

Abstract
Transformation of Chlamydia trachomatis should greatly advance the chlamydial research. However, significant progress has been hindered by the failure of C. trachomatis to induce clinically relevant pathology in animal models. Chlamydia muridarum, which naturally infects mice, can induce hydrosalpinx in mice, a tubal pathology also seen in women infected with C. trachomatis. We have developed a C. muridarum transformation system and confirmed Pgp1, -2, -6, and -8 as plasmid maintenance factors, Pgp3, -5, and -7 as dispensable for in vitro growth, and Pgp4 as a positive regulator of genes that are dependent on plasmid for expression. More importantly, we have discovered that Pgp5 can negatively regulate the same plasmid-dependent genes. Deletion of Pgp5 led to a significant increase in expression of the plasmid-dependent genes, suggesting that Pgp5 can suppress the expression of these genes. Replacement of pgp5 with a mCherry gene, or premature termination of pgp5 translation, also increased expression of the plasmid-dependent genes, indicating that Pgp5 protein but not its DNA sequence is required for the inhibitory effect. Replacing C. muridarum pgp5 with a C. trachomatis pgp5 still inhibited the plasmid-dependent gene expression, indicating that the negative regulation of plasmid-dependent genes is a common feature of all Pgp5 regardless of its origin. Nevertheless, C. muridarum Pgp5 is more potent than C. trachomatis Pgp5 in suppressing gene expression. Thus, we have uncovered a novel function of Pgp5 and developed a C. muridarum transformation system for further mapping chlamydial pathogenic and protective determinants in animal models.
INTRODUCTION
Urogenital tract infection with Chlamydia trachomatis is a leading cause of sexually transmitted bacterial infection. Although the initial infection in the female lower genital tract accompanies no apparent clinic symptoms; if untreated, the lower genital tract infection can lead to ascending infection, causing long-term pathology such as hydrosalpinx, resulting in tubal factor infertility (1). However, the precise pathogenic mechanisms of C. trachomatis infection remain unknown, and there is still no licensed anti-C. trachomatis vaccine.
Almost all C. trachomatis clinical isolates contain a highly conserved plasmid (2–4). The plasmid may play a significant role in C. trachomatis pathogenesis since plasmid-free trachoma serovar A organisms failed to cause pathology in primate ocular tissues (5). This observation validated the finding that plasmid-free C. muridarum organisms were highly attenuated in mouse genital tract (6). The plasmid in C. trachomatis serovar L2 is known to regulate expression of more than 20 chromosomal genes at the transcription level (7). These chromosomal genes have been designated plasmid-dependent genes, including those involved in glycogen metabolism such as glgA and hypothetical open reading frames (ORFs) such as CTL0397 (a homolog of CT142 in serovar D of C. trachomatis and TC0419 in C. muridarum). The expression of some of these plasmid-dependent genes was significantly reduced in plasmid-free C. trachomatis L2 organisms. As a result, plasmid-deficient C. trachomatis L2 organisms lacked glycogen accumulation in the inclusions (7). Most of the plasmid-dependent genes are upregulated by the plasmid, although a few genes, including cpaf, were modestly increased in plasmid-free organisms (7).
The recent success in transforming C. trachomatis (8–17) has the potential to greatly advance our understanding of chlamydial pathogenic mechanisms and promote the development of chlamydial vaccines. This technology enables chlamydiologists to genetically manipulate chlamydial organisms. After the successful transformation with the shuttle vector pGFP::SW2 by Wang et al. (8), Song et al. used a pBRCT shuttle vector, which is similar to pGFP::SW2 but is based on the L2 plasmid instead of SW2 and also lacks the green fluorescent protein (GFP) gene, for generating plasmid open reading frame (pORF) deletion mutants. The pORF deletion analysis has revealed that Pgp1, -2, -6, and -8 are critical for plasmid maintenance, whereas Pgp3, -5, and -7 are dispensable for chlamydial growth in cell culture (9). Pgp4 appears to be a major transcriptional regulator of chlamydial gene expression since Pgp4 deletion resulted in >5-fold reduction in the expression of the plasmid-encoded pgp3 and plasmid-dependent chromosomal genes glgA (CTL0167 or CT798 in serovar D of C. trachomatis and TC0181 in C. muridarum) and hypothetical genes CTL0305, CTL0339, CTL0397, CTL0398, and CTL0399 (CT049, CT084, CT142, CT143, and CT144 or TC0319, TC0357, TC0419, TC0420, and TC0421, respectively). As a result, Pgp4 deletion dramatically reduced glycogen synthesis (9). Thus, many phenotypes of the plasmid-free organisms were reproduced by deletion of Pgp4 in C. trachomatis L2. These observations were validated by using the pGFP::SW2 vector (10–12). In addition, Gong et al. (10) demonstrated that it was the pgp8 coding DNA sequence and not the Pgp8 protein that was required for the maintenance of C. trachomatis plasmid. This is because stable transformants were obtained using a plasmid with multiple premature termination codons in pgp8, while the plasmid depleted of pgp8 ORF failed to transform C. trachomatis L2. Gong et al. further used an mCherry gene to replace pgp3, pgp4, and pgp5, respectively (10), suggesting that the transformation system can be used to introduce multiple genetically codable structures into chlamydial organisms. The transformation system has continuously been improved, including the introduction of alternative selectable markers (13, 15) and inducible expression cassettes (16, 17). Despite these achievements, more significant progress has been hindered by the failure of C. trachomatis to induce clinically relevant pathology in animal models.
Although C. muridarum causes no known human diseases, these organisms have been extensively used to study the mechanisms of C. trachomatis pathogenesis and immunity (18, 19). This is because intravaginal infection of mice with C. muridarum organisms can lead to upper genital tract pathology such as hydrosalpinx, a surrogate marker for tubal factor infertility in both mice (19, 20) and women (21, 22). Thus, developing a transformation system for C. muridarum should greatly facilitate chlamydial pathogenesis studies.
We sought here to develop and characterize a transformation system for C. muridarum. We constructed a C. muridarum plasmid-based shuttle vector designated pGFP::CM by using the C. muridarum plasmid pCM or pMoPn (23) to replace the C. trachomatis serovar E plasmid SW2 portion in the pGFP::SW2 shuttle vector. We then transformed a plasmid-free C. muridarum CMUT3 clone with pGFP::CM and reproducibly obtained stable transformants. Using this transformation system, we have both confirmed the roles of individual pORFs in plasmid maintenance and discovered a novel function of Pgp5. Pgp5 protein, but not the DNA coding sequence, was necessary for inhibiting the expression of the plasmid-dependent genes. Interestingly, the Pgp5-mediated inhibition of gene expression was more obvious during infection with C. muridarum than C. trachomatis, which might explain why this novel function of Pgp5 was overlooked during the characterization of the C. trachomatis pORFs. In the present study, we have not only developed a C. muridarum transformation system for further mapping the chlamydial pathogenic and protective determinants in animal models but also uncovered a novel function of Pgp5. Together with the previous findings of Pgp4 mainly as a positive regulator of plasmid-dependent gene expression (9, 10), the discovery of Pgp5 for negatively regulating the same set of genes has allowed us to hypothesize that the chlamydial plasmid may help chlamydial organisms to modulate chlamydial gene expression in response to environmental cues.
MATERIALS AND METHODS
Chlamydial organisms and infection.
Chlamydia muridarum (Nigg) organisms (originally from Robert Brunham's lab at the University of Manitoba, Manitoba, Canada [R. Brunham is now at the UBC CDC, Vancouver, Canada]) were propagated, purified, divided into aliquots, and stored as described previously (24). The HeLa (human cervical epithelial carcinoma cells, catalog no. CCL2), L929 (mouse fibroblast cell, catalog no. CCL-1), and McCoy (mouse fibroblast cell, CRL-1696) cells used here were purchased from the American Type Culture Collection (Manassas, VA). For chlamydial infection in cell culture, cells grown in 24-well plates with or without coverslips or six-well plates containing Dulbecco modified Eagle medium (DMEM; Sigma, St. Louis, MO) with 10% fetal bovine serum (FBS; Gemini Bio-products, West Sacramento, CA) at 37°C in an incubator supplied with 5% CO2 were inoculated with chlamydial organisms as described previously (24). Plasmid-free Chlamydia muridarum organisms (clone CMUT3) were generated by using novobiocin (catalog no. 74675; Sigma-Aldrich, St. Louis, MO) and isolated via a standard plaque assay as described elsewhere (25). The plaque-isolated clones were identified by using an anti-Pgp3 antibody-based immunofluorescence assay (3; C. Chen et al. unpublished data).
Constructing recombinant plasmids for transformation.
The pGFP::CM shuttle vector was constructed by replacing the SW2 portion of the pGFP::SW2 (kindly provided by Ian Clark [8]) with the C. muridarum plasmid pCM or pMoPn (3, 23, 26) using an in-fusion cloning technique as described previously (10). The three corresponding DNA fragments were amplified using pGFP::SW2 and pCM as the templates and primers listed in Table S1 in the supplemental material. PCR products were purified and digested with the FastDigest DpnI to remove the template DNA. An in-fusion HD cloning kit (Clontech Laboratories Inc., Mountain View, CA) was used to fuse the PCR products according to the manufacturer's instruction. The fusion plasmids were amplified in bacteria. The final plasmid after DNA sequencing validation was designated pGFP::CM and used for further experiments.
To make pORF deletion mutants, the primer pairs listed in Table S2 in the supplemental material were used to amplify DNA fragments lacking corresponding pORFs by PCR using AccuPrime Pfx SuperMix (Life Technologies, Grand Island, NY) and pGFP::CM as the template. To replace pgp5 with the mCherry gene, an additional PCR product containing the mCherry gene was generated using the appropriate primers listed in Table S3 in the supplemental material. The corresponding PCR products were generated and processed as described above. The desired PCR products were fused to produce the appropriate plasmids using the in-fusion HD cloning kit as described above. The final plasmids were extracted and subjected to DNA sequencing analyses and used for chlamydial transformation.
To construct a pGFP::CM plasmid with premature termination in Pgp5 translation, a stop codon was introduced into the 11th cysteine residue codon position of pgp5 gene using a QuikChange II site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. Briefly, pGFP::CM plasmid was amplified with the appropriate primers (see Table S3 in the supplemental material) that incorporate the desired nucleotide substitution using PCR with AccuPrime Pfx SuperMix. PCR products were purified and digested with the FastDigest DpnI to remove the template DNA. The digested PCR products were purified, recombined, and transformed into Escherichia coli XL1-Blue competent cells. Plasmids with the correct sequence were designated pGFP::CMpgp5C11stop that was used for transforming chlamydial organisms.
Transforming plasmid-free C. muridarum CMUT3 organisms.
All resultant C. muridarum plasmid-based shuttle vectors were used to transform plasmid-free C. muridarum clone CMUT3 in the form of purified elementary bodies (EBs) using a procedure as described previously (10). Briefly, 10 μl of CMUT3 organisms (107 inclusion-forming units [IFU]) and 10 μl of plasmid DNA (∼7 μg) were mixed in a total volume of 200 μl of CaCl2 buffer for 45 min at room temperature. Freshly trypsinized L929 cells (6 × 106 cells) resuspended in 200 μl of CaCl2 buffer were added to the EB/plasmid mixture, followed by incubation for a further 20 min at room temperature with occasional mixing. Each 70 μl of the final mixture was plated to a single well of a six-well plate together with 1.5 ml of prewarmed DMEM plus 10% FBS. The cells were allowed to adhere to the culture plate by incubating at 37°C in 5% CO2 for 12 h without cycloheximide or ampicillin. Then, cultures were replenished with fresh DMEM plus 10% FBS containing cycloheximide (2 μg/ml) and ampicillin (5 μg/ml) and incubated an additional 20 h. Inclusions positive for green fluorescence were identified under a fluorescence microscope and transferred to fresh monolayers of HeLa cells in the presence of ampicillin (20 μg/ml). The resultant inclusions remaining positive for green fluorescence were defined as generation 2 and were passaged for four to five additional generations to enrich fluorescence-positive organisms that were finally plaque cloned as described previously (25) for further experiments.
Live cell culture microscopy and indirect immunofluorescence assay.
An IX-81 inverted fluorescence microscope (Olympus, Center Valley, PA) was used to visualize live cells without or with infection by C. muridarum, CMUT3, or various CMUT3 transformants as described previously (10, 13). The immunofluorescence assay was carried out as described previously (24, 27). Immunofluorescence images were acquired by using an Olympus AX-70 fluorescence microscope equipped with multiple filter sets and Simple PCI imaging software as described previously (28).
Iodine staining.
HeLa cells with or without chlamydial infection were fixed with ice-cold methanol for 10 min and stained with 5% iodine stain (5% potassium iodide and 5% iodine in 50% ethanol) for 40 min. Individual coverslips were mounted in 50% glycerol containing 5% potassium iodide and 5% iodine. Images were acquired by using Olympus CH-30 microscope equipped with a Canon EOS Rebel T3i Digital SLR camera and processed using Adobe Photoshop.
PCR and quantitative real-time RT-PCR (qRT-PCR) and Western blotting.
Chlamydial organisms were lysed with 0.1% sodium dodecyl sulfate (SDS), and lysates were used as PCR templates after dilution at 1:1,000. Gene-specific primers used are listed in Table S4 in the supplemental material.
To quantitate transcripts, HeLa cells grown in six-well plates (106/well) were infected with EBs at a multiplicity of infection (MOI) of 2. At 20 h (C. muridarum) or 28 h (C. trachomatis L2) after infection, the cells were harvested using TRIzol reagent (Life Technologies), and the total RNA from each sample was extracted with a Direct-Zol RNA miniprep kit (Zymo Research, Irvine, CA). RNA preparations were used for cDNA synthesis with random hexamer primers via a ThermoScript RT-PCR System (Life Technologies). TaqMan RT-PCR assays were then performed using a CFX96 Touch-Deep-Well real-time PCR detection system (Bio-Rad, Hercules, CA) with iQ Supermix (Bio-Rad). The gene-specific primers used (see Table S5 in the supplemental material) included unlabeled primers and double-quenched probe (5′FAM/ZEN/3′IBFQ; Integrated DNA Technologies, Coralville, IA). Transcript copy numbers for a given gene from triplicate samples were calculated based on a standard plasmid DNA prep and further normalized to the copy numbers of chlamydial lpdA mRNA in the corresponding samples.
The Western blot assay was carried out as described elsewhere (29, 30). Infected cultures, as described above, were resolved in SDS-polyacrylamide gels. The resolved protein bands were transferred to nitrocellulose membranes for antibody detection. The primary antibodies included mouse polyclonal antiserum against GlgA (30) or Pgp3 (3), monoclonal antibody (MAb) MC22 against chlamydial major outer membrane protein (MOMP [24]), and MAb BC7.1 against HSP60 (unpublished data). The anti-MOMP and anti-HSP60 antibodies were used to ensure that all lanes with chlamydial-organism-containing samples had equivalent amounts of the organisms loaded. The primary antibody binding was probed with a horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (Jackson ImmunoResearch) and visualized with an enhanced chemiluminescence kit (Santa Cruz Biotech).
RESULTS
Characterization of C. muridarum plasmid pORFs.
The pGFP::SW2 plasmid (8, 10) was modified by replacing the SW2 portion with plasmid pCM from C. muridarum. The resultant plasmid, designated pGFP:CM, was then used to transform plasmid-free C. muridarum clone CMUT3 as shown in Fig. 1. L929 cells were used as the host cells for supporting the replication of the transformed CMUT3-pGFP::CM. We grew the cultures for 12 h without ampicillin and then with ampicillin for another 20 h. GFP-positive inclusions (defined as generation 1) were passaged to new cell cultures in the presence of ampicillin selection. In most cases, when the selection and passage were repeated for six rounds (generation 6), most inclusions remained GFP positive. A single clone was finally isolated via a plaque assay and purified for subsequent experiments.
FIG 1.

Transformation of plasmid-free C. muridarum (clone CMUT3) with a C. muridarum plasmid-based shuttle plasmid pGFP::CM. (A) The pGFP::CM shuttle vector was constructed by replacing the SW2 portion of the pGFP::SW2 with the C. muridarum plasmid pCM using an in-fusion cloning technique. (B) The resultant C. muridarum plasmid-based shuttle plasmid pGFP::CM was used to transform a plasmid-free C. muridarum clone CMUT3 in the form of purified elementary bodies (EBs). L929 cells were used as the host cells. The culture was incubated for 12 h without ampicillin and then with ampicillin for another 20 h. GFP-positive inclusions (defined as generation 1) were picked up and grown in the presence of ampicillin selection. (C) Selection and passage were repeated for six rounds (generation 6). Most inclusions were GFP positive, and a single clone was isolated via a plaque assay and purified for further studies.
To characterize the eight pORFs in the shuttle vector pGFP::CM, we created pORF deletion mutants designated pGFP::CMΔPgp1 to pGFP::CMΔPgp8, respectively, for transforming CMUT3 (Fig. 2). Stable CMUT3 transformants were only obtained with pGFP::CMΔPgp3, pGFP::CMΔPgp4, pGFP::CMΔPgp5, and pGFP::CMΔPgp7, respectively. The plasmids with deletions of pgp1, pgp2, pgp6, and pgp8, respectively, failed to stably transform CMUT3. We further monitored Pgp3 and GlgA protein expression, as well as glycogen synthesis, by these stable transformants (Fig. 3). Although Pgp3, GlgA, and glycogen were all detected in the cultures infected with wild-type (wt) C. muridarum, the plasmid-free CMUT3-infected cultures lacked the corresponding signals. Importantly, both GlgA expression and glycogen accumulation were restored in CMUT3 transformed with the parent plasmid pGFP::CM or pORF deletion plasmids pGFP::CMΔPgp3, pGFP::CMΔPgp5, or pGFP::CMΔPgp7 but not pGFP::CMΔPgp4. The Pgp3 protein was also significantly reduced in the CMUT3-pGFP::CMΔPgp4 transformant. These results were largely consistent with previous reports that Pgp4 is necessary for upregulating the expression of Pgp3 and GlgA, as well as the synthesis of glycogen (9, 10).
FIG 2.

Construction of pORF deletion mutants. (A) Each of the individual eight ORFs from the shuttle vector pGFP::CM was deleted with the deletion region indicated. (B) The deletion mutants designated pGFP::CMΔPgp1 to pGFP::CMΔPgp8, together with the parent plasmid (pGFP::CM), as listed on top of the gel image were detected for the presence of each of the 8 pORFs (from top to bottom rows) by PCR. (C) The eight plasmids with each deleted of one pORF were transformed into plasmid-free (pf) CMUT3 organisms. However, stable CMUT3 transformants were only obtained with four of the eight plasmids, and they were CMUT3 transformed with pGFP::CMΔPgp3, pGFP::CMΔPgp4, pGFP::CMΔPgp5, and pGFP::CMΔPgp7.
FIG 3.
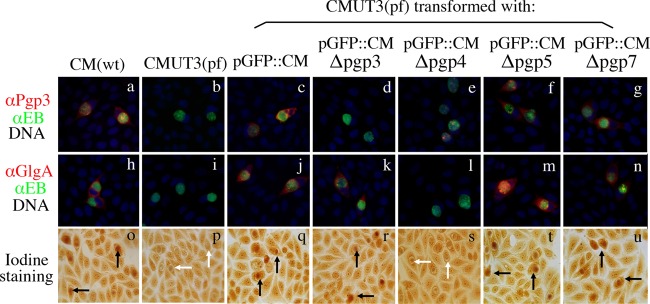
Effect of pORF deletion on Pgp3 and GlgA protein expression, as well as glycogen synthesis. The five stable transformants along with wild-type (wt) C. muridarum (CM) and plasmid-free (pf) CMUT3 organisms were used to infect HeLa cells. The infected cells were used for triple immunofluorescence labeling for Pgp3 (red, panels a to g) and GlgA (red, h to n) proteins, chlamydial organisms (green) and DNA (blue) and iodine staining of glycogen accumulation (o to u). Inclusions positive for glycogen staining were marked with black arrows, while inclusions lacked glycogen with white arrows. Note that both GlgA expression and glycogen accumulation were restored in CMUT3 transformed with pGFP::CM (panels j and q) and plasmids with deletion of pgp3 (panels k and r), pgp5 (panels m and t), or pgp7 (panels n and u) but not pgp4 (panels l and s). The Pgp3 protein was also significantly reduced in pgp4 deletion mutant (panel e).
Since C. trachomatis Pgp4 has been known to regulate chromosomal genes that are dependent on plasmid for expression (7, 9, 10), we further evaluated the effects of the C. muridarum pORF deletions on plasmid-dependent gene expression (Fig. 4). We focused on six plasmid-dependent chromosomal genes each with >5-fold differences in expression levels between plasmid-free and wt or plasmid-transformed C. trachomatis organisms (7, 9). The six plasmid-dependent genes were glgA (TC0181) and hypothetical genes TC0319, TC0357, TC0419, TC0420, and TC0421. In addition, both ompA (independent of plasmid regulation) and cpaf (slightly increased in plasmid-free organism-infected cultures) were included as controls. As expected, cultures infected with plasmid-free CMUT3 organisms displayed the lowest expression levels of the plasmid-dependent genes glgA (TC0181), TC0319, TC0419, TC0420, and TC0421 in addition to lacking the plasmid-borne genes. The expression of plasmid-dependent genes was restored by transformation with the parental plasmid pGFP::CM or pORF deletion plasmids pGFP::CMΔPgp3, pGFP::CMΔPgp5, or pGFP::CMΔPgp7 but not pGFP::CMΔPgp4. The plasmid-dependent gene expression was significantly lower in the pGFP::CMΔPgp4 transformant compared to the wild-type C. muridarum or other transformants. These observations demonstrated that Pgp4, but not Pgp3, -5, or -7, is required for upregulating the plasmid-dependent genes. Furthermore, the obvious reduction in pgp3 expression and the increase in pgp2 gene expression in the pGFP::CMΔPgp4-transformed cultures were also noted. However, only pgp3 reduction was maintained in the culture infected with a plasmid carrying a premature stop codon in pgp4 (data not shown), suggesting that Pgp4 protein is also required for optimal expression of pgp3. These results are consistent with what was found in the C. trachomatis system (7, 9, 10).
FIG 4.

Effect of pORF deletion on the expression of plasmid-encoded and -regulated genes. Wild-type (wt) C. muridarum and six plasmid-free C. muridarum organisms were used to infect HeLa cells. Cultures were harvested at 20 h postinfection for quantitative RT-PCR detection of transcripts of the eight plasmid-encoded (panels a to h) and eight genome-encoded (panels i to p) ORFs. The plasmid-free C. muridarum organisms were transformed without (CMUT3) or with pGFP::CM (Intact) or with deletion of pORFpgp3 (Δpgp3), pgp4 (Δpgp4), pgp5 (Δpgp5), and pgp7 (Δpgp7), respectively, as listed along the x axis. The transcript copy numbers normalized to lpdA mRNA for each of 16 measured ORFs are shown along the y axis. Note that pgp5 deletion significantly increased expression of pgp6 (panel f), TC0319 (homologue of CT049, panel j), TC0181 (homologue of CT798, panel k), TC0419 (homologue of CT142, panel l), TC0420 (homologue of CT143, panel m), TC0421 (homologue of CT144, panel n), and TC0357 (homologue of CT084, panel o). Stars (*) indicate a significant increase compared to transformants that carry the intact pGFP::CM, while “#” symbols indicate a significant decrease in gene expression compared to wild-type C. muridarum (CM). The transcript profiles were also compared after normalization with genome copies (see Fig. S1 in the supplemental material), and similar results were obtained.
Surprisingly, in the cultures infected with CMUT3-pGFP::CMΔPgp5, the expression levels of the six plasmid-dependent genes were dramatically increased compared to the cultures infected with CMUT3-pGFP::CM. This finding suggests that Pgp5 may be able to downregulate these genes during C. muridarum infection. However, the role of Pgp5 in downregulating these plasmid-dependent genes was not noted during C. trachomatis infection (9, 10). Nevertheless, the more relevant and immediate question to be addressed is whether the pgp5 ORF deletion-altered gene expression is dependent on Pgp5 protein or pgp5 coding DNA sequence or as a result of plasmid DNA structural or copy number changes.
Pgp5 protein, but not its DNA sequence, is required for suppressing plasmid-dependent genes.
As shown in Fig. 5A, all transformants contained similar number of plasmids per genome with the lowest copy in CMUT3-pGFP::CM and the highest copy in CMUT3-pGFPΔPgp3. Nevertheless, all transformants displayed significantly higher plasmid copy numbers than the wt C. muridarum. We further used a Western blot to compare the protein levels of Pgp3 and the plasmid-dependent GlgA (Fig. 5B). Despite the similar plasmid copy numbers among all transformants, the CMUT3-pGFPΔPgp5 transformant expressed the highest level of GlgA protein.
FIG 5.
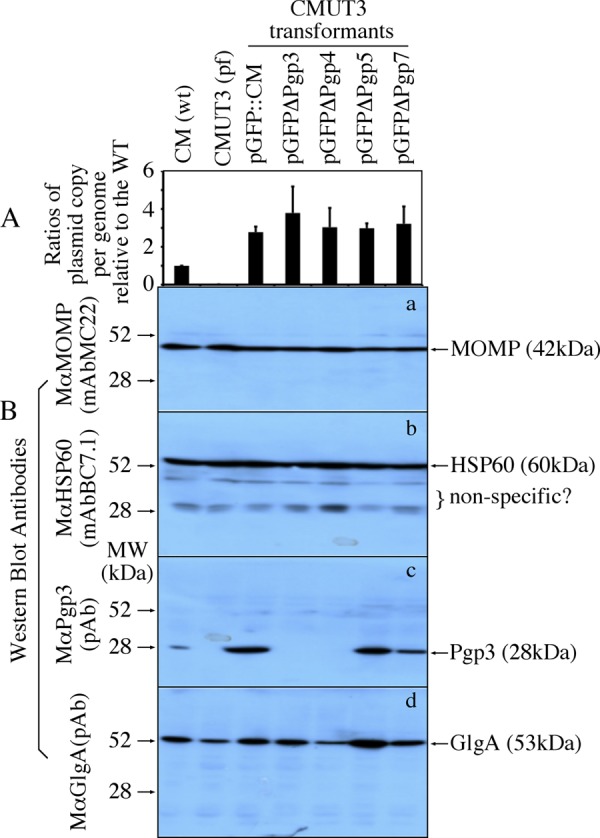
Plasmid copy number and GlgA expression among CMUT3 transformants. HeLa cell cultures infected with wild-type (wt) C. muridarum (CM), plasmid-free (pf) CMUT3, or various CMUT3 transformants as indicated on top of the figure were harvested for quantitating genome and plasmid copies using real-time PCR, as well as various chlamydial protein levels, as listed on the right side of the figure. The ratios were calculated based on plasmid copy number per genome from each culture sample as displayed along y axis (A). Please note that although all transformants displayed significantly higher plasmid copy numbers per genome than the wt CM, there was no significant difference among the transformants. (B) Both the chlamydial major outer membrane protein (MOMP) and heat shock protein (HSP) 60 (detected by MAbs) were used as control for equal sample loading among different lanes of the Western blot. Note that although all cultures expressed similar levels of MOMP and HSP60, the CMUT3 or CMUT3 transformed with pGFPDPgp3 or DPgp4 failed to express Pgp3. More importantly, the pGFPΔPgp5 transformed CMUT3 expressed the highest level of GlgA.
To reduce the impact of the genetic manipulation on plasmid DNA structure, we introduced a premature stop codon to the pgp5 gene by replacing the cysteine residue codon at the 11th position with a stop codon. The resultant plasmid was designated pGFP::CMpgp5C11stop. In parallel, we also used a mCherry gene (with a similar length to pgp5 gene) to replace pgp5, and the resultant plasmid was designated pGFP::CMmCherryRpgp5. We found that all transformants with either pGFP::CMΔpgp5, pGFP::CMpgp5C11stop, or pGFP::CMmCherryRpgp5 exhibited increased levels of GlgA protein (Fig. 6), suggesting that the increased expression of GlgA protein was not due to the varied alterations in the plasmid DNA sequence caused by different mutation methods. Possibly, Pgp5 protein—but not its DNA coding sequence—may be necessary for downregulating GlgA protein expression. When the levels of both plasmid-encoded and plasmid-dependent chromosomal genes were carefully compared (Fig. 7), the CMUT3-pGFP::CMmCherryRpgp5 transformant increased the expression of pgp3 (panel c), pgp4 (panel d), and pgp8 (panel h) but not pgp6 (panel f), while the CMUT3-pGFP::CMΔpgp5 transformants increased the expression of pgp6. However, the CMUT3-pGFP::CMpgp5C11stop transformant did not exhibit any of these alterations, suggesting that the varied plasmid gene expression may not be due to loss of Pgp5 protein or function. Importantly, despite the varied effects on plasmid gene expression among the pgp5 mutants, all three versions of the pgp5 mutants consistently increased the expression of the plasmid-dependent chromosomal genes. This result demonstrated that Pgp5-mediated downregulation of the plasmid-dependent genes is dependent on Pgp5 protein and not Pgp5 DNA coding sequence.
FIG 6.

Effect of pgp5 replacement with mCherry or premature termination on GlgA protein expression and glycogen synthesis. Wild-type (wt) C. muridarum (CM) or plasmid-free (pf) CMUT3 organisms with or without transformation were used to infect HeLa cells. Cultures were monitored for live cell imaging for GFP (green, panels a to f) or for mCherry (red, g to l) or used for triple immunofluorescence labeling (m to r) for GlgA protein (red), chlamydial organisms (green), and DNA (blue), or for iodine staining of glycogen accumulation (s to x). Plasmid-free CMUT3 organisms were transformed with intact pGFP::CM plasmid (pGFP::CM, panels c, i, o, and u) or pGFP::CM with pgp5 deletion (pGFP::CMΔpgp5, panels d, j, p, and v), pgp5 replacement by mCherry gene (pGFP::CMmCherryRpgp5, panels e, k, q, and w) or premature stop codon installed at the 11th cysteine residue position (pGFP::CMpgp5C11stop, panels f, l, r, and x). Inclusions positive for glycogen staining were marked with black arrows, while inclusions that lacked glycogen staining were marked with white arrows. Note that GlgA expression was increased in the cultures infected with CMUT3-pGFP::CMΔpgp5 (panel p), CMUT3-pGFP::CMmCherryRpgp5 (panel q), and CMUT3-pGFP::CMpgp5C11stop (panel r) organisms, respectively.
FIG 7.

Effect of pgp5 replacement with mCherry or premature stop on the expression of plasmid-encoded and -regulated genes. Wild-type (wt) C. muridarum and five plasmid-free C. muridarum organisms were used to infect HeLa cells. Cultures were harvested at 20 h postinfection for quantitative RT-PCR detection of transcripts of the eight plasmid-encoded (panels a to h) and eight genome-encoded (panels i to p) ORFs. Plasmid-free C. muridarum organisms were transformed without (CMUT3) or with pGFP::CM plasmid (Intact) or with deletion of pORFpgp5 (Δpgp5), pgp5 replacement with mCherry gene (mCherry), or premature stop codon at the 11th cysteine residue position (pgp5S) as listed along the x axis. The transcript copy numbers normalized to lpdA mRNA for each of 16 measured ORFs are shown along the y axis. Note that pgp5 deletion, replacement with mCherry, or premature stop all significantly increased the expression of TC0319 (homologue of CT049, panel j), TC0181 (homologue of CT798, panel k), TC0419 (homologue of CT142, panel l), TC0420 (homologue of CT143, panel m), TC0421 (homologue of CT144, panel n), and TC0357 (homologue of CT084, panel o); only pgp5 deletion increased the expression of pgp6 (panel f). Stars (*) indicate a significant increase of the gene expression compared to transformants that carry the intact pGFP::CM.
Inhibition of plasmid-dependent genes is a common property of Pgp5 from both C. muridarum and C. trachomatis.
The finding that the loss of Pgp5 protein resulted in upregulation of plasmid-dependent chromosomal genes was a surprise since this phenomenon was not noted during C. trachomatis infection (9, 10). We wondered whether the inhibition of gene expression is unique to C. muridarum Pgp5. To address this question, we monitored plasmid-dependent gene expression levels during C. muridarum infection after C. trachomatis pgp5 was used to replace C. muridarum pgp5 (and vice versa, during C. trachomatis infection after the C. muridarum pgp5 was used to replace C. trachomatis pgp5) (Fig. 8). We found that C. trachomatis pgp5 was as competent as C. muridarum pgp5 in suppressing the expression of the plasmid-dependent genes during C. muridarum infection, demonstrating that inhibition of the plasmid-dependent chromosomal genes is a common property of Pgp5 protein from different chlamydial species. However, during C. trachomatis infection, pgp5 deletion did not result in any significant increase in expression of the plasmid-dependent genes. The plasmid-dependent genes maintained similar expression levels in L2 organisms transformed with either the intact plasmid or plasmid with pgp5 deletion. Interestingly, both premature termination and mCherry gene replacement of pgp5 resulted in a significant increase in plasmid-dependent gene expression. This observation is consistent with the concept that a whole pORF deletion imposes much more dramatic alterations to the plasmid DNA structure than either the premature termination or replacement with an unrelated but similar length gene does. Thus, the finding observed from the latter two constructs has demonstrated that Pgp5 protein can significantly suppress the plasmid-dependent gene expression during C. trachomatis L2 infection. Furthermore, replacement of L2 Pgp5 with C. muridarum Pgp5 resulted in significant inhibition of the plasmid-dependent gene expression during C. trachomatis infection, confirming that the Pgp5-mediated inhibition of plasmid-dependent genes is conserved among different chlamydial species. Nevertheless, it is worth noting that the extent of Pgp5-mediated negative regulation varies between different chlamydial species. For example, neither the increase in the plasmid-dependent gene expression caused by disruption of pgp5 nor the inhibition of the plasmid-dependent gene expression by Pgp5 during C. trachomatis infection was as obvious as that observed during C. muridarum infection, which might explain why the pgp5-mediated suppression of plasmid-dependent gene expression was previously overlooked (10). It was the C. muridarum transformation system that enabled us to recognize the novel function of Pgp5.
FIG 8.

Comparison of the effects of Pgp5 from C. muridarum versus C. trachomatis on the expression of plasmid-regulated genes. Wild-type (wt) C. muridarum and four plasmid-free C. muridarum (CMUT3) organisms (panels a to g) or wild-type (wt) C. trachomatis L2 and six plasmid-free L2 [L2(R)] organisms (panels h to n) were used to infect HeLa cells. Cultures were harvested at 20 h (C. muridarum) or 28 h (C. trachomatis L2) postinfection for quantitative RT-PCR detection of transcripts of one control gene (TC0052, homologue of CT681, panels a and h) and six plasmid-regulated genes (TC0319, homologue of CT049, panels b and I; TC0181, homologue of CT798, panels c and j; TC0419, homologue of CT142, panels d and k; TC0420, homologue of CT143, panels e and l; TC0421, homologue of CT144, panels f and m; and TC0357, homologue of CT084, panels g and n). As shown along the x axis, CMUT3 organisms were transformed with intact pGFP::CM plasmid or with pgp5 replacement by mCherry (mCherry) or by C. trachomatis L2 pgp5 (L2pgp5), whereas the L2(R) organisms were transformed with intact pGFP::SW2 plasmid or with pgp5 deletion (Δpgp5), premature termination (pgp5S), or replacement by mCherry (mCherry) or by C. muridarum pgp5 (CMpgp5). Transcript copy numbers normalized to lpdA mRNA for each of seven measured ORFs are shown along the y axis. Note that both the replacements of C. muridarum pgp5 with C. trachomatis pgp5 and of C. trachomatis pgp5 with C. muridarum pgp5 maintained the suppression of the six plasmid-regulated genes in corresponding cultures. A number sign (#) indicates significant inhibition of gene expression compared to transformants carrying the mCherry replacement (b to g and i to n) or premature termination (i to n) constructs. Stars (*) indicate significant increase in the gene expression compared to transformants that carry the intact pGFP::SW2. Unexpectedly, deletion of pgp5 failed to increase the expression of the six plasmid-dependent genes during C. trachomatis L2 infection (i to n).
DISCUSSION
The roles of plasmids in the pathogenesis of both C. trachomatis and C. muridarum infections have been demonstrated using plasmid-free organisms (5, 6). The successful transformation of plasmid-free C. trachomatis has allowed chlamydiologists to determine the molecular basis of C. trachomatis plasmid-dependent pathogenicity in cell culture model systems (8–11). Pgp1, -2, -6, and -8 were determined as plasmid maintenance factors, Pgp3, -5, and -7 as dispensable for chlamydial growth in vitro, and Pgp4 as a transcriptional regulator. These findings were confirmed during C. muridarum infection in the present study by using a C. muridarum plasmid-based shuttle vector (pGFP::CM) to transform a plasmid-free C. muridarum strain (CMUT3). Both plasmid-free C. trachomatis and C. muridarum organisms have been characterized as deficient in glycogen accumulation due to the reduced expression of GlgA and the reduction in expression of other chromosomal hypothetical genes. Many of these plasmid-dependent genes are dependent on Pgp4 for expression (9, 10; the present study). Thus, Pgp4 has been identified as a positive regulator of the plasmid-dependent genes during both C. trachomatis and C. muridarum infection. During revision of the present manuscript, Song et al. reported that transformation of C. muridarum was only restricted to shuttle vector that contained the C. muridarum plasmid backbone and Pgp4 remained to be a positive regulator for C. muridarum plasmid-dependent gene expression (14). However, how these plasmid-dependent genes were negatively regulated during infection with either C. trachomatis or C. muridarum was still unknown.
In the present study, using the C. muridarum transformation system, we unexpectedly discovered that Pgp5 protein negatively regulated the plasmid-dependent chromosomal genes, including glgA (TC0181) and the hypothetical genes TC0319, TC0357, TC0419, TC0420, and TC0421. First, deletion of the pgp5 ORF resulted in significant increases in expression of these genes, indicating the necessity of pgp5 for suppressing these genes. Second, despite the similar plasmid copy numbers among all CMUT3 transformants, the CMUT3-pGFPΔPgp5 transformant expressed the highest level of GlgA protein, indicating that the increased expression of the plasmid-dependent genes in the Pgp5-deficient mutant was not due to alteration in plasmid copy numbers. Third, although pgp5 ORF deletion also resulted in increased expression of plasmid gene pgp6 and mCherry replacement of Pgp5 increased expression of pgp3, pgp4, and pgp8, both versions of the pgp5 mutation consistently increased the expression of the plasmid-dependent chromosomal genes. The varied effects on the plasmid genes may be due to varied alterations to the plasmid DNA sequences caused by the manipulation of the pgp5 ORF. Despite the varied effects on plasmid genes, the consistent effect on the plasmid-dependent genes demonstrates that it is the loss of pgp5 function that causes the increase in the expression of the plasmid-dependent chromosomal genes. Fourth, premature termination of Pgp5 protein synthesis resulted in increased expression of only plasmid-dependent chromosomal genes but not any plasmid genes, indicating that loss of the Pgp5 protein is responsible for the increased expression of the chromosomal genes and that the Pgp5 function does not involve any other plasmid genes. Fifth, the expression of Pgp5 from C. trachomatis in C. muridarum organisms also led to the suppression of expression of the plasmid-dependent chromosomal genes, indicating that the gene regulation function is a conserved property of all Pgp5 regardless of its origin. Finally, during C. trachomatis L2 infection, either mCherry replacement or premature termination of pgp5 resulted in significant increase in the expression of the plasmid-dependent genes, and the increased expression was inhibited by C. trachomatis and C. muridarum pgp5, respectively.
Although Pgp4 as a positive regulator of chlamydial plasmid-dependent chromosomal genes was obviously noted in both C. trachomatis (9, 10) and C. muridarum (the present study) systems, the finding that Pgp5 negatively regulated the same set of genes was obvious only in the present C. muridarum study. This discrepancy is not likely due to variation in Pgp5 protein function since Pgp5 proteins from both C. trachomatis and C. muridarum similarly inhibited expression of the plasmid-dependent genes, although they both exhibited greater ability to negatively regulate the plasmid-dependent genes during C. muridarum infection compared to C. trachomatis infection. The reduced, yet still significant inhibition of the plasmid-dependent genes by Pgp5 during C. trachomatis infection suggests that the C. trachomatis species may have evolved other mechanism(s) for negatively regulating the plasmid-dependent genes. Pgp5 is predicted to be a MinD that participates in genome segregation/partitioning. Both C. trachomatis (31) and C. muridarum (23) genomes encode MinD molecules presumably required for chromosomal segregation. Is it possible that the genomic MinD from C. trachomatis is more functionally relevant to Pgp5 than the genomic MinD from C. muridarum? Such a difference may explain why Pgp5 knockout resulted in more dramatic increase in the plasmid-dependent gene expression during C. muridarum infection compared to C. trachomatis infection. However, this explanation is not supported by the amino acid sequence homology analyses. The genomic MinDs from C. trachomatis and C. muridarum share ∼80% identity and Pgp5 from both species also shared ∼80% amino acid sequence identity. However, the homology between any genomic MinD and any Pgp5 is only ca. 30%. These analyses suggest that the genomic minD and Pgp5 from both species are highly conserved and MinD and Pgp5 may have diverged enough from each other. It is unlikely the genomic minD from one species is functionally more relevant to Pgp5 than the other. The Pgp5 DNA coding sequence also contains noncoding RNAs (32, 33), and these ncRNAs may contribute to the negative regulation of the plasmid-dependent gene expression. However, the fact that premature termination of Pgp5 without altering the expression of ncRNA resulted in increased expression of plasmid-dependent genes suggests that the Pgp5 protein, but not the DNA sequence nor ncRNA encoded in the region, is responsible for the gene regulation function.
It is now clear that the chlamydial plasmid encodes both positive (Pgp4) and negative (Pgp5) regulators for modulating plasmid-dependent chromosomal gene expression. The next question is how these opposing regulators work. Pgp4 is a Chlamydia-specific protein with 102 amino acids but with no known function. Secondary structural analyses reveal that Pgp4 has a putative helix-loop-helix domain and may be a novel member of the helix-loop-helix family of transcriptional regulators. However, evidence for Pgp4 binding to regulator elements of the plasmid-dependent genes is still lacking. Pgp5 is a minD protein with 239 amino acids. MinD is known to bind to ATP and participate in plasmid/chromosome segregation/partitioning. Clearly, the chlamydial plasmid-encoded minD is not essential for segregation of either the plasmid or chromosome since deletion of pgp5 permitted replication of both chlamydial organism and plasmid. Like Pgp4, it remains unknown how Pgp5 selectively suppresses the expression of the plasmid-dependent genes. It is likely that additional factors may be involved in both Pgp4- and Pgp5-mediated regulation of plasmid-dependent genes. Efforts are under way to map functional domains and to identify partners of both Pgp4 and Pgp5. Regardless of how Pgp4 and Pgp5 work, the discovery of Pgp5 being able to negatively regulate the same set of genes positively regulated by Pgp4 has allowed us to hypothesize that the chlamydial plasmid may help chlamydial organisms to modulate chlamydial gene expression in response to environmental cues. This hypothesis is consistent with the fact that plasmid is dispensable for chlamydial replication and both Pgp4 and Pgp5 are dispensable for plasmid maintenance and replication. Thus, Chlamydia may be able to afford to use Pgp4 and Pgp5 for responding to the environmental factors from the host and vaginal microbiome and coinfection.
Supplementary Material
Footnotes
Published ahead of print 20 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01161-13.
REFERENCES
- 1.Sherman KJ, Daling JR, Stergachis A, Weiss NS, Foy HM, Wang SP, Grayston JT. 1990. Sexually transmitted diseases and tubal pregnancy. Sex. Transm. Dis. 17:115–121. 10.1097/00007435-199007000-00001 [DOI] [PubMed] [Google Scholar]
- 2.Ricci S, Ratti G, Scarlato V. 1995. Transcriptional regulation in the Chlamydia trachomatis pCT plasmid. Gene 154:93–98. 10.1016/0378-1119(94)00825-D [DOI] [PubMed] [Google Scholar]
- 3.Li Z, Chen D, Zhong Y, Wang S, Zhong G. 2008. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect. Immun. 76:3415–3428. 10.1128/IAI.01377-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seth-Smith HM, Harris SR, Persson K, Marsh P, Barron A, Bignell A, Bjartling C, Clark L, Cutcliffe LT, Lambden PR, Lennard N, Lockey SJ, Quail MA, Salim O, Skilton RJ, Wang Y, Holland MJ, Parkhill J, Thomson NR, Clarke IN. 2009. Co-evolution of genomes and plasmids within Chlamydia trachomatis and the emergence in Sweden of a new variant strain. BMC Genomics 10:239. 10.1186/1471-2164-10-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc. Natl. Acad. Sci. U. S. A. 108:7189–7193. 10.1073/pnas.1102229108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Connell CM, Ingalls RR, Andrews CW, Jr, Scurlock AM, Darville T. 2007. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J. Immunol. 179:4027–4034 http://www.jimmunol.org/content/179/6/4027.long [DOI] [PubMed] [Google Scholar]
- 7.Carlson JH, Whitmire WM, Crane DD, Wicke L, Virtaneva K, Sturdevant DE, Kupko JJ, III, Porcella SF, Martinez-Orengo N, Heinzen RA, Kari L, Caldwell HD. 2008. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect. Immun. 76:2273–2283. 10.1128/IAI.00102-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog. 7:e1002258. 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song L, Carlson JH, Whitmire WM, Kari L, Virtaneva K, Sturdevant DE, Watkins H, Zhou B, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 2013. Chlamydia trachomatis plasmid-encoded Pgp4 is a transcriptional regulator of virulence-associated genes. Infect. Immun. 81:636–644. 10.1128/IAI.01305-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gong S, Yang Z, Lei L, Shen L, Zhong G. 2013. Characterization of Chlamydia trachomatis plasmid-encoded open reading frames. J. Bacteriol. 195:3819–3826. 10.1128/JB.00511-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Persson K, Bjartling C, Clarke IN. 2013. Genetic transformation of a clinical (genital tract), plasmid-free isolate of Chlamydia trachomatis: engineering the plasmid as a cloning vector. PLoS One 8:e59195. 10.1371/journal.pone.0059195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Cutcliffe LT, Skilton RJ, Persson K, Bjartling C, Clarke IN. 2013. Transformation of a plasmid-free, genital tract isolate of Chlamydia trachomatis with a plasmid vector carrying a deletion in CDS6 revealed that this gene regulates inclusion phenotype. Pathog. Dis. 67:100–103. 10.1111/2049-632X.12024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ding H, Gong S, Tian Y, Yang Z, Brunham R, Zhong G. 2013. Transformation of sexually transmitted infection-causing serovars of Chlamydia trachomatis using blasticidin for selection. PLoS One 8:e80534. 10.1371/journal.pone.0080534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song L, Carlson JH, Zhou B, Virtaneva K, Whitmire WM, Sturdevant GL, Porcella SF, McClarty G, Caldwell HD. 11 November 2013. Plasmid-mediated transformation tropism of chlamydial biovars. Pathog. Dis. 10.1111/2049-632X.12104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu S, Battaglia L, Bao X, Fan H. 2013. Chloramphenicol acetyltransferase as a selection marker for chlamydial transformation. BMC Res. Notes 6:377. 10.1186/1756-0500-6-377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agaisse H, Derre I. 2013. A Chlamydia trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8:e57090. 10.1371/journal.pone.0057090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wickstrum J, Sammons LR, Restivo KN, Hefty PS. 2013. Conditional gene expression in Chlamydia trachomatis using the tet system. PLoS One 8:e76743. 10.1371/journal.pone.0076743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrison RP, Caldwell HD. 2002. Immunity to murine chlamydial genital infection. Infect. Immun. 70:2741–2751. 10.1128/IAI.70.6.2741-2751.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex. Transm. Dis. 32:49–56. 10.1097/01.olq.0000148299.14513.11 [DOI] [PubMed] [Google Scholar]
- 20.de la Maza LM, Pal S, Khamesipour A, Peterson EM. 1994. Intravaginal inoculation of mice with the Chlamydia trachomatis mouse pneumonitis biovar results in infertility. Infect. Immun. 62:2094–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strandell A, Waldenstrom U, Nilsson L, Hamberger L. 1994. Hydrosalpinx reduces in-vitro fertilization/embryo transfer pregnancy rates. Hum. Reprod. 9:861–863 [DOI] [PubMed] [Google Scholar]
- 22.Vasquez G, Boeckx W, Brosens I. 1995. Prospective study of tubal mucosal lesions and fertility in hydrosalpinges. Hum. Reprod. 10:1075–1078 [DOI] [PubMed] [Google Scholar]
- 23.Read TD, Brunham RC, Shen C, Gill SR, Heidelberg JF, White O, Hickey EK, Peterson J, Utterback T, Berry K, Bass S, Linher K, Weidman J, Khouri H, Craven B, Bowman C, Dodson R, Gwinn M, Nelson W, DeBoy R, Kolonay J, McClarty G, Salzberg SL, Eisen J, Fraser CM. 2000. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 28:1397–1406. 10.1093/nar/28.6.1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong G, Fan P, Ji H, Dong F, Huang Y. 2001. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J. Exp. Med. 193:935–942. 10.1084/jem.193.8.935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumoto A, Izutsu H, Miyashita N, Ohuchi M. 1998. Plaque formation by and plaque cloning of Chlamydia trachomatis biovar trachoma. J. Clin. Microbiol. 36:3013–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas NS, Lusher M, Storey CC, Clarke IN. 1997. Plasmid diversity in Chlamydia. Microbiology 143(Pt 6):1847–1854. 10.1099/00221287-143-6-1847 [DOI] [PubMed] [Google Scholar]
- 27.Zhong G, Reis e Sousa C, Germain RN. 1997. Production, specificity, and functionality of monoclonal antibodies to specific peptide-major histocompatibility complex class II complexes formed by processing of exogenous protein. Proc. Natl. Acad. Sci. U. S. A. 94:13856–13861. 10.1073/pnas.94.25.13856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan T, Lu H, Hu H, Shi L, McClarty GA, Nance DM, Greenberg AH, Zhong G. 1998. Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187:487–496. 10.1084/jem.187.4.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong G, Castellino F, Romagnoli P, Germain RN. 1996. Evidence that binding site occupancy is necessary and sufficient for effective major histocompatibility complex (MHC) class II transport through the secretory pathway redefines the primary function of class II-associated invariant chain peptides (CLIP). J. Exp. Med. 184:2061–2066. 10.1084/jem.184.5.2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu C, Lei L, Peng B, Tang L, Ding H, Gong S, Li Z, Wu Y, Zhong G. 2013. Chlamydia trachomatis GlgA is secreted into host cell cytoplasm. PLoS One 8:e68764. 10.1371/journal.pone.0068764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. 1998. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282:754–759. 10.1126/science.282.5389.754 [DOI] [PubMed] [Google Scholar]
- 32.Ricci S, Cevenini R, Cosco E, Comanducci M, Ratti G, Scarlato V. 1993. Transcriptional analysis of the Chlamydia trachomatis plasmid pCT identifies temporally regulated transcripts, antisense RNA and sigma 70-selected promoters. Mol. Gen. Genet. 237:318–326 [DOI] [PubMed] [Google Scholar]
- 33.Abdelrahman YM, Rose LA, Belland RJ. 2011. Developmental expression of noncoding RNAs in Chlamydia trachomatis during normal and persistent growth. Nucleic Acids Res. 39:1843–1854. 10.1093/nar/gkq1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.