

Abstract
HIV Vpr induces a cell-cycle arrest at the G2-to-M transition through a poorly understood mechanism. In a recent issue of Cell, Laguette et al. (2014) demonstrate that untimely activation of the structure-specific endonuclease regulator SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing.
Early innate and cell-intrinsic responses are vital to protect host cells from invading pathogens. In turn, viruses have developed sophisticated mechanisms to establish productive infections by counteracting host innate immune responses. HIV-1, HIV-2, and other primate lentiviruses encode a group of accessory proteins comprised of Vif, Vpr, Vpx, Vpu, and Nef, whose function is to promote immune evasion by modulating, among other things, intrinsic antiviral factors such as APOBEC3G (counteracted by Vif), Tetherin (counteracted by Vpu/Nef), and SAMHD1 (counteracted by Vpx) (Blanco-Melo et al., 2012; Yan and Chen, 2012). Of these viral proteins, HIV-1 Vpr (Viral Protein R), has remained one of the least understood in terms of its functional role and mode of action during viral infection. Vpr is a small phosphorylated nuclear protein that is conserved across all primate lentiviruses. The protein is packaged into progeny virions through an interaction with the structural protein Gag and, following viral entry, is associated with the preintegration complex, suggesting an early role during the virus life cycle. While multiple biological activities have been ascribed to HIV-1 Vpr, the most widely studied are the induction of a cell-cycle arrest at the G2-to-M transition in dividing cells and enhancing infection in terminally differentiated myeloid cells, such as monocyte-derived macrophages. The functional relevance of Vpr-induced G2/M arrest has remained a long-standing question in the HIV field since Vpr is dispensible for HIV-1 replication in dividing CD4+ T cells in cell culture. Nevertheless, the critical importance of Vpr for HIV infection and pathogenesis in vivo is underlined by the fact that chimpanzees (and at least one reported case in human) infected with Vpr-defective HIV-1 strains developed revertant mutants (Blanco-Melo et al., 2012; Malim and Emerman, 2008).
With respect to Vpr-induced G2/M arrest, one key advance in the field was the finding that the mechanism absolutely required the engagement of the DDB1-Cullin4A-VPRBP E3 ubiquitin ligase through a direct interaction between Vpr and the substrate specificity receptor VPRBP (also called DCAF1) (Romani and Cohen, 2012). Indeed, several lines of evidence suggested that the recruitment of the E3 ubiquitin ligase complex by Vpr was required to establish an intracellular environment that mimicked a DNA stress/damage response initiated by the DNA lesion-sensing Ataxia Telangiectasia and Rad3-related (ATR) protein kinase, which usually results in the activation of a G2/M checkpoint pathway (Dehart and Planelles, 2008). While the induction of a G2/M arrest in dividing cells could indeed be the function that Vpr was evolved to fulfill, another possibility was that Vpr-mediated G2/M arrest could be the result of the destruction/modification of host proteins that played one role in cell-cycle regulation and another in host-mediated defense against viruses (Malim and Emerman, 2008). However, the identity of the host proteins targeted by the Vpr/DDB1-Cullin4A-VPRBP complex has remained elusive, thus limiting our understanding of the molecular mechanism underlying Vpr-induced G2/M arrest and, importantly, its functional role during HIV infection.
In a recent issue of Cell, Laguette and colleagues (Laguette et al., 2014) report that Vpr interacts with the structure-specific endonuclease (SSE) regulator SLX4 complex (SLX4com) via a direct interaction with the C terminus of the SLX4 scaffold protein. Interestingly, they further show that VPRBP interacts directly with the SLX4com and that Vpr enhances this interaction. SLX4, the newly identified Fanconi anemia protein, has been implicated in the modulation of multiple DNA repair pathways and in Holliday junction resolution by regulating appropriate nucleases (Cybulski and Howlett, 2011; Svendsen and Harper, 2010). The multidomain protein interacts with the SSEs MUS81-EME1, ERCC1-ERCC4XPF, and SXL1, which are involved in the resolution of DNA replication and repair intermediates that are usually branched or multistranded DNA structures. Interaction of SLX4 with these SSEs stimulates their enzymatic activities. Interestingly, time course experiments revealed that expression of Vpr induced a remodeling of the SLX4com prior to G2/M arrest. Indeed, in the presence of Vpr, the authors observed an increased recruitment of polo-like kinase 1 (PLK1) and its kinase active form (pPLK1) to the SLX4com with concomitant enhancement in the phosphorylation of EME1 within the heteromeric MUS81-EME1 endonuclease subunit (Figure 1). Because SSEs, such as MUS81-EME1, act as the “Swiss army knives” of DNA interstrand crosslink repair in the nucleus, they are under strict regulatory control in that they are recruited to DNA only at specific phases of the cell cycle and activated only when needed (Minocherhomji and Hickson, 2013). Indeed, in yeast, MUS81-EME1 activation is mostly confined to the G2/M transition through PLK1 activation of EME1. In human cells, EME1 is phosphorylated by both cyclin-dependent kinases (CDKs) and PLK1, and this modification is directly correlated with increased MUS81 cleavage activity in vitro. In that regard, the study by Laguette and colleagues establishes that pPLK1 and MUS81-EME1 associate with SLX4 during mitosis in mammalian cells and that Vpr expression induces premature activation of SLX4-bound MUS81-EME1 prior to G2/M. This condition likely leads to replication stress and abnormal processing of stalled replication forks, events that are known to trigger DNA damage responses and G2/M arrest. Consistent with its importance in Vpr-mediated cell-cycle arrest, VPRBP was found to be required for the activation of the SLX4-associated MUS81-EME1. Furthermore, Vpr induced the ubiquitination of MUS81 and decreased its levels by a process that relied on its interaction with VPRBP (Figure 1). Therefore, it appears that VPRBP recruitment to the SLX4com is required not only for Vpr-mediated activation of SLX4-bound MUS81-EME1 but also for the regulation of MUS81 levels. Deregulation of the heteromeric MUS81-EME1 complex has been shown to lead to accumulation of damaged DNA and subsequent genomic instability (Minocherhomji and Hickson, 2013). Consistently, the authors found a marked increase in Fanconi anemia group D2 protein (FANCD2) foci in the presence of Vpr. Accumulation of FANCD2 foci is usually a hallmark of ongoing replication stress and persistence of unresolved replication intermediates. Lastly, the link between premature activation of the SLX4com by Vpr and the induction of G2/M arrest was demonstrated by the use of G2/M arrest defective Vpr mutants, which failed to interact with SLX4 or to induce recruitment of MUS81 and PLK1. Furthermore, silencing of the SLX4com subunits, including SLX4, MUS81, or EME1, inhibited Vpr-induced G2 arrest. Altogether, these results support the notion that VPRBP is important in the regulation of MUS81-EME1 activity and that SLX4com activation is required for Vpr-mediated G2/M arrest (Figure 1).
Figure 1. HIV-1 Vpr Subverts the SLX4 Complex to Promote G2/M Arrest and Escape from Innate Immune Sensing.
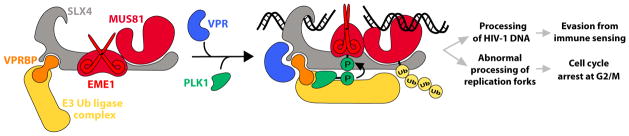
Because of the potentially damaging effect of the structure-specific endonucleases bound to SLX4, the SLX4com is kept inactive during the G1 and S phases of the cell cycle. Direct interaction of Vpr with SLX4 increases binding of VPRBP to the complex and induces the recruitment of kinase active PLK1 to the SLX4com. The resulting Vpr-mediated remodeling of the SLX4com causes the phosphorylation of EME1 and the ubiquitination of MUS81. These modifications trigger the activation of the heteromeric EME1-MUS81 endonuclease complex and lead to the processing of HIV-1 DNA, a condition that promotes evasion from innate immune sensing. In parallel, untimely activation of the SLX4com by Vpr prior to G2/M results in replication stress and abnormal processing of replication forks, which in turn trigger signaling pathways that ultimately arrest cells at the G2-to-M transition.
The results presented by Laguette and colleagues raise many new questions about the mechanism underlying Vpr-induced G2/M arrest. First, does Vpr-mediated ubiquitination of MUS81 represent a critical step in the activation of the SLX4com, or is it involved in regulating MUS81 levels? Second, what is the precise sequence of events from premature activation of the SLX4com to G2/M arrest? Finally, would targeting of the SLX4com explain other biological activities of Vpr, including the enhancement of viral infection in macrophages, or whether Vpr has the ability to target or/and modulate other host factors?
Another important finding from this study is that cells in which the SLX4 subunits were knocked down resulted in enhanced transcription of the type 1 interferon genes (ifna and ifnb) and interferon-stimulated genes, as demonstrated by induction of the antiviral protein MxA. The data suggest that the SLX4com is also used to cleave endogenous DNA fragments that would otherwise trigger innate immune sensing. By recruiting this complex to HIV DNA, Vpr could avoid innate immune detection. Indeed, infection of model cell lines with Vpr-defective virus apparently resulted in increased IFNα and IFNβ expression compared to infection with wild-type HIV-1. Interestingly, in that context, Vpr was shown to recruit the SLX4com to HIV DNA during reverse transcription, and silencing of SLX4 led to accumulation of viral DNA intermediates. These findings raise the possibility that like the TREX exonuclease (Yan and Chen, 2012), the MUS81-EME1 endonucleases within the SLX4com contribute to the removal of cytosolic nonproductive reverse-transcribed HIV DNA that would otherwise be sensed by cytosolic innate sensors (Figure 1). Future investigations will need to examine these processes in more physiological HIV target cells and assess the relative contribution and sequence of events mediated by TREX and active SLX4com to avoid triggering innate immune sensing. Clearly, the study by Laguette and colleagues marks only the beginning of a fascinating story that will shed new light not only on the functional role of Vpr during HIV infection but also on an important but still poorly understood aspect of the interaction between the DNA repair machinery and innate immunity.
Acknowledgments
The author thanks Dr. Mariana G. Bego, Dr. Tram N.Q. Pham, and Dr. Bizhan Romani for comments on the manuscript. The author also thanks Dr. Mariana G. Bego for the graphical artwork.
References
- Blanco-Melo D, Venkatesh S, Bieniasz PD. Immunity. 2012;37:399–411. doi: 10.1016/j.immuni.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski KE, Howlett NG. Cell Cycle. 2011;10:1757–1763. doi: 10.4161/cc.10.11.15818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehart JL, Planelles V. J Virol. 2008;82:1066–1072. doi: 10.1128/JVI.01628-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguette N, Brégnard C, Hue P, Basbous J, Yatim A, Larroque M, Kirchhoff F, Constantinou A, Sobhian B, Benkirane M. Cell. 2014;156:134–145. doi: 10.1016/j.cell.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Malim MH, Emerman M. Cell Host Microbe. 2008;3:388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Minocherhomji S, Hickson ID. Trends Cell Biol. 2013 doi: 10.1016/j.tcb.2013.11.007. in press. Published online December 19, 2013 http://dx.doi.org/10.1016/j.tcb.2013.11.007. [DOI] [PubMed]
- Romani B, Cohen EA. Curr Opin Virol. 2012;2:755–763. doi: 10.1016/j.coviro.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen JM, Harper JW. Genes Dev. 2010;24:521–536. doi: 10.1101/gad.1903510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Chen ZJ. Nat Immunol. 2012;13:214–222. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]