

Abstract
Despite the longstanding importance of polyketide natural products in human medicine, nearly all commercial polyketide-based drugs are prepared through fermentation or semi-synthesis. The paucity of manufacturing routes involving de novo chemical synthesis reflects the inability of current methods to concisely address the preparation of these complex structures. Direct alcohol C-H bond functionalization via “C-C bond forming transfer hydrogenation” provides a powerful, new means of constructing type I polyketides that bypasses stoichiometric use of chiral auxiliaries, premetallated C-nucleophiles, and discrete alcohol-to-aldehyde redox reactions. Using this emergent technology, the total syntheses of 6-deoxyerythronolide B, bryostatin 7, trienomycins A and F, cyanolide A, roxaticin, and formal syntheses of rifamycin S and scytophycin C, were accomplished. These syntheses represent the most concise routes reported to any member of these respective natural product families.
1. Introduction
Polyketides are a broad class of secondary metabolites that display a diverse range of biological properties and are used extensively in human medicine.1,2 Approximately 20% of the top-selling small molecule drugs are polyketides,2,3 and it is estimated that polyketides are five times more likely to possess drug activity compared to other natural product families.3 The first commercial polyketide drug, erythromycin A, a macrolide antibiotic isolated from soil bacteria from the Philippines, was marketed by Eli Lilly in 1952.4 Later in 1955, amphotericin B, an important anti-fungal drug was discovered in soil samples taken from Venezuela,5 and shortly thereafter, rifamycin B, which vanquished drug-resistant tuberculosis in the 1960s,6 was isolated from soil samples taken from the south of France (Figure 1). While these and many other medicinally relevant polyketides derive from soil bacteria, it is estimated that less than 5% of soil bacteria are amenable to culture, and those that are do not represent the complete phylogenetic diversity of these organisms, as many bacterial phyla have eluded culture.7 As methods for the culture of soil bacteria improve, the role of polyketide natural products in human medicine will surely broaden, as will the need for manufacturing routes to these complex structures and their derivatives.
Figure 1.

Representative polyketide natural products isolated from soil bacteria used in human medicine.
Nearly all commercial polyketides are prepared through fermentation or semi-synthesis.8 Polyketide construction by de novo chemical synthesis offers entry to otherwise inaccessible functional analogues; however, despite enormous strides, the current ensemble of synthetic methods does not permit concise access to such complex structures. To address this deficiency, our laboratory has developed a broad, new suite of catalytic processes that promote C-C bond formation via direct alcohol C-H functionalization.9 In these processes, hydrogen exchange between alcohols and π-unsaturated reactants generates aldehyde-organometal pairs that combine to form products of carbonyl addition. These methods streamline synthesis as they bypass discrete alcohol-to-aldehyde redox reactions, and the stoichiometric use of chiral auxiliaries or premetallated C-nucleophiles. Applications of this technology to the total syntheses of 6-deoxyerythronolide B, bryostatin 7, trienomycins A and F, cyanolide A, roxaticin are described in this review, as are formal syntheses of rifamycin S and scytophycin C. These syntheses represent the most concise routes to any member of these respective natural products families.
2. A New Lexicon of Catalytic Methods
Enantioselective carbonyl allylation and crotylation are important gateways to polyketide natural products.10 Traditional methods for carbonyl allylation are not well suited for implementation on scale due to the frequent requirement of cryogenic conditions, multi-step synthesis of the requisite allylmetal reagents, generation of stoichiometric byproducts, the need for discrete alcohol-to-aldehyde oxidation, and the configurational and chemical lability of chiral α-stereogenic aldehydes. To address these shortcomings, our laboratory developed a series of ortho-cyclometallated iridium catalysts for the enantioselective C-H allylation of primary alcohols employing allyl acetate as the allyl donor.11 Our initial studies employed cyclometallated iridium catalysts generated in situ from allyl acetate, 3-nitrobenzoic acid and a chiral bis-phosphine ligand.11a,b Aliphatic, allylic and benzylic alcohols couple with allyl acetate to form the corresponding homoallylic alcohols with consistently high levels of enantioselectivity. Under otherwise identical conditions, but in the presence of isopropanol, aldehydes are transformed to an equivalent set of homoallylic alcohols (Scheme 1).
Scheme 1.
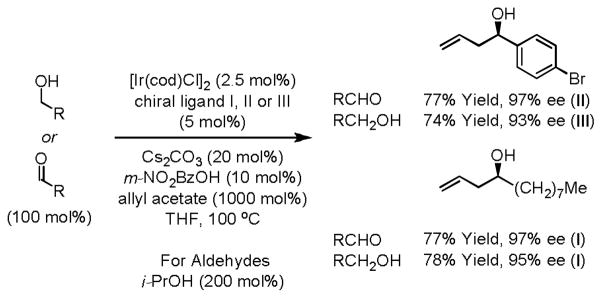
Enantioselective iridium catalyzed alcohol C-H allylation.a
aI = (R)-Cl,MeO-BIPHEP, II = (R)-TMBTP, III = (R)-BINAP
This method was applied to the iterative elongation of 1,3-polyols to form polyacetate substructures evident in oxo-polyene macrolides.11d Complete levels of catalyst directed stereoselectivity were observed. Using the cyclometallated iridium complex purified by conventional silica gel flash chromatography, chemo- and stereoselective iridium catalyzed alcohol C-H allylation of polyols is achieved in the absence of protecting groups.11f Secondary alcohols present in the reactant and product remain unchanged due to a pronounced kinetic preference for primary alcohol dehydrogenation. Propane diol engages in two-directional chain elongation through successive generation and capture of transient mono-aldehydes to provide C2-symmetric adducts in good yield.11c The minor enantiomer of the mono-adduct is transformed to the meso-stereoisomer, amplifying the enantiomeric purity of the product.12 The ability to engage polyols directly in a site-selective manner streamlines synthesis, as it removes steps otherwise required for protecting group installation-removal and it avoids discrete alcohol-to-aldehyde oxidation (Scheme 2).
Scheme 2.

Generation of polyacetate substructures via 1- and 2-directional chain elongation.
The cyclometallated π-allyliridium 3,4-dinitro-C,O-benzoate complex modified by (R)- or (S)-Cl,MeO-BIPHEP promotes the transfer hydrogenative coupling of allyl acetate to chiral β-stereogenic alcohols with good to excellent levels of catalyst-directed diastereoselectivity to furnish homoallylic alcohols that possess stereotriads found in numerous type I polyketides.11e,13 This protocol bypasses discrete generation of configurationally labile chiral α-stereogenic aldehydes, which are used routinely in polyketide construction, yet often cannot be stored or subjected to silica gel chromatography without erosion of enantiomeric purity (Scheme 3).14
Scheme 3.

Asymmetric iridium catalyzed C-H allylation of chiral β-stereogenic alcohols.
Related alcohol C-H crotylations employing α-methyl allyl acetate as the crotyl donor initially were achieved using the ortho-cyclometallated iridium C,O-benzoate prepared in situ from [Ir(cod)Cl]2, α-methyl allyl acetate, 4-cyano-3-nitrobenzoic acid and the chiral phosphine ligand (S)-SEGPHOS.15a Although in situ generation of the catalyst is convenient and exceptional enantioselectivities were observed, moderate levels of anti-diastereoselectivity were observed (5:1 – 11:1 dr). It was found that the cyclometallated π-allyliridium complex can be purified by conventional silica gel flash chromatography, and that the chromatographically purified catalyst functions at lower temperature (60 °C), resulting in enhanced levels of anti-diastereo- and enantioselectivity (Scheme 4).15b
Scheme 4.

Enantioselective iridium catalyzed alcohol C-H crotylation.
A general catalytic mechanism has been proposed (Figure 2).9a,11b Mechanistic studies corroborate turnover limiting carbonyl addition.11b The pronounced influence of remote electron withdrawing groups at the 4-position of the C,O-benzoate moiety of the catalyst can be interpreted on this basis. For example, in C-H allylations of chiral β-stereogenic alcohols (Scheme 3), remote electron withdrawing groups were shown to minimize racemization of the transient chiral α-stereogenic aldehyde. Here, enhanced Lewis acidity at iridium may accelerate turnover limiting carbonyl addition with respect to aldehyde epimerization. Additionally, in the C-H crotylation of primary alcohols, kinetic selectivity observed in the generation of the (E)-σ-crotyliridium intermediate, which reacts stereospecifically to deliver the anti-diastereomer, is preserved when the rate of carbonyl addition is accelerated with respect to isomerization to the (Z)-σ-crotyliridium intermediate, which would react to form the syn-diastereomer. Increased Lewis acidity at iridium also may facilitate catalysis by strengthening the agostic interaction between iridium and the carbinol C-H bond to facilitate alcohol dehydrogenation. Deprotonation of the iridium(III) hydride obtained upon alcohol dehydrogenation and product-to-reactant alkoxide exchange also may be accelerated by enhanced Lewis acidity at iridium. As revealed in single crystal X-ray diffraction data of a series of π-allyliridium C,O-benzoate complexes,11e more electron deficient C,O-benzoates possess longer C-Ir, O-Ir, and P-Ir bonds, suggesting enhanced Lewis acidity (Figure 2).
Figure 2.

General catalytic mechanism and survey of selected bond lengths from a series of π-allyliridium C,O-benzoate complexes.
Using these catalytic methods for enantioselective alcohol C-H allylation and crotylation, a total synthesis of the oxo-polyene macrolide (+)-roxaticin was accomplished in 20 steps (LLS) from 1,3-propanediol.16f In this approach, nine of ten C-C bonds formed in the longest linear sequence are made via metal catalysis, including 7 C-C bonds formed via iridium catalyzed alcohol C-C coupling. Notably, the present synthesis, which represents the most concise preparation of any oxo-polyene macrolide reported to date, is achieved in the absence of chiral reagents, chiral auxiliaries and premetallated C-nucleophiles (Scheme 5).
Scheme 5.
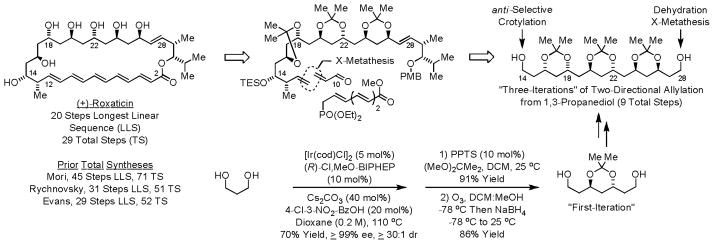
Total synthesis of the oxo-polyene macrolide (+)-roxaticin.
The rapid generation of polyacetate substructures via iterative double allylation of 1,3-propanediol prompted an investigation into related double crotylations of 2-methyl-1,3-propanediol.15c In the event, the chromatographically purified iridium precatalyst modified by (R)-SEGPHOS delivers the product of double crotylation as predominantly 1 of 16 possible stereoisomers due to the aforementioned amplification effect.12 The resulting pseudo-C2-symmetric polypropionate stereoquintet possesses a chirotopic nonstereogenic center at the central carbon atom. Iodoetherification defines this stereocenter and differentiates the two terminal olefin and two secondary alcohol moieties. With the iodoether in hand, the C19–C27 stereoheptad of rifamycin S was rapidly assembled (Scheme 6).18 In a similar fashion, the indicated iodoether was converted to a scytophycin C substructure in nearly half the number of steps previously required (Scheme 6).19
Scheme 6.

Asymmetric double anti-crotylation of 2-methyl-1,3-propanediol: formal syntheses of rifamycin S and scytophycin C.
Our initial efforts to develop syn-diastereo- and enantioselective alcohol C-H crotylations relied on the use of 2-silyl-substituted butadienes prepared from chloroprene.20a Exposure of alcohols to such dienes in the presence of ruthenium complexes modified by (R)-SEGPHOS or (R)-DM-SEGPHOS triggers generation of geometrically defined crotylruthenium-aldehyde pairs that engage in stereospecific carbonyl addition to form products of carbonyl crotylation in the absence of stoichiometric byproducts. In the presence of isopropanol under otherwise identical conditions, aldehydes are converted to an equivalent set of adducts (Scheme 7). Using this method for syn-crotylation, the triene-containing C17-benzene ansamycins trienomycins A and F were prepared in 16 steps (longest linear sequence, LLS) and 28 total steps.21 The present approach is 14 steps shorter (LLS) than the prior syntheses of trienomycins A and F, and 8 steps shorter (LLS) than any prior synthesis of a triene-containing C17-benzene ansamycins (Scheme 8).
Scheme 7.

Ruthenium catalyzed syn-diastereo- and enantioselective crotylation via hydrohydroxyalkylation of 2-silyl-butadienes.
a Chiral Ligand = (R)-DM-SEGPHOS. b Chiral Ligand = (R)- SEGPHOS.
Scheme 8.

Total syntheses of trienomycins A and F via ruthenium catalyzed syn-diastereo- and enantioselective hydrohydroxyalkylation of 2-silyl-butadienes.
Ideally, it would be desirable to perform direct regio- and stereoselective hydrohydroxyalkylations of butadiene itself. Ultimately, it was found that ruthenium catalysts modified by a chiral BINOL-derived phosphate counterion catalyze anti-diastereo- and enantioselective butadiene hydrohydroxyalkylation with benzylic alcohols.20b The chiral counterion is conveniently installed through the acid-base reaction of H2Ru(CO)(PPh3)3 and the corresponding BINOL-derived phosphoric acid. Remarkably, diastereoselectivity is inverted upon use of a TADDOL-derived phosphate counterion in combination with a chiral phosphine ligand, enabling syn-diastereo- and enantioselective butadiene hydrohydroxyalkylation with aliphatic alcohols (Scheme 9).20c
Scheme 9.

Inversion of diastereoselectivity in enantioselective ruthenium catalyzed crotylation via butadiene hydrohydroxyalkylation.
Using both iridium and ruthenium catalyzed alcohol C-H crotylation methodologies, a synthesis of 6-deoxyerythronolide B was undertaken (Scheme 10).22f Double anti-diastereo- and enantioselective crotylation of 2-methyl-1,3-propanediol is used to define the 5-contiguous stereogenic centers spanning C2–C6 of the indicated carboxylic acid fragment. Ruthenium catalyzed hydrohydroxyalkylation of butadiene with n-propanol is used to establish the C12 and C13 stereocenters found in the indicated acetylenic alcohol. Assembly of 6-deoxyerythronolide B from these two fragments was accomplished through esterification followed by ring-closing enyne metathesis to form the 14-membered macrolide. In this way, 6-deoxyerythronolide B was prepared in 14 steps (LLS) and 20 total steps. This route represents the most concise construction of any erythromycins family member reported, to date.22
Scheme 10.

Total synthesis of 6-deoxyerythronlide B via diastereo- and enantioselective alcohol C-H crotylation.
Geminal dimethyl groups appear ubiquitously in type I polyketides. Such structural motifs are typically generated through the agency of SAM-dependent C-methyltransferases within PKS modules, which doubly methylate β-ketoacyl thioester intermediates. Using the indicated iridium C,O-benzoate complex modified by (S)-SEGPHOS,15a which was isolated by precipitation, hydrogen transfer from primary alcohols to 1,1-dimethylallene triggers generation of aldehyde-n-prenyliridium pairs that combine with allylic inversion to form products of tert-prenylation.23 Notably, the enantiofacial selectivity in these processes is opposite to that observed in corresponding allylations and crotylations. Under otherwise identical conditions, but in the presence of isopropanol, aldehydes are transformed to an equivalent set of homoallylic alcohols (Scheme 11).
Scheme 11.
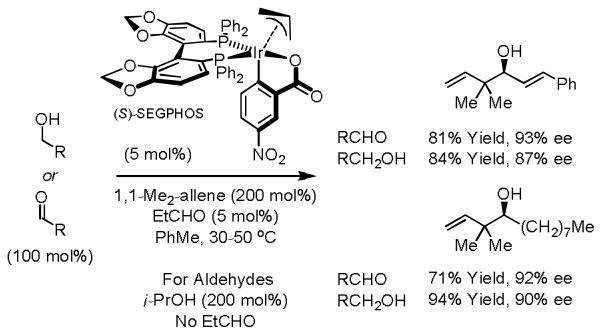
Enantioselective iridium catalyzed alcohol C-H reverse prenylation.
A total synthesis of marine macrolide bryostatin 724g was accomplished using a convergent assembly of the core from Fragments A and B employing the Keck-Yu pyran annulation25 and Yamaguchi macrolactonization (Scheme 12).26 For the synthesis of Fragment A, the indicated hydrogen-mediated glyoxal-enyne reductive coupling was strategic,27,28 as the key C20–C21 bond is formed with control of the C20 carbinol stereochemistry and C21 olefin geometry. The synthesis of Fragment B, which incorporates the A-ring,29 takes advantage of three transfer hydrogenative processes: enantioselective double allylation of 1,3-propanediol to form the C2-symmetric diol,11c subsequent aldehyde tert-prenylation23 to establish the C7 carbinol stereochemistry and install the C8 gem-dimethyl moiety and, finally, allylation11a,b at C9 to introduce the C11 aldehyde. Using these methods, bryostatin 7 was prepared in 20 steps (LLS) and 36 total steps.
Scheme 12.

Total synthesis of bryostatin 7 via C-C bond forming hydrogenation and transfer hydrogenation.
Through sequential application of the double allylation of 1,3-glycols11c and Fuwa’s cascading cross-metathesis-oxa-Michael cyclization,30 neopentyl glycol is transformed to the pyran core found in the cyanolide31 and clavosolide32 natural product families in only 2 steps (Scheme 13). This approach enabled the total synthesis of the C2-symmetric macrodiolide cyanolide A in 6 steps (LLS) and 10 total steps.31g Notably, the synthesis is accomplished in fewer than half the steps of any prior approach in the absence of any protecting groups, chiral auxiliaries or premetallated C-nucleophiles.
Scheme 13.

Total synthesis of cyanolide A via enantioselective double allylation of neopentyl glycol.
3. Conclusions
Whereas first-generation methods and strategies for the synthesis of polyketide natural products involving carbonyl addition by way of stoichiometric chiral auxiliaries and premetallated reagents represent important advances, they are clearly interim solutions to the challenge of de novo polyketide construction. As organic molecules are defined as compounds composed of carbon and hydrogen, the terminal solutions to this challenge will encompass stereoselective and atom-efficient methods for skeletal assembly involving the addition, removal or redistribution of hydrogen in the absence of protecting groups, discrete oxidation level adjustment, or non-constructive functional group interconversions.33 As illustrated in syntheses of diverse type I polyketide natural products, the merged redox and C-C bond construction events developed in our laboratory have availed a step-function change in efficiency. This technology has been embraced by several other research groups engaged in the total synthesis of polyketide natural products.34 Future studies will focus on the development of related alcohol C-H functionalizations, including the regio- and stereoselective hydrohydroxyalkylation of α-olefins.
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) are acknowledged for partial support of this research.
References
- 1.For selected reviews on polyketide natural products, see: O’Hagan D. The Polyketide Metabolites. Ellis Horwood; Chichester: 1991. Rimando AM, Baerson SR. ACS Symposium Series 955. American Chemical Society; 2007. Polyketides Biosynthesis, Biological Activity, and Genetic Engineering.
- 2.(a) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; (b) Newman DJ, Grothaus PG, Cragg GM. Chem Rev. 2009;109:3012. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Rohr J. Angew Chem Int Ed. 2000;39:2847. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 4.Isolation of erythromycin A: McGuire JM, Bunch RL, Anderson RC, Boaz HE, Flynn EH, Powell HM, Smith JW. Antibiot Chemother. 1952;2:281.
- 5.Isolation of amphotericin B: Vandeputte J, Watchtel JL, Stiller ET. Antibior Annu. 1956:587.X-ray structure: Mechinski W, Shaffner CP, Ganis P, Avitabile G. Tetrahedron Lett. 1970;11:3873.Ganis P, Avitabile G, Mechinski W, Shaffner CP. J Am Chem Soc. 1971;93:4560. doi: 10.1021/ja00747a037.
- 6.Isolation of rifamycin B: Sensi P, Margalith P, Timbal MT. Farmaco Ed Sci. 1959;14:146.Sensi P, Greco AM, Ballotta R. Antibiot Chemotherapy. 1960:262.Margalith P, Beretta G. Mycopathol Mycol Appl. 1960;13:21.Margalith P, Pagani H. Appl Microbiol. 1961;9:325. doi: 10.1128/am.9.4.325-334.1961.
- 7.Sait M, Hugenholtz P, Janssen PH. Environ Microbiol. 2002;4:654. doi: 10.1046/j.1462-2920.2002.00352.x. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 8.A notable exception is eribulin, a truncated derivative of Halichondrin. The current manufacturing route to Eribulin requires 33 steps (longest linear sequence) and 65 total steps: Chase CE, Fang FG, Lewis BM, Wilkie GD, Schnaderbeck MJ, Zhu X. Synlett. 2013:323.Austad BC, Benayoud F, Calkins TL, Campagna S, Chase CE, Choi HW, Christ W, Costanzo R, Cutter J, Endo A, Fang FG, Hu Y, Lewis BM, Lewis MD, McKenna S, Noland TA, Orr JD, Pesant M, Schnaderbeck MJ, Wilkie GD, Abe T, Asai N, Asai Y, Kayano A, Kimoto Y, Komatsu Y, Kubota M, Kuroda H, Mizuno M, Nakamura T, Omae T, Ozeki N, Suzuki T, Takigawa T, Watanabe T, Yoshizawa K. Synlett. 2013:327.Austad BC, Calkins TL, Chase CE, Fang FG, Horstmann TE, Hu Y, Lewis BM, Niu X, Noland TA, Orr JD, Schaderbeck MJ, Zhang H, Asakawa N, Asai N, Chiba H, Hasebe T, Hoshino Y, Ishizuka H, Kajima T, Kayano A, Komatsu Y, Kubota M, Kuroda H, Miyazawa M, Tagami K, Watanabe T. Synlett. 2013:333.
- 9.For recent reviews of C-C bond forming hydrogenation and transfer hydrogenation, see: Han SB, Kim IS, Krische MJ. Chem Commun. 2009:7278. doi: 10.1039/b917243m.Bower JF, Krische MJ. Top Organomet Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5.Hassan A, Krische MJ. Org Proc Res Devel. 2011;15:1236. doi: 10.1021/op200195m.Moran J, Krische MJ. Pure Appl Chem. 2012;84:1729. doi: 10.1351/PAC-CON-11-10-18.
- 10.For selected reviews on enantioselective carbonyl allylation and crotylation, see: Yamamoto Y, Asao N. Chem Rev. 1993;93:2207.Ramachandran PV. Aldrichim Acta. 2002;35:23.Kennedy JWJ, Hall DG. Angew Chem Int Ed. 2003;42:4732. doi: 10.1002/anie.200301632.Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yu CM, Youn J, Jung HK. Bull Korean Chem Soc. 2006;27:463.Marek I, Sklute G. Chem Commun. 2007:1683. doi: 10.1039/b615042j.Hall DG. Synlett. 2007:1644.Li J, Menche D. Synthesis. 2009:2293.Leighton JL. Aldrichim Acta. 2010;43:3.Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 11.For iridium catalyzed allylation of alcohol C-H bonds, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Lu Y, Kim IS, Hassan A, Del Valle DJ, Krische MJ. Angew Chem Int Ed. 2009;48:5018. doi: 10.1002/anie.200901648.Hassan A, Lu Y, Krische MJ. Org Lett. 2009;11:3112. doi: 10.1021/ol901136w.Schmitt DC, Dechert-Schmitt AMR, Krische MJ. Org Lett. 2012;14:6302. doi: 10.1021/ol3030692.Dechert-Schmitt AMR, Schmitt DC, Krische MJ. Angew Chem Int Ed. 2013;52:3195. doi: 10.1002/anie.201209863.
- 12.(a) Kogure T, Eliel EL. J Org Chem. 1984;49:576. [Google Scholar]; (b) Midland MM, Gabriel J. J Org Chem. 1985;50:1144. [Google Scholar]
- 13.For a review of polyketide stereotetrads in natural products, see: Koskinen AMP, Karisalmi K. Chem Soc Rev. 2005;34:677. doi: 10.1039/b417466f.
- 14.Attempts to purify the Roche aldehyde [ROCH2CH(Me)CHO] by silica gel chromatography result in 5–7% racemization: Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348.
- 15.For stereoselective iridium catalyzed crotylation of alcohol C-H bonds, see: Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350. doi: 10.1021/jo200068q.Gao X, Han H, Krische MJ. J Am Chem Soc. 2011;133:12795. doi: 10.1021/ja204570w.
- 16.For total syntheses of (+)-roxaticin, see: Rychnovsky SD, Hoye RC. J Am Chem Soc. 1994;116:1753.Mori Y, Asai M, Kawade J, Okumura A, Furukawa H. Tetrahedron Lett. 1994;35:6503.Mori Y, Asai M, Okumura A, Furukawa H. Tetrahedron. 1995;51:5299.Mori Y, Asai M, Kawade J, Furukawa H. Tetrahedron. 1995;51:5315.Evans DA, Connell BT. J Am Chem Soc. 2003;125:10899. doi: 10.1021/ja027638q.Han SB, Hassan A, Kim IS, Krische MJ. J Am Chem Soc. 2010;132:15559. doi: 10.1021/ja1082798.
- 17.For isolation of (+)-roxaticin, see: Maehr H, Yang R, Hong LN, Liu CM, Hatada MH, Todaro LJ. J Org Chem. 1992;57:4793.
- 18.For the total synthesis of rifamycin S, see: Nagaoka H, Rutsch W, Schmid G, Iio H, Johnson MR, Kishi Y. J Am Chem Soc. 1980;102:7962.Iio H, Nagaoka H, Kishi Y. J Am Chem Soc. 1980;102:7965.Kishi Y. Pure Appl Chem. 1981;53:1163.Nagaoka H, Kishi Y. Tetrahedron. 1981;37:3873.
- 19.For the total syntheses of scytophycins, see: Paterson I, Watson C, Yeung KS, Wallace PA, Ward RA. J Org Chem. 1997;62:452. doi: 10.1021/jo962189w.Paterson I, Yeung KS, Watson C, Ward RA, Wallace PA. Tetrahedron. 1998;54:11935.Paterson I, Watson C, Yeung KS, Ward RA, Wallace PA. Tetrahedron. 1998;54:11955.Nakamura R, Tanino K, Miyashita M. Org Lett. 2003;5:3579. doi: 10.1021/ol035227o.Nakamura R, Tanino K, Miyashita M. Org Lett. 2003;5:3583. doi: 10.1021/ol0352299.
- 20.For stereoselective ruthenium catalyzed crotylation of alcohol C-H bonds, see: Zbieg JR, Moran J, Krische MJ. J Am Chem Soc. 2011;133:10582. doi: 10.1021/ja2046028.Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274.McInturff EL, Yamaguchi E, Krische MJ. J Am Chem Soc. 2012;134:20628. doi: 10.1021/ja311208a.
- 21.For total and formal syntheses of triene-containing C17-benzene ansamycins, see: trienomycins A and F and thiazinotrienomycin E: Smith AB, III, Barbosa J, Wong W, Wood JL. J Am Chem Soc. 1995;117:10777.Smith AB, III, Barbosa J, Wong W, Wood JL. J Am Chem Soc. 1996;118:8316.Smith AB, III, Wan Z. Org Lett. 1999;1:1491. doi: 10.1021/ol991049g.Smith AB, III, Wan Z. J Org Chem. 2000;65:3738. doi: 10.1021/jo991958j.Del Valle DJ, Krische MJ. J Am Chem Soc. 2013;135:10986. doi: 10.1021/ja4061273.mycotrienol I and mycotrienin I: Panek JS, Masse CE. J Org Chem. 1997;62:8290. doi: 10.1021/jo971793j.Masse CE, Yang M, Solomon J, Panek JS. J Am Chem Soc. 1998;120:4123.cytotrienin A: Hayashi Y, Shoji M, Ishikawa H, Yamaguchi J, Tamura T, Imai H, Nishigaya Y, Takabe K, Kakeya H, Osada H. Angew Chem Int Ed. 2008;47:6657. doi: 10.1002/anie.200802079.
- 22.For total syntheses of 6-deoxyerythronolide B, see: Masamune S, Hirama M, Mori S, Ali SA, Garvey DS. J Am Chem Soc. 1981;103:1568.Myles DC, Danishefsky SJ, Schulte G. J Org Chem. 1990;55:1636.Evans DA, Kim AS. Tetrahedron Lett. 1997;38:53.Evans DA, Kim AS, Metternich R, Novack VJ. J Am Chem Soc. 1998;120:5921.Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351.Gao X, Woo SK, Krische MJ. J Am Chem Soc. 2013;135:4223. doi: 10.1021/ja4008722.
- 23.Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2009;131:6916. doi: 10.1021/ja902437k.For a correction, see: Han SB, Kim IS, Han H, Krische MJ. J Am Chem Soc. 2010;132:12517.
- 24.For total syntheses of the bryostatins, see: bryostatin 1: Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA. J Am Chem Soc. 2011;133:744. doi: 10.1021/ja110198y.bryostatin 2: Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew Chem Int Ed. 1998;37:2354. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9.Evans DA, Carter PH, Carreira EM, Charette AB, Prunet JA, Lautens M. J Am Chem Soc. 1999;121:7540.bryostatin 3: Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew Chem Int Ed. 2000;39:2290.Ohmori K. Bull Chem Soc Jpn. 2004;77:875.bryostatin 7: Kageyama M, Tamura T, Nantz M, Roberts JC, Somfai P, Whritenour DC, Masamune S. J Am Chem Soc. 1990;112:7407.Lu Y, Woo SK, Krische MJ. J Am Chem Soc. 2011;133:13876. doi: 10.1021/ja205673e.bryostatin 9: Wender PA, Schrier AJ. J Am Chem Soc. 2011;133:9228. doi: 10.1021/ja203034k.bryostatin 16: Trost BM, Dong G. Nature. 2008;456:485. doi: 10.1038/nature07543.
- 25.(a) Keck GE, Covel JA, Schiff T, Yu T. Org Lett. 2002;4:1189. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yu CM, Lee JY, So B, Hong J. Angew Chem Int Ed. 2002;41:161. doi: 10.1002/1521-3773(20020104)41:1<161::aid-anie161>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 26.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Soc Chem Jpn. 1979;52:1989. [Google Scholar]
- 27.For our model Bryostatin C-ring synthesis, see: Cho CW, Krische MJ. Org Lett. 2006;8:891. doi: 10.1021/ol052976s.
- 28.For related enantioselective hydrogen-mediated reductive couplings of conjugated alkynes to vicinal dicarbonyl compounds, see: Kong JR, Ngai MY, Krische MJ. J Am Chem Soc. 2006;128:718. doi: 10.1021/ja056474l.Cho CW, Krische MJ. Org Lett. 2006;8:3873. doi: 10.1021/ol061485k.Hong YT, Cho CW, Skucas E, Krische MJ. Org Lett. 2007;9:3745. doi: 10.1021/ol7015548.Liu P, Krische MJ, Houk KN. Chem Eur J. 2011;17:4021. doi: 10.1002/chem.201002741.
- 29.For our model bryostatin A-ring synthesis, see: Lu Y, Krische MJ. Org Lett. 2009;11:3108. doi: 10.1021/ol901096d.
- 30.(a) Fuwa H, Noto K, Sasaki M. Org Lett. 2010;6:1636. doi: 10.1021/ol100431m. [DOI] [PubMed] [Google Scholar]; (b) Fuwa H, Noto K, Sasaki M. Org Lett. 2011;7:1820. doi: 10.1021/ol200333p. [DOI] [PubMed] [Google Scholar]; (c) Fuwa H, Ichinokawa N, Noto K, Sasaki M. J Org Chem. 2012;77:2588. doi: 10.1021/jo202179s. [DOI] [PubMed] [Google Scholar]; (d) Fuwa H, Noguchi T, Noto K, Sasaki M. Org Biomol Chem. 2012;10:8108. doi: 10.1039/c2ob26189h. [DOI] [PubMed] [Google Scholar]
- 31.For total and formal syntheses of cyanolide A, see: Kim H, Hong J. Org Lett. 2010;12:2880. doi: 10.1021/ol101022z.Hajare AK, Ravikumar V, Khaleel S, Bhuniya D, Reddy DS. J Org Chem. 2011;76:963. doi: 10.1021/jo101782q.Yang Z, Xie X, Jing P, Zhao G, Zheng J, Zhao C, She X. Org Biomol Chem. 2011;9:984. doi: 10.1039/c0ob00971g.Pabbaraja S, Satyanarayana K, Ganganna B, Yadav JS. J Org Chem. 2011;76:1922. doi: 10.1021/jo102401v.Gesinski MR, Rychnovsky SD. J Am Chem Soc. 2011;133:9727. doi: 10.1021/ja204228q.Sharpe RJ, Jennings MP. J Org Chem. 2011;76:8027. doi: 10.1021/jo201210u.Waldeck AR, Krische MJ. Angew Chem Int Ed. 2013;52:4470. doi: 10.1002/anie.201300843.
- 32.For total and formal syntheses of clavosolide A (revised structure), see: Son JB, Kim SN, Kim NY, Lee DH. Org Lett. 2006;8:661. doi: 10.1021/ol052851n.Simov V, Smith AB., III Org Lett. 2006;8:3315. doi: 10.1021/ol0611752.Barry CS, Elsworth JD, Seden PT, Bushby N, Harding JR, Alder RW, Willis CL. Org Lett. 2006;8:3319. doi: 10.1021/ol0611705.Chakraborty TK, Reddy VR, Gajula PK. Tetrahedron. 2008;64:5162.Carrick JD, Jennings MP. Org Lett. 2009;11:769. doi: 10.1021/ol8028302.Peh G, Floreancig PE. Org Lett. 2012;14:5614. doi: 10.1021/ol302744t.
- 33.“The ideal synthesis creates a complex skeleton… in a sequence only of successive construction reactions involving no intermediary refunctionalizations, and leading directly to the structure of the target, not only its skeleton but also its correctly placed functionality.” Hendrickson JB. J Am Chem Soc. 1975;97:5784.
- 34.For applications of our methodology to syntheses of polyketide natural product in other laboratories, see: goniothalamin: Harsh P, O’Doherty GA. Tetrahedron. 2009;65:5051. doi: 10.1016/j.tet.2009.03.097.atorvastatin: Sawant P, Maier ME. Tetrahedron. 2010;66:9738.Sawant P, Maier ME. Eur J Org Chem. 2012:6576.fluvastatin: Kim A, Kim IS. Bull Korean Chem Soc. 2011;32:3748.pederin, psymberin: Wan S, Wu F, Rech JC, Green ME, Balachandran R, Horne WS, Day BW, Floreancig PE. J Am Chem Soc. 2011;133:16668. doi: 10.1021/ja207331m.neopeltolide: Yang Z, Zhang B, Zhao G, Yang J, Xie X, She X. Org Lett. 2011;13:5916. doi: 10.1021/ol2025718.Raghavan S, Samanta PK. Org Lett. 2012;14:2346. doi: 10.1021/ol3007698.psymberin (irciniastatin A): Feng Y, Jiang X, De Brabander JK. J Am Chem Soc. 2012;134:17083. doi: 10.1021/ja3057612.cryptophycin analogues: Bolduc KL, Larsen SD, Sherman DH. Chem Comm. 2012;48:6414. doi: 10.1039/c2cc32417b.rhizopodin: Dieckmann M, Rudolph S, Dreisigacker S, Menche D. J Org Chem. 2012;77:10782. doi: 10.1021/jo302134y.Kretschmer M, Menche D. Org Lett. 2012;14:382. doi: 10.1021/ol203130b.Kretschmer M, Dieckmann M, Li P, Rudolph S, Herkommer D, Troendlin J, Menche D. Chem Eur J. 2013;19:15993. doi: 10.1002/chem.201302197.leupyrrin A1: Debnar T, Wang T, Menche D. Org Lett. 2013;15:2774. doi: 10.1021/ol401110x.trans-aerangis Lactone: Kim A, Sharma S, Kwak JH, Kim IS. Bull Korean Chem Soc. 2013;34:75.peloruside A: Raghavan S, Kumar VV. Tetrahedron. 2013;69:4835.aspercyclide A: Sejberg JJP, Smith LD, Leatherbarrow RJ, Beavil AJ, Spivey AC. Tetrahedron Lett. 2013;54:4970.