
Figure 1.
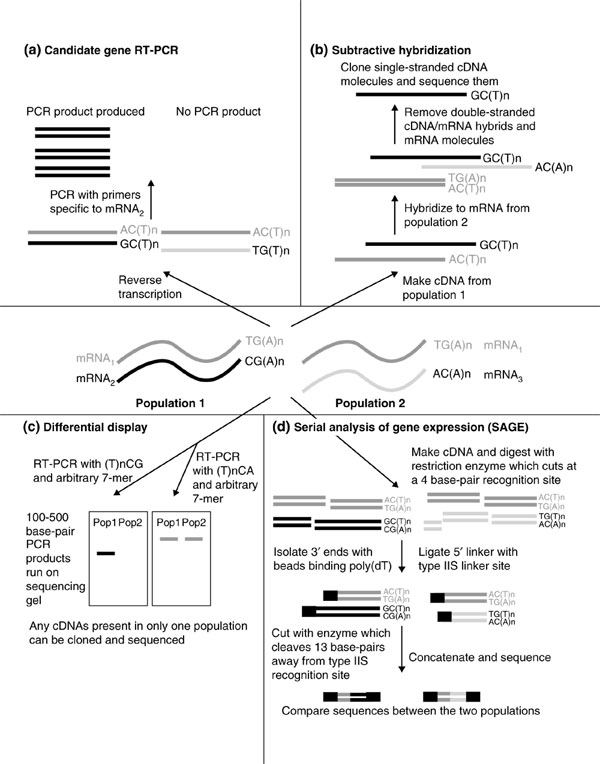
Four established techniques that are used to identify transcription-factor targets. These methods all compare mRNAs extracted from two populations of cells, one of which has the transcription factor in question overexpressed or knocked out. (a) Differences in the levels of specific candidate target genes in the two populations can be analyzed by reverse-transcriptase-coupled (RT-)PCR (for example, see [1,40]). (b) Any mRNAs that are equally expressed in both populations are subtracted, or removed, by cDNA-RNA hybridization. The remaining cDNAs are derived from mRNAs that are differentially expressed in one of the populations, and these can then be cloned and sequenced [3]. (c) With differential display, partial cDNA sequences are amplified from mRNA pools by RT-PCR. One primer - (T)nNN - binds to the polyadenylated tail of a subset of mRNAs that is defined by the two bases immediately 53 to the tail. The other binds to short sequences (6 or 7 base-pairs) that will occur with moderate frequency within the transcriptome. The products are radiolabeled and analyzed by polyacrylamide gel electrophoresis. Short cDNAs present in only one population can be isolated and sequenced [41,42]. (d) In serial analysis of gene expression (SAGE), cDNA is synthesized from mRNA and cleaved by a restriction enzyme that recognizes a 4 nucleotide sequence. The 33 end of the cleaved cDNA is isolated using beads that bind to oligo-dT, and 53 linkers are ligated to the restriction sites. These linkers contain type-IIS restriction sites, which are recognized by endonucleases that cleave a defined distance away (up to 20 base-pairs). This produces short DNA tags whose sequence and position are sufficient to identify the original transcript, provided cDNA sequences or expressed sequence tags (ESTs) are already known. The tags can be concatenated and sequenced, providing quantitative analysis of many transcripts simultaneously [43].