

Abstract
The misfolding and aggregation of proteins into amyloid has been linked to a variety of age-related diseases. Aggregation of proteins, such as Aβ in Alzheimer’s disease and Islet Amyloid Polypeptide (IAPP, amylin) in type 2 diabetes, appears to lead to the formation of toxic assemblies. These assemblies range in size from small oligomers (2–8 proteins) to large fibrils (thousands of proteins). It remains unclear how these amyloidogenic proteins misfold and form toxic species, but growing evidence suggests that inhibiting the aggregation of these proteins could slow, if not prevent altogether, the progression of these diseases. We describe the use of small peptides (<43 amino acids) as inhibitors of amyloid-based aggregation. These peptides, often short complementary segments of the amyloid proteins, can be useful (i) for identifying the aggregation-prone regions of the amyloid proteins (ii) as models for drug discovery and (iii) as potential therapeutic agents themselves.
Keywords: Amyloid Inhibition, Peptide Libraries, ABeta, Amylin, IAPP
INTRODUCTION
Many proteins are known to adopt unwanted, misfolded structures that can be linked to a variety of age-related diseases [1–3]. Proteins such as Islet Amyloid Polypeptide (IAPP, amylin) in type 2 diabetes and Aβ42 in Alzheimer’s disease are known to misfold and aggregate into toxic oligomers and fibers. In these amyloid diseases, proteins misfold and self-assemble into a variety of structures. It remains unknown if these misfolded proteins are the cause of their respective diseases or a side-effect. However, evidence is accumulating to suggest that the aggregation of amyloid proteins plays an important role in the development and progression of these diseases.
The amyloid diseases represent a serious threat to our overall global health. Within the United States, the economic and social costs associated with Alzheimer’s disease and type 2 diabetes have been well documented over the past decade. The Alzheimer’s Association estimates that 5.1 million Americans are currently afflicted with Alzheimer’s disease [4]. Likewise, the American Diabetes Association estimates that 23.6 million children and adults in the United States have diabetes [5]. Because the prevalence of these diseases increases with age, the number of people afflicted with Alzheimer’s disease and type 2 diabetes will increase as the population ages. If no treatments are found to slow these diseases, it is estimated that 11 to 16 million Americans will be afflicted with Alzheimer’s disease by 2050 [4] and as many as 100 million Americans could have diabetes by that time [6]. The economic costs of just these two amyloidogenic diseases is estimated to be more than $148 billion annually for Alzheimer’s disease and $174 billion for type 2 diabetes.
While the amyloidogenic proteins differ in many ways (length, organismal localization, amino acid compositions), they all form well-ordered fibrils and appear to be toxic to living cells [7–14]. These similarities are not solely shared by naturally occurring proteins, but have been identified in designed proteins expressed in mammalian cells [15]. These results indicate that the formation of protein aggregates, whether they are small oligomers or large fibrils, plays a central role in cell toxicity and disease pathology. These studies suggest that the mechanism of this cellular toxicity is likely conserved from one amyloid protein to the next. These results raise the hope that if a strategy is uncovered to prevent amyloid formation for one disease, the same strategy may lead to therapies for many, if not all, of the remaining amyloid diseases.
Many strategies are currently being assessed for preventing the formation of amyloid and toxic oligomers. Some strategies rely on preventing the production of the amyloid protein in the first place (for instance, inhibiting γ-secretase activity in Aβ production). Some strategies rely on removing the amyloid protein after it has been produced (such as using antibodies to remove Aβ). Both of these strategies hold great potential for slowing and/or preventing the onset of amyloid-based diseases. However, it remains to be determined if the amyloid proteins themselves serve a useful purpose to the organism. There may be a benefit to having the protein that has not yet been determined. With this in mind, there have been some recent successes made in slowing the rate of aggregation of amyloid proteins without having to remove the protein entirely. Several classes of small molecules have been identified as showing some activity toward inhibiting the formation of amyloid. [16–27]. While small-molecule aggregation inhibitors have been reported, none have been developed into a clinically useful therapeutic.
Since 2002, we have seen a considerable increase in the number of reports identifying short peptides as inhibitors of amyloid formation. While none of these peptides have been developed into a therapeutic, the potential does exist. Unlike small-molecule inhibitors of amyloid aggregation, short peptides can yield information regarding the amyloid aggregates themselves. Many of the peptide inhibitors are truncated versions of the full-length amyloid proteins. By identifying the sequence regions that inhibit aggregation, we can infer that those are the regions responsible for driving amyloid aggregation. In this review, we will focus on the short peptide sequences comprised of naturally occurring amino acids that have been identified for their ability to inhibit the aggregation of the amyloidogenic proteins Aβ (Alzheimer’s disease) and IAPP (type 2 diabetes).
PEPTIDE INHIBITION OF ABETA AGGREGATION
Peptides showing an ability to inhibit the aggregation of Aβ can be organized into two major groups; rationally designed peptides (which are typically similar in sequence to that of wild-type Aβ) and randomly generated peptides (those which are identified from peptide libraries that may, or may not, show sequence similarity to wild-type Aβ). The advances made in molecular biology and biotechnology in the past decade have made it possible to construct and screen large peptide libraries to identify those sequences that interact with Aβ. These library-screening experiments, which were either impossible or impractical just a few years ago, have successfully identified several amyloid inhibitors.
Rationally Designed Peptide Inhibitors of Aβ Aggregation
The earliest attempts to identify Aβ aggregation inhibitors came from the sequence of Aβ itself (Table 1). Many of the first peptide inhibitors were designed to target the aggregation-prone hydrophobic core region of Aβ. This region (LVFFA) is considered by many to be the primary aggregation-prone site of Aβ and is often termed the self-recognition site [28–30]. Many rationally designed peptides retain the LVFFA sequence, with the hope that the inhibitor peptide will selectively bind to the analogous sequence of natural Aβ42 [28, 31–34]. What often differs among these peptides are the N and C –terminal amino acids surrounding the LVFFA core. These surrounding residues are thought to act as aggregation blockers (termed β-sheet breakers), preventing additional amyloid peptides from adhering to the Aβ-peptide complex [35]. Many of these LVFFA peptides, and their later-generation variants, have been shown to inhibit Aβ aggregation at stoichiometric concentrations and rescue living cells from the toxic effects of Aβ42. For example, Garno and coworkers recently synthesized and studied 15 different variants having KLVFF as the core sequence (additional residues were added to the N-terminus and/or the C-terminus of this core sequence) [31]. From Thioflavin T and transmission electron microscopy experiments, nearly all of these peptides possessed some ability to inhibit aggregation at a 1:1 molar ratio (40 μM peptide to 40 μM Aβ40). Several peptides inhibited aggregation by approximately 80% compared to samples lacking the inhibitory peptides. Atomic force microscopy was used to identify the aggregation products of Aβ40 alone, the peptides alone and Aβ40 in the presence of the peptide inhibitors. Several peptides were found to self-aggregate into spherical assemblies, yet still showed inhibitory capacity. They found that placing positively charged residues at the N-terminal end of the KLVFF sequence were less likely to inhibit than if positive amino acids were placed at the C-terminal end. The authors also concluded that the peptides with the highest affinity to Aβ40 showed the greatest ability to inhibit amyloid-based aggregation. These results seem to be typical of the KLVFF core peptide inhibitors. These peptides demonstrate the potential of rationally designed peptide inhibitors as amyloid blocking agents.
Table 1.
Peptide Inhibitors of Aβ42

|
The hydrophobic core of Aβ42 is not the only region targeted for inhibition of aggregation. Aβ42 aggregation has been inhibited using C-terminal fragment (CTF) sequences of various lengths [36–38]. The best of these inhibitors spanned the final 13, 11 and 4 amino acids of Aβ42 [38], indicating that the length of the inhibitor peptide did not necessarily correlate with inhibitory propensity. Instead, the best correlation between CTF and their physical properties appeared to rely on the ability of the CTF to form a coil-turn conformation [38]. This conformation seems to appear in other inhibitory models, such as the peptide hairpin inhibitor of Teplow and coworkers [39]. While this peptide does contain one D-amino acid (D-Proline), the polypeptide is believed to form a hairpin turn held together by a disulfide bridge.
The rationally designed peptides have proven to be a strong class of inhibitory agents for Aβ42. These peptides continue to be modified with the ultimate goal to increase their ability to inhibit amyloid aggregation. Many of these peptides have been found to protect living cells from toxic amyloid aggregates. Perhaps future iterations of these peptides will show even greater ability to protect cellular viability.
Peptide Inhibitors of Aβ Aggregation Selected from Combinatorial Libraries
Advances in molecular biology, coupled with the decreased cost of DNA synthesis, have led to a greater ability to generate high-quality gene/protein libraries [40]. Libraries that once took months to construct, can now be done in a matter of weeks. While the ability to construct gene libraries has greatly evolved over the past decade, the ability to select for those variants that show a desirable activity has not. There are very few techniques in the literature that describe screens to select for peptides that inhibit amyloid aggregation and protect living cells from toxic protein aggregates. Despite this paucity of screening techniques, those screens that have been utilized have proven to be successful at identifying multiple amyloid-inhibiting peptides. We will discuss each of these screening techniques and the inhibitory peptides they have identified.
Peptides Identified Using Phage-Display
Phage display has proven to be an excellent method for screening large libraries of peptides to select for those that bind to an epitope of interest [41]. Many of the libraries screened by phage display have targeted large peptides/proteins. These libraries of larger peptides have generated sequences that inhibit Aβ aggregation, but may prove difficult to convert into a therapeutically useful drug. For example, several papers have been published recently that describe antibody-based, affibody, and other scaffold-based proteins selected by phage display [42–52]. Many of these selected proteins have been shown to inhibit aggregation, but their therapeutic potential remains untested.
Kamijo and coworkers recently screened a randomized heptapeptide library against Aβ42 [53]. Their goal was to identify peptides that could bind Aβ42, but did not necessarily possess sequence similarities to Aβ42. They identified several peptides, rich in arginines, that showed in vitro ability to inhibit Aβ42 aggregation. Size-exclusion chromatography and SDS-PAGE gel-shift assays indicated that several of these selected peptides kept Aβ42 from forming soluble oligomers [53]. Additional experiments will need to be performed to assess how these peptides inhibit aggregation. Of specific interest is why arginine-rich sequences were highly selected in this screen.
Not all peptides identified with phage display have been found to inhibit aggregation. Kiessling and coworkers identified several peptides that could bind to different aggregation states of Aβ40 [54]. While several peptides were identified that could bind to Aβ40, none slowed the rate of aggregation, and many increased aggregation. Therapeutically, this increase in aggregation could be beneficial if, in fact, aggregated Aβ is less toxic than small oligomers. These aggregation-enhancing peptides may function to help sequester Aβ40 into less toxic fibrils rather than the more toxic soluble oligomers.
Peptides Identified Using Aβ42-EGFP
Hecht and coworkers described the use of a GFP-based screen to assess the in vivo aggregation propensity of Aβ42 in E. coli cells [29, 55]. This screen has been used to assess the aggregation potential of Aβ mutants [55–57] as well as to screen for small molecules that can inhibit aggregation [58]. We recently used this screen, replacing GFP with enhanced GFP (EGFP) to select for mutants of IAPP that resisted aggregation [59]. In this screen, the amyloid protein (such as Aβ42 or IAPP) is genetically fused to the reporter protein EGFP. When expressed in E. coli, the Amyloid-EGFP fusion proteins produce virtually no green color or fluorescence due to the propensity of the amyloid proteins to aggregate (Fig. 1). However, when the amyloid proteins are prevented from aggregating (either by mutagenesis or in the presence of an aggregation inhibitor), the fused EGFP folds and fluoresces brightly.
Fig. (1).

(A) Amyloidogenic peptides, such as Aβ42 and IAPP self-assemble, ultimately leading to the formation of amyloid. (B) When linked to an amyloid peptide, EGFP does not fold or fluoresce. The aggregation of the amyloid peptides prevents proper folding and fluorescence of EGFP. (C) Inhibitors that prevent the self-assembly of the amyloid peptides allow the EGFP to fold and fluoresce.
We have recently used this screen to select peptides from different combinatorially randomized libraries for their ability to inhibit Aβ42 aggregation [60]. Combinatorially diverse library peptides were co-expressed in E. coli with the Aβ42-EGFP fusion protein. Peptides that prevented the aggregation of the Aβ42 allowed EGFP to fold and fluoresce. Individual E. coli colonies expressing both a library peptide and amyloid-EGFP were screened to select for those colonies that showed the greatest fluoresce. Using this screen, we identified three short peptides capable of inhibiting Aβ42 aggregation [60]. We believe this screen can be used to select for increasingly potent inhibitors of Aβ42 by improving the selection conditions and the combinatorial library design.
Gene libraries can be easily constructed using a variety of techniques. Gene libraries can be designed to encode for short peptides targeted to anneal to, and disrupt aggregation of, the amyloidogenic Aβ42 peptide. For example, Fig (2) shows two peptide libraries targeted to mimic each of the two hydrophobic regions of Aβ42. Both gene libraries were constructed using synthetic single-stranded oligos* (Table 3) and pieced together using oligo overlap and extension (Fig. 3). Gene variability was introduced to the gene libraries by using degenerate codons encoding for combinatorial mixtures of amino acids (Table 3).
Fig. (2).
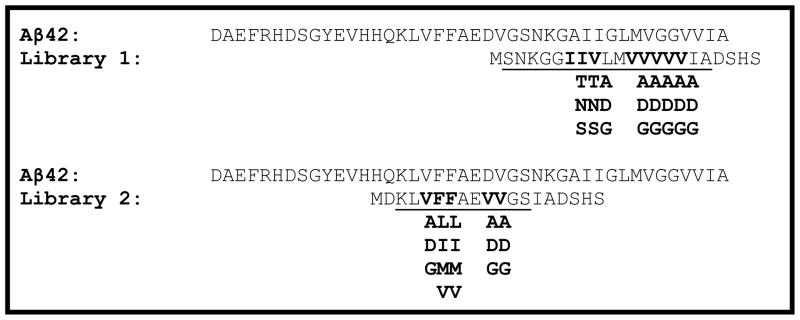
Amino Acid Sequences of Aβ42 and Library 1 and Library 2 Peptides. The amino acid sequence for Aβ42 is shown. Library 1 was constructed to have mixtures of amino acids in the bold-faced positions. Degenerate gene construction: Codon ANT encodes for an equal mixture of I, T, N and S. Codon GNT encodes for an equal mixture of V, A, D and G. Codon NTN encodes for a mixture of F, L, I, M and V.
Table 3.
Oligos Used for the Construction of Gene Libraries 1 and 2
| Library 1 Forward | 5′-CTAGCTGT CAT ATG TCT AAC AAA GGC GCG ANT ANT GNT CTG ATG GNT GNT GNT GNT GNT ATT GCG GAT AGT CAT AGT TAA-3′ |
| Library 2 Forward | 5′-CTAGCTGT CAT ATG CAG AAA CTG GNT DTN DTN GCG GAA GNT GNT GGC TCT AAC AAA TAA ATT GCG GAT AGT CAT AGT TAA-3′ |
| Reverse | 5′-CGATAGGAC GAATTC CAG TGG TAG CTT GTG TGC CAA CAG TAG TTA ACG GTA GAA CCG TTA ACT ATG ACT ATC CGC AAT -3′ |
Degenerate codons are underlined. Complementary regions are colored blue. All oligos are written from 5′-3′. N: equal mixture of A, T, G and C. D: Equal mixture of A, G and T.
Fig. (3).
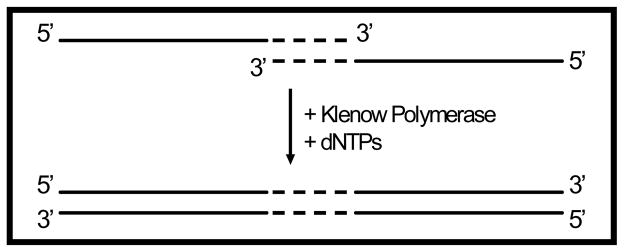
Oligo overlap and extension: Single-stranded DNA oligos were designed having complementary 3′ overhang regions (dashed lines). When mixed, the complementary regions anneal and act as templates for Klenow Fragment catalyzed DNA synthesis. Nucleotides not involved in annealing (solid lines) can be explicitly designed base by base or combinatorially varied.
Both peptide libraries were designed to mimic the hydrophobic patches of Aβ42, with the intention of producing peptides that could anneal to the monomeric form of Aβ42. Also designed into the peptide libraries are strongly polar and charged aspartic acid residues. These acidic residues are intended to incorporate a strongly charged “bump” on the library peptides that prevents the aggregation of additional Aβ42 proteins to an Aβ42-library peptide complex. The library peptides are designed to act as two-sided patches; one side being sticky and annealing to monomeric Aβ42, and the other side presenting a repulsive residue (or residues) that prevent additional Aβ42 peptides from annealing.
The gene library is cloned into one plasmid and cotrans-formed with the amyloid-EGFP plasmid into a suitable E. coli expression host (Fig. 4). The resultant colonies are transferred to plates that can induce protein expression. With this system, thousands of transformed colonies can be screened to identify those few that show elevated levels of EGFP fluorescence.
Fig. (4).

Construction and selection of peptide libraries. Cotransformed cells were plated on nitrocellulose absorbed on LB plates containing Ampicillin and Streptomycin. After incubation, resulting colonies are transferred to LB plates containing IPTG. Green-colored colonies are selected and analyzed.
PEPTIDE INHIBITORS OF IAPP AGGREGATION
In type II diabetes the amyloid-forming peptide is islet amyloid polypeptide (IAPP, amylin). This 37 amino acid polypeptide misfolds and forms toxic aggregates within the pancreas. This misfolding into toxic aggregates, such as small soluble oligomers or large fibers, is believed to contribute to the loss of pancreatic β-cells. A great debate continues within the scientific community to identify the toxic form (or forms) of IAPP. While the exact role of IAPP in type II diabetes is unclear, it is known that IAPP is found as extracellular deposits of amyloid in approximately 90% of patients afflicted with type II diabetes [61–63]. This peptide is highly amyloidogenic, finding any substance to inhibit its aggregation could be a potent weapon in the fight against type 2 diabetes.
It is well established that rodent IAPP (rIAPP) is nonamyloidogenic and nontoxic. It is also established that rats and mice are not known to develop type II diabetes [64], strengthening the argument that inhibiting IAPP aggregation could help to prevent type 2 diabetes. Rodent IAPP, which differs from human IAPP by six amino acids, has been demonstrated to be capable of inhibiting human IAPP aggregation [65].
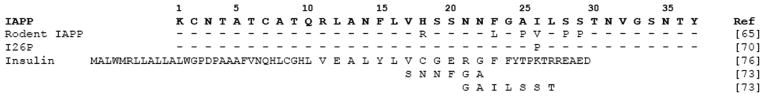
Five of the substitutions known to convert amyloidogenic human IAPP into non-amyloidogenic rodent IAPP occur in the 20–29 region of the peptide (Table 2). Because of these substitutions the 20–29 region has long been described as the amyloidogenic region of IAPP [32, 66, 67]. Recent studies have indicated that several mutations outside of the 20–29 region can hinder fibrillogenesis, and are likely important for aggregation [59, 68–74]. Several short peptides targeting this aggregation-prone region have been identified [73]. Additionally, a single mutation in the middle of this aggregation region, I26P, has been shown to act as a potent inhibitor of IAPP aggregation [70].
Table 2.
Peptide Inhibitors of IAPP
|
Insulin is an additional peptide that is known to inhibit IAPP aggregation [75]. Insulin is co-secreted with IAPP from pancreatic B-cells. While the exact role these two peptides play in the development of type 2 diabetes remains to be determined, it is not surprising that this metabolism-driving hormone is important in preventing IAPP aggregation. Raleigh and coworkers were able to determine the crystal structure of IAPP fused to maltose binding protein [76]. Based on the structure of IAPP in this fusion protein, they were able to model the possible binding interaction between IAPP and the B-chain of insulin, showing the importance of IAPP Phe15 in this aggregation (Table 2 shows the sequence of insulin as well as the residues of insulin suggested as interacting with IAPP).
In one of the few articles describing short peptide inhibitors of IAPP aggregation, Fraser and coworkers tested a series of hexapeptides, each overlapping with a segment of IAPP [73]. When co-incubated with IAPP, several of the peptides, such as GAILSST appeared to decrease the density of fiber formation. More importantly, one peptide SNNFGA (Table 2) had the ability to rescue cultured cells from IAPP toxicity.
OUTLOOK
Over the past decade, we have witnessed a great increase in the number of articles describing short peptides as inhibitors of amyloid aggregation. Advances in molecular biology (which allows for the inexpensive synthesis of DNA libraries) and peptide synthesis (which have lowered the costs of chemically synthesizing peptides) have made it possible to both identify and synthesize specific peptides capable of inhibiting aggregation. While it remains to be seen whether short peptides can be used as therapeutic agents in preventing the amyloid diseases, these short peptide inhibitors can aid in identifying the aggregation-prone regions of the amyloid proteins. These short peptide inhibitors can also be useful in identifying the important characteristics of potential therapeutic agents in amyloid inhibition. Perhaps future iterations of peptides will be identified that can protect cells from amyloid aggregates as well as resist our body’s natural protein degradation machinery.
Acknowledgments
We thank Luiza A. Nogaj for critical reading of this manuscript. Funding for this work was from the National Institutes of Health (1R15AG032582).
ABBREVIATIONS
- IAPP
Islet amyloid polypeptide
- Aβ
Amyloid β-protein
- EGFP
Enhanced green fluorescent protein
Footnotes
Library 1: 5′-CTAGCTGT CAT ATG TCT AAC AAA GGC GCG ANT ANT GNT CTG ATG GNT GNT GNT GNT GNT ATT GCG GAT AGT CAT AGT TAA-3′, Library 2: 5′-CTAGCTGT CAT ATG CAG AAA CTG GNT DTN DTN GCG GAA GNT GNT GGC TCT AAC AAA TAA ATT GCG GAT AGT CAT AGT TAA-3′, Reverse sequence for both libraries: 5′-CGATAGGAC GAATTC CAG TGG TAG CTT GTG TGC CAA CAG TAG TTA ACG GTA GAA CCG TTA ACT ATG ACT ATC CGC AAT -3′.
CONFLICT OF INTEREST
None declared.
References
- 1.Bucciantini M, Giannoni E, Chiti F, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Murphy RM, Kendrick BS. Protein misfolding and aggregation. Biotech Progress. 2007;23:548–52. doi: 10.1021/bp060374h. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer’s Disease Facts and Figures. Alzheimer’s Association; 2007. Available from: http://www.alz.org/ [Google Scholar]
- 5.Type 2 Diabetes Facts and Figures. American Diabetes Association; 2007. Available from: http://www.diabetes.org. [Google Scholar]
- 6.CDC Revise Estimates, Predicts 100 Million Americans Could be Diabetic by 2050. Centers for Disease Control and Prevention; 2010. [Google Scholar]
- 7.Ritzel RA, Meier JJ, Lin CY, Veldhuis JD, Butler PC. Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes. 2007;56:65–71. doi: 10.2337/db06-0734. [DOI] [PubMed] [Google Scholar]
- 8.Yan LM, Velkova A, Tatarek-Nossol M, Andreetto E, Kapurniotu A. LAPP mimic blocks A beta cytotoxic self-assembly: Cross-suppression of amyloid toxicity of A beta and IAPP suggests a molecular link between Alzheimer’s disease and type II diabetes. Angew Chem-Int Ed. 2007;46:1246–52. doi: 10.1002/anie.200604056. [DOI] [PubMed] [Google Scholar]
- 9.Kapurniotu A, Schmauder A, Tenidis K. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J Mol Biol. 2002;315:39–50. doi: 10.1006/jmbi.2001.5244. [DOI] [PubMed] [Google Scholar]
- 10.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–89. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 11.Fezoui Y, Hartley DM, Walsh DM, Selkoe DJ, Osterhout JJ, Teplow DB. A de novo designed helix-turn-helix peptide forms non-toxic amyloid fibrils. Nat Struct Biol. 2000;7:1095–9. doi: 10.1038/81937. [DOI] [PubMed] [Google Scholar]
- 12.Wakabayashi M, Matsuzaki K. Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett. 2009;583:2854–8. doi: 10.1016/j.febslet.2009.07.044. [DOI] [PubMed] [Google Scholar]
- 13.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA. 2011;108:9460–5. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bucciantini M, Calloni G, Chiti F, et al. Prefibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem. 2004;279:31374–82. doi: 10.1074/jbc.M400348200. [DOI] [PubMed] [Google Scholar]
- 15.Olzscha H, Schermann SM, Woerner AC, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 16.Ritchie CW, Bush AI, Mackinnon A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 17.Ritchie CW, Bush AI, Masters CL. Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin Investig Drugs. 2004;13:1585–92. doi: 10.1517/13543784.13.12.1585. [DOI] [PubMed] [Google Scholar]
- 18.Wood SJ, MacKenzie L, Maleeff B, Hurle MR, Wetzel R. Selective inhibition of Abeta fibril formation. J Biol Chem. 1996;271:4086–92. doi: 10.1074/jbc.271.8.4086. [DOI] [PubMed] [Google Scholar]
- 19.Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ. New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer’s disease. J Biol Chem. 2002;277:42881–90. doi: 10.1074/jbc.M206593200. [DOI] [PubMed] [Google Scholar]
- 20.Talaga P. Beta-amyloid aggregation inhibitors for the treatment of Alzheimer’s disease: dream or reality? Mini Rev Med Chem. 2001;1:175–86. doi: 10.2174/1389557013407098. [DOI] [PubMed] [Google Scholar]
- 21.Cohen T, Frydman-Marom A, Rechter M, Gazit E. Inhibition of amyloid fibril formation and cytotoxicity by hydroxyindole derivatives. Biochemistry. 2006;45:4727–35. doi: 10.1021/bi051525c. [DOI] [PubMed] [Google Scholar]
- 22.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 23.Porat Y, Mazor Y, Efrat S, Gazit E. Inhibition of islet amyloid polypeptide fibril formation: a potential role for heteroaromatic interactions. Biochemistry. 2004;43:14454–62. doi: 10.1021/bi048582a. [DOI] [PubMed] [Google Scholar]
- 24.Saengkhae C, Salerno M, Ades D, et al. Ability of carbazole salts, inhibitors of Alzheimer beta-amyloid fibril formation, to cross cellular membranes. Eur J Pharm. 2007;559:124–31. doi: 10.1016/j.ejphar.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Riviere C, Richard T, Quentin L, Krisa S, Merillon JM, Monti JP. Inhibitory activity of stilbenes on Alzheimer’s beta-amyloid fibrils in vitro. Bioorg Med Chem. 2007;15:1160–7. doi: 10.1016/j.bmc.2006.09.069. [DOI] [PubMed] [Google Scholar]
- 26.Hull RL, Zraika S, Udaya-Sankar J, Kisilevsky R, Szarek WA, Kahn SE. Inhibition of islet amyloid formation in vitro by a small molecule inhibitor that reduces heparan sulfate proteoglycan (HSPG) synthesis. Diabetes. 2006;55:A372. [Google Scholar]
- 27.Hamaguchi T, Ono K, Yamada M. Anti-amyloidogenic therapies: strategies for prevention and treatment of Alzheimer’s disease. Cell Mol Life Sci. 2006;63:1538–52. doi: 10.1007/s00018-005-5599-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estrada LD, Soto C. Inhibition of protein misfolding and aggregation by small rationally-designed peptides. Curr Pharma Des. 2006;12:2557–67. doi: 10.2174/138161206777698792. [DOI] [PubMed] [Google Scholar]
- 29.Wurth C, Kim W, Hecht MH. Combinatorial approaches to probe the sequence determinants of protein aggregation and amyloidogenicity. Protein Pept Lett. 2006;13:279–86. doi: 10.2174/092986606775338506. [DOI] [PubMed] [Google Scholar]
- 30.Esler WP, Stimson ER, Ghilardi JR, et al. Point substitution in the central hydrophobic cluster of a human beta-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry. 1996;35:13914–21. doi: 10.1021/bi961302+. [DOI] [PubMed] [Google Scholar]
- 31.Bett CK, Serem WK, Fontenot KR, Hammer RP, Garno JC. Effects of peptides derived from terminal modifications of the A beta central hydrophobic core on A beta fibrillization. ACS Chem Neurosci. 2010;1:661–78. doi: 10.1021/cn900019r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazit E. Mechanisms of amyloid fibril self-assembly and inhibition. FEBS J. 2005;272:5971–8. doi: 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- 33.Orner BP, Liu L, Murphy RM, Kiessling LL. Phage display affords peptides that modulate beta-amyloid aggregation. J Am Chem Soc. 2006;128:11882–9. doi: 10.1021/ja0619861. [DOI] [PubMed] [Google Scholar]
- 34.Austen BM, Paleologou KE, Ali SAE, Qureshi MM, Allsop D, El-Agnaf OMA. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s beta-amyloid peptide. Biochemistry. 2008;47:1984–92. doi: 10.1021/bi701415b. [DOI] [PubMed] [Google Scholar]
- 35.Poduslo JF, Curran GL, Kumar A, Frangione B, Soto C. Beta-sheet breaker peptide inhibitor of Alzheimer’s amyloidogenesis with increased blood-brain barrier permeability and resistance to proteolytic degradation in plasma. J Neurobiol. 1999;39:371–82. [PubMed] [Google Scholar]
- 36.Li HY, Monien BH, Lomakin A, et al. Mechanistic investigation of the inhibition of A beta 42 assembly and neurotoxicity by A beta 42 C-terminal fragments. Biochemistry. 2010;49:6358–64. doi: 10.1021/bi100773g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monien BH, Fradinger EA, Spring SM, Bernstein SL, Bowers MT, Bitan G. A novel approach to Alzheimer’s disease therapy: Inhibition of A beta 42 oligomerization by C-terminal A beta 42 fragments. J Pept Sci. 2006;12:147–7. [Google Scholar]
- 38.Li HY, Monien BH, Fradinger EA, Urbanc B, Bitan G. Biophysical characterization of A beta 42 C-terminal fragments: inhibitors of A beta 42 neurotoxicity. Biochemistry. 2010;49:1259–67. doi: 10.1021/bi902075h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamin G, Ruchala P, Teplow DB. A peptide hairpin inhibitor of amyloid beta-protein oligomerization and fibrillogenesis. Biochemistry. 2009;48:11329–31. doi: 10.1021/bi901325g. [DOI] [PubMed] [Google Scholar]
- 40.Moffet DA, Hecht MH. De novo proteins from combinatorial libraries. Chem Rev. 2001;101:3191–203. doi: 10.1021/cr000051e. [DOI] [PubMed] [Google Scholar]
- 41.Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 42.Habicht G, Haupt C, Friedrich RP, et al. Directed selection of a conformational antibody domain that prevents mature amyloid fibril formation by stabilizing A beta protofibrils. Proc Natl Acad Sci USA. 2007;104:19232–7. doi: 10.1073/pnas.0703793104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Nuallain B, Allen A, Ataman D, Weiss DT, Solomon A, Wall JS. Phage display and peptide mapping of an immunoglobulin light chain fibril-related conformational epitopet. Biochemistry. 2007;46:13049–58. doi: 10.1021/bi701255m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frenkel D, Solomon B, Benhar I. Modulation of Alzheimer’s beta-amyloid neurotoxicity by site-directed single-chain antibody. J Neuroimmunol. 2000;106:23–31. doi: 10.1016/s0165-5728(99)00232-5. [DOI] [PubMed] [Google Scholar]
- 45.Manoutcharian K, Acero G, Munguia ME, et al. Amyloid-beta peptide-specific single chain Fv antibodies isolated from an immune phage display library. J Neuroimmunol. 2003;145:12–17. doi: 10.1016/j.jneuroim.2003.08.038. [DOI] [PubMed] [Google Scholar]
- 46.Lindgren J, Wahlstrom A, Danielsson J, et al. N-terminal engineering of amyloid-beta-binding Affibody molecules yields improved chemical synthesis and higher binding affinity. Protein Sci. 2010;19:2319–29. doi: 10.1002/pro.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoyer W, Gronwall C, Jonsson A, Stahl S, Hard T. Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc Natl Acad Sci USA. 2008;105:5099–104. doi: 10.1073/pnas.0711731105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith TJ, Stains CI, Meyer SC, Ghosh I. Inhibition of beta-amyloid fibrillization by directed evolution of a beta-sheet presenting miniature protein. J Am Chem Soc. 2006;128:14456–7. doi: 10.1021/ja065557e. [DOI] [PubMed] [Google Scholar]
- 49.Funke SA, Willbold D. Mirror image phage display-a method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol Biosyst. 2009;5:783–6. doi: 10.1039/b904138a. [DOI] [PubMed] [Google Scholar]
- 50.van Groen T, Wiesehann K, Funke SA, Kadish I, Nagel-Steger L, Willbold D. Reduction of Alzheimer’s disease amyloid plaque load in transgenic mice by D3, a D-enantiomeric peptide identified by mirror image phage display. Chem Med Chem. 2008;3:1848–52. doi: 10.1002/cmdc.200800273. [DOI] [PubMed] [Google Scholar]
- 51.Funke SA, van Groen T, Kadish I, et al. Oral treatment with the D-enantiomeric peptide D3 improves the pathology and behavior of Alzheimer’s disease transgenic mice. ACS Chem Neurosci. 2010;1:639–48. doi: 10.1021/cn100057j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu HM, Funke SA, Willbold D. Transport of Alzheimer disease amyloid-beta-binding D-amino acid peptides across an in vitro blood-brain barrier model. Rejuvenation Res. 2010;13:210–13. doi: 10.1089/rej.2009.0926. [DOI] [PubMed] [Google Scholar]
- 53.Kawasaki T, Onodera K, Kamijo S. Selection of peptide inhibitors of soluble A beta (1–42) oligomer formation by phage display. Biosci Biotech Biochem. 2010;74:2214–9. doi: 10.1271/bbb.100388. [DOI] [PubMed] [Google Scholar]
- 54.Orner BP, Liu L, Murphy RM, Kiessling LL. Phage display affords peptides that modulate beta-amyloid aggregation. J Am Chem Soc. 2006;128:11882–9. doi: 10.1021/ja0619861. [DOI] [PubMed] [Google Scholar]
- 55.Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer’s Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–90. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 56.de Groot NS, Aviles FX, Vendrell J, Ventura S. Mutagenesis of the central hydrophobic cluster in A beta 42 Alzheimer’s pepticle - Side-chain properties correlate with aggregation propensities. FEBS J. 2006;273:658–68. doi: 10.1111/j.1742-4658.2005.05102.x. [DOI] [PubMed] [Google Scholar]
- 57.Kim W, Hecht MH. Mutations enhance the aggregation propensity of the Alzheimer’s A beta peptide. J Mol Biol. 2008;377:565–74. doi: 10.1016/j.jmb.2007.12.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim W, Kim Y, Min J, Kim DJ, Chang YT, Hecht MH. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol. 2006;1:461–9. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 59.Fox A, Snollaerts T, Casanova CE, Calciano A, Nogaj LA, Moffet DA. Selection for nonamyloidogenic mutants of islet amyloid polypeptide (IAPP) identifies an extended region for amyloidogenicity. Biochemistry. 2010;49:7783–9. doi: 10.1021/bi100337p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baine M, Georgie DS, Shiferraw EZ, Nguyen TPT, Nogaj LA, Moffet DA. Inhibition of A beta 42 aggregation using peptides selected from combinatorial libraries. J Pept Sci. 2009;15:499–503. doi: 10.1002/psc.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Apostolidou M, Jayasinghe SA, Langen R. Structure of alpha-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J Biol Chem. 2008;283:17205–10. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89:3629–43. doi: 10.1210/jc.2004-0405. [DOI] [PubMed] [Google Scholar]
- 63.Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241–53. doi: 10.2337/diabetes.48.2.241. [DOI] [PubMed] [Google Scholar]
- 64.Matveyenko AV, Butler PC. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006;47:225–33. doi: 10.1093/ilar.47.3.225. [DOI] [PubMed] [Google Scholar]
- 65.Cao P, Meng F, Abedini A, Raleigh DP. The ability of rodent islet amyloid polypeptide to inhibit amyloid formation by human islet amyloid polypeptide has important implications for the mechanism of amyloid formation and the design of inhibitors. Biochemistry. 2010;49:872–81. doi: 10.1021/bi901751b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Westermark P, Engstrom U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide - pinpointing amino-acid-residues linked to amyloid fibril formation. Proc Natl Acad Sci USA. 1990;87:5036–40. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nanga RP, Brender JR, Xu J, Veglia G, Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP(1–19) in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–97. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abedini A, Raleigh DP. Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: Is there a critical amyloidogenic domain in human IAPP? J Mol Biol. 2006;355:274–81. doi: 10.1016/j.jmb.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 69.Marek P, Abedini A, Song BB, et al. Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry. 2007;46:3255–61. doi: 10.1021/bi0621967. [DOI] [PubMed] [Google Scholar]
- 70.Abedini A, Meng FL, Raleigh DP. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrillization inhibitor. J Am Chem Soc. 2007;129:11300. doi: 10.1021/ja072157y. [DOI] [PubMed] [Google Scholar]
- 71.Abedini A, Raleigh DP. The role of His-18 in amyloid formation by human islet amyloid polypeptide. Biochemistry. 2005;44:16284–91. doi: 10.1021/bi051432v. [DOI] [PubMed] [Google Scholar]
- 72.Koo BW, Hebda JA, Miranker AD. Amide inequivalence in the fibrillar assembly of islet amyloid polypeptide. Protein Eng Des Select. 2008;21:147–54. doi: 10.1093/protein/gzm076. [DOI] [PubMed] [Google Scholar]
- 73.Scrocchi LA, Chen Y, Waschuk S, et al. Design of peptide-based inhibitors of human islet amyloid polypeptide fibrillogenesis. J Mol Biol. 2002;318:697–706. doi: 10.1016/S0022-2836(02)00164-X. [DOI] [PubMed] [Google Scholar]
- 74.Tracz SM, Abedini A, Driscoll M, Raleigh DP. Role of aromatic interactions in amyloid formation by peptides derived from human Amylin. Biochemistry. 2004;43:15901–8. doi: 10.1021/bi048812l. [DOI] [PubMed] [Google Scholar]
- 75.Larson JL, Miranker AD. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J Mol Biol. 2004;335:221–31. doi: 10.1016/j.jmb.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 76.Wiltzius JJW, Sievers SA, Sawaya MR, Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009;18:1521–30. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]