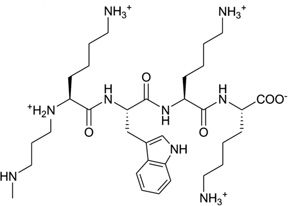 |
5′-GCTAGCGAGTCC-3′ 3′-CGATCGCTCAGG-5′ |
2KV6 | 76 |
| 18 | Pentamidine | 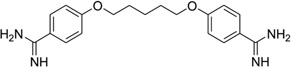 |
5′-(ATATATATAT)2-3′ | 3EY0 | 78 |
| 19 | Hoechst 33258 | 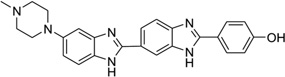 |
5′-(CGCGGATfUCGCG)2-3′ | 3AJK | 80 |
| 20 | DB921 |  |
5′-(CGAATTCGCG)2-3′ | 2B0K | 84 |
| 21 | DB1055 | 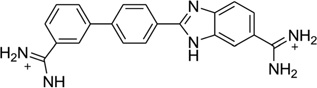 |
5′-(CGCGAATTCGCG)2-3′ | 2I5A | 85 |
| 22 | Netropsin | 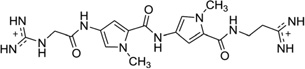 |
5′-(CTTAATTCGAATTAAG)2 | 1ZTT | 86 |
| 23 | CD27 | 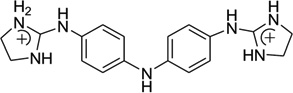 |
5′-(CTTAATTCGAATTAAG)2 | 3FSI | 87 |
| 24 | NCSi-gb | 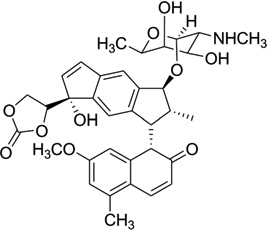 |
5′-CCCGATGC-3′ 3′-GGGCTTAACG-5′ |
1KVH | 98 |
| 25 | NCSi-glu | 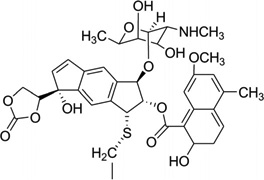 |
5′-(GCCAGAGAGC)2-3′ | 1MP7 | 99 |
| 26 | Neomycin-Hoechst- NH1 |
 |
5′-(CGCAAATTTGCG)2-3′ | Model | 103 |
| 27 | Neomycin-Hoechst- NH2 |
 |
5′-(CGCAAATTTGCG)2-3′ | Model | 103 |
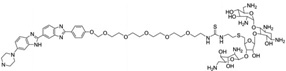
| 28 | Neomycin-Hoechst- pyrene (NHP) |
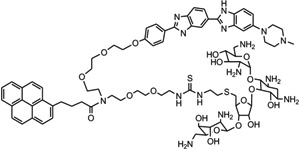 |
5′-(CGCAAATTTGCG)2-3′ | Model | 104 |
| (C) Intercalators | |||||
| 29 | Daunomycin | 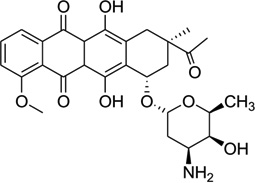 |
5′-(dC)(rG)d(ATCG)2-3′ | 1D10 | 124 |
| 30 | Adriamycin | 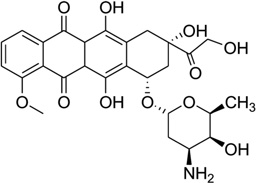 |
5′-d(CGATCG)2-3′ | 1D12 | 118 |
| 31 | Ditercalinium | 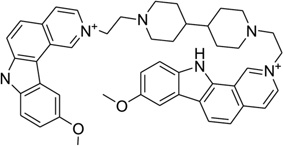 |
5′-d(CGCG)2-3′ | 1D32 | 131 |
| 32 | Echinomycin | 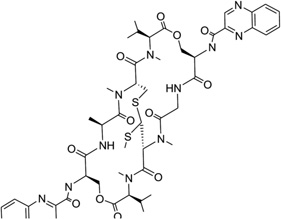 |
5′-d(GCGTACGC)2-3′ | 1PFE | 140 |
| 33 | Echinomycin | 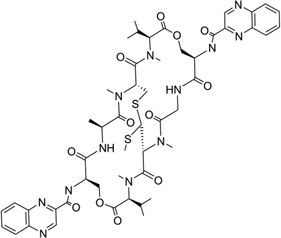 |
5′-(ACGTACGT)2-3′ | 2ADW | 141 |
| 34 | Cryptolepine | 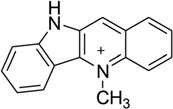 |
5′-(CGTACG)2-3′ | 1K9G | 143 |
| (D) Multimode binders | |||||
| 35 | PBD-BIMZ | 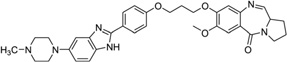 |
5′-(AACAATTGTT)2-3′ | 2KTT | 147 |
| 36 | PDB-naphthalimide | 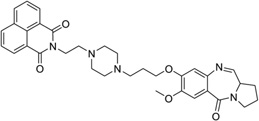 |
5′-d(AACAATTGTT)2-3′ | 2KY7 | 148 |
| (E) Quadruplex binders | |||||
| 37 | Daunomycin | 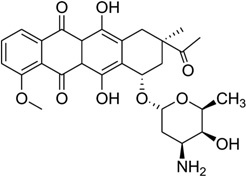 |
5′-d(TGGGGT)4-3′ | 1O0K | 163 |
| 38 | Distamycin A | 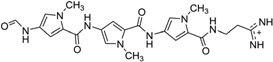 |
5′-d(TGGGGT)4-3′ | 2JT7 | 166 |
| (F) Bulge, junctions and phosphate backbone binders | |||||
| 39 | SCA-α2 | 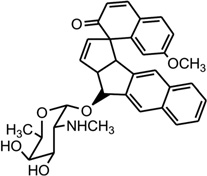 |
5′-(CCATCGTCTACCTTTGGT AGGATGG)2-3′ |
2OEY | 99 |
| 40 | DDI | 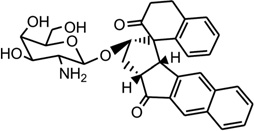 |
5′-CACGCAGTTCGGAC-3′ 5′-GTCCGATGCGTG-3′ |
1P96 | 171 |
| 41 | ent-DDI | 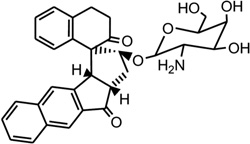 |
5′-CACGCAGTTCGGAC-3′ 5′-GTCCGATGCGTG-3′ |
1R4E | 172 |
| 42 | [Rh(bpy)2chrysi]3+ | 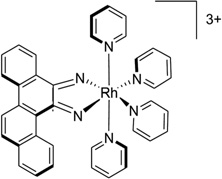 |
5′-(CGGAAATTCCCG)2-3′ | 2O1I | 173 |
| 43 | Naphtyridine- azaquinoline (NA) |
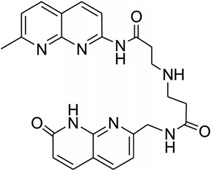 |
5′-CTAACAGAATG-3′ 5′-CATTCAGTTAG-3′ |
1 × 26 | 174 |
| 44 | NPM | 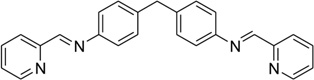 |
5′-(CGTACG)2-3′ | 2ET0 | 174 |
| 45 | A4C |  |
5′-(TCGGTACCGA)2-3′ | 2GWA | 182 |
| 46 | TriplatinNC |  |
5′-(CGCGAATTCGCG-3′)2 | 2DYW | 183 |
2. STRATEGY THROUGH CHEMICAL BOND FORMATION
A. Pt-containing Compounds Forming Covalent Bonds with DNA
Cisplatin [or cis-diamminedichloroplatinum (II), Fig. 1A] has become one of the most widely used and effective anticancer drugs today to treat testicular, ovarian, cervical, head and neck, esophageal, and nonsmall cell lung cancers, since it was approved by FDA in 1978.6–8 However, the chemotherapeutical treatment by cisplatin always causes severe side effects, including nausea, vomiting, and ear damage, because cisplatin attacks both cancer cells and normal ones. It is therefore important to elucidate the details of the cisplatin action modes in order to design new cisplatin analogs that can specifically target cancer cells. There are tremendous progresses in studying cisplatin mechanisms over the past three decades, including the exploration of its possible biological targets, the elucidation of its mechanisms, and the design of the novel derivatives. Although cisplatin can interact with many different components in cells, such as phospholipids, phosphatidylserine, DNA, and RNA,9, 10 it is generally accepted that DNA is the primary target of cisplatin.11 Cisplatin can form stable adducts with DNA either in a 1,2-intrastrand cross-links between two adjacent bases (GG or GA), or a 1,3-intrastrand cross-link between two guanines separated by another base.12, 13 The resulting Pt-DNA adducts can hamper and inhibit a series of downstream biological processes, including replication and transcription, cell cycle arrest, and attempted repair of damaged nucleotides, which eventually lead to cell death.14
Figure 1.

(A) Chemical structure of cisplatin. (B) Overall structure of DNA duplex (5′-CCTCTG*G*TCTCC-3′ and 5′-GGAGACCAGAGG-3′) in complex with cisplatin (PDB ID: 1AIO). (C) Close view of G*G* platination sites. The base pairs are propeller-twisted, but retain the hydrogen-bonding interactions. (D) Superimposition of the low-resolution structure (PDB: 1AIO) and high-resolution structure (PDB: 3LPV) of DNA duplexes modified with a 1,2-cis-{Pt(NH3)2}2+-d(GpG) cross-links. The DNA is shown as sticks, the N and Pt atoms are shown as the cyan and red spheres in 1AIO, and the yellow and red spheres in 3LPV, respectively.
Extensive structural studies have been performed on cisplatin-DNA adducts by NMR, molecular mechanics calculation, and X-ray crystallography. The early crystallographic information of these adducts is mainly limited to short single-stranded DNAs, and it has been concluded that cisplatin reacted with DNA by covalently linking to the N7 positions of the adjacent G or A. The milestone in this research area appeared in 1995, when Lippard and his co-workers determined the first X-ray crystallographic complex structure of a double-stranded DNA dodecamer with cisplatin.15 The 12mer DNA sequence is d(CCTCTG*G*TCTCC)• d(GGAGACCAGAGG), where the G*G* means the cisplatin cross-linked intrastrand GG dimer at N7 positions. As shown in Figure 1B and C, the base pairs are propeller-twisted, but retain hydrogen-bonding interactions. Very recently, the same group improved the resolution of the complex structure (PDB 3LPV) to 1.77 Å.16 The higher resolution structure clearly shows the amine ligands and the cross-linked geometry of both platinum center and nucleobases, although their overall structures are virtually identical (Fig. 1D). The deoxyribose sugar puckers are also better resolved, allowing reexamination of the global structure of this platinum-modified DNA. In addition, this new structure reveals four octahedral [Mg(H2O)6]2+ ions associated with bases in the DNA major groove and 124 highly ordered water molecules participating in hydrogen-bonding interactions with either the nucleic acid or the diammine-platinum(II) moiety.
As shown in the Figure 1B and D, the major groove of the platinated DNA is compressed and the minor groove is widened, making the double-stranded DNA ready for structure-specific recognition by proteins, such as the high-mobility group domain (HMG). It has also been known for a long time that the platinated DNA duplex structure can attract HMG and other proteins.17 To further investigate its molecular basis for this recognition, Lippard and co-workers accomplished another milestone by solving the X-ray crystal structure of a 1:1 cisplatin-modified DNA/HMG-domain complex, as showed in Figure 2.18 It is revealed from the structure that domain A of the HMG1 protein binds to the widened minor groove of a 16mer DNA duplex containing the cisplatin adduct, and the DNA is probably further bent in order to better interact with the protein.
Figure 2.

Overall structure of the HMG in complex with platinated DNA duplex (PDB ID: 1CKT). The protein is shown in cartoon mode and the DNAs are shown in sticks with the cis-[Pt(NH3)2{d(GpG)-N7(G8),-N7(G9)}] intrastrand adduct with the N and Pt atoms in spheres.
Besides HMG, it has been shown that two special polymerases (Pol η and Pol ζ) can also interact with cisplatin during cellular replication to bypass the platinum lesion, which reduces the therapeutic effect of cisplatin and contributes to tumor resistance.19 Very recently, Carell and co-workers20 reported a crystal structure of yeast Pol η pairing the Pt–GTG lesion with two nucleobases, which revealed the mechanism of the partial bypass reaction. As shown in Figure 3, the low-fidelity polymerase Pol η can place two nucleobases opposite the Pt–GTG lesion and the flipped-out central T of the Pt–GTG cross-link cannot be positioned in the active site, thus slowing down movement of the polymerase through the primer–template complex.
Figure 3.

The overall crystal structure of Pol η in complex with a DNA primer and template containing a cisplatin–(1,3-GTG) lesion (PDB ID: 2WTF). Protein is shown as cartoon. DNAs are shown as sticks outlined with cartoon backbone. Pt and N atoms are shown as spheres. The central flipped-out dT is indicated by arrow.
Additional understanding of how proteins like HMG and polymerases bind to cisplatin-modified DNAs has provided a platform for SBDD and optimization in order to develop better therapeutics. So far, over 3000 cisplatin analogues, including the Pt-containing prodrugs,21 have been screened for further clinical trials to overcome certain side effects, such as high toxicity and intrinsic or acquired resistance.22 In addition, special drug delivery systems have been developed to deliver cisplatin to tumor cells in more efficient ways.23
B. Organic Compounds Forming Covalent Bonds with DNA
In addition to the platinum compounds, other types of small molecules have been found to covalently bind DNA, including nitrogen mustard drugs (melphalan, uramustine, chlorambucil, and bendamustine, etc.), mitomycin C and psoralen. The detailed adduct formation and repairing mechanisms of these compounds in cells have been well summarized.24 We focus on a few 3D structures to illustrate their modes of molecular recognition and interaction with DNA.
Nitrogen mustard compounds (Fig. 4A) have been used as effective anticancer drugs for a long time. Their cytotoxicity is mainly caused by the nonspecific DNA cross-linking, which prevents DNA duplex separation through the formation of a N7-dG/N7-dG interstrand cross-linking. To obtain more structural insight into this interaction, Stone, Egli, and their co-workers inserted the 3-aminopropyl-2′-dU and 7-deaza-dG into the Dickerson–Dew dodecamer.25 The crystal structure of this modified DNA duplex (Fig. 4B) suggests a potential mechanism of interstrand N + 2 DNA cross-link formation by nitrogenmustard compounds without bending DNA.
Figure 4.

(A) Chemical structures of the typical nitrogen mustard compounds HN1, HN2, and HN3. (B) Duplex structure of the Dickerson–Dew dodecamer containing 3-aminopropyl-2′-dU and 7-deaza-dG (PDB ID: 2QEF).
Mitomycin C (Fig. 5A) is a potential antitumor drug that works through alkylation of guanine in the d(C-G)•d(C-G) steps of the DNA duplex.26 Patel, Tomasz, and their co-workers solved a NMR solution structure of a 9mer DNA duplex containing this monoalkylated mitomycin C. As shown in Figure 5B, the mitomycin ring is located in the minor groove with its indoloquinone aromatic ring system at a 45° angle relative to the helix axis. As a result, conformational perturbation has been observed in this modified DNA structure, presumably through widening of its minor groove in order to stack the indoloquinone ring over the neighboring bases.27 Interestingly, the mitomycin alkylated DNA presents B-form sugar puckers and A-form base stacking patterns. The major metabolite of mitomycin C in cells, 2,7-diaminomitosene (2,7-DAM, Fig. 5C) has been identified and studied structurally to provide new insight into the interaction with DNA. The NMR structure of this drug–DNA complex (2:1 ratio) (Fig 5D) contains a self-complementary DNA dodecamer d(GTGG*TATACCAC) with G4 modified with mitosene at its N7 position. Surprisingly, 2,7-DAM is located in the major groove of DNA and oriented 3′ to the modified guanine, which explains why the overall conformation does not change significantly after the bulky modification.28
Figure 5.

(A) Chemical structure of mitomycin C. (B) NMR complex structure of DNA-9mer duplex with mitomycin C, the red stick representing the mitomycin–monoalkylateddG (PDB ID: 199D). (C) Chemical structure of the 2,7-diaminomitosene (2,7-DAM). (D) NMR complex structure of DNA-12mer duplex with 2,7-DAM, the red stick representing the mitosene–monoalkylateddG (PDB ID: 1JO1).
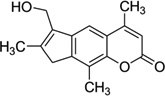
Psoralens, a class of photomutagenic and photochemotherapeutic agents, have been widely used for the treatment of vitiligo, psoriasis, and other cutaneous diseases. It is believed that they play these roles through interstrand cross-linking with DNA and RNA, specifically through the 5,6 double bond in pyrimidines, thereby blocking replication, transcription, and other DNA activities. 4′-(Hydroxymethyl)-4,5′,8-trimethylpsoralen (HMT, Fig. 6A) is one of the major synthetic psoralen derivatives that enhances its solubility in water and form higher yields of DNA adducts. Wemmer and co-workers reported a solution structure of HMT-monoadduct and cross-linked DNA duplex [5′-(GCGTACGC)2-3′], as shown in Figure 6B. It has been observed that the HMT molecule spans across both the major and minor grooves by attaching with 5,6 double bonds in the two T4 bases, while maintaining the overall conformation. The aromatic rings of HMT have obvious stacking interactions with the surrounding four bases, which form van der Waals interactions with the HMT methyl groups. The cross-linking, base stacking, and hydrophobic interactions result in significant enhancement of the duplex stability. The X-ray crystallographic investigation of the interaction between HMT and DNA by Ho and co-workers revealed that the HMT-d(CCGCTAGCGG)2 adduct can form a psoralen-induced four-way Holliday junction (Fig. 6C). Surprisingly, the HMT-d(CCGGTACCGG)2 adduct with a single switched base pair forms a distorted junction (Fig. 6D).29 This sequence-dependent interaction might also indicate a role of this important class of chemotherapeutics in initiating repair of psoralen lesions in mammalian DNA. In both structures, the assembly of four DNA strands forms a junction with two strands crossing over to connect the stacked helical arms. The two stacked DNA helical arms are related by crystallographic twofold symmetry, so that the two cross-linked strands define the asymmetric unit in the crystal. Both duplexes are highly distorted at the thymine bases linked to the six-member pyrone ring of the drug.
Figure 6.

(A) Chemical structure of 4′-(hydroxymethyl)-4,5′,8-trimethylpsoralen (HMT). (B) NMR structure of DNA-8mer [5′-(GCGTACGC)2-3′] complexed with HMT, with two psoralens alkylating two thymidines (PDB ID: 204D). (C) Crystal structure of the Holliday junction induced in the HMT-d(CCGCTAGCGG)2 complex. (D) Crystal structure of the distorted junction induced in the HMT-d(CCGGTACCGG)2 complex.
Aflatoxin B1 (AFB) is involved in mutation of the p53 tumor suppressor gene and the etiology of human liver cancer.30 The metabolism of AFB includes oxidation to AFB-exo-8,9-epoxide by cytochrome P450.31 This epoxide can react with duplex DNA efficiently to yield the N7 adducts of guanosine, a cationic adduct. The cationic species undergoes hydrolysis of the imidazole ring to form the AFB form amidopyrimidine adduct (AFB-FAPY, Fig. 7A).32 Recently, Stone and co-workers solved an NMR structure of the DNA adduct containing the α form of this AFB-FAPY moiety (Fig. 7B).33 This structure study indicated that the AFB moiety intercalates on the 5′-face of pyrimidine at the damaged nucleotides, causing obvious backbone torsion and overall duplex perturbation.
Figure 7.

(A) Structures of the FAPY adduct following the base-catalyzed ring opening of the AFB-N7-dG cationic adduct. (B) Overall structure of a AFB-FAPY-G containing DNA duplex {5′-d[CTAT(AFB-FAPY-G)ATTCA]-3′ and 5′-d(TGAATCATAG)-3′ (PDB ID: 2KH3)}.
As discussed above, these covalent binders can cross-link to DNAs, causing modifications and perturbations in the DNA structures. The modified DNAs can no longer interact with their protein partners, thereby blocking the replication or transcription processes. This is probably the main mechanism of these covalent binders during the chemotherapy treatments. Due to relatively low selectivity, however, these agents are of high toxicity. Moreover, cells can develop different strategies to identify and repair the inter- or intrastrand DNA cross-links, which could play major roles in the drug resistance of certain cancers to chemotherapeutic treatments. In order to enhance the selectivity and reduce drug resistance, a clear understanding of the molecular mechanisms involved in the recognition and repair of cross-linked DNA will be especially important. From a structural point of view, characterization of various types of DNA adducts can largely facilitate the study in this field. These structural studies can also provide direction toward molecular design and synthesis. For example, a synthetic N4C-ethyl-N4C interstrand cross-linked DNA was structurally characterized by Miller, Kielkopf, and co-workers as both a platform for drug design and a model system for DNA repair study. As shown in Figure 8, the detailed structure studies revealed that the ethyl cross-link in the CpG major groove does not significantly disrupt the B-form DNA helix and can selectively stabilize a preexisting conformation, although it can result in subtle differences in the roll of base pairs and loss of some divalent cation binding sites.34 Moreover, Stone and co-workers solved two solution structures of DNA duplexes containing the cross-linked trimethylene moiety with the sequence contexts of GpC and CpG.35 The structures indicated that both duplexes can maintain B-form geometry as well as the hydrogen bonds. However, the stacking between the cross-linked bases and the neighboring ones is quite different in these two structures (Fig. 9). The interaction in the GpC-containing duplex is greatly reduced, which is also consistent with the duplex stability result. The same research group also studied a DNA duplex structure with the hydroxytrimethylene interstrand linkage at the two N2 positions of guanosine using 15N NOESY-HSQC NMR (Fig. 10), providing evidence for the stereochemical preference of this type of linkage and its thermal stability in duplex DNA.36 Very recently, the same group also investigated a similar DNA structure modified with trans-4-hydroxynonenal (HNE) moiety (Fig. 11).37
Figure 8.

(A) and (B) Overall X-ray crystal structure of DNA [d(CCAAC*GTTGG)2] bound by the N4C-ethyl-N4C interstrand cross-link between the cytosines of the central CpG (*). The ethyl cross-linked cytosine bases are in the middle of duplex. (PDB ID: 2OKS). (C) Chemical structure of the N4C-ethyl-N4C interstrand cross-link.
Figure 9.

(A) Overall NMR structure of the CG-containing DNA duplex [(5′-TCCG*CGGA-3′)2; PDB ID: 2KNL)] with the N2G-trimethylene–N2G interstrand cross-link in the middle of the duplex. (B) Overall NMR structure of the GC-containing DNA duplex [(5′-AGGCG*CCT-3′)2; PDB ID: 2KNK] with the N2G-trimethylene–N2G interstrand cross-link in the middle of the duplex. (C) The chemical structure of the N2G-trimethylene–N2G interstrand cross-link.
Figure 10.

(A) The N15-labeled DNA duplex cross-linked with hydroxytrimethylene. (B) Local 3D structure of the cross-linked DNA duplex.
Figure 11.

(A) The DNA duplex linked with the trans-4-hydroxynonenal (HNE) moiety. (B) Local 3D structure of the cross-linked DNA duplex.
3. TARGETING DNA WITH MINOR AND MAJOR GROOVE BINDERS
A. Minor Groove Binders
To disrupt the specific gene expression, the minor groove binding offers the highest sequence specificity between a small molecule and DNA. Classic minor groove binders are polyamides or heterocyclic dications, such as DAPI, berenil, Hoechst 33258, netropsin, distamycin, and pentamidine (Fig. 12). These groove binders were originally developed to prefer AT-rich regions and later further developed to expand to all A/T, T/A, G/C, and C/G pairs.38, 39 Due to their capability to interact with DNA in a more specific manner, minor groove binders hold a great promise to treat the diseases at the DNA level. Actually, these minor groove-binding agents have been extensively studied as a class of potential therapeutics with antibacterial, antiprotozoal, antitumor, and antiviral activities.40–42 Dickerson, Sundaralingam, Dervan, Neidle, Wilson, and their co-workers have pioneered this research area by revealing the 3D structures of DNA complexed with minor groove binders,43–47 and some of the investigations in this research field have been summarized recently.48–50 The shapes of the groove-binding molecules typically match the curve of the minor groove, thus allowing a perfect fit via interactions such as van der Waals contacts and hydrogen bonding. Here we listed some of the typical complex structures.51–56 The structure of the DNA in complex with Hoechst 33528 is shown in Figure 13. In the structure, the ligand is clamped by the phosphate backbone of the DNA in the minor grove and forms extensive van der Waals contacts with the DNA. Although the hydrogen bonds (total of 2) do not play an essential role in the recognition between Hoechst 33528 and DNA, H-bonds do play an essential role in the cases of the netropsin–DNA and distamycin–DNA interactions (Fig. 14). In the netropsin–DNA structure (Fig. 14A), eight hydrogen bonds have formed between the ligand and DNA. In the distamycin–DNA structure (Fig. 14B), even more hydrogen bonds (total of ten) have been observed. In these structures, the ratios between the DNA and ligands are different. In the 1:1-binding mode, the minor groove of the DNA duplex is compressed, and the major groove is widened, which gives the access to other molecules binding to the major groove of the DNA duplex. In the 1:2-binding mode, the DNA minor groove binder works through the opposite way, that is, widening the minor grove and compressing the major grove. Other similar drug–DNA complexes, such as imidazole-pyrole-hydroxypyrole polyamide,57 benzimidazole,58, 59 and tribenzimidazole60, have been extensively discussed and summarized previously.
Figure 12.

Chemical structures of the typical DNA minor groove-binding agents: DAPI, berenil, pentamidine, Hoechst 33258, netropsin, and distamycin.
Figure 13.

Overall structure of the Hoechst 33258 (structure showed in Fig. 12) in complex with DNA [5′-d(CGCGAATTCGCG)2-3′] (PDB ID: 8BNA). The DNA is shown in the surface view. Hoechst molecule is shown as sticks. The two hydrogen bonds are indicated by the black dashed lines.
Figure 14.

(A) The chemical structure of netropsin (left) and its crystal structure in complex with DNA [5′-d(C5-BrCCCCIIIII)2-3′; PDB ID: 358D]. (B) The chemical structure of distamycin (left) and its crystal structure in complex with DNA [5′-d(GTATATAC)2-3′; PDB ID: 378D]. The structures are presented in two different formats to show the van der Waals interaction (middle) and hydrogen bonding (right, indicated by black dashed lines). Some H-bonds are obscured at the current view angle.
The classic polyamides, composed of amide monomers linked by peptide bonds, can bind many different DNA sequences, permeate the cellular membrane, access chromatin, and disrupt interactions between the target DNA and transcriptional factors, such as hypoxia-inducible factor, androgen receptor (AR), and glucocorticoid receptors (GRs).61–63 Their hairpin derivatives containing two (R)-β-amino-γ-turns (Fig. 15A) have been demonstrated to have favorable DNA-binding affinities and better gene regulating ability.64–66 The binding location and orientation of the polyamides have been widely studied through several complex structures.57, 67–69 The high-resolution structures provide valuable insights into their molecular interactions with the DNAs and DNA–protein complexes, which serves as the foundation for further drug optimization. Recently, Dervan and co-workers reported a complex structure of an 8-ring cyclic Py-Im polyamide and a DNA duplex [d(CCAGGCCTGG)]2 at 1.18 Å resolution (Fig. 15B). The determined structure shows that the overall duplex conformation has been dramatically perturbed. The minor groove is widened by 4 Å, while the major groove is compressed by ~4 Å .Moreover, the helix axis is bent toward the major groove by more than 18°, compared to the native counterpart.66 These large distortions provide a molecular basis for supporting the idea of allosteric perturbation of the DNA helix by small molecules as minor groove binders, thereby chemically and selectively regulating gene expression.66 In addition, this complex structure elucidates the source of sequence-specific recognition. The interactions between DNAs and ligands include hydrophobic packing, specific hydrogen bonding of the connecting amides to the groove floor before and after the turn, water-mediated hydrogen bonding of the α-amine to the groove floor, and a conformational pucker that minimizes steric interactions of α-amino substituents. From this structure, it is also clear that the exocyclic amine of the guanine base under the turn will cause a large steric interference with the β-methylene of the amino turn.66
Figure 15.

(A) Chemical structure of the 8-ring pyrole–imidazole cyclic polyamide. (B) Overall structure (PDB ID: 3I5L) of the DNA duplex [d(CCAGGCCTGG)2] complexed with the Py-Im ligand (in the middle). (C) The hydrogen-bonding interactions between the Py/Im and G/C pair. (D) The hydrophobic packing between the O4′ atom and the amide ring systems. (E) The direct and water-mediated hydrogen-bonding interactions between the turn and the A/T pair.
To further address the issue of sequence recognition specificity, Dervan and co-workers reported another high-resolution complex structure (0.95 Å resolution) containing the cyclic polyamide with two antiparallel ImPyPyPy strands capped by (R)-β-amino-γ-turns (Fig. 16). In the complex structure, the DNA is a 10-mer DNA duplex [(5′-CCAGTACTGG-3′)2], where the middle TA replaced the GC of the previous sequence.70 Very similar to the previous structure, a large perturbation is observed following ligand binding. The minor groove is widened by more than 4 Å, while the major groove is also compressed by more than 4 Å. The narrow major groove is too small to accommodate the typical proteins, such as transcription factors. The helix is also bent toward the major groove by over 15°. To better understand the impact of the major groove compression on the binding of transcription factors, they further analyzed the previously reported structures of AR and GR in complex with ds DNA.71, 72 From the structural comparison, it turns out that after ligand binding, the major groove is no longer compatible with AR and GR binding, which may be taken as a potential mechanism to control gene expression chemically. As shown in Figure 16C, the A/T pair forms two hydrogen bonds with two Py groups of the ligand, suggesting that Py/Py can selectively interact with T/A pair. Moreover, the structure information allowed the chemical modification to achieve more specific T/A pair recognition. As shown in Figure 17, Hp forms one extra hydrogen bond with T, due to the addition of OH group at the C3 position. The space-filling model also shows that the Hp/Py pair fits tightly with the T/A pair and will sterically overlap with the A/T pair.
Figure 16.

(A) Chemical structure of the 8-ring pyrrole–imidazole cyclic polyamide. (B) Overall structure of the DNA duplex [d(5′-CCAGTACTGG-3′)2] complexed with the ligand (0.95 Å resolution and PDB ID: 3OMJ). (C) The hydrogen-bonding interactions between the Py/Py and A/T pair. (D) The direct and water-mediated hydrogen-bonding interactions between the turn and the A/T pair.
Figure 17.

(A) The hydrogen-bonding interactions between the HP/Py and T/A pair observed in the DNA duplex (5′-CCAGTACTGG-3′)2 complexed with ImHpPyPy (PDB ID: 407D). (B) The space-filling model showing the tight fitting between the Hp/Py pair and T/A pair.
Different from the prevention of DNA–protein binding interactions, some chemicals, following exposure to ionizing radiation or UV light, will result in the formation of cellular DNA–protein conjugates, thereby interfering with both DNA replication and transcription.73 One of the repair mechanisms involves the formation of DNA–peptide conjugates as substrates for nucleotide excision repair.74, 75 Recently, Stone and co-workers linked the amino-terminal lysine of the peptide KWKK to the exocyclic amino group of dG in the DNA duplex (5′-GCTAGCG*AGTCC-3′•5′-GGACTCGCTAGC-3′) through a trimethylene linkage (Fig. 18A).76 Strictly speaking, this work should be catalogued into covalent binding, since the small peptide ligand is covalently bound with DNA. But it was observed from the complex structure (Fig. 18B) that the amino acids are in the minor groove, which provides the 3D interaction information of the DNA and its ligand, and serves as a good model for designing novel minor groove binders. This study also indicated that the small peptide conjugation can stabilize the DNA duplex.
Figure 18.

(A) Chemical structure of the DNA–KWKK conjugate. The trimethylene moiety was named as residue X25. (B) An expanded view of the average structure of the peptide-conjugated DNA duplex (PDB ID: 2KV6). The peptide is in the middle.
It has been well known that alternating AT sequences, which may contain Hoogsteen base pairs and form coiled coil structures, are extremely abundant in most eukaryotic genomes. For example, the (AT)5 sequence occurs over a million times in the human genome.77 However very little structural data is available to help understand its cellular functions, probably due to the difficulty of crystallization. Subirana, Campos, and their co-workers have recently studied this type of sequence with minor groove-binding agents.78 The X-ray complex structure (Fig. 19A and B) of a DNA duplex [d(ATATATATAT)2] with pentamidine was found to be able to stabilize a left-handed coiled coil arrangement through formation of cross-links between neighboring duplexes (Fig. 19C). The DNA duplex exhibits a mixture of Watson–Crick and Hoogsteen base pairs (in the bottom base pair). Unlike the previous complexes, pentamidine is not entirely bound within the minor groove. Instead, only the central portion of pentamidine resides inside the groove, leaving the positively charged ends detached from the DNA and free to interact with phosphate groups from adjacent duplexes in the crystal. This novel binding mode has potential implications for the antiprotozoal and antibacterial activities of pentamidine as anti-HIV drug.
Figure 19.

(A) Complex structure of pentamidine bound with the DNA duplex [d(ATATATATAT)2]. (PDB: 3EY0). The drug is in the minor groove at the center of sequence. The water molecules interact with the DNA and drug through hydrogen bonding (dashed lines). (B) Structure of pentamidine and its electron-density map (2Fo-Fc, contoured at 1 σ level). (C) Stereo view of one and a half turns of the coiled coil structure.
5-Formyluracil (fU) is one of the major damaged thymidine derivatives, and is caused by the attack of hydroxyl radicals, inducing pyrimidine transition during replication.79 In order to obtain structural information regarding this mutagenesis, Takenaka and co-workers solved the X-ray structure of a DNA duplex [d(CGCGGATfUCGCG)2] containing a fU-G wobble pair and complexed with the minor groove-binding dye Hoechst33258.80 The main purpose of the ligand was to stabilize the DNA duplex and obtain high-quality crystals. Despite no direct interaction between the fU residue and ligand, the structure still provided a potential starting point in the design of drugs to treat fU-caused diseases. It was revealed that the overall duplex structure did not have any obvious changes compared to the unmodified DNA. However, analysis of minor groove widths indicated that the minor groove of the fU-G base pair is wider, compared with other base pairs in the same DNA duplex. The crystal structure clearly indicated that the widening is caused by the binding of the ligand. The complex structure and its comparison with the corresponding native Dickerson–Drew DNA (12mer) are shown in Figure 20A and B.
Figure 20.

(A) Overall crystal structure of the DNA duplex [d(CGCGGATfUCGCG)2] bound with Hoechst33258 (PDB ID: 3AJK). (B) Superimposition of the ligand-bound DNA duplex (the ligand is not shown.) with the native Dickerson–Drew DNA 12mer [d(CGCGAATTCGCG)2] (PDB ID: 5BNA). The rms is 0.28 Å.
As one of the traditional design rules, most of the classical models for minor groove binding emphasize the importance of the ligand curves in order to fit the groove shape and achieve high binding affinity.81–83 However, one famous exception is compound DB921 (Fig. 21A), which is a linear heterocyclic diamidine able to strongly bind the DNA minor groove much stronger than similar and curved compounds.84 For example, the crystallographic analysis of DB921 bound to AATT indicates its indirect contacts with the base through bound water molecules, resulting in an induced structural change in DB921 by forming a curve that perfectly matches the shape of minor groove (Fig. 21B).84 Wilson, Neidle, and co-workers have carried out systematic studies for the role of water molecules in recognizing both the ligand and DNA minor groove. This represents an alternative recognition concept and has a great potential to guide novel drug design.40 Very recently, through a series of biophysical and crystallographic investigations, the same groups of scientists have systematically modified the DB921 structure and further studied the role of bound water molecules bridging the DNA and ligand.85 The complex crystal structure of dodecamer DNA (CGCGAATTCGCG)2 with DB1055, which was derivatized from DB921 by shifting the amidine at the phenyl group to the meta position (Fig. 21C), has been presented and compared with the DB921-bound DNA structure. The structure confirmed that the ligand binds in the AT region of DNA minor groove in a typical interaction mode. Both amidine groups interact with the O2 atom of the neighboring cytosine bases through the hydrogen-bonding network formed by the two highly conserved water molecules in the minor groove. (Fig. 21D) These studies provide the structural basis for designing sequence-specific minor groove binders, which interacts with DNA through the water-mediated hydrogen binding.
Figure 21.

(A) Chemical structure of DB921. (B) Water network observed in the structure of DNA in complex with of DB921 (PDB: 2B0K). (C) Chemical structure of DB1055. (D) Local hydrogen-bonding interaction in the DB1055-DNA complex structure. Water molecules are shown as spheres. (PDB: 2I5E).
It is also worth mentioning that in addition to the drug–DNA binary complex systems, Georgiadis and co-workers have developed a novel host–guest complex system for studying the drug–DNA interaction. They have used the N-terminal fragment of Moloney murine leukemia virus reverse transcriptase (MMLV-RT) as a host and a DNA 16mer d(CTTAATTCGAATTAAG)2 as a guest.86 As an example, they co-crystallized netropsin (Fig. 12) with this DNA–protein system and determined the structure at 1.85 Å resolution. The crystal asymmetric unit contains one protein molecule and half of the 16mer DNA bound with one netropsin molecule (Fig. 22). The guanidinium moiety of netropsin binds in the minor groove, while the amidinium is bound to the widest region of the enlarged minor groove. By comparing this structure to other netropsin–DNA structures, different hydrogen-bonding patterns were found, and the orientation of netropsin within the groove was attributed to the asymmetry of the minor groove in the AATT site.86 This work not only revealed the possible antiviral mechanism of the DNA groove binders but also established a powerful platform to study ligand–DNA interactions, although the DNA length is the major limitation for complex crystallization.
Figure 22.

The crystal structure (PDB ID:1ZTT) of an RT fragment–DNA–netropsin complex. The DNA sequence is 5′-d(CTTAATTCGAATTAAG)-3′. Netropsin is shown as spheres.
Recently, this host–guest approach has been further applied to solve the crystal structure of a DNA duplex [d(CTTAATTCGAATTAAG)2] in complex with CD27 [4,4′-bis(imidazolinylamino)diphenylamine], a potent antitrypanosomal agents, which binds to its preferred AATT site.87 Although the therapeutic effects of this compound have been explored previously,88 the further development and optimization of this compound series requires more detailed structural and binding information. In the structure determined at 1.75Å resolution (Fig. 23), a planar crescent shape of CD27 was observed upon binding to the DNA minor groove, although it was predicted to be highly twisted in its energy-minimal state. The crystal structure reveals that CD27 interacts with the DNA by forming bifurcated hydrogen bonds with AATT, which results in the minor groove widening and the formation of favorable van der Waals interaction. This induced fit with conformational changes in both the ligand and DNA has provided another rationalization and base for designing effective minor groove binders.
Figure 23.

(A) Chemical structure of CD27. (B) The overall structure (PDB ID: 3FSI) of a RT fragment in complex with DNA duplex and CD27. The DNA duplex is d(CTTAATTCGAATTAAG)2.
B. Major Groove Binders
Compared to minor groove binding, interactions between small molecules and the DNA major groove have not been extensively explored. There are also very few structural reports on complexes of DNA and major groove binders, probably due to the requirement of much larger molecules and the difficulties to conduct the complex structure studies. Carbohydrates represent a class of agents capable of binding the major groove due to their size and intrinsic distribution of hydrophobic and hydrophilic moieties in their molecular structures, although the majority of them still show significant binding with the DNA minor groove. To date, the carbohydrate compounds that can bind within the major groove include neocarzinostatin, nogalamycin, and several aminoglycoside (neomycin) conjugates (Fig. 24).89
Figure 24.

Chemical structures of neocarzinostatin (A), nogalamycin (B), and a neomycin-groove binder conjugate (C).
As a representative antitumor agent, the neocarzinostatin chromophore (NCS-chrom) and its derivatives have been studied for more than four decades.90, 91 It has been well-known that this small molecule exists by forming a complex with a protein in its natural form and causes DNA damage by hydrogen abstraction from DNA strands through a diradical intermediate, while the drug itself forms the postactivated derivatives.90, 92 However, the molecular recognition and mechanism of this complex system have yet to be clearly elucidated through structural investigation. The previous structure study revealed that this type of drug interacts with a specific sequence of DNA through both minor groove binding and intercalation.93–95 Another interaction mode between NCS-chrom and DNA contains a bulge-forming structure, which leads to the single strand cleavage.96, 97 In this bulge structure, the drug molecule works as a major groove binder and its ring systems fill the bulge cavity and interact with DNA aromatic bases.98 To better understand the molecular interaction and mechanism of this unusual complex, Goldberg and co-workers analyzed and compared the detailed NMR structures (Fig. 25) of the NCSi-gb-bulge-DNA, bulge-free DNA, as well as the NCSi-glu-DNA.98 As shown in Figure 25C, conformational changes in both DNA and ligand are observed. The NCSi-gb mainly binds at the major groove and the two ring systems of the drug are brought closer to each other in the complex. More interestingly, this binding promotes the formation of a bulge pocket and this was not found in the unbound DNA structure. The bulge formation represents the potential transition of DNA conformation.98 On the contrary, NCSi-glu, which is generated by glutathione induced activation, binds with the DNA duplex in the minor groove (Fig. 25D). Considering that these two NCS derivatives have the similar composition and structure, the striking differences observed from their DNA complexes provided important insight into DNA recognition and specificity, which are valuable for drug design and discovery. Later on, the same research group designed a new spirocyclic helical ligand, which is similar to the NCSi-gb structure without the bulky cyclocrabonate moiety, and they solved the solution structure of a two-base bulge-containing DNA bound with this new ligand.99 However, the structure revealed that this ligand binds to the minor groove. This example also indicates the flexibility of DNA recognition and interaction with small molecules and the importance in studying individual complex structures at high resolution.
Figure 25.

(A) Chemical structure of NCSi-gb, formed by the postactivated NCS-chrom under basic conditions. (B) Chemical structure of NCSi-glu in presence of a thiol agent. (C) Induced formation of a DNA bulge structure (PDB ID: 1KVH) by binding of NCSi-gb on the DNA major groove. (D) Complex structure (PDB ID: 1MP7) of a DNA duplex bound by NCSi-glu.
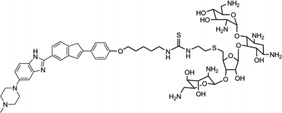
Arya and co-workers pioneered the design and development of a novel neomycin-Hoechst 33258 conjugate (Fig. 26) for probing the binding effect of an aminoglycoside antibiotic (neomycin, Fig. 26A) on the B-DNA structure.100–102 Neomycin is known to bind A-like conformations of nucleic acids including DNA and RNA, and is able to stabilize triplex formation. Since A-form DNA has a narrow major groove, it is suspected that neomycin binds in the major groove of these structures,100 although no direct structure evidence is available. It is also known that neomycin itself cannot bind to the major groove of B-form DNA. Therefore, in order to examine whether this type of aminoglycoside can bind to the major groove of B-from DNA, Arya and co-workers covalently attached neomycin with a strong minor groove-binding moiety (i.e., Hoechst 33258, Fig. 26A), which has preference to bind AT-rich DNA sequences and is used to block the minor groove space.103 Systematic biophysical studies, such as UV melting, isothermal fluorescence titration, differential scanning calorimetry, circular dichroism, and computational modeling, have indicated that the neomycin moiety interacts with the DNA major groove (Fig. 26B and C), while the Hoechst moiety binds to the minor groove. It is also worth mentioning that the linker connecting these two moieties can also be adjusted for better binding efficiency and sequence selectivity. Although X-ray crystallographic or NMR studies of these complexes are not yet available to confirm this binding model, the design of such dual-binding ligands opens up great potential to develop novel conjugates for selective nucleic acid targeting and gene expression inactivation.
Figure 26.

(A) The chemical structure of Hoechst 33258, neomycin, and their two conjugates (NH1 and NH2). (B) and (C) Computation models of NH1- and NH2-bound DNA duplex (CGCAAATTTGCG)2.
Recently, along this line of the covalent binder conjugation, the same group further included an intercalative moiety as a third party of the complex system.104 For better comparison, they synthesized a neomycin-Hoechst 33258-pyrene conjugate (NHP, Fig. 27A) and studied DNA binding with this triple-recognition ligand. The computational modeling of the interaction between this ligand and a DNA duplex (CGCAAATTTGCG)2 (Fig. 27B) clearly suggested a “clamp” model: the Hoechst moiety fits very well with the minor groove curve, and the neomycin moiety binds tightly and selectively with the major groove through hydrogen-bonding interactions. Moreover, the pyrene moiety intercalates into stacked base pairs by enlarging their distance at its binding site. Clearly, the high-resolution structure study will provide invaluable information to confirm the biophysical data and the binding model.
Figure 27.

(A) The chemical structure of the NHP conjugate containing the neomycin, Hoechst 33258, and pyrene moieties. (B) Computer model of NHP binding to a DNA duplex (CGCAAATTTGCG)2.
Other than the small molecule ligands, some typical additives in crystallization conditions can also bind DNA in the major groove. Lattman and co-workers reported these interactions during their study of transcriptional inhibitor P4N with the Sp1 consensus sequence.105 The structural study revealed specific interactions between polyethylene glycol (PEG) molecules and the major groove of the Sp1-DNA recognition site. The DNA adopts the A-conformation and contains three curved PEG molecules. In addition, a cobalt hexamine ion per DNA duplex binds to the major groove. It is likely that these PEG molecules open the major groove by approximately 1 Å, compared to the modeled A-form DNA. The specific binding of these small ligands to the DNA major groove opens many opportunities to design novel small molecules for major groove recognition and interaction.
Although these DNA groove-binding agents hold the great promise in modulating specific gene expression, in terms of clinical applications, the successful use of these compounds is largely limited by cell uptake and nuclear localization. They are dependent on a wide range of molecular determinants, such as the Py/Im content of polyamide compounds, the number and location of charges, composition and the attaching position of linkers. Recently, various chemical modifications have been introduced into small molecule ligands, and some compounds have showed improved biological activity. For example, experimental data suggested that the polyamides derivatized with isophthalic acid (IPA) at the C-terminus have improved activities in He La (cervical cancer) and U251 (glioma) cell lines.106 This enhanced potency by IPA modification was also observed in other cell lines.107 A more recent study suggested that polyamide potency can also be enhanced by other modifications at the C-terminus, such as a C3-aliphatic linker tethered to an aromatic group through an oxime linkage. This modification resulted in approximately 20-fold enhancement of the potency of the polyamides targeting the androgen response element in LNCaP cells.108 In general, the C-terminus modification may be a common strategy to overcome the problems in cellular uptake and nuclear localization. Solving these problems will significantly broaden the applications of the polyamides in biology and human medicine.
4. INTERCALATING INTO DNA
Intercalation is another binding mode of small molecule drugs with nucleic acids. It involves the insertion of a planar molecule between DNA base pairs, thereby interrupting DNA functions. Intercalators bind to the DNA duplex well, normally with association constants of 105–1011 M−1. This is due to several favorable forces, including hydrophobic, ionic, hydrogen-bonding, and van der Waals interactions.109 The intercalation decreases the DNA helical twist and lengthens the DNA.110, 111 Traditionally, it has been thought that only molecules with fused-ring structures could be good intercalators. But it was later found that the presence of basic, cationic, or electrophilic functional groups is often necessary for a compound to be a good intercalator, while the fused-ring structures are not always required.112, 113
DNA intercalators, such as adriamycine, daunomycin, daunorubicin, dactinomycin, echinomycin, and ditercalinium, have been extensively used as antitumor, antineoplastic, antimalarial, antibiotic, and antifungal agents, as well as the chemotherapeutic agents to inhibit the growth of cancer cells. Unfortunately, the requirement for a ligand to function as an intercalator is relatively low. Therefore, although thousands of different intercalators have been identified, most of them were eliminated from clinical application due to their high toxicity and nonspecificity.114 Again, the structure-based research of these DNA–drug complexes is very important, not only for biochemical and biophysical studies, but also to shed new light on further design and optimization of novel compounds with better target specificity and low toxicity. So far, many crystal structures are available for typical intercalators. Herein we will discuss complex structures of DNA molecules with several important intercalators, such as daunomycin, adriamycine, echinomycin, and ditercalinium, as well as their derivatives.
Daunomycin, an anthracycline-type antibiotic, consists of a planar aromatic ring system, a fused cyclohexane ring, and an amino sugar moiety (Fig. 28A). It is a naturally occurring agent that is widely used in chemotherapy treatment of cancer. It is also the starting model for thousands of potential drugs.115 Extensive studies, including high-resolution crystallographic and NMR investigations, have been performed to understand the structural, kinetic, and energetic contribution of this drug upon interaction with nucleic acids.116–122 Most of these structure studies have revealed that two drug molecules intercalate between the GC steps with the sugar moiety placed in the minor groove in each duplex unit (Fig 28B), though the DNA duplexes are usually short (hexamers or octamers). Through sequence selectivity investigation by different biophysical methods, it was revealed that this ligand prefers a purine-pyrimidine step123 and the DNA molecules are usually elongated or twisted after binding. A complementary computational modeling by Liedl and co-workers described a possible 1:1 binding mode in its complex with a 14mer DNA duplex.124 By comparing the two binding modes with different ligand ratios, they found that the sugar ring was highly flexible in the minor groove. Furthermore, this intercalation increased duplex stability by reducing the flexibility of phosphate groups at DNA backbone.
Figure 28.

(A) Chemical structure of daunomycin. (B) Overall structure (PDB ID: 1D10) of the daunomycin–DNA complex (2:1 ratio). The duplex is a B-form DNA/RNA chimera (dC)(rG)d(ATCG). (C) Chemical structure of adriamycin. (D) Overall structure (PDB ID: 1D12) of the adriamycin–DNA complex (2:1 ratio). The DNA duplex is 5′-d(CGATCG)2-3′. Water molecules are shown as spheres.
Adriamycin is the closest derivative of daunomycin with only an extra hydroxyl group at the C14 position (Fig. 28C), but their biomedical functions are significantly different. Daunomycin is most effective in the treatment of leukemias, while adriamycin is more effective in solid tumor treatment.118 Rich, Wang, and co-workers pioneered the structural determination of these anthracycline compounds in complex with DNA. To better understand a more detailed relationship between the structure and function, they also compared the binding of these two compounds to the same DNA duplex d(CGATCG)2. Only very subtle differences were found in terms of the binding mode. However, additional solvent interactions were observed for the extra hydroxyl group in the adriamycin–DNA complex (Fig. 28D). Moreover, they found that the conformations of spermine in these two complexes were different. These changes may be the reason for the different clinical activities of these important drugs.
Ditercalinium, a dimer of 7H-pyridocarbozole (Fig. 29A), is a widely used synthetic anticancer drug that plays its role by binding to DNA through bis-intercalation and inducing DNA repair in both prokaryotic and eukaryotic cells.125–127 This noncovalent binding with DNA is recognized by the cellular repair system as a covalent interaction, therefore activating the repair process until the cell death. This is a novel mechanism for cancer treatment. Many chemical derivatives and their biomedical effects have been evaluated.128–130 The complex structure (Fig. 29B) indicated that ditercalinium bis-intercalates at the two CpG steps of a short DNA (CGCG)2, and the DNA retains an underwound and right-handed double helix with the linker of drug molecule located in the major groove. The positively charged nitrogen atoms of ditercalinium are located near the bottom of the DNA major groove, making charge–charge interactions with the DNA groove that prove to be another important factor in the complex stability.131 Upon binding, the DNA is bent toward the minor groove at about 15°. These aforementioned structural properties provide further direction in the optimization of these types of drugs.
Figure 29.

(A) Chemical structure of ditercalinium. (B) Overall structure (PDB ID: 1D32) of the ditercalinium–DNA complex (1:1 ratio). The DNA duplex is 5′-d(CGCG)2-3′, and the darker structure is ditercalinium.
Echinomycin (Fig. 30A) is a quinoxaline antibiotic that also binds to DNA duplexes by bis-intercalation and interferes with both replication and transcription.132 These types of compounds have great potential as anticancer agents.133 Therefore, the investigation of the interaction between echinomycin and DNA will be very helpful for drug optimization and novel design. Several NMR structure studies have been reported for echinomycin-bound DNA complexes.134, 135 Crystal structure studies of its closely related derivative (i.e., triostin A) bound DNA complexes were reported as well.138, 139 In 2005, Sheldrick and co-workers reported three complex structures of echinomycin with different DNA sequences and revealed binding details for this compound. It was found that the complex structures of echinomycin and DNA are very similar to that of the triostin A-bound DNA structure.140 It is shown in Figure 30B that one complex structure of DNA d(GCGTACGC)2 bound two drug molecules intercalating between two CpG steps in each duplex. The main moiety of the drug is located in the minor groove, with the quinoxalines protruding into the major groove. Except for the Watson–Crick base pairs in the binding sites, all other base pairs are Hoogsteen base paired. In addition, the base stacking of the DNA is obviously extended, imposing more rigidity on the structure. Moreover, several hydrogen bonds between the drug and DNA have also been observed, which are consistent with previous studies. It is also worth pointing out that, from an energetic point of view, binding of the first intercalating group will increase the entropy, allowing the second intercalator to bind much easier than the first one. Therefore, bis-intercalators often bind much stronger than a single intercalator. Although only the Hoogsteen pairs were found outside the echinomycin binding site, a complex structure solved later by the same group (Fig. 30C) demonstrated that Watson–Crick base pairs can also exist outside and close to the bis-intercalation site.141 Clearly, these structures indicate that the drug acts as a rigid body with a conformation independent of the presence of DNA and promotes DNA structure distortions. This and other structural investigations provide novel structures that can be investigated for drug discovery.
Figure 30.

(A) Chemical structure of echinomycin. (B) Overall structure (PDB ID: 1PFE) of the echinomycin–DNA complex (2:1 ratio). The DNA duplex is 5′-d(GCGTACGC)2-3′, and the darker structure is echinomycin. (C) Overall structure (PDB ID: 2ADW) of the echinomycin–DNA complex (2:1 ratio). DNA duplex is 5′-d(ACGTACGT)2-3′, and the darker structure is echinomycin.
Generally speaking, the binding specificity of DNA intercalators is relatively low, which is the reason why most of the intercalators are too toxic to be used as drugs. Occasionally, new molecules with unexpected sequence preferences can still be discovered as candidates for novel therapeutic development. For example, cryptolepine (5-methyl indolo[2,3b]-quinoline, Fig. 31A), a naturally occurring indoloquinoline alkaloid isolated from the roots of Cryptolepis triangularis collected in Kisantu, has been found to have many pharmacological effects, especially as an antimalarial and cytotoxic drug as well as a potent topoisomerase II inhibitor.142 This drug was found to intercalate preferentially with CpG-rich sequences and go to the cytosine–cytosine sites, instead of CG steps.143 The crystal structure solved by Aymami and co-workers showed, for the first time, how a drug interacts with the d(CpC)-d(GpG) site of a DNA duplex [d(CCTAGG)2] in a base-stacking intercalation mode (Fig. 31B). This binding to nonalternating bases is unusual among the nearly 100 intercalator–DNA complexes, most of which have a CG intercalation site, a few with GC sites, and a few with TG sites. Revealed by the structure, the drug molecule is sandwiched between two consecutive C-G base pairs, with the six-member ring stacking between two cytosines and the fused aromatic rings between two guanines. As a result, cryptolepine is slightly bent, with a 6.8° angle between the two aromatic rings, which probably formed due to the five-membered ring between the two aromatic functionalities. In addition, the positive charge interactions between the drug and DNA can enhance stability of the duplex. The intercalation parallel to the base pairs and the asymmetry of the drug molecule are both key factors for such a nonalternating site preference, which enhances its accommodation in nonalternating sites (CC)-(GG) by allowing a distinct stacking interaction with either (CC) or (GG), without hydrogen-bonding interactions.144
Figure 31.

(A) Chemical structure of cryptolepine. (B) Overall structure (PDB ID: 1K9G) of the cryptolepine–DNA complex (4:1 ratio). The DNA duplex is 5′-d(CGTACG)2-3′, and the darker structure is cryptolepine.
5. TARGETING DNA THROUGH MULTIFUNCTIONALIZED LIGANDS
On the basis of the discussed binding modes, it is rational to design small molecule ligands with multiple interacting elements, which will presumably improve the stability and specificity of the target recognition. Several compounds designed using this strategy have been examined.49, 145, 146 Actually, most of the covalently bound complex structures always contain a second interacting element. Several previously discussed examples (e.g., the major groove binder, neomycin-Hoechst 33258-pyrene, in Fig. 27) also fit in this category. Other than those, very limited progress in structure study has been made in this area, including a few NMR solution structures with multiple binding modes. For example, Weisz and co-workers solved a complex structure containing a pyrrolo[2,1-c][1,4]benzodiazepine-benzimidazole hybrid (PBD-BIMZ) (Fig. 32A) that was covalently linked to a guanine base at its exocyclic 2-amino group in a DNA duplex [(5′-AACAATTGTT-3′)2]. These hybrids have significant anticancer activity in a number of cancer cell lines. This NMR structure indicated that the ligand binds to one of the two symmetric guanine bases in a special configuration and is oriented in the minor groove with its benzimidazole moiety toward the 5′-end of the modified guanine (Fig. 32B). The overall DNA structure is still the B-form, and only the local region with the covalent binding site has certain helical distortions. In contrast, the terminal N-methylpiperazine ring appears to be more flexible with various conformations.147 More recently, the same research group also solved the structure of a PBD-naphthalimide derivative in complex with the same DNA decamer (Fig. 32C) by 1H and 31P 2D-NMR. This bis-functional hybrid covalently links to a guanine within the minor groove and the naphthalimide moiety inserts between the A-T and A-T base pair step. The intercalation results in an opposite buckling of the base pairs at the intercalation site and duplex unwinding at adjacent internucleotide steps (Fig. 32D).148
Figure 32.

(A) Chemical structure of the PBD-BIMZ conjugate. (B) Overall structure (PDB ID: 2KTT) of the DNA decamer complexed with PBD-BIMZ. The DNA duplex is 5′-d(AACAATTGTT)2-3′, and the darker structure is PBD-BIMZ. (C) Chemical structure of the PBD-naphthalimide conjugate. (D) Overall structure (PDB ID: 2KY7) of the DNA decamer with PBD-naphthalimide. The DNA duplex is 5′-d(AACAATTGTT)2-3′, and the darker structure is PBD-naphthalimide.
Likewise, recently several other minor groove binders or intercalating agents have also been covalently linked with DNA at specific positions.33, 149 It was revealed that this covalent bonding can greatly stabilize DNA–ligand interactions, and the duplex conformation can also be altered. More importantly, since most molecules only have weak preference for a specific sequence and binding to several alternative sites often happens, these combination modes thus provide a convenient way of “site-specific DNA-targeting.”150–153 This combination of binding elements opens up huge opportunities for synthetic and medicinal chemists to design, synthesize, and evaluate a variety of novel compounds with multiple functions. This exploration will in turn stimulate structural studies in this field,154 especially high-resolution crystal structural analysis, which will further provide insight into these combined binding modes and reveal more structural details.
6. TARGETING THE DNA QUADRUPLEX
Telomere recognition and regulation have been recognized as an important potential strategy for cancer therapy. The 3′-end of eukaryotic DNA (telomere) contains 100–200 copies of a G-rich sequence, which can fold into G-quadruplex structures intermolecularly or intramolecularly. The formation of G-quadruplex structures can inhibit and regulate telomerase activity, which is critical in maintaining telomere length in cancer cells during replication. Besides telomeres, G-quadruplex-forming sequences have also been found in the promoter regions of many other genes, such as c-MYC, chicken β-globin gene, human ubiquitin-ligase RFP2, and the protooncogenes: c-KIT, BCL2, VEGF, H-ras, and N-ras. Structural studies have clearly revealed the conformational diversity of these G-quadruplex structures. These structures or conformations are highly dynamic and sensitive to cations, proteins, flanking sequences, and even concentrations. Biological studies have further confirmed that small molecules stabilizing G-quadruplex structures can also inhibit telomerase activity as well as telomere-binding proteins, which are often overexpressed in most cancer cells.155–157 Therefore, the G-quadruplex structures, as novel anticancer targets, have naturally become a focus of novel drug discovery.
Since G-quadruplex structures mainly contain stacked guanine tetrads with several planar layers, a large number of small molecules, especially compounds with electron-deficient aromatic systems (such as the amidoanthraquinone derivatives), have been examined for binding.158 It has been demonstrated that molecules like daunomycin and distamycin A, which are known to intercalate into duplex DNA, are also capable of binding to these quadruplex structures. But whether the planar ligands are intercalated in the center of the G-quadruplex structure or stacked on the exterior needs to be further investigated.159, 160 Despite their low selectivity, G-quadruplex recognition by anthracyclines (e.g., doxorubicin and daunomycin) has been biochemically investigated.161, 162 Later on, Neidle and co-workers presented the first complex structure of parallel G-quadruplex and daunomycin.163 The asymmetric unit includes a quadruplex molecule formed by four parallel d(TGGGGT) strands, and three end-stacking daunomycin molecules associated with three sodium cations (Fig. 33A). The G-quadruplex complex structure is very similar to the native counterpart. And the daunomycin moiety has interactions with the quadruplex grooves through hydrogen bonding and van der Waals contacts. This structure indicates that the complex might be more stable with the ligand stacking on the end compared to the intercalation, since the surface around the terminal is large.164 These results are consistent with another complex structure containing a disubstituted acridine, where aG-quadruplex was formed by two strands of d(GGGGTTTTGGGG) and the ligand was also bound at the end of the G-quartet.165 In addition, these binding modes provide clues on how the molecules may interfere with telomere modification and maintenance by related proteins.
Figure 33.

(A) Overall structure (PDB ID: 1O0K) of the G-quadruplex [d(TGGGGT)4] complexed with daunomycin (the darker structures). (B)Overall structure (PDB ID: 2JT7) of the G-quadruplex [d(TGGGGT)4] complexed with distamycin A (the darker structures). The ligand molecules are in black. The dark spheres represent the sodium atoms.
Similarly, distamycin A is known to bind side by side to the minor groove of duplex DNA. The complex solution structure of a parallel quadruplex DNA [d(TGGGGT)4] and distamycin A has been accomplished recently by Randazzo and co-workers. The structure shows that drug binding is also similar to DNA minor groove binding, and two drug dimers bind two opposite sites of the quadruplex structure (Fig. 33B).166 Because the minor groove environment of quadruplexes differs from each other, this structure provides a fine starting point for designing quadruplex groove-specific binders. It was also found that the affinity between distamycin A and the G-quadruplex was enhanced approximately tenfold when the ratio of ligand to DNA was increased.
These studies also imply that DNA duplex-binding ligands should be reevaluated in G-quadruplex systems. In addition to the typical duplex DNA-binding agents, new types of compounds that can specifically bind to quadruplex DNA have been found and characterized by a series of biochemical and structural studies. These new compounds include BRACO 19, RHPS4, TMPyP4, and telomestatin (Fig. 34). These biochemical and structural investigations not only revealed more details about the interaction between the G-quadruplex DNA and ligands, such as the effects of external loop rearrangement, hydration patterns, and strands conformations, but also offered new platforms for further drug discovery to target G-quadruplex structures and regulate gene expression. The progresses in this area have been summarized recently by Neidle.167
Figure 34.

Chemical structures of several DNA quadruplex-binding ligands.
7. TARGETING UNMATCHED BULGE, DNA JUNCTIONS, AND PHOSPHATE BACKBONE
Besides the discussed binding modes, several other interesting “non-standard” interactions have also been observed between DNA and small molecules with therapeutic potential. A few examples of small molecules, which target the unmatched bulge, DNA junction, and phosphate backbone and have the DNA-complex structure information, are discussed here.
Mismatched and bulged DNAs can happen in hairpins or be caused by mutations of either of the paired bases, especially at extended triplet-repeat sequences in the genome of human diseases.168 Therefore, specific recognition of these structures may result in novel therapies. Several studies have shown that these types of local topology of nucleic acids can indeed be recognized by small compounds, following the study of a complex structure of an aminoglycoside antibiotic and a small ribosomal RNA.169, 170 Goldberg and co-workers solved a solution structure of a designed spirocyclic helical ligand (SCA-alpha2) (Fig. 35A) bound at the two-base bulge site in a DNA duplex [d(5′-CCATCGTCTACCTTTGGTAGGATGG-3′)2].99 SCA-alpha2 is a close mimic of the previously discussed NCSi-gb binder to the major groove. As shown in Figure 35B, the two rigid aromatic rings of the ligand stack on the DNA bulge bases and form a right-handed helical wedge that can penetrate the bulge-binding pocket and immobilize the two bulge residues (GT), which point toward the minor groove. The amino sugar anchors in the DNA major groove and points toward the 3′-bulge-flanking base pair.99 These details suggest that both shape and space are important in designing novel ligand targeting the duplex bulge site. The same group also solved the complex structure of a DNA duplex containing a GT bulge (5′-CACGCAGTTCGGAC•5′-GTCCGATGCGTG) and DDI (NCSi-gb synthetic mimic, Fig. 35C). As shown in Figure 35D, the benzindanone moiety stacks with neighboring G and A bases in the bulge site. The dihydronaphthalenone and aminoglycoside moieties are aligned to each other and are positioned in the minor groove, preventing its intercalation into the bulge site. In this complex, the unpaired guanosine is intrahelical and stacks with the intercalating moiety of DDI, while the unpaired thymine is extrahelical.171 Later, the ent-DDI (the DDI enantiomer, Fig. 35A) has also been examined in the same DNA duplex with the two-base bulge. It was found that ligand binding in the bulge area was opposite due to the opposite spirocycle chirality.172 The complex structure formed by binding of the synthetic agent to the two-base bulged DNA reveals a binding mode that differs in important details from that of the natural product, explaining the different binding specificity for the DNA bulge sites. Both structures indicated a large conformation change of the DNA upon the complex formation. This information at the molecular level provides a platform for designing stereochemically controlled drugs that recognize bulged DNA.
Figure 35.

Structures of SCA-α2, DDI, ent-DDI, and their DNA complexes. (A) Overall structure (PDB ID: 2OEY) of SCA-α2-bound DNA duplex with a two-base bulge [d(5′-CCATCGTCTACCTTTGGTAGGATGG-3′)2]. (B) Overall structure (PDB ID: 1P96) of DDI-bound DNA duplex with a two-base bulge. (C) Overall structure (PDB ID: 1R4E) of ent-DDI-bound DNA duplex with a two-base bulge. The duplex sequences for C and D are 5′-CACGCAGTTCGGAC-3′ and 5′-GTCCGATGCGTG-3′. The ligands are showed as black spheres.
A conventional strategy to recognize the mismatched bases is to use an intercalator to replace the mismatched base, while avoiding the phosphate backbone adjustment. For example, in a complex structure of a rhodium compound ([Rh(bpy)2chrysi]3+, Fig. 36A), there are several intercalating moieties (or ligands) capable of binding to a bulged DNA duplex. The ligand intercalates at the mismatch site, ejecting the mismatched base and replacing this pair with the aromatic four-ring system (Fig. 36B). The ejected bases can stay in both the minor and major groove.173 This interaction mode can be expected to displace a protein (such as DNA repair enzymes), thus making the damaged DNA unrepaired.
Figure 36.

(A) Chemical structure of rhodium compound ([Rh(bpy)2chrysi]3+. (B) Overall structure (PDB ID: 2O1I) of the rhodium complex (red sticks) bound to the DNA duplex [d(5′-CGGAAATTCCCG-3′)2], with the bulge bases (purple sticks) ejected. (C) Chemical structure of naphtyridine-azaquinoline conjugate (NA). (D) Overall structure of NA (red sticks) bound with a DNA duplex (5′-CTAACAGAATG-3′•5′-CATTCAGTTAG-3′), ejecting the two mismatched cytosine bases (purple sticks), PDB: 1 × 26.
Recently, a naphtyridine-azaquinoline (NA) derivative (Fig. 36C), containing two aromatic ring systems connected by a spacer, was found to bind to DNA with triplet-repeat sequences, especially CAG repeats.174 As shown in Figure 36D, the two intercalating moieties of the ligand simultaneous pair with the mismatched A and the adjacent-G. Both of the A and G nucleobases are still located in the duplex structure, while the C paring with the adjacent G was pushed away from the original position. It is also observed that the spacers of the ligand molecules are parallel to each other in the major groove. Using this structure as a starting point, it is possible to design compounds that can mimic base pairs and recognize the mismatched DNA structure.
It is well known that DNA can form other higher ordered structures, such as the three-way junction and Holliday junction, under certain conditions. Three-way junctions have been found to be present in diseases, such as Huntington’s disease175 and myotonic dystrophy176, in viruses and in replicating DNAs, especially during the rapid growth of cancers. Small molecules have been developed to target three-way junctions.177, 178 Similar to bulge DNA targeting, these compounds usually contain two intercalating groups connected by a rigid linker, which forces the compounds to adopt elongated conformation. Recently, a complex structure of three-way junction bound to a small molecule was determined (Fig. 37A). In the structure, the ligand forms extensive interactions with the DNA. Its arms wrapped around the central iron atoms and form π–π stacking interactions with the central DNA bases. The shapes of the ligand and the DNA central cavity are complementary to each other. The DNA in the structure adopts an open and Y-shaped conformation with three arms extended away from the ligand-binding cavity. The DNA minor grooves interact with the pyrimidine rings of the ligand, which dominate the interactions between the DNA and ligand. The DNA major grooves are open for accepting more interactions to form tighter and more specific binding.
Figure 37.

(A) Structure of the three-way junction-binding ligand and the overall complex structure (PDB 2ET0) with the DNA three-way junction. (B) Structure of the Holiday junction-binding ligand and the overall complex structure (PDB 2GWA) with the Holiday junction.
Moreover, homologous recombination is a biological process for repair of a collapsed replication fork, which involves strand invasion, branch migration, and resolution of the Holliday junction formed by two adjacent duplexes.179 Homologous recombination-deficient cells are sensitive to poly(ADP-ribose) polymerase (PARP) inhibitors.180, 181 Inhibition of Holliday junction resolution may lead to general PARP1 sensitivity in replicating tumor cells. Recently, one Holiday junction structure in complex with an acridine derivative has been determined.182 In this structure (Fig. 37B), the two acridine moieties of the ligand functionalize as intercalators. The spacer has an appropriate length, which places the two acridine moieties at the correct distance for insertion at two distant sites. In addition to the four direct hydrogen bonds, π–π stacking interactions were also observed between DNA and ligand. These unique interactions in the structure can guide and facilitate design of new ligands that specifically bind the Holiday junction. To develop Holiday junction-specific binders, it is essential to avoid the molecular scaffold that binds duplex DNA. It is expected that more effective Holiday junction inhibitors will be discovered, when more Holiday junction structures complexed with ligands become available.
To target DNA with high specificity, most of the work in this area focuses on the nucleobases, which of course play the central role in the function of DNA through both hydrogen bonding and base stacking. Targeting other parts of the DNA structure (e.g., ribose and backbone) for specificity has not attracted much attention. In principle, it is also essential to target these components, since each DNA sequence presents a unique array of nucleobases, sugars, and backbone phosphates with subtle differences, which can result in high specificity in the DNA recognition. A recent study by Williams and co-workers described the targeting of the phosphate backbone by a novel type of platinum compound through a binding mode named “phosphate clamps.”183 In the complex structure of the ligand and DNA at 1.2 Å high resolution, a double-stranded Dickerson dodecamer DNA [(CGCGAATTCGCG)2] is recognized by a cytotoxic platinum (II) complex (Triplatin NC). This Pt-compound, without the covalent bonding potential, consists of three cationic Pt(II) centers and hydrogen-bonding functionalities linked by a flexible hydrophobic linkage. As shown in Figure 38, this ligand associates with the backbone by binding with the phosphate oxygen atoms, and presents its hydrophobic linkages over the minor groove. Interestingly, the ligand appears to prefer O2P over O1P atoms. Recognition of the subtle differences on the backbone may offer a new potential to target DNA sequence specifically, especially in combination with other recognizing elements.
Figure 38.

(A) Chemical structure of triplatinNC. (B) Overall structure (PDB ID: 2DYW) of triplatinNC (darker sticks) bound to DNA duplex [d(5′-CGCGAATTCGCG-3′)2]. The spheres represent the platinum atoms.
8. TARGETING DNA BY TRIPLEX FORMATION
As another type of DNA-major groove binder, single-stranded DNA can also bind to a DNA duplex sequence specifically through Hoogsteen or reverse Hoogsteen-type interactions. These are called triplex-formation oligonucleotides (TFOs).184 This research subject has been extensively reviewed recently.185, 186 As this review focuses mainly on the interactions of nucleic acids and small molecules, we will just briefly discuss the TFO strategy here. The binding of TFOs can block the DNA major groove and prevent access of other molecules, such as proteins, thereby inhibiting transcription.187 The resulting overall conformation of the triplex is similar, but different from the B-form. DNA triplex structures can form either intermolecularly through binding of an exogenously applied oligonucleotide to a target duplex sequence,188 or intramolecularly by the naturally existing endogenous mirror-repeat sequences.189 Currently, the intermolecular triplexes have gained much more attention because of their potential clinical applications in treating cancer and other human diseases. The only requirement of this binding mode is the existence of purine-rich tracts in the target duplex in order to gain high-binding affinity and specificity.190, 191
In theory, the TFO should be a good strategy target DNA, but there are several practical drawbacks preventing them from being a useful therapeutic tool, such as nuclease sensitivity, tendency to form self-associating structures, poor discrimination of DNA from RNA, triplex instability, and difficult cellular uptake.144 However, a lot of research efforts have been made in order to overcome these drawbacks and make it feasible for therapeutic use, including the introduction of chemical modifications such as threose nucleic acid (TNA), peptide nucleic acid (PNA), and locked nucleic acid (LNA). Furthermore, detailed structural studies of these systems have facilitated advancement in this area. Since the TFO naturally exists in biological systems, therefore, the TFO itself can also serve as a therapeutic target for novel drug development. Actually, most of the previously mentioned DNA intercalators can also bind and stabilize the triplex system due to their planar structure,192 though the selectivity is expected to be low. Moreover, Arya and co-workers have presented an alternative method to recognize the DNA triplex using aminosugar compounds. For example, neomycin, a major groove binder, can stabilize both the TAT triplex and base-mixed DNA triplexes without affecting the DNA duplex. This selectivity was attributed to its potential and shape complementarity to the triplex Watson–Hoogsteen groove, which was supported by biophysical and computational studies.193 Based on that, intercalator–neomycin conjugates have also been developed, similar to the major groove binders. Among them, pyrene–neomycin194 and BQQ–neomycin195 are the two most effective conjugates. These principles of dual recognition should be applicable to design ligands that can bind any nucleic acid target with high affinity and selectivity. The associated structure is yet to be determined, which will demonstrate the proposed binding modes and serve as a platform for further ligand design and drug development.
9. PERSPECTIVES
Drug discovery is a very complicated process and involves many different steps, such as target selection, target site identification, lead compound selection, and optimization. Integrated efforts are required from structural biology, computational modeling, and biophysical studies. The majority of currently marketed drugs target proteins, such as G-protein-coupled receptors, nuclear hormone receptors, and other proteins involved in disease pathway. Due to the accessibility of proteins, drug design, and discovery have being predominantly focused on this class of macromolecule since the beginning. The therapeutic potential of other macromolecules, such as nucleic acids as drug targets, has been largely ignored mainly due to their apparent chemical composition and similarity. For example, only about 2% of drugs targeted DNA and RNA molecules in 2003. Over the past few years, significant advances have been made in the nucleic acid research field both functionally and structurally. Functionally, it has been realized that besides genetic information storage and transfer, nucleic acids also play critical roles in many other biological processes, such as cell growth, development, and neoplastic transformation. Structurally, it has been revealed that in addition to forming duplex structures, nucleic acids can fold into higher ordered structures such as triplexes, quadruplexes, riboswitches, and ribozymes. When combined with proteins, nucleic acids can assemble into much more complicated structures, such as the ribosome.
Nucleic acids functioning as the primary molecular targets of some clinical drugs, such as anticancer drugs and antibiotics, have been experimentally confirmed. It is also clear that small molecule drugs can affect the function of nucleic acids through different mechanisms, such as altering the nucleic acid conformation or competing with the endogenous molecule regulators of nucleic acids. Unfortunately, many drug candidates have failed before or during the clinical trails due to their low sequence specificity or high toxicity. Thus, to speed up the process of drug discovery, scientists have developed many different strategies, including virtual screening, molecular modeling, and SBDD. Compared to other methods, SBDD, which depends on the availability of the target structure, especially the high-resolution crystal structures, is more rational. By summarizing many different DNA–ligand complex structures, we show in this review that small molecules can interact with nucleic acids through various interactions, such as hydrogen bonding, stacking, van de Waals interaction, salt bridge, solvent network, and shape complementary. These interactions and principles will undoubtedly stimulate the design of novel drugs targeting nucleic acids. Moreover, on the basis of extensive structure and function studies, it is expected that the proper arrangement and combination of multiple interacting elements will significantly increase target specificity as well as affinity.
Compared with determined protein structures, the number of determined nucleic acid structures is very limited (approximately 4% of protein structures), which is one of the main reasons for the lagging development of drugs targeting nucleic acids. Structure determination of nucleic acids is hindered by two main bottleneck problems, including crystallization and phase determination. Similar to protein, the phase problem of nucleic acid structure determination can be solved by molecular replacement, multi wavelength anomalous dispersion (MAD) and single wavelength anomalous dispersion (SAD) methods. Conventionally, heavy metal atoms such as iodine or bromide atoms are either soaked or chemically introduced into the nucleic acid structures for SAD and/or MAD.196–198 Recently, our laboratory has pioneered and developed a novel technology, the selenium derivatization of nucleic acids (SeNA).50, 199–207 Our experimental results have shown that the Se atom can be stably introduced to various positions of nucleic acids and significantly facilitate structural and functional studies of nucleic acids and protein–nucleic acid complexes. SeNA has great potentials to facilitate phase determination as well as the crystallization of nucleic acids, their small molecule complexes, and their protein complexes. Drug discovery to target nucleic acids has advanced significantly over the past several years in treating various human diseases, such as cancer, inflammation, and viral infections. Despite the many challenges (such as toxicity and sequence nonspecificity) that remain to be solved, continuing research on nucleic acid structure and function will rapidly advance our understanding of nucleic acid–ligand interactions as well as novel drug discovery. It is clear that nucleic acid targeting is an emergent area in both therapeutic and diagnostic development.
ACKNOWLEDGMENT
This work was financially supported by the Georgia Cancer Coalition (GCC) Distinguished Cancer Clinicians and Scientists and NIH (R01GM095881).
Biographies
Jia Sheng obtained his MS degree in organic chemistry in East China University of Science and Technology (Shanghai, China) with Prof. Limin Wang, where he studied the rare-earth catalyzed organic reactions. He went to USA in 2005 and obtained his Ph.D. degree in bioorganic chemistry at Georgia State University (Atlanta, Georgia) in 2009, under the supervision of Prof. Zhen Huang, where his focus was nucleic acid derivatization with selenium and tellurium for macromolecule structure and function studies. He is currently a research scientist in the same lab with Prof. Zhen Huang, further expanding the research in nucleic acid-small molecule interactions and protein–nucleic acid complex structure determination.
Jianhua Gan obtained his Ph.D. degree, in 2002, in organic chemistry in Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Zongxiang Xia, where he studied the protein crystallography. In 2003, he went to USA and joined Prof. Xinhua Ji’s group in National Cancer Institute (NIH, USA), studying the function and structure of ribonucleases III. Currently, he is a research scientist in Prof. Zhen Huang’s lab at Georgia State University, focusing on nucleic acid–protein interaction and structure determination.
Zhen Huang obtained his BS degree from Sichuan University in 1984, under the supervision of Prof. Shuling Chen, and his MS degree from Peking University in 1987, under the supervision of Prof. Zhong Wen. He obtained his Ph.D. degree from Swiss Federal Institute of Technology at Zurich (ETH) in 1994, under the supervision of Prof. Steven A. Benner. In the same year, he joined Department of Genetics at Harvard Medical School as Research Fellow, under the supervision of Prof. Jack W. Szostak. He was hired by Brooklyn College, the City University of New York, in 1998, as Assistant Professor, and was promoted to Associate Professor in 2004. In the same year, Dr. Huang was recruited to Chemistry Department at Georgia State University, and was promoted to Full Professor of Chemistry & Chemical Biology in 2009. His current research interests are in medicinal chemistry, anticancer research, synthesis and structure-and-function study of selenium-derivatized DNAs and RNAs, nucleic acid X-ray crystallography, structure and function studies of protein–nucleic acid complexes, development of novel RNA microchip technology for pathogen direct detection and gene expression profiling, nucleic acid-based cancer diagnosis, nanomedicine, and drug discovery.
REFERENCES
- 1.Kan CC. Impact of recombinant DNA technology and protein engineering on structure-based drug design: Case studies of HIV-1 and HCMV proteases. Curr Top Med Chem. 2002;2:247–269. doi: 10.2174/1568026023394218. [DOI] [PubMed] [Google Scholar]
- 2.Blount KF, Uhlenbeck OC. The structure-function dilemma of the hammerhead ribozyme. Annu Rev Biophys Biomol Struct. 2005;34:415–440. doi: 10.1146/annurev.biophys.34.122004.184428. [DOI] [PubMed] [Google Scholar]
- 3.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 4.Egli M, Pallan PS. Insights from crystallographic studies into the structural and pairing properties of nucleic acid analogs and chemically modified DNA and RNA oligonucleotides. Annu Rev Biophys Biomol Struct. 2007;36:281–305. doi: 10.1146/annurev.biophys.36.040306.132556. [DOI] [PubMed] [Google Scholar]
- 5.Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- 6.Keys HM, Bundy BN, Stehman FB, Muderspach LI, Chafe WE, Suggs CL, 3rd, Walker JL, Gersell D. Cisplatin, radiation, and adjuvant hysterectomy compared with radiation and adjuvant hysterectomy for bulky stage IB cervical carcinoma. N Engl J Med. 1999;340:1154–1161. doi: 10.1056/NEJM199904153401503. [DOI] [PubMed] [Google Scholar]
- 7.Morris M, Eifel PJ, Lu J, Grigsby PW, Levenback C, Stevens RE, Rotman M, Gershenson DM, Mutch DG. Pelvic radiation with concurrent chemotherapy compared with pelvic and para-aortic radiation for high-risk cervical cancer. N Engl J Med. 1999;340:1137–1143. doi: 10.1056/NEJM199904153401501. [DOI] [PubMed] [Google Scholar]
- 8.Rose PG, Bundy BN, Watkins EB, Thigpen JT, Deppe G, Maiman MA, Clarke-Pearson DL, Insalaco S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N Engl J Med. 1999;340:1144–1153. doi: 10.1056/NEJM199904153401502. [DOI] [PubMed] [Google Scholar]
- 9.Wang K, Lu J, Li R. The events that occur when cisplatin encounters cells. Coord Chem Rev. 1996;151:53–58. [Google Scholar]
- 10.Hostetter AA, Osborn MF, Derose VJ. RNA-Pt adducts following cisplatin treatment of Saccharomyces cerevisiae. ACS Chem Biol. 2012;7:218–225. doi: 10.1021/cb200279p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lippard SJ. New chemistry of an old molecule: cis-[Pt(NH3)2Cl2] Science. 1982;218:1075–1082. doi: 10.1126/science.6890712. [DOI] [PubMed] [Google Scholar]
- 12.Jamieson ER, Lippard SJ. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev. 1999;99:2467–2498. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- 13.Egger AE, Hartinger CG, Ben Hamidane H, Tsybin YO, Keppler BK, Dyson PJ. High resolution mass spectrometry for studying the interactions of cisplatin with oligonucleotides. Inorg Chem. 2008;47:10626–10633. doi: 10.1021/ic801371r. [DOI] [PubMed] [Google Scholar]
- 14.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 15.Takahara PM, Rosenzweig AC, Frederick CA, Lippard SJ. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature. 1995;377:649–652. doi: 10.1038/377649a0. [DOI] [PubMed] [Google Scholar]
- 16.Todd RC, Lippard SJ. Structure of duplexDNAcontaining the cisplatin 1,2-{Pt(NH3)2}2+-d(GpG) cross-link at 1.77 A resolution. J Inorg Biochem. 2010;104:902–908. doi: 10.1016/j.jinorgbio.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zlatanova J, Yaneva J, Leuba SH. Proteins that specifically recognize cisplatin-damaged DNA: A clue to anticancer activity of cisplatin. FASEB J. 1998;12:791–799. doi: 10.1096/fasebj.12.10.791. [DOI] [PubMed] [Google Scholar]
- 18.Ohndorf UM, Rould MA, He Q, Pabo CO, Lippard SJ. Basis for recognition of cisplatin-modified DNA by high-mobility-group proteins. Nature. 1999;399:708–712. doi: 10.1038/21460. [DOI] [PubMed] [Google Scholar]
- 19.Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T, Geacintov N, Livneh Z. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009;28:383–393. doi: 10.1038/emboj.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reissner T, Schneider S, Schorr S, Carell T. Crystal structure of a cisplatin-(1,3-GTG) cross-link within DNA polymerase eta. Angew Chem Int Ed Engl. 2010;49:3077–3080. doi: 10.1002/anie.201000414. [DOI] [PubMed] [Google Scholar]
- 21.Hall MD, Mellor HR, Callaghan R, Hambley TW. Basis for design and development of platinum( IV) anticancer complexes. J Med Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 22.Bruijnincx PC, Sadler PJ. New trends formetal complexes with anticancer activity. CurrOpin Chem Biol. 2008;12:197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacDiarmid JA, Mugridge NB, Weiss JC, Phillips L, Burn AL, Paulin RP, Haasdyk JE, Dickson KA, Brahmbhatt VN, Pattison ST, James AC, Al Bakri G, Straw RC, Stillman B, Graham RM, Brahmbhatt H. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11:431–445. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chem Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang F, Li F, Ganguly M, Marky LA, Gold B, Egli M, Stone MP. A bridging water anchors the tethered 5-(3-aminopropyl)-2’-deoxyuridine amine in the DNA major groove proximate to the N+2 C.G base pair: Implications for formation of interstrand 5′-GNC-3′ cross-links by nitrogen mustards. Biochemistry. 2008;47:7147–7157. doi: 10.1021/bi800375m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyer VN, Szybalski W. A molecular mechanism of mitomycin action: Linking of complementary DNA strands. Proc Natl Acad Sci USA. 1963;50:355–362. doi: 10.1073/pnas.50.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sastry M, Fiala R, Lipman R, Tomasz M, Patel DJ. Solution structure of the monoalkylated mitomycin C-DNA complex. J Mol Biol. 1995;247:338–359. doi: 10.1006/jmbi.1994.0143. [DOI] [PubMed] [Google Scholar]
- 28.Subramaniam G, Paz MM, Suresh Kumar G, Das A, Palom Y, Clement CC, Patel DJ, Tomasz M. Solution structure of a guanine-N7-linked complex of the mitomycin C metabolite 2,7- diaminomitosene and DNA. Basis of sequence selectivity. Biochemistry. 2001;40:10473–10484. doi: 10.1021/bi010965a. [DOI] [PubMed] [Google Scholar]
- 29.Eichman BF, Mooers BH, Alberti M, Hearst JE, Ho PS. The crystal structures of psoralen crosslinked DNAs: Drug-dependent formation of Holliday junctions. J Mol Biol. 2001;308:15–26. doi: 10.1006/jmbi.2001.4567. [DOI] [PubMed] [Google Scholar]
- 30.Mace K, Aguilar F, Wang JS, Vautravers P, Gomez-Lechon M, Gonzalez FJ, Groopman J, Harris CC, Pfeifer AM. Aflatoxin B1-induced DNA adduct formation and p53 mutations in CYP450- expressing human liver cell lines. Carcinogenesis. 1997;18:1291–1297. doi: 10.1093/carcin/18.7.1291. [DOI] [PubMed] [Google Scholar]
- 31.Shimada T, Guengerich FP. Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver. ProcNatl Acad Sci USA. 1989;86:462–465. doi: 10.1073/pnas.86.2.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hertzog PJ, Smith JR, Garner RC. Characterisation of the imidazole ring-opened forms of trans- 8,9-dihydro-8,9-dihydro-8-(7-guanyl)9-hydroxy aflatoxin B1. Carcinogenesis. 1982;3:723–725. doi: 10.1093/carcin/3.6.723. [DOI] [PubMed] [Google Scholar]
- 33.Brown KL, Voehler MW, Magee SM, Harris CM, Harris TM, Stone MP. Structural perturbations induced by the alpha-anomer of the aflatoxin B(1) formamidopyrimidine adduct in duplex and single-strand DNA. J Am Chem Soc. 2009;131:16096–16107. doi: 10.1021/ja902052v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swenson MC, Paranawithana SR, Miller PS, Kielkopf CL. Structure of a DNA repair substrate containing an alkyl interstrand cross-link at 1.65 A resolution. Biochemistry. 2007;46:4545–4553. doi: 10.1021/bi700109r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang H, Dooley PA, Harris CM, Harris TM, Stone MP. Differential base stacking interactions induced by trimethylene interstrand DNA cross-links in the 5′-CpG-3′ and 5′-GpC-3′ sequence contexts. Chem Res Toxicol. 2009;22:1810–1816. doi: 10.1021/tx900225c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang H, Kim HY, Kozekov ID, Cho YJ, Wang H, Kozekova A, Harris TM, Rizzo CJ, Stone MP. Stereospecific formation of the (R)-gamma-hydroxytrimethylene interstrand N2-dG:N2-dG cross-link arising from the gamma-OH-1,N2-propano-2’-deoxyguanosine adduct in the 5′-CpG-3′ DNA sequence. J Am Chem Soc. 2009;131:8416–8424. doi: 10.1021/ja809543j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang H, Wang H, Kozekova A, Rizzo CJ, Stone MP. Formation of a N(2)-dG:N(2)-dG carbinolamine DNA cross-link by the trans-4-hydroxynonenal-derived (6S,8R,11S) 1,N(2)-dG adduct. J Am Chem Soc. 2011;133:16101–16110. doi: 10.1021/ja205145q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wartell RM, Larson JE, Wells RD. Netropsin. A specific probe for A-T regions of duplex deoxyribonucleic acid. J Biol Chem. 1974;249:6719–6731. [PubMed] [Google Scholar]
- 39.Zimmer C. Effects of the antibiotics netropsin and distamycin A on the structure and function of nucleic acids. Prog Nucleic Acid Res Mol Biol. 1975;15:285–318. doi: 10.1016/s0079-6603(08)60122-1. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen B, Neidle S, Wilson WD. A role for water molecules in DNA-ligand minor groove recognition. Acc Chem Res. 2009;42:11–21. doi: 10.1021/ar800016q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Antiparasitic compounds that target DNA. Biochimie. 2008;90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arafa RK, Ismail MA, Munde M, Wilson WD, Wenzler T, Brun R, Boykin DW. Novel linear triaryl guanidines, N-substituted guanidines and potential prodrugs as antiprotozoal agents. Eur J Med Chem. 2008;43:2901–2908. doi: 10.1016/j.ejmech.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schultz PG, Dervan PB. Distamycin and penta-N-methylpyrrolecarboxamide binding sites on native DNA. A comparison of methidiumpropyl-EDTA-Fe(II) footprinting and DNA affinity cleaving. J Biomol Struct Dyn. 1984;1:1133–1147. doi: 10.1080/07391102.1984.10507508. [DOI] [PubMed] [Google Scholar]
- 44.Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. The molecular origin of DNA-drug specificity in netropsin and distamycin. Proc Natl Acad Sci USA. 1985;82:1376–1380. doi: 10.1073/pnas.82.5.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nunn CM, Garman E, Neidle S. Crystal structure of the DNA decamer d(CGCAATTGCG) complexed with the minor groove binding drug netropsin. Biochemistry. 1997;36:4792–4799. doi: 10.1021/bi9628228. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, Mitra SN, Rao ST, Sekar K, Sundaralingam M. A novel end-to-end binding of two netropsins to the DNA decamers d(CCCCCIIIII)2, d(CCCBr5CCIIIII)2and d(CBr5CCCCIIIII)2. Nucleic Acids Res. 1998;26:5464–5471. doi: 10.1093/nar/26.23.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tevis DS, Kumar A, Stephens CE, Boykin DW, Wilson WD. Large, sequence-dependent effects on DNA conformation by minor groove binding compounds. Nucleic Acids Res. 2009;37:5550–5558. doi: 10.1093/nar/gkp558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egli M, Pallan PS. The many twists and turns of DNA: Template, telomere, tool, and target. Curr Opin Struct Biol. 2010;20:262–275. doi: 10.1016/j.sbi.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, Romagnoli R. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev. 2004;24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]
- 50.Egli M. Nucleic acid crystallography: Current progress. Curr Opin Chem Biol. 2004;8:580–591. doi: 10.1016/j.cbpa.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Kopka ML, Yoon C, Goodsell D, Pjura P, Dickerson RE. Binding of an antitumor drug to DNA, netropsin and C-G-C-G-A-A-T-T-BrC-G-C-G. J Mol Biol. 1985;183:553–563. doi: 10.1016/0022-2836(85)90171-8. [DOI] [PubMed] [Google Scholar]
- 52.Larsen TA, Goodsell DS, Cascio D, Grzeskowiak K, Dickerson RE. The structure of DAPI bound to DNA. J Biomol Struct Dyn. 1989;7:477–491. doi: 10.1080/07391102.1989.10508505. [DOI] [PubMed] [Google Scholar]
- 53.Pjura PE, Grzeskowiak K, Dickerson RE. Binding of Hoechst 33258 to theminor groove of B-DNA. J Mol Biol. 1987;197:257–271. doi: 10.1016/0022-2836(87)90123-9. [DOI] [PubMed] [Google Scholar]
- 54.Brown DG, Sanderson MR, Skelly JV, Jenkins TC, Brown T, Garman E, Stuart DI, Neidle S. Crystal structure of a berenil-dodecanucleotide complex: The role of water in sequence-specific ligand binding. EMBO. 1990;9:1329–1334. doi: 10.1002/j.1460-2075.1990.tb08242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitra SN, Wahl MC, Sundaralingam M. Structure of the side-by-side binding of distamycin to d(GTATATAC)2. Acta Crystallogr D Biol Crystallogr. 1999;55:602–609. doi: 10.1107/s0907444998012475. [DOI] [PubMed] [Google Scholar]
- 56.Edwards KJ, Jenkins TC, Neidle S. Crystal structure of a pentamidine-oligonucleotide complex: Implication for DNA-binding properties. Biochemistry. 1992;31:7104–7109. doi: 10.1021/bi00146a011. [DOI] [PubMed] [Google Scholar]
- 57.Kielkopf CL, White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB, Rees DC. A structural basis for recognition of A.T and T.A base pairs in the minor groove of B-DNA. Science. 1998;282:111–115. doi: 10.1126/science.282.5386.111. [DOI] [PubMed] [Google Scholar]
- 58.Squire CJ, Baker LJ, Clark GR, Martin RF, White J. Structures ofm-iodo Hoechst-DNAcomplexes in crystals with reduced solvent content: Implications for minor groove binder drug design. Nucleic Acids Res. 2000;28:1252–1258. doi: 10.1093/nar/28.5.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neidle S, Mann J, Rayner E, Baron A, Boahen Y, Simpson IJ, Smith NJ, Fox KR, Hartley JA, Kelland LR. Symmetric bis-benzimidazoles: New sequence-selective DNA-binding molecules. Chem Commun. 1999:929–930. [Google Scholar]
- 60.Aymami J, Nunn CM, Neidle S. DNA minor groove recognition of a non-self-complementary AT-rich sequence by a tris-benzimidazole ligand. Nucleic Acids Res. 1999;27:2691–2698. doi: 10.1093/nar/27.13.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu CF, Dervan PB. Quantitating the concentration of Py-Im polyamide-fluorescein conjugates in live cells. Bioorg Med Chem Lett. 2008;18:5851–5855. doi: 10.1016/j.bmcl.2008.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edayathumangalam RS, Weyermann P, Gottesfeld JM, Dervan PB, Luger K. Molecular recognition of the nucleosomal “supergroove”. Proc Natl Acad Sci USA. 2004;101:6864–6869. doi: 10.1073/pnas.0401743101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crowley KS, Phillion DP, Woodard SS, Schweitzer BA, Singh M, Shabany H, Burnette B, Hippenmeyer P, Heitmeier M, Bashkin JK. Controlling the intracellular localization of fluorescent polyamide analogues in cultured cells. Bioorg Med Chem Lett. 2003;13:1565–1570. doi: 10.1016/s0960-894x(03)00152-5. [DOI] [PubMed] [Google Scholar]
- 64.Dose C, Farkas ME, Chenoweth DM, Dervan PB. Next generation hairpin polyamides with (R)- 3,4-diaminobutyric acid turn unit. J Am Chem Soc. 2008;130:6859–6866. doi: 10.1021/ja800888d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harki DA, Satyamurthy N, Stout DB, Phelps ME, Dervan PB. In vivo imaging of pyrrole-imidazole polyamides with positron emission tomography. Proc Natl Acad Sci USA. 2008;105:13039–13044. doi: 10.1073/pnas.0806308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chenoweth DM, Dervan PB. Allosteric modulation of DNA by small molecules. Proc Natl Acad Sci USA. 2009;106:13175–13179. doi: 10.1073/pnas.0906532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kielkopf CL, Baird EE, Dervan PB, Rees DC. Structural basis for G.C recognition in the DNA minor groove. Nat Struct Biol. 1998;5:104–109. doi: 10.1038/nsb0298-104. [DOI] [PubMed] [Google Scholar]
- 68.Hawkins CA, Baird EE, Dervan PB, Wemmer DE. Analysis of hairpin polyamide complexes having DNA binding sites in close proximity. J Am Chem Soc. 2002;124:12689–12696. doi: 10.1021/ja020335b. [DOI] [PubMed] [Google Scholar]
- 69.Zhang Q, Dwyer TJ, Tsui V, Case DA, Cho J, Dervan PB, Wemmer DE. NMR structure of a cyclic polyamide-DNA complex. J Am Chem Soc. 2004;126:7958–7966. doi: 10.1021/ja0373622. [DOI] [PubMed] [Google Scholar]
- 70.Chenoweth DM, Dervan PB. Structural basis for cyclic Py-Im polyamide allosteric inhibition of nuclear receptor binding. J Am Chem Soc. 2010;132:14521–14529. doi: 10.1021/ja105068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT. Structural basis of androgen receptor binding to selective androgen response elements. Proc Natl Acad Sci USA. 2004;101:4758–4763. doi: 10.1073/pnas.0401123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barker S, Weinfeld M, Murray D. DNA-protein crosslinks: Their induction, repair, and biological consequences. Mutat Res. 2005;589:111–135. doi: 10.1016/j.mrrev.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 74.Truglio JJ, Croteau DL, Skorvaga M, DellaVecchia MJ, Theis K, Mandavilli BS, Van Houten B, Kisker C. Interactions between UvrA and UvrB: The role of UvrB’s domain 2 in nucleotide excision repair. EMBO J. 2004;23:2498–2509. doi: 10.1038/sj.emboj.7600263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Minko IG, Zou Y, Lloyd RS. Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. ProcNatlAcad SciUSA. 2002;99:1905–909. doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang H, Kozekov ID, Kozekova A, Rizzo CJ, McCullough AK, Lloyd RS, Stone MP. Minor groove orientation of the KWKK peptide tethered via the N-terminal amine to the acroleinderived 1,N2-gamma-hydroxypropanodeoxyguanosine lesion with a trimethylene linkage. Biochemistry. 2010;49:6155–6164. doi: 10.1021/bi100364f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Subirana JA, Messeguer X. Structural families of genomic microsatellites. Gene. 2008;408:124–132. doi: 10.1016/j.gene.2007.10.025. [DOI] [PubMed] [Google Scholar]
- 78.Moreno T, Pous J, Subirana JA, Campos JL. Coiled-coil conformation of a pentamidine-DNA complex. Acta Crystallogr D Biol Crystallogr. 2010;66:251–257. doi: 10.1107/S0907444909055693. [DOI] [PubMed] [Google Scholar]
- 79.Halliwell B, Aruoma OI. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991;281:9–19. doi: 10.1016/0014-5793(91)80347-6. [DOI] [PubMed] [Google Scholar]
- 80.Tsunoda M, Sakaue T, Naito S, Sunami T, Abe N, Ueno Y, Matsuda A, Takenaka A. Insights into the structures ofDNAdamaged by hydroxyl radical: Crystal structures ofDNAduplexes containing 5-formyluracil. J Nucleic Acids. 2010;2010:1–10. doi: 10.4061/2010/107289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dervan PB, Burli RW. Sequence-specific DNA recognition by polyamides. Curr Opin Chem Biol. 1999;3:688–693. doi: 10.1016/s1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 82.Lajiness J, Sielaff A, Mackay H, Brown T, Kluza J, Nguyen B, Wilson WD, Lee M, Hartley JA. Polyamide curvature and DNA sequence selective recognition: Use of 4-aminobenzamide to adjust curvature. Med Chem. 2009;5:216–226. doi: 10.2174/157340609788185945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buchmueller KL, Staples AM, Howard CM, Horick SM, Uthe PB, Le NM, Cox KK, Nguyen B, Pacheco KA, Wilson WD, Lee M. Extending the language of DNA molecular recognition by polyamides: Unexpected influence of imidazole and pyrrole arrangement on binding affinity and specificity. J Am Chem Soc. 2005;127:742–750. doi: 10.1021/ja044359p. [DOI] [PubMed] [Google Scholar]
- 84.Miao Y, Lee MP, Parkinson GN, Batista-Parra A, Ismail MA, Neidle S, Boykin DW, Wilson WD. Out-of-shape DNA minor groove binders: Induced fit interactions of heterocyclic dications with the DNA minor groove. Biochemistry. 2005;44:14701–14708. doi: 10.1021/bi051791q. [DOI] [PubMed] [Google Scholar]
- 85.Liu Y, Kumar A, Depauw S, Nhili R, David-Cordonnier MH, Lee MP, Ismail MA, Farahat AA, Say M, Chackal-Catoen S, Batista-Parra A, Neidle S, Boykin DW, Wilson WD. Water-mediated binding of agents that target the DNA minor groove. J Am Chem Soc. 2011;133:10171–10183. doi: 10.1021/ja202006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goodwin KD, Long EC, Georgiadis MM. A host-guest approach for determining drug-DNA interactions: An example using netropsin. Nucleic Acids Res. 2005;33:4106–4116. doi: 10.1093/nar/gki717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Glass LS, Nguyen B, Goodwin KD, Dardonville C, Wilson WD, Long EC, Georgiadis MM. Crystal structure of a trypanocidal 4,4’-bis(imidazolinylamino)diphenylamine bound toDNA. Biochemistry. 2009;48:5943–5952. doi: 10.1021/bi900204w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dardonville C, Brun R. Bisguanidine, bis(2-aminoimidazoline), and polyamine derivatives as potent and selective chemotherapeutic agents against Trypanosoma brucei rhodesiense. Synthesis and in vitro evaluation. J Med Chem. 2004;47:2296–2307. doi: 10.1021/jm031024u. [DOI] [PubMed] [Google Scholar]
- 89.Willis B, Arya DP. An expanding view of aminoglycoside-nucleic acid recognition. Adv Carbohydr Chem Biochem. 2006;60:251–302. doi: 10.1016/S0065-2318(06)60006-1. [DOI] [PubMed] [Google Scholar]
- 90.Goldberg IH. Mechanism of neocarzinostatin action: role of DNA microstructure in determination of chemistry of bistranded oxidative damage. Acc Chem Res. 1991;24:191–198. [Google Scholar]
- 91.Kappen LS, Goldberg IH. Neocarzinostatin acts as a sensitive probe of DNA microheterogeneity: Switching of chemistry from C-1’ to C-4’ by a G.T mismatch 5′ to the site of DNA damage. Proc Natl Acad Sci USA. 1992;89:6706–6710. doi: 10.1073/pnas.89.15.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Myers AG, Proteau PJ, Handel TM. Stereochemical assignment of neocarzinostatin chromophore. Structures of neocarzinostatin chromophore-methyl thioglycolate adducts. J Am Chem Soc. 1988;110:7212–7214. [Google Scholar]
- 93.Gao X, Stassinopoulos A, Rice JS, Goldberg IH. Structural basis for the sequence-specific DNA strand cleavage by the enediyne neocarzinostatin chromophore. Structure of the post-activated chromophore-DNA complex. Biochemistry. 1995;34:40–49. doi: 10.1021/bi00001a006. [DOI] [PubMed] [Google Scholar]
- 94.Gao X, Stassinopoulos A, Gu J, Goldberg IH. NMR studies of the post-activated neocarzinostatin chromophore-DNA complex. Conformational changes induced in drug and DNA. Bioorg Med Chem. 1995;3:795–809. doi: 10.1016/0968-0896(95)00063-m. [DOI] [PubMed] [Google Scholar]
- 95.Stassinopoulos A, Ji J, Gao X, Goldberg IH. Solution structure of a two-baseDNAbulge complexed with an enediyne cleaving analog. Science. 1996;272:1943–1946. doi: 10.1126/science.272.5270.1943. [DOI] [PubMed] [Google Scholar]
- 96.Kappen LS, Goldberg IH. DNA conformation-induced activation of an enediyne for site-specific cleavage. Science. 1993;261:1319–1321. doi: 10.1126/science.8362243. [DOI] [PubMed] [Google Scholar]
- 97.Hensens OD, Chin DH, Stassinopoulos A, Zink DL, Kappen LS, Goldberg IH. Spontaneous generation of a biradical species of neocarzinostatin chromophore: Role in DNA bulge-specific cleavage. Proc Natl Acad Sci USA. 1994;91:4534–4538. doi: 10.1073/pnas.91.10.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gao X, Stassinopoulos A, Ji J, Kwon Y, Bare S, Goldberg IH. Induced formation of a DNA bulge structure by a molecular wedge ligand-postactivated neocarzinostatin chromophore. Biochemistry. 2002;41:5131–5143. doi: 10.1021/bi012112o. [DOI] [PubMed] [Google Scholar]
- 99.Zhang N, Lin Y, Xiao Z, Jones GB, Goldberg IH. Solution structure of a designed spirocyclic helical ligand binding at a two-base bulge site in DNA. Biochemistry. 2007;46:4793–4803. doi: 10.1021/bi602599d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arya DP, Xue L, Willis B. Aminoglycoside (neomycin) preference is for A-form nucleic acids, not just RNA: Results from a competition dialysis study. J Am Chem Soc. 2003;125:10148–10149. doi: 10.1021/ja035117c. [DOI] [PubMed] [Google Scholar]
- 101.Arya DP, Willis B. Reaching into the major groove of B-DNA: Synthesis and nucleic acid binding of a neomycin-Hoechst 33258 conjugate. J Am Chem Soc. 2003;125:12398–12399. doi: 10.1021/ja036742k. [DOI] [PubMed] [Google Scholar]
- 102.Willis B, Arya DP. Recognition of B-DNA by neomycin–Hoechst 33258 conjugates. Biochemistry. 2006;45:10217–10232. doi: 10.1021/bi0609265. [DOI] [PubMed] [Google Scholar]
- 103.Tok JB, Cho J, Rando RR. Aminoglycoside antibiotics are able to specifically bind the 5′- untranslated region of thymidylate synthase messenger RNA. Biochemistry. 1999;38:199–206. doi: 10.1021/bi9819428. [DOI] [PubMed] [Google Scholar]
- 104.Willis B, Arya DP. Triple recognition of B-DNA by a neomycin-Hoechst 33258-pyrene conjugate. Biochemistry. 2010;49:452–469. doi: 10.1021/bi9016796. [DOI] [PubMed] [Google Scholar]
- 105.Dohm JA, Hsu MH, Hwu JR, Huang RC, Moudrianakis EN, Lattman EE, Gittis AG. Influence of ions, hydration, and the transcriptional inhibitor P4N on the conformations of the Sp1 binding site. J Mol Biol. 2005;349:731–744. doi: 10.1016/j.jmb.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 106.Nickols NG, Jacobs CS, Farkas ME, Dervan PB. Improved nuclear localization of DNA-binding polyamides. Nucleic Acids Res. 2007;35:363–370. doi: 10.1093/nar/gkl1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequencespecific DNA-binding polyamide. Proc Natl Acad Sci USA. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jacobs CS, Dervan PB. Modifications at the C-terminus to improve pyrrole-imidazole polyamide activity in cell culture. J Med Chem. 2009;52:7380–7388. doi: 10.1021/jm900256f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chaires JB. Energetics of drug-DNA interactions. Biopolymers. 1997;44:201–215. doi: 10.1002/(SICI)1097-0282(1997)44:3<201::AID-BIP2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 110.Richards AD, Rodger A. Synthetic metallomolecules as agents for the control of DNA structure. Chem Soc Rev. 2007;36:471–483. doi: 10.1039/b609495c. [DOI] [PubMed] [Google Scholar]
- 111.Lerman LS. Structural considerations in the interaction of DNA and acridines. J Mol Biol. 1961;3:18–30. doi: 10.1016/s0022-2836(61)80004-1. [DOI] [PubMed] [Google Scholar]
- 112.Snyder RD. Assessment of atypical DNA intercalating agents in biological and in silico systems. Mutat Res. 2007;623:72–82. doi: 10.1016/j.mrfmmm.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 113.Snyder RD, McNulty J, Zairov G, Ewing DE, Hendry LB. The influence of N-dialkyl and other cationic substituents on DNA intercalation and genotoxicity. Mutat Res. 2005;578:88–99. doi: 10.1016/j.mrfmmm.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 114.Tse WC, Boger DL. Sequence-selective DNA recognition: Natural products and nature’s lessons. Chem Biol. 2004;11:1607–1617. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 115.Weiss RB. The anthracyclines:Willwe ever find a better doxorubicin? SeminOncol. 1992;19:670–686. [PubMed] [Google Scholar]
- 116.Wang AH, Ughetto G, Quigley GJ, Rich A. Interactions between an anthracycline antibiotic and DNA: Molecular structure of daunomycin complexed to d(CpGpTpApCpG) at 1.2-A resolution. Biochemistry. 1987;26:1152–1163. doi: 10.1021/bi00378a025. [DOI] [PubMed] [Google Scholar]
- 117.Wang AH, Gao YG, Liaw YC, Li YK. Formaldehyde cross-links daunorubicin andDNAefficiently: HPLC and X-ray diffraction studies. Biochemistry. 1991;30:3812–3815. doi: 10.1021/bi00230a002. [DOI] [PubMed] [Google Scholar]
- 118.Frederick CA, Williams LD, Ughetto G, van derMarel GA, vanBoom JH, Rich A, Wang AH. Structural comparison of anticancer drug-DNA complexes: Adriamycin and daunomycin. Biochemistry. 1990;29:2538–2549. [PubMed] [Google Scholar]
- 119.Moore MH, Hunter WN, d’Estaintot BL, Kennard O. DNA-drug interactions. The crystal structure of d(CGATCG) complexed with daunomycin. J Mol Biol. 1989;206:693–705. doi: 10.1016/0022-2836(89)90577-9. [DOI] [PubMed] [Google Scholar]
- 120.Nunn CM, Van Meervelt L, Zhang SD, Moore MH, Kennard O. DNA-drug interactions.The crystal structures of d(TGTACA) and d(TGATCA) complexed with daunomycin. J Mol Biol. 1991;222:167–177. doi: 10.1016/0022-2836(91)90203-i. [DOI] [PubMed] [Google Scholar]
- 121.Leonard GA, Brown T, Hunter WN. Anthracycline binding to DNA. High-resolution structure of d(TGTACA) complexed with 4’-epiadriamycin. Eur J Biochem. 1992;204:69–74. doi: 10.1111/j.1432-1033.1992.tb16606.x. [DOI] [PubMed] [Google Scholar]
- 122.Davies DB, Eaton RJ, Baranovsky SF, Veselkov AN. NMR investigation of the complexation of daunomycin with deoxytetranucleotides of different base sequence in aqueous solution. J Biomol Struct Dyn. 2000;17:887–901. doi: 10.1080/07391102.2000.10506577. [DOI] [PubMed] [Google Scholar]
- 123.Frezard F, Garnier-Suillerot A. Comparison of the binding of anthracycline derivatives to purified DNA and to cell nuclei. Biochim Biophys Acta. 1990;1036:121–127. doi: 10.1016/0304-4165(90)90023-p. [DOI] [PubMed] [Google Scholar]
- 124.Trieb M, Rauch C, Wellenzohn B, Wibowo F, Loerting T, Mayer E, Liedl KR. Daunomycin intercalation stabilizes distinct backbone conformations ofDNA. J Biomol Struct Dyn. 2004;21:713–724. doi: 10.1080/07391102.2004.10506961. [DOI] [PubMed] [Google Scholar]
- 125.Lambert B, Roques BP, Le Pecq JB. Induction of an abortive and futile DNA repair process in E. coli by the antitumorDNAbifunctional intercalator, ditercalinium:Role in polA in death induction. Nucleic Acids Res. 1988;16:1063–1078. doi: 10.1093/nar/16.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lambert B, Jones BK, Roques BP, Le Pecq JB, Yeung AT. The noncovalent complex between DNA and the bifunctional intercalator ditercalinium is a substrate for the UvrABC endonuclease of Escherichia coli. Proc Natl Acad Sci USA. 1989;86:6557–6561. doi: 10.1073/pnas.86.17.6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lambert B, Segal-Bendirdjian E, Esnault C, Le Pecq JB, Roques BP, Jones B, Yeung AT. Recognition by the DNA repair system of DNA structural alterations induced by reversible drug-DNA interactions. Anticancer Drug Des. 1990;5:43–53. [PubMed] [Google Scholar]
- 128.Pelaprat D, Delbarre A, Le Guen I, Roques BP, Le Pecq JB. DNA intercalating compounds as potential antitumor agents. 2. Preparation and properties of 7H-pyridocarbazole dimers. J Med Chem. 1980;23:1336–1343. doi: 10.1021/jm00186a010. [DOI] [PubMed] [Google Scholar]
- 129.Leon P, Garbay-Jaureguiberry C, Barsi MC, Le Pecq JB, Roques BP. Modulation of the antitumor activity by methyl substitutions in the series of 7H-pyridocarbazole monomers and dimers. J Med Chem. 1987;30:2074–2080. doi: 10.1021/jm00394a024. [DOI] [PubMed] [Google Scholar]
- 130.Garbay-Jaureguiberry C, Laugaa P, Delepierre M, Laalami S, Muzard G, Le Pecq JB, Roques BP. DNA bis-intercalators as new anti-tumour agents: Modulation of the anti-tumour activity by the linking chain rigidity in the ditercalinium series. Anticancer Drug Des. 1987;1:323–335. [PubMed] [Google Scholar]
- 131.Gao Q, Williams LD, Egli M, Rabinovich D, Chen SL, Quigley GJ, Rich A. Drug-induced DNA repair: X-ray structure of a DNA-ditercalinium complex. Proc Natl Acad Sci USA. 1991;88:2422–2426. doi: 10.1073/pnas.88.6.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Waring MJ. DNA modification and cancer. Annu Rev Biochem. 1981;50:159–192. doi: 10.1146/annurev.bi.50.070181.001111. [DOI] [PubMed] [Google Scholar]
- 133.Park JY, Park SJ, Shim KY, Lee KJ, Kim YB, Kim YH, Kim SK. Echinomycin and a novel analogue induce apoptosis of HT-29 cells via the activation of MAP kinases pathway. Pharmacol Res. 2004;50:201–207. doi: 10.1016/j.phrs.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 134.Gilbert DE, Feigon J. The DNA sequence at echinomycin binding sites determines the structural changes induced by drug binding: NMR studies of echinomycin binding to [d(ACGTACGT)]2 and [d(TCGATCGA)]2. Biochemistry. 1991;30:2483–2494. doi: 10.1021/bi00223a027. [DOI] [PubMed] [Google Scholar]
- 135.Gilbert DE, Feigon J. Proton NMR study of the [d(ACGTATACGT)]2–2echinomycin complex: Conformational changes between echinomycin binding sites. Nucleic Acids Res. 1992;20:2411–2420. doi: 10.1093/nar/20.10.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gao XL, Patel DJ. NMR studies of echinomycin bisintercalation complexes with d(A1-C2-G3- T4) and d(T1-C2-G3-A4) duplexes in aqueous solution: Sequence-dependent formation of Hoogsteen A1.T4 and Watson–Crick T1.A4 base pairs flanking the bisintercalation site. Biochemistry. 1988;27:1744–1751. doi: 10.1021/bi00405a054. [DOI] [PubMed] [Google Scholar]
- 137.Gao XL, Patel DJ. Antitumour drug-DNA interactions: NMR studies of echinomycin and chromomycin complexes. Q Rev Biophys. 1989;22:93–138. doi: 10.1017/s0033583500003814. [DOI] [PubMed] [Google Scholar]
- 138.Quigley GJ, Ughetto G, van der Marel GA, van Boom JH, Wang AH, Rich A. Non-Watson-Crick G.C and A.T base pairs in a DNA-antibiotic complex. Science. 1986;232:1255–1258. doi: 10.1126/science.3704650. [DOI] [PubMed] [Google Scholar]
- 139.Wang AH, Ughetto G, Quigley GJ, Hakoshima T, van der Marel GA, van Boom JH, Rich A. The molecular structure of a DNA-triostin A complex. Science. 1984;225:1115–1121. doi: 10.1126/science.6474168. [DOI] [PubMed] [Google Scholar]
- 140.Cuesta-Seijo JA, Sheldrick GM. Structures of complexes between echinomycin and duplex DNA. Acta Crystallogr D Biol Crystallogr. 2005;61:442–448. doi: 10.1107/S090744490500137X. [DOI] [PubMed] [Google Scholar]
- 141.Cuesta-Seijo JA, Weiss MS, Sheldrick GM. Serendipitous SAD phasing of an echinomycin- (ACGTACGT)2 bisintercalation complex. Acta Crystallogr D Biol Crystallogr. 2006;62:417–424. doi: 10.1107/S0907444906003763. [DOI] [PubMed] [Google Scholar]
- 142.Bonjean K, De Pauw-Gillet MC, Defresne MP, Colson P, Houssier C, Dassonneville L, Bailly C, Greimers R, Wright C, Quetin-Leclercq J, Tits M, Angenot L. TheDNAintercalating alkaloid cryptolepine interferes with topoisomerase II and inhibits primarily DNA synthesis in B16 melanoma cells. Biochemistry. 1998;37:5136–5146. doi: 10.1021/bi972927q. [DOI] [PubMed] [Google Scholar]
- 143.Lisgarten JN, Coll M, Portugal J, Wright CW, Aymami J. The antimalarial and cytotoxic drug cryptolepine intercalates into DNA at cytosine-cytosine sites. Nat Struct Biol. 2002;9:57–60. doi: 10.1038/nsb729. [DOI] [PubMed] [Google Scholar]
- 144.Boer DR, Canals A, Coll M. DNA-binding drugs caught in action: The latest 3D pictures of drug-DNA complexes. Dalton Trans. 2009:399–414. doi: 10.1039/b809873p. [DOI] [PubMed] [Google Scholar]
- 145.Bailly C, Henichart JP. DNA recognition by intercalator-minor-groove binder hybrid molecules. Bioconjug Chem. 1991;2:379–393. doi: 10.1021/bc00012a001. [DOI] [PubMed] [Google Scholar]
- 146.Banerjee D, Pal SK. Simultaneous binding of minor groove binder and intercalator to dodecamer DNA: Importance of relative orientation of donor and acceptor in FRET. J Phys Chem B. 2007;111:5047–5052. doi: 10.1021/jp0715427. [DOI] [PubMed] [Google Scholar]
- 147.Rettig M, Weingarth M, Langel W, Kamal A, Kumar PP, Weisz K. Solution structure of a covalently bound pyrrolo[2,1-c][1,4]benzodiazepine-benzimidazole hybrid to a 10mer DNA duplex. Biochemistry. 2009;48:12223–12232. doi: 10.1021/bi901655t. [DOI] [PubMed] [Google Scholar]
- 148.Rettig M, Langel W, Kamal A, Weisz K. NMR structural studies on the covalent DNA binding of a pyrrolobenzodiazepine-naphthalimide conjugate. Org Biomol Chem. 2010;8:3179–3187. doi: 10.1039/c001893g. [DOI] [PubMed] [Google Scholar]
- 149.Wang Y, Schnetz-Boutaud NC, Kroth H, Yagi H, Sayer JM, Kumar S, Jerina DM, Stone MP. 3′-intercalation of a N2-dG 1R-trans-anti-benzo[c]phenanthrene DNA adduct in an iterated (CG)3 repeat. Chem Res Toxicol. 2008;21:1348–1358. doi: 10.1021/tx7004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Siegmund K, Maheshwary S, Narayanan S, Connors W, Riedrich M, Printz M, Richert C. Molecular details of quinolone-DNA interactions: Solution structure of an unusually stable DNA duplex with covalently linked nalidixic acid residues and non-covalent complexes derived from it. Nucleic Acids Res. 2005;33:4838–4848. doi: 10.1093/nar/gki795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, Margulis LA, Live D, Geacintov NE, Broyde S, et al. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc Natl Acad Sci USA. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang F, DeMuro NE, Elmquist CE, Stover JS, Rizzo CJ, Stone MP. Base-displaced intercalated structure of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme, a hotspot for-2 bp deletions. J Am Chem Soc. 2006;128:10085–10095. doi: 10.1021/ja062004v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang F, Elmquist CE, Stover JS, Rizzo CJ, Stone MP. DNA sequence modulates the conformation of the food mutagen 2-amino-3-methylimidazo[4,5-f]quinoline in the recognition sequence of the NarI restriction enzyme. Biochemistry. 2007;46:8498–8516. doi: 10.1021/bi700361u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.David-Cordonnier MH, Hildebrand MP, Baldeyrou B, Lansiaux A, Keuser C, Benzschawel K, Lemster T, Pindur U. Design, synthesis and biological evaluation of new oligopyrrole carboxamides linked with tricyclic DNA-intercalators as potential DNA ligands or topoisomerase inhibitors. Eur J Med Chem. 2007;42:752–771. doi: 10.1016/j.ejmech.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 155.Heald RA, Modi C, Cookson JC, Hutchinson I, Laughton CA, Gowan SM, Kelland LR, Stevens MF. Antitumor polycyclic acridines. 8.(1) Synthesis and telomerase-inhibitory activity of methylated pentacyclic acridinium salts. J Med Chem. 2002;45:590–597. doi: 10.1021/jm011015q. [DOI] [PubMed] [Google Scholar]
- 156.Riou JF, Guittat L, Mailliet P, Laoui A, Renou E, Petitgenet O, Megnin-Chanet F, Helene C, Mergny JL. Cell senescence and telomere shortening induced by a newseries of specific G-quadruplex DNA ligands. Proc Natl Acad Sci USA. 2002;99:2672–2677. doi: 10.1073/pnas.052698099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Harrison RJ, Gowan SM, Kelland LR, Neidle S. Human telomerase inhibition by substituted acridine derivatives. Bioorg Med Chem Lett. 1999;9:2463–2468. doi: 10.1016/s0960-894x(99)00394-7. [DOI] [PubMed] [Google Scholar]
- 158.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 159.Fedoroff OY, Salazar M, Han H, Chemeris VV, Kerwin SM, Hurley LH. NMR-Based model of a telomerase-inhibiting compound bound to G-quadruplex DNA. Biochemistry. 1998;37:12367–12374. doi: 10.1021/bi981330n. [DOI] [PubMed] [Google Scholar]
- 160.Gavathiotis E, Heald RA, Stevens MF, Searle MS. Recognition and stabilization of quadruplex DNA by a potent new telomerase inhibitor: NMR studies of the 2:1 complex of a pentacyclic methylacridinium cation with d(TTAGGGT)(4)We thank the EPSRC of the UK and AstraZeneca for financial support to E.G. M.F.G.S and R.A.H. are supported by the CancerResearch Campaign of the UK. Angew Chem Int Ed Engl. 2001;40:4749–4751. doi: 10.1002/1521-3773(20011217)40:24<4749::aid-anie4749>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 161.Elmore LW, Rehder CW, Di X, McChesney PA, Jackson-Cook CK, Gewirtz DA, Holt SE. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–35515. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 162.Ren J, Chaires JB. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry. 1999;38:16067–16075. doi: 10.1021/bi992070s. [DOI] [PubMed] [Google Scholar]
- 163.Clark GR, Pytel PD, Squire CJ, Neidle S. Structure of the first parallel DNA quadruplex-drug complex. J Am Chem Soc. 2003;125:4066–4067. doi: 10.1021/ja0297988. [DOI] [PubMed] [Google Scholar]
- 164.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 165.Haider SM, Parkinson GN, Neidle S. Structure of a G-quadruplex-ligand complex. J Mol Biol. 2003;326:117–125. doi: 10.1016/s0022-2836(02)01354-2. [DOI] [PubMed] [Google Scholar]
- 166.Martino L, Virno A, Pagano B, Virgilio A, Di Micco S, Galeone A, Giancola C, Bifulco G, Mayol L, Randazzo A. Structural and thermodynamic studies of the interaction of distamycin A with the parallel quadruplex structure [d(TGGGGT)]4. J Am Chem Soc. 2007;129:16048–16056. doi: 10.1021/ja075710k. [DOI] [PubMed] [Google Scholar]
- 167.Neidle S. The structures of quadruplex nucleic acids and their drug complexes. Curr Opin Struct Biol. 2009;19:239–250. doi: 10.1016/j.sbi.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 168.Turner DH. Bulges in nucleic acids. Curr Opin Struct Biol. 1992;2:334–337. [Google Scholar]
- 169.Kondo J, Francois B, Urzhumtsev A, Westhof E. Crystal structure of the Homo sapiens cytoplasmic ribosomal decoding site complexed with apramycin. Angew Chem Int Ed Engl. 2006;45:3310–3314. doi: 10.1002/anie.200600354. [DOI] [PubMed] [Google Scholar]
- 170.Kondo J, Pachamuthu K, Francois B, Szychowski J, Hanessian S, Westhof E. Crystal structure of the bacterial ribosomal decoding site complexed with a synthetic doubly functionalized paromomycin derivative: A new specific binding mode to an a-minor motif enhances in vitro antibacterial activity. ChemMedChem. 2007;2:1631–1638. doi: 10.1002/cmdc.200700113. [DOI] [PubMed] [Google Scholar]
- 171.Hwang GS, Jones GB, Goldberg IH. Solution structure of a wedge-shaped synthetic molecule at a two-base bulge site in DNA. Biochemistry. 2003;42:8472–8483. doi: 10.1021/bi030072d. [DOI] [PubMed] [Google Scholar]
- 172.Hwang GS, Jones GB, Goldberg IH. Stereochemical control of small molecule binding to bulged DNA: Comparison of structures of spirocyclic enantiomer-bulged DNA complexes. Biochemistry. 2004;43:641–650. doi: 10.1021/bi035824i. [DOI] [PubMed] [Google Scholar]
- 173.Pierre VC, Kaiser JT, Barton JK. Insights into finding a mismatch through the structure of a mispaired DNA bound by a rhodium intercalator. Proc Natl Acad Sci USA. 2007;104:429–434. doi: 10.1073/pnas.0610170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nakatani K, Hagihara S, Goto Y, Kobori A, Hagihara M, Hayashi G, Kyo M, Nomura M, Mishima M, Kojima C. Small-molecule ligand induces nucleotide flipping in (CAG)n trinucleotide repeats. Nat Chem Biol. 2005;1:39–43. doi: 10.1038/nchembio708. [DOI] [PubMed] [Google Scholar]
- 175.Sinden RR, Potaman VN, Oussatcheva EA, Pearson CE, Lyubchenko YL, Shlyakhtenko LS. Triplet repeatDNAstructures and human genetic disease: Dynamicmutations fromdynamicDNA. J Biosci. 2002;27:53–65. doi: 10.1007/BF02703683. [DOI] [PubMed] [Google Scholar]
- 176.Pearson CE, Sinden RR. Alternative structures in duplex DNA formed within the trinucleotide repeats of the myotonic dystrophy and fragile X loci. Biochemistry. 1996;35:5041–5053. doi: 10.1021/bi9601013. [DOI] [PubMed] [Google Scholar]
- 177.Oleksy A, Blanco AG, Boer R, Uson I, Aymami J, Rodger A, Hannon MJ, Coll M. Molecular recognition of a three-way DNA junction by a metallosupramolecular helicate. Angew Chem Int Ed Engl. 2006;45:1227–1231. doi: 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]
- 178.Cerasino L, Hannon MJ, Sletten E. DNA three-way junction with a dinuclear iron(II) supramolecular helicate at the center: A NMR structural study. Inorg Chem. 2007;46:6245–6251. doi: 10.1021/ic062415c. [DOI] [PubMed] [Google Scholar]
- 179.Ortiz-Lombardia M, Gonzalez A, Eritja R, Aymami J, Azorin F, Coll M. Crystal structure of a DNA Holliday junction. Nat Struct Biol. 1999;6:913–917. doi: 10.1038/13277. [DOI] [PubMed] [Google Scholar]
- 180.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 181.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 182.Brogden AL, Hopcroft NH, Searcey M, Cardin CJ. Ligand bridging of theDNA Holliday junction: Molecular recognition of a stacked-X four-way junction by a small molecule. Angew Chem Int Ed Engl. 2007;46:3850–3854. doi: 10.1002/anie.200603760. [DOI] [PubMed] [Google Scholar]
- 183.Komeda S, Moulaei T, Woods KK, Chikuma M, Farrell NP, Williams LD. A third mode of DNA binding: Phosphate clamps by a polynuclear platinum complex. J Am Chem Soc. 2006;128:16092–16103. doi: 10.1021/ja062851y. [DOI] [PubMed] [Google Scholar]
- 184.Le Doan T, Perrouault L, Praseuth D, Habhoub N, Decout JL, Thuong NT, Lhomme J, Helene C. Sequence-specific recognition, photocrosslinking and cleavage of the DNA double helix by an oligo-[alpha]-thymidylate covalently linked to an azidoproflavine derivative. Nucleic Acids Res. 1987;15:7749–7760. doi: 10.1093/nar/15.19.7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nielsen PE. Sequence-selective targeting of duplex DNA by peptide nucleic acids. Curr Opin Mol Ther. 2010;12:184–191. [PubMed] [Google Scholar]
- 186.Jain A, Wang G, Vasquez KM. DNA triple helices: Biological consequences and therapeutic potential. Biochimie. 2008;90:1117–1130. doi: 10.1016/j.biochi.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 188.Malkov VA, Soyfer VN, Frank-Kamenetskii MD. Effect of intermolecular triplex formation on the yield of cyclobutane photodimers in DNA. Nucleic Acids Res. 1992;20:4889–4895. doi: 10.1093/nar/20.18.4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mirkin SM, Frank-Kamenetskii MD. H-DNA and related structures. Annu Rev Biophys Biomol Struct. 1994;23:541–576. doi: 10.1146/annurev.bb.23.060194.002545. [DOI] [PubMed] [Google Scholar]
- 190.Fichou Y, Ferec C. The potential of oligonucleotides for therapeutic applications. Trends Biotechnol. 2006;24:563–570. doi: 10.1016/j.tibtech.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 191.Rhee S, Han Z, Liu K, Miles HT, Davies DR. Structure of a triple helicalDNAwith a triplex-duplex junction. Biochemistry. 1999;38:16810–16815. doi: 10.1021/bi991811m. [DOI] [PubMed] [Google Scholar]
- 192.Sandstrom K, Warmlander S, Bergman J, Engqvist R, Leijon M, Graslund A. The influence of intercalator binding on DNA triplex stability: Correlation with effects on A-tract duplex structure. J Mol Recognit. 2004;17:277–285. doi: 10.1002/jmr.665. [DOI] [PubMed] [Google Scholar]
- 193.Arya DP, Micovic L, Charles I, Coffee RL, Jr, Willis B, Xue L. Neomycin binding to Watson- Hoogsteen (W-H) DNA triplex groove: A model. J Am Chem Soc. 2003;125:3733–3744. doi: 10.1021/ja027765m. [DOI] [PubMed] [Google Scholar]
- 194.Xue L, Charles I, Arya DP. Pyrene-neomycin conjugate: Dual recognition of a DNA triple helix. Chem Commun (Camb) 2002:70–71. doi: 10.1039/b108171c. [DOI] [PubMed] [Google Scholar]
- 195.Arya DP, Xue L, Tennant P. Combining the best in triplex recognition: Synthesis and nucleic acid binding of a BQQ-neomycin conjugate. J Am Chem Soc. 2003;125:8070–8071. doi: 10.1021/ja034241t. [DOI] [PubMed] [Google Scholar]
- 196.Xiong Y, Sundaralingam M. Crystal structure of a DNA.RNA hybrid duplex with a polypurine RNA Rgaagaagag) and a complementary polypyrimidine DNA d(CTCTTCTTC) Nucleic Acids Res. 2000;28:2171–2176. doi: 10.1093/nar/28.10.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Pley HW, Flaherty KM, McKay DB. Three-dimensional structure of a hammerhead ribozyme. Nature. 1994;372:68–74. doi: 10.1038/372068a0. [DOI] [PubMed] [Google Scholar]
- 198.Scott WG, Finch JT, Klug A. The crystal structure of an all-RNA hammerhead ribozyme: A proposed mechanism for RNA catalytic cleavage. Cell. 1995;81:991–1002. doi: 10.1016/s0092-8674(05)80004-2. [DOI] [PubMed] [Google Scholar]
- 199.Carrasco N, Buzin Y, Tyson E, Halpert E, Huang Z. Selenium derivatization and crystallization of DNA and RNA oligonucleotides for X-ray crystallography using multiple anomalous dispersion. Nucleic Acids Res. 2004;32:1638–1646. doi: 10.1093/nar/gkh325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Carrasco N, Ginsburg D, Du Q, Huang Z. Synthesis of selenium-derivatized nucleosides and oligonucleotides for X-ray crystallography. Nucleosides Nucleotides Nucleic Acids. 2001;20:1723–1734. doi: 10.1081/NCN-100105907. [DOI] [PubMed] [Google Scholar]
- 201.Carrasco N, Huang Z. Enzymatic synthesis of phosphoroselenoateDNA using thymidine 5′-(alpha- P-seleno)triphosphate and DNA polymerase for X-ray crystallography via MAD. J Am Chem Soc. 2004;126:448–449. doi: 10.1021/ja0383221. [DOI] [PubMed] [Google Scholar]
- 202.Caton-Williams J, Huang Z. Biochemistry of selenium-derivatized naturally occurring and unnatural nucleic acids. Chem Biodivers. 2008;5:396–407. doi: 10.1002/cbdv.200890040. [DOI] [PubMed] [Google Scholar]
- 203.Jiang J, Sheng J, Carrasco N, Huang Z. Selenium derivatization of nucleic acids for crystallography. Nucleic Acids Res. 2007;35:477–485. doi: 10.1093/nar/gkl1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Serganov A, Keiper S, Malinina L, Tereshko V, Skripkin E, Hobartner C, Polonskaia A, Phan AT, Wombacher R, Micura R, Dauter Z, Jaschke A, Patel DJ. Structural basis for Diels-Alder ribozyme-catalyzed carbon-carbon bond formation. Nat Struct Mol Biol. 2005;12:218–224. doi: 10.1038/nsmb906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sheng J, Jiang J, Salon J, Huang Z. Synthesis of a 2’-Se-thymidine phosphoramidite and its incorporation into oligonucleotides for crystal structure study. Org Lett. 2007;9:749–752. doi: 10.1021/ol062937w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sheng J, Huang Z. Selenium derivatization of nucleic acids for X-ray crystal-structure and function studies. Chem Biodivers. 2010;7:753–785. doi: 10.1002/cbdv.200900200. [DOI] [PubMed] [Google Scholar]
- 207.Lin L, Sheng J, Huang Z. Nucleic acid X-ray crystallography via direct selenium derivatization. Chem Soc Rev. 2011;40:4591–4602. doi: 10.1039/c1cs15020k. [DOI] [PubMed] [Google Scholar]