

ABSTRACT
Virus emergence is a complex phenomenon, which generally involves spread to a new host from a wild host, followed by adaptation to the new host. Although viruses account for the largest fraction of emerging crop pathogens, knowledge about their emergence is incomplete. We address here the question of whether Pepino Mosaic Virus (PepMV) emergence as a major tomato pathogen worldwide could have involved spread from wild to cultivated plant species and host adaptation. For this, we surveyed natural populations of wild tomatoes in southern Peru for PepMV infection. PepMV incidence, genetic variation, population structure, and accumulation in various hosts were analyzed. PepMV incidence in wild tomatoes was high, and a strain not yet reported in domestic tomato was characterized. This strain had a wide host range within the Solanaceae, multiplying efficiently in most assayed Solanum species and being adapted to wild tomato hosts. Conversely, PepMV isolates from tomato crops showed evidence of adaptation to domestic tomato, possibly traded against adaptation to wild tomatoes. Phylogenetic reconstructions indicated that the most probable ancestral sequence came from a wild Solanum species. A high incidence of PepMV in wild tomato relatives would favor virus spread to crops and its efficient multiplication in different Solanum species, including tomato, allowing its establishment as an epidemic pathogen. Later, adaptation to tomato, traded off against adaptation to other Solanum species, would isolate tomato populations from those in other hosts.
IMPORTANCE Virus emergence is a complex phenomenon involving multiple ecological and genetic factors and is considered to involve three phases: virus encounter with the new host, virus adaptation to the new host, and changes in the epidemiological dynamics. We analyze here if this was the case in the recent emergence of Pepino Mosaic Virus (PepMV) in tomato crops worldwide. We characterized a new strain of PepMV infecting wild tomato populations in Peru. Comparison of this strain with PepMV isolates from tomato crops, plus phylogenetic reconstructions, supports a scenario in which PepMV would have spread to crops from wild tomato relatives, followed by adaptation to the new host and eventually leading to population isolation. Our data, which derive from the analysis of field isolates rather than from experimental evolution approaches, significantly contribute to understanding of plant virus emergence, which is necessary for its anticipation and prevention.
INTRODUCTION
Emergent diseases often have a high socioeconomic impact. As described by Woolhouse and Dye (1), an emergent disease can be defined as that “whose incidence is increasing following its first introduction into a new host population, or whose incidence is increasing in an existing host population as a result of long-term changes in its underlying epidemiology.” Viruses account for the largest fraction of emerging diseases in humans, animals, and plants (2, 3). Virus emergence is a complex phenomenon involving multiple ecological and genetic factors, which act during three different phases: in the first phase the virus encounters the new host, often by spread from a wild host; in the second phase the virus adapts to the new host, which involves genetic changes; and in the third phase epidemiological dynamics adapt to the new environment, most often by increasing the between-host transmission rates (3, 4). It is necessary to understand these processes to anticipate and prevent virus emergence. However, understanding plant virus emergence in crops may be limited by a lack of knowledge on the occurrence of virus species and strains in wild plant species from which cross-species spread to crops might occur and on the potential for adaptation to crops of wild-plant-infecting viruses (5, 6).
Pepino Mosaic Virus (PepMV) (genus Potexvirus) is an important pathogen of tomato crops worldwide and a typical example of an emergent plant virus (6, 7). PepMV has flexuous rod-like particles, which encapsidate a messenger-sense, single-stranded RNA (ssRNA) genome of about 6.4 kb. The PepMV genome encodes five proteins: a protein involved in virus replication (RdRp); three proteins involved in cell-to-cell movement, whose overlapping genes are organized into a triple gene block (proteins TGBp1, TGBp2, and TGBp3); and the coat protein (CP) (8–10). PepMV is transmitted by plant-to-plant contact at high rates, and it is also transmitted through the seed (11, 12), which may have been relevant in its long-distance dispersal.
PepMV was first isolated in 1974 in Peru from pepino (Solanum muricatum Ait.) plants showing symptoms of yellow mosaic (10). It was not reported as a pathogen of tomato (Solanum lycopersicum L.) until 1999, in greenhouses in The Netherlands (13, 14). Shortly after, it became a major pathogen of tomato worldwide, with its epidemic expansion being reported first in Europe and then in North America (6–9, 15–18). Isolates from the initial epidemic in Europe were highly similar (99%) to each other, constituting the European tomato strain (EU) (10). EU isolates are closely related (96% similarity) to the strain named Peruvian or LP (7, 10), which includes the original pepino isolate (SM.74) (10) and an isolate from wild Solanum peruvianum L. in Peru (19). Since 2005, new PepMV isolates sharing only 80% sequence similarity with the EU or LP strains have been characterize. These isolates were reported from the United States (US1 and US2) (18) and from tomato seeds produced in Chile (CH1 and CH2) (17). Full characterization of these isolates showed that US2 was a recombinant between US1 and CH1, and currently four PepMV strains are recognized (20): the original Peruvian isolate (LP), the European tomato strain (EU), and the American (US1) and Chilean (CH2) strains. PepMV epidemics have been characterized by the replacement of strains. Thus, in Europe EU isolates have been replaced by CH2 isolates (6, 20, 21), with mixed infection between both strains and recombination among them playing a role in the virus evolutionary dynamics (6, 10, 20–22). The US1 genotype has also been found in the Canary Islands (23), and finally, in North America the EU strain became prevalent (24, 25), to be later replaced by the CH2 strain (26).
Characterized isolates from the LP strain are asymptomatic in tomato, and in this host they accumulate to lower levels than the related EU isolates (13, 27, 28), which suggests that emergence of the EU strain could be due to spread of LP isolates to tomato from pepino or wild Solanum species, followed by adaptation to tomato. Also, the origin of isolates reported in the United States from tomato seed produced in Chile (CH1 and CH2) (15) is again suggestive of an origin in spread from wild Solanum in the areas of seed production. Since the initial emergence of PepMV in tomato crops has been followed by the emergence of new strains causing similar or different symptoms (6, 7, 29), it seems that there is a continued risk of across-host spread and adaptation of new PepMV strains resulting in new emergences.
To test the hypothesis that emergence of PepMV could be due to spread from wild hosts followed by adaptation to tomato, we made a survey for PepMV infection in natural populations of wild tomatoes in southern Peru and analyzed its genetic variation and population structure. We report the occurrence in wild tomatoes of a new PepMV strain, not yet reported in domestic tomato, and characterized this strain for the traits that may be relevant to evaluate its potential for emergence in tomato crops.
MATERIALS AND METHODS
Field surveys, plant collections, and PepMV detection.
Plants of several species of wild tomatoes [Solanum chilense (Dunal) Reiche, S. peruvianum, Solanum pimpinellifolium L., and Solanum pinnatifidum Ruiz & Pav.] were sampled in March 2008 at different sites over the distribution of these species in the provinces of Arequipa and Moquegua in southern Peru (Table 1). A total of 12 populations were sampled in different habitats. Relevant information on these populations appears in Table 1. At the time of sampling, plants of all populations were at a similar phenological stage, at flowering but before fruit set. Plants were sampled so that one plant out of every x plants was sampled along fixed itineraries, with itinerary length and x (0 < x ≤ 4) depending on the population size. For populations consisting of fewer than 10 plants, all individuals were sampled. Samples consisted of 1 to 3 young branches with fresh leaves per plant. Samples were taken to the laboratory, and infection by PepMV was assessed by two complementary procedures: (i) double-antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) using PepMV-specific commercial antibodies (Loewe, Germany) and (ii) reverse transcription-PCR (RT-PCR) using primers PepMVCP-F and PepMVCP-R (sequences are available upon request), which amplified the CP gene.
TABLE 1.
Description of wild tomato populations
| Population | Altitude, m | Latitude/longitude | Habitat | PepMV incidence, %a |
|---|---|---|---|---|
| Yura 1 (YUR1) | 2,636 | 16°17′S/71°39′W | Mountain short grass | 50.0 (3/6) |
| Yura 2 (YUR2) | 2,423 | 16°14′S/71°42′W | Mountain short grass | 0 (0/5) |
| Cerro Verde 1 (CVD1) | 2,236 | 16°28′S/71°37′W | Mountain short grass | 0 (0/5) |
| Cerro Verde 2 (CVD2) | 2,115 | 16°27′S/71°42′W | Mountain short grass | 16.7 (1/6) |
| Chiguata (CHI1) | 2,757 | 16°25′S/71°27′W | Mountain short grass | 31.2 (5/16) |
| Chiguata (CHI2) | 2,847 | 16°25′S/71°27′W | Mountain short grass | 45.5 (5/11) |
| Mollendo (MOL1) | 601 | 16°57′S/72°3′W | Coastal desert | 33.3 (2/6) |
| Mollendo (MOL2) | 544 | 16°57′S/72°3′W | Coastal desert | 25.0 (2/8) |
| Torata (TOR) | 2,170 | 17°6′ S/70°52′W | Mountain short grass | 25.0 (4/16) |
| Samegua (SAM1) | 1,653 | 17°10′S/70°52′W | Coastal desert | 0 (0/6) |
| Samegua (SAM2) | 1,712 | 17°10′S/70°52′W | Coastal desert | 50.0 (3/6) |
| Cerro el Baul (CEB) | 1,992 | 17°7′S/70°50′W | Coastal desert | 23.1 (3/13) |
| Avg | 26.9 (28/104) |
Percentage of infected plants. The number of infected plants/total number of sampled plants are shown in parentheses.
Nucleotide sequence determination.
For all PepMV-positive field plants, RT-PCR amplification of the coat protein (CP) gene was attempted. For this, total RNA extracts were prepared from leaves of field-infected samples using the TRIzol reagent (Life Technologies, CA, USA) following the manufacturer's instructions, and primers PepMVCP-F and PepMVCP-R were used.
The nucleotide sequence of the genomic RNA was determined after transfer of PepMV isolates from a subset of PepMV-positive samples to tomato plants and multiplication. For this, 15-day-old plants of tomato (S. lycopersicum cv. Rutgers) were inoculated at the cotyledons with sap of field-infected plants in 0.01 M sodium phosphate buffer, pH 7.0. Inoculated plants were maintained in a growth chamber at 25 or 15°C (for day and night, respectively) and with 15 h of light. The infection status of these plants was checked at 30 days postinoculation (dpi) by DAS-ELISA as described above. To minimize the effect of isolate passage in a different host, leaves from all tomato plants infected with the same isolate were pooled, virus particles were purified as described by Aguilar et al. (8) and disrupted in 4% sodium dodecyl sulfate–0.1 M Tris-HCl (pH 9.0)–1 mg/ml benthonite, and genomic RNA was extracted as described by Pagán et al. (10). The complete nucleotide sequence of the genomic RNA was obtained for three PepMV isolates. To do so, the sequence at the 5′ untranslated region (UTR) was determined using the degenerate primer UTR5′RSL and rapid amplification of cDNA ends (RACE) adapters. The 3′ UTR sequence was determined using oligo(dT) and UTR3′FSL. For the rest of the genome, 8 pairs of degenerate primers were used for RT-PCR amplification (Pep1F-Pep1R to Pep8F-Pep8R; sequences are available upon request). Sequences were confirmed on amplicons obtained using specific primers designed on previously sequenced regions (not shown). All amplifications were carried out using the high-yield reverse transcriptase SuperScript III (Life Technologies) and PCR SuperMix high-fidelity polymerase (Life Technologies).
Analyses of PepMV nucleotide genomic sequences.
Sequence alignments were constructed using MUSCLE 3.7 (30) and adjusted manually using Se-Al (31). Genetic diversities were estimated using the general time-reversible substitution model with invariant sites and a gamma distribution of among-site rate variation (GTR+I+Γ4) as implemented in MEGA 5 (32). This was the best-fitted nucleotide substitution model selected by the corrected Akaike information criterion as determined by jModelTest 0.1.1 (33). Standard errors of each genetic diversity measure were based on 1,000 replicates bootstrap using MEGA 5. Alternative nucleotide substitution models (Kimura 2-parameter model and Tamura-Nei model) yielded similar results. For phylogenetic analyses, the occurrence of recombination in the utilized sequences was determined using four methods based on different assumptions (34), and implemented in RDP3 (http://darwin.uvigo.es/rdp/rdp.html) (RDP, BOOTSCAN/RECSCAN, Siscan, and Chimaera, with default parameters) (35). Only recombination signals detected by all methods (P < 0.05) were considered positive. Maximum-likelihood (ML) phylogenetic trees were inferred with PhyML (36), using the subtree pruning regrafting (SPR) method and incorporating the GTR+I+Γ4 substitution model.
For reconstruction of the ancestral state of host species and PepMV strains and the time scale of virus evolution, the presence of temporal and state-related phylogenetic signals in the analyzed data set is essential to an accurate estimation. Thus, we first assessed the strength of the temporal signal in a data set with 34 complete genomic PepMV sequences. To do so, we constructed an ML tree as described above, and clock-like behavior of the data set was then assessed by regressing the root-to-tip distance in the ML tree against the date of sampling of each sequence using Path-O-Gen v1.4 (http://tree.bio.ed.ac.uk/software/pathogen/). To confirm the presence of a temporal signal, the BEAST analyses described below were repeated on data sets in which sampling times were randomized so that the temporal structure was disrupted. Randomizations were repeated 10 times, and substitution rates for randomized and real data were compared. Randomized and real values were considered significantly different when the 95% highest posterior density (HPD) values for all of the randomized controls excluded the mean substitution rates estimated for the real data, indicating the presence of a temporal signal. Second, we analyzed the presence of a phylogenetic signal by calculating the number of steps required for parsimony reconstruction of PepMV strain/host over the maximum clade credibility tree of 1,000 trees (rescaled to reflect median node heights for the contained clades) extracted from the BEAST analysis described below and comparing this number of steps to that for the same character reshuffled 1,000 times, while keeping the proportion of states constant, utilizing Mesquite v.2.75 (http://mesquiteproject.org/mesquite/). The null hypothesis of phylogenetic random distribution was rejected if the observed state distribution was outside the 95% confidence interval (CI) of the randomized state distribution, which indicated the presence of a phylogenetic signal.
The ancestral states of the host species and PepMV strain were then inferred using the Bayesian Markov chain Monte Carlo (MCMC) method available in BEAST v.1.7.5 (37). The utilized data set was run using the GTR+I+Γ4 model. A relaxed (uncorrelated, log-normal) molecular clock, which allows rate variation among branches in the phylogeny, was assumed. Finally, a flexible Bayesian skyline model as a coalescent prior was used, as estimating demographic parameters was not the aim of this study. The BEAST analysis was run until all relevant parameters converged, with 10% of the MCMC chains discarded as burn-in. TreeAnnotator v.1.7.5 (37) was used to construct a maximum clade credibility tree, which was assumed in estimating posterior distributions for ancestral states. Tree topology and ancestral state posterior probabilities were drawn from 2,000 trees generated in the BEAST analysis. Ancestral state reconstructions using ML methods as implemented in Mesquite yielded similar results. For simplicity, only Bayesian reconstructions are presented.
Construction of biologically active cDNA clones of PepMV isolates.
Biologically active clones of PepMV isolates Chi2.9 and Tor9 were obtained essentially as described by Hasiów-Jaroszewska et al. (38) The complete genome was amplified by two-step RT-PCR using the SuperScript III high-yield reverse transcriptase and a SuperMix high-fidelity DNA polymerase with primers UTR5F and UTR3R. Primer UTR5F had in its 5′ end the sequence of the T7 promoter, and primer UTR3R had in its 3′ end a NotI restriction site. The resulting amplicon was cloned in the pCR-XL-Topo vector (Life Technologies). Thus, plasmids pChi2.9 and pTor9 were obtained. Infectious RNA was obtained from these plasmids after linearization with NotI and transcription with T7 RNA polymerase in the presence of a Cap analogue. RNA was inoculated into Nicotiana benthamiana Domin plants, infection was checked by DAS-ELISA, and virus particles were purified as described above for bioassays.
Biological characterization of PepMV isolates.
The host ranges of isolates Chi2.9 and Tor9 derived from infectious clones were explored by inoculating 10 μl of a 20-mg/ml suspension of virus particles in phosphate buffer into the cotyledons or first leaves of a panel of 14 species from the Solanaceae. At least four plants per species were inoculated. At 15 and 30 dpi, infection in the inoculated or systemically infected leaves, respectively, was analyzed by DAS-ELISA. Systemic symptoms at 60 dpi were rated.
Within-host multiplication was estimated in some host plant species as virus accumulation. Virus accumulation was quantified in each plant at 30 dpi in pools of all systemically infected leaves by quantitative real-time RT-PCR of total nucleic acid extracts that were obtained using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. For each sample, 58 to 129 ng of total RNA was utilized with Brilliant III Ultra-Fast SYBR green QRT-PCR Master Mix according to the manufacturer's recommendations (Agilent Technologies, CA, USA) in a final volume of 10 μl. Assays were performed in triplicate on a LightCycler 480 II real-time PCR system (Roche, IN, USA). Relative levels of viral RNA were deduced from standard curves produced using a set of serial dilutions of plant material and purified viral RNAs of isolate Tor9 (PES) and reference isolates Mu07-20 (EU) and Al08-66 (CH2), obtained by us from single-infected tomato plants in southeastern Spain and characterized on the basis of nucleotide sequence determination of various genomic regions (not shown). In order to quantify virus accumulation in the various samples, the following primers were designed: qPCRPESF (TCAGTGGCTACCCCAACTGAA) and qPCRPESR (CGATCAAATTGTGCAGCTAGG), amplifying 202 nucleotides (nt) of the CP gene of PES isolates (GenBank accession no. HG000317); qPCRCH2F (TGCTGAAATTGAGGCCCTTGG) and qPCRCH2R (AGTGCACGTCTAGACAAAGCA), amplifying 170 nt of the CP gene of CH2 isolates (GenBank accession no. DQ000985); and qPCREUF (GCAAAATCTTCACCGCTATGG) and qPCREUR (TGATTCGGTCAAACTGAGCAG), amplifying 173 nt of the CP gene of EU isolates (GenBank accession no. AJ438767). Reactions with mixtures including no template and no reverse transcriptase were included in each trial. Thermal parameters for RT-PCR amplification were 50°C for 10 min, 95°C for 3 min, and 40 cycles of 95°C for 5 s and 60°C for 10 s. Dissociation curves were generated to ascertain that only a single product was produced in each case.
Statistical analyses.
Incidences of PepMV according to population, location, or host plant species were compared by association tests (χ2). Analysis of aggregation of infected plants in each population was performed by the run test for dichotomized data (39). Generalized linear models (GLM) assuming a binary logistic distribution were used to test differences in the efficiency of mechanical inoculation between PepMV isolates. GLM were used to analyze the differences in virus accumulation in the different host plant species of the assayed virus isolates. The presence of outliers, which potentially introduce a bias in GLM analyses, was detected by calculating the studentized residual for each data point, dividing the residual by its standard deviation. Values outside the 95% confidence interval of the Student t distribution drawn with all of the studentized residuals were considered outliers (39). Host species and virus isolate were considered fixed factors in the GLM analyses. To determine whether values of analyzed traits were significantly different among classes within each factor, least significant difference (LSD) analyses were employed in all cases (39). Statistical analyses were performed using the statistical software package SPSS 17.0 (SPSS Inc., IL, USA).
Nucleotide sequence accession numbers.
The CP gene nucleotide sequences obtained in this work are available from the EMBL data bank under accession numbers HG000306 to HG000320. Full-length nucleotide sequences of the three sequenced isolates are available from the EMBL data bank under accession numbers HG313805 to HG313807.
RESULTS
Incidence of PepMV infection in natural populations of wild tomatoes in Peru.
In March 2008 we conducted a survey for PepMV infection in natural populations of wild tomato relatives (Solanum spp.) in the provinces of Arequipa and Moquegua in southern Peru. Twelve populations at nine different locations, spanning the distribution of different Solanum species and different ecological environments, were visited (Table 1). Populations varied considerably in size, from about 1,000 plants (Chiguata 1) to fewer than 20 plants (e.g., Mollendo 1 and 2, Cerro Verde 1, and Torata), and showed a highly aggregated spatial distribution. A total of 104 plants were sampled, belonging to four species: S. peruvianum (74 plants), S. chilense (14 plants), S. pimpinellifolium (14 plants), and S. pinnatifidum (2 plants). The PepMV incidence was about 27% (28/104 infected plants) (Table 1). The incidence did not vary significantly according to population (χ211 = 7.44, P = 0.767), host plant species, or location (χ2 < 0.33, P > 0.882) (Table 1). The small size of the populations did not allow the analysis of the spatial distribution of infected plants except for the Chiguata 1 and Chiguata 2 populations, in which this distribution showed aggregation ([E(U)] > U, with P < 0.05 in ordinary run analyses).
Molecular characterization and genetic variation of PepMV isolates from wild tomatoes in southern Peru.
The CP gene was successfully RT-PCR amplified for 15 field-infected plants, and its nucleotide sequence was determined (Table 2). Sequences were highly similar for all 15 isolates, with only three polymorphic sites at positions 468, 495, and 712 of the CP gene and two other polymorphic sites at positions 725 and 726 (in the 3′ UTR for most isolates). All mutations were silent except the mutation U→A at position 712, which resulted in the change Stop→K and in the addition of four amino acids to the 268 of the PepMV CP. Population diversity measured as nucleotide diversity was very low, about 0.001. Nucleotide polymorphisms defined five haplotypes. Haplotype H1 represented 73% of the population (11/15 analyzed plants) and was found in all locations and in different host species. Haplotypes H2 to H5 occurred only once each in the sample and were found in different populations (Table 2).
TABLE 2.
Nucleotide polymorphisms at the coat protein gene and distribution of haplotypes in the PepMV population
| Isolate | Origin |
Nucleotide at position: |
Haplotype | |||
|---|---|---|---|---|---|---|
| Population | Host species | 468 | 495 | 712 | ||
| Chi1.8 | Chiguata 1 | S. peruvianum | U | U | U | H1 |
| Chi2.5 | Chiguata 2 | S. peruvianum | U | U | U | H1 |
| Cvd2.6 | Cerro Verde 2 | S. pimpinellifolium | U | U | U | H1 |
| Mol1.3 | Mollendo 1 | S. chilense | U | U | U | H1 |
| Mol2.2 | Mollendo 2 | S. chilense | U | U | U | H1 |
| Yur1.2 | Yura 1 | S. peruvianum | U | U | U | H1 |
| Yur1.5 | Yura 1 | S. peruvianum | U | U | U | H1 |
| Sam2.5 | Samegua 2 | S. peruvianum | U | U | U | H1 |
| Sam2.6 | Samegua 2 | S. peruvianum | U | U | U | H1 |
| Ceb3 | Cerro El Baúl | S. peruvianum | U | U | U | H1 |
| Tor9 | Torata | S. peruvianum | U | U | U | H1 |
| Ceb6 | Cerro el Baul | S. peruvianum | U | C | U | H2 |
| Chi2.9 | Chiguata 2 | S. pimpinellifolium | C | U | U | H3 |
| Mol2.8 | Mollendo 2 | S. chilense | U | U | A | H4 |
| Tor13 | Torata | S. peruvianum | C | U | A | H5 |
Three out of the 15 PepMV isolates with sequenced CPs were transferred to tomato plants for multiplication and molecular characterization. These isolates were selected because they were collected from different host plant species and different regions in southern Peru and represented different CP haplotypes. Isolates Yur1.5 and Tor9 (both H1) were originally from S. peruvianum plants collected from the Yura 1 and Torata populations, respectively, and isolate Chi2.9 (H3) was originally from an S. pimpinellifolium plant collected from the Chiguata 2 population. The nucleotide sequences of their full genomes were determined and showed a high pairwise sequence similarity of above 99%. Similarity was much lower with representative isolates of the four recognized strains: about 86% with US1 and between 78% and 82% with EU, LP, and CH2. When the different genomic regions were compared separately, similarity between isolates Chi2.9, Yur1.5, and Tor9, and the previously recognized strains was highest at the 3′ UTR with US1 (94.2%) and at the TGB2 and TGB3 open reading frames (ORFs) with the US1 and EU strains (86 to 92%). Values for the 5′ UTR and for the three other ORFs were similar to those for the whole genome (not shown). An analysis to identify possible recombination events between isolates Chi2.9, Yur1.5, and Tor9 and reported full-length sequenced isolates of the four recognized PepMV strains failed to detect any recombination breakpoint (not shown). Thus, isolates Chi2.9, Yur1.5, and Tor9 belong to a new strain of PepMV that we named the PES strain, after the southern Peru region of isolation. Phylogenetic analyses showed that the new PES strain clustered significantly with US1 and that strains PES and US1 grouped together with CH2 isolates. The EU and LP sequences formed a separate cluster (Fig. 1). A phylogenetic tree obtained using the 15 CP sequences plus representative isolates of all other PepMV strains mimicked the topology obtained using complete nucleotide genomic sequences (not shown). Hence, the sampled PepMV population in wild tomato relatives in southern Peru consisted only of isolates of the PES strain and was genetically a single, undifferentiated, highly homogeneous population.
FIG 1.

Maximum-likelihood phylogenetic tree of Pepino Mosaic Virus isolates, showing five recognized strains. Significance of nodes in a bootstrap analysis with 1,000 replicates is indicated. All isolates for which the complete sequence of the genome has been reported were included in the analysis. The tree was midpoint rooted.
Biological characterization of isolates of PepMV strain PES.
As there is no known local lesion host for the biological cloning of PepMV isolates (6) and the sampled field-infected plants could be mixed infected with other viruses, for the biological characterization of the new PES strain of PepMV, full-length, biologically active cDNA clones were obtained for the above-described isolates Chi2.9 and Tor9. Host range tests using a panel of 14 species from the Solanaceae showed that PES PepMV isolates had a host range very similar to that of isolates Mu07-20 and Al08-66, representative of the EU and CH2 strains, respectively (Table 3), with the efficiency of mechanical inoculation in each host species being not significantly different between PepMV isolates (χw2 < 1.77, P > 0.184). All assayed isolates infected systemically and with high efficiency Datura stramonium L., S. lycopersicum, S. muricatum, S. pimpinellifolium, Solanum melongena L., Nicotiana clevelandii A. Gray, and N. occidentalis. Systemic infection also occurred in the wild tomato species S. peruvianum, S. chilense, and S. habrochaites, except that isolate Al08-66 (CH2) did not infect the last species. Efficiency of mechanical inoculation was lower in the wild Solanum species than in the previous group of hosts (χw2 = 6.45, P = 0.011). No isolate systemically infected Capsicum annuum or Physalis floridiana Rydb. Infection of the systemic hosts was mostly asymptomatic or resulted in mild symptoms of chlorosis, leaf distortion, or leaf curl. Symptoms were generally more severe in cultivated than in wild Solanum host species (Table 3).
TABLE 3.
Host range and symptomatology of PepMV isolates
| Host species | % of plants infected or symptomatic with isolate (strain)a: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Chi2.9 (PES) |
Tor9 (PES) |
Mu07-20 (EU) |
Al08-66 (CH2) |
|||||
| Infection | Symptoms | Infection | Symptoms | Infection | Symptoms | Infection | Symptoms | |
| Solanum lycopersicum | 100 | 100 C | 100 | 100 C | 100 | 25 AS, 75 LC | 100 | 25 AS, 75 C/LC |
| Solanum peruvianum | 50 | 50 AS | 50 | 50 AS | 100 | 100 AS | 25 | 25 AS |
| Solanum pimpilellifolium | 100 | 100 C/N | 100 | 100 C/N | 100 | 100 C | 100 | 100 C |
| Solanum chilense | 0 | 50 | 50 AS | 0 | 25 | 25 AS | ||
| Solanum habrochaites | 25 | 25 AS | 50 | 50 AS | 25 | 25 AS | 0 | |
| Solanum muricatum | 100 | 50 C/W, 50 C | 75 | 25 LC/W, 50 C | 75 | 50 C, 25 N | 100 | 50 C, 50 C/LC |
| Solanum melongena | 100 | 100 N | 100 | 50 AS, 50 W | 100 | 75 AS, 25 C | 100 | 100 AS |
| Nicotiana benthamiana | 25 | 25 C/LC | 0 | 75 | 75 C/LC | 50 | 50 AS, 50 LC | |
| Nicotiana clevelandii | 100 | 25 C, 75 LC | 100 | 100 C/LC | 100 | 100 C/LC | 75 | 75 C/LC |
| Nicotiana occidentalis | 100 | 100 LC/N | 100 | 100 C/LC | 100 | 100 LC | 100 | 100 C/N |
| Nicotiana tabacum | 50 | 50 AS | 0 | 25 | 25 AS | 0 | ||
| Capsicum annuum | 0 | 0 | 0 | 0 | ||||
| Datura stramonium | 100 | 25 C/LC, 25 C/W, 25 C, 25 C/LC/W | 100 | 100 C/LC/W | 100 | 100 C/LC | 100 | 100 C/N |
| Physalis floridiana | 0 | 0 | 0 | 0 | ||||
Numbers are the percentage of infected or symptomatic plants over four plants inoculated. Symptoms: AS, asymptomatic; C, chlorosis; LC, leaf curl; N, necrosis; W, wilting.
Differential accumulation of PepMV isolates in wild and domestic tomatoes.
Virus accumulation of PepMV isolates Chi2.9, Tor9, Mu07-20, and Al08-66 in three wild tomato species (S. peruvianum, S. chilense, and S. pimpinellifolium) and in two cultivars of domestic tomato (S. lycopersicum cv. Marglobe and S. lycopersicum cv. Moneymaker) was determined via real-time RT-PCR (Table 4). GLM analyses indicated that virus accumulation significantly depended on the host species (F4,158 = 6.56, P < 1 × 10−4), on the virus isolate (F3,158 = 38.38, P < 1 × 10−4), and on the species-per-isolate interaction (F11,158 = 4.25, P < 1 × 10−4). Since we chose Solanum species to represent wild and domesticated hosts, we also performed GLM analyses with nesting of host species to host status (i.e., wild or domesticated). Again, virus accumulation varied according to virus isolate (F3,158 = 9.35, P = 2 × 10−3) and to the interaction virus isolate per host species (F11,158 = 4.25, P < 1 × 10−4), but it did not vary according to host species (F4,158 = 1.11, P = 0.384).
TABLE 4.
Accumulation of PepMV isolates in different Solanum species
| Solanum species and cultivar | μg viral RNA/g total RNA for isolatea: |
|||
|---|---|---|---|---|
| Mu07-20 (EU) | Al08-66 (CH2) | Chi2.9 | Tor9 | |
| S. lycopersicum | ||||
| Marglobe | 0.269 ± 0.076 | 0.049 ± 0.011 | 0.509 ± 0.046 | 0.316 ± 0.058 |
| Moneymaker | 0.415 ± 0.056 | 0.168 ± 0.052 | 0.615 ± 0.117 | 0.613 ± 0.127 |
| S. pimpinellifolium | 0.284 ± 0.064 | 0.103 ± 0.029 | 1.592 ± 0.276 | 1.121 ± 0.149 |
| S. chilense | 0.117 ± 0.026 | 0.016 ± 0.006 | 1.402 ± 0.272 | 0.337 ± 0.124 |
| S. peruvianum | 0.455 ± 0.075 | 0.018 ± 0.005 | 0.954 ± 0.149 | 0.849 ± 0.146 |
Data are means ± standard errors for at least 5 plants.
As we observed an interaction between virus strain and Solanum species on PepMV multiplication, even when correcting for the effect of plant domestication status, we analyzed next if there was evidence of PepMV adaptation in the wild and/or the domestic hosts. For this analysis, two classes of virus isolates were considered: (i) isolates from wild tomatoes (isolates Chi2.9 and Tor9) and (ii) isolates from tomato crops (isolates Mu07-20 and Al08-66). Also, two classes of hosts were considered: (i) wild tomato species (i.e., S. peruvianum, S. chilense, and S. pimpinellifolium) and (ii) the cultivars of S. lycopersicum (Marglobe and Moneymaker) as representatives of the major crop (tomato) in which PepMV has recently emerged as an important pathogen. Analyses of the data presented in Fig. 2 showed that accumulation of PepMV isolates from wild hosts was higher in wild than in domestic tomatoes (F1,83 = 14.28, P < 1 × 10−4), while tomato isolates showed a nonsignificant trend toward higher accumulation in cultivated than in wild Solanum species (F1,75 = 2.59, P = 0.112). Also, accumulation of PepMV isolates from domestic tomato was lower than accumulation of PepMV isolates from wild tomatoes in both wild (F1,99 = 65.17, P < 1 × 10−4) and domestic (F1,59 = 18.63, P < 1 × 10−4) tomatoes. These analyses show strong evidence of host adaptation for PepMV isolates from wild hosts, and they suggest adaptation to tomato of tomato isolates. Also, they suggest that there is a trade-off between virus fitness in wild tomato species and in domestic tomato.
FIG 2.
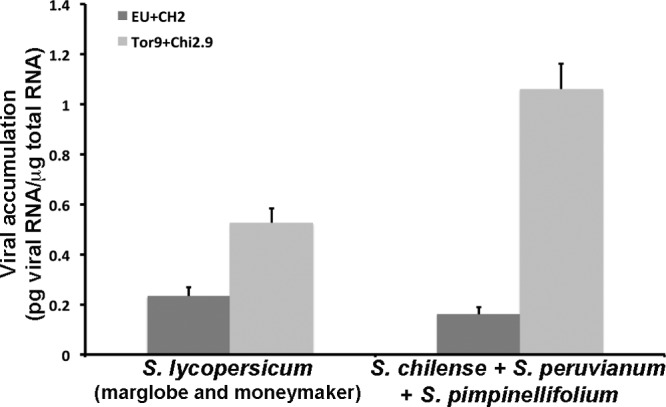
Accumulation of Pepino Mosaic Virus isolates in different host plant species. The accumulation of isolates from tomato crops in Spain (dark gray bars) or from wild Solanum spp. in Peru (light gray bars) was compared in two different host types: tomato (S. lycopersicum cv. Marglobe and cv. Moneymaker) and wild tomato (S. peruvianum, S. pimpinellifolium, and S. chilense). Data are means and standard errors from at least 5 infected plants.
Host ancestral state reconstruction of PepMV isolates.
Our results on PepMV incidence and virus multiplication as well as previously reported epidemiological data are compatible with PepMV emergence in tomato being associated with spread from wild Solanum species. To further analyze this possibility, we performed an ancestral state reconstruction considering as state the PepMV strain, since LP and PES strains are largely associated with wild hosts and EU and CH2 with domestic tomatoes. To do so, complete genomic sequences of PepMV PES, CH2, and EU isolates (Fig. 3) (i.e., of those strains that we had compared for host range and within-host multiplication) and LP isolates that contained the oldest known PepMV sequence were used.
FIG 3.

Phylogenetic tree with ancestral state reconstruction of PepMV isolates. Branch tip times reflect the times of viral sampling. The tree is automatically rooted through the use of a relaxed molecular clock, and the total depth of the tree is the time to the most recent common ancestor. Nodes are labeled with the most probable state, and its posterior probability is shown in parenthesis. Tip names indicate the GenBank accession number. The two columns on the right indicate the year of isolation and country of origin of the PepMV isolate that originated each sequence. All sequences were obtained from PepMV isolates infecting S. lycopersicum, with the exceptions of sequences AJ606361, Yur1.5, and Tor9 (obtained from S. peruvianum), sequence Chi2.9 (obtained from S. pimpinellifolium), and sequence AM109896 (obtained from S. muricatum).
As the PepMV ancestral state reconstruction considered the time scale of virus evolution, we first assessed the strength of temporal and state-related phylogenetic signals in the utilized data set, which is necessary for a meaningful analysis. A significant root-to-tip correlation of sampling time versus genetic distance (r = 0.60; P = 1 × 10−4) was observed. Analyses performed using only those sequences collected between 1999 and 2010 yielded similar results (r = 0.58; P = 1 × 10−4). Therefore, the utilized PepMV data set contained sufficient temporal structure for reliable estimation of the PepMV evolutionary time scale. This was confirmed by the significantly smaller mean and larger HPDs of the substitution rate estimates obtained for randomized data sets compared with those from the real data (not shown). In addition, the null hypothesis of phylogenetic random distribution of PepMV strain state was rejected by parsimony reconstruction of trait evolution because the observed distribution (4 steps) was outside the 95% CI of the randomized state distribution (mean = 12.8, median = 17, CILower = 8.2, CIUpper = 21.3), indicating the presence of a PepMV state-related phylogenetic signal.
Phylogenetic trees annotated with the node state (i.e., the PepMV strain with the highest posterior probability [PP] at each node) indicated that the most probable ancestor of all PepMV sequences belonged to the LP strain (PP = 0.55), and was thus from a wild host, with the PP of any other strain being lower than 0.15. Interestingly, the cluster grouping sequences of PES and CH2 isolates had a common ancestor from the PES strain, again associated with wild hosts (PP = 0.74) (Fig. 3), with the PP of any other strain being lower than 0.07. As expected, the ancestral state reconstruction using host species as state yielded comparable results. Importantly, ancestral state reconstruction based on ML methods yielded similar results. In addition, the Bayesian node-annotated trees showed topologies similar to those of trees obtained by ML, either midpoint rooted or rooted using the Narcissus mosaic virus type sequence (trees are available upon request). Thus, our phylogenetic reconstructions appear to be robust regardless of the methodology or the rooting method utilized.
DISCUSSION
Emergent viral diseases of plants often have severe negative impacts in agriculture and forestry and may result in dramatic changes in the species composition and dynamics of wild plant communities. Human activities resulting in the alteration of natural ecosystems, agriculture intensification and spread, or increased connectivity among plant populations due to trade, as well as global climatic change, are considered to presently increase the risk of virus emergence (2, 3, 5, 40–43). It is thus necessary to understand how multiple ecological, evolutionary and epidemiological factors contribute to plant viral disease emergence (2–4). With this aim, we have attempted to analyze the causes leading to the emergence of a virus that became a worldwide threat to tomato production in the recent past, PepMV (6, 7). Most work aimed at understanding plant virus emergence has focused on either the a posteriori identification of ecological and epidemiological causes or the experimental evolutionary analysis of host adaptation (3, 4). We followed a different approach: we evaluated the ecological and evolutionary risk of emergence of a PepMV strain as yet unreported in crops, we compared the relevant traits of this strain and of strains infecting tomato crops, and we attempted to trace the origin of the PepMV epidemic by comparative genomics analyses.
Spread from wild hosts is often the first step in virus emergence (43–45). This could also have been the case in PepMV emergence, as (i) the virus was detected infecting wild tomato species in central and southern Peru (28), (ii) Peru and Chile are important regions of seed production for seed companies (46), and (iii) some strains of PepMV were first identified in tomato seed produced in this region (17). Thus, we surveyed for PepMV infection in natural populations of several wild tomato species in different habitats in southern Peru, finding a high prevalence of infection by a new strain of PepMV that we named PES. Note that the prevalence reported here is likely a lower threshold, as PepMV detection was probably inefficient due to suboptimal survival of samples. The high prevalence of infection at all locations, including in quite small plant populations, and the aggregated distribution of infected plants observed in large host populations are compatible with reported seed and contact transmission of PepMV (6, 7, 13). A high incidence of infection and a large host range within the Solanaceae family of PepMV strain PES isolates would favor virus spread to local solanaceous crops, such as pepino or tomato, either directly or through weeds such as local species of Datura or Nicotiana (47, 48). It should be pointed out that wild tomato species also occur frequently as weeds in irrigated crops in southern Peru (49), a factor further favoring virus spread to crops. Importantly, our phylogenetic reconstruction of the PepMV ancestral state indicated that the deepest node in the phylogeny and those defining the LP-EU and the PES-CH2 clades were associated with viruses infecting wild hosts. Therefore, the analyses performed with the current sampled diversity of PepMV support the hypothesis of an origin of the PepMV epidemic as a consequence of virus spread from a wild host. Obviously, we cannot discard that there is still undiscovered PepMV genetic diversity, which is a clear limitation of our phylogenetic analyses. Future surveys will allow a finer reconstruction of PepMV emergence.
After spread from wild hosts has resulted in the infection of a few individuals of the new host, viruses must adapt to establish a productive infection for successful transmission in the new host population. Host adaptation is facilitated if the original virus population has enough genetic variation to contain variants with a positive fitness in the new host, without the need of generating these variants anew through mutation (4, 50). The PepMV PES population infecting wild tomatoes would not fulfill this condition, as it showed very low genetic variation, a trait shared with single-strain populations of PepMV infecting tomato crops elsewhere (10, 20, 21). However, low genetic diversity should not be a hindrance for adaptation to tomato or pepino crops: PepMV PES isolates appear to be generalists, being able to successfully infect different wild and domestic species of at least two genera of the subfamily Solanoidea, i.e., Solanum and Datura (Table 3). Hence, emergence in pepino and/or tomato would not involve a host range expansion, as is often the case for emerging plant pathogens (2, 3). Finally, the characterized PepMV PES isolates multiplied to high levels in both wild and domestic tomatoes, which could favor their future emergence in tomato crops. Thus, all our observations point to a high risk of an emergence of PepMV PES as a crop pathogen.
The more efficient multiplication in wild than in cultivated hosts of PepMV PES isolates supports a scenario of adaptation to their wild hosts (Fig. 2). Conversely, tomato isolates of PepMV strains EU and CH2 show a trend toward more efficient multiplication in tomato than in wild Solanum species, which is also suggestive of a process of host adaptation. Note also that the phylogeny of PepMV, in which isolates clustered mostly according to their host species (Fig. 1 and 3), may also be indicative of host adaptation. We cannot discard other scenarios. For instance, the ancestor of the EU or CH2 strains may have had high fitness on wild tomato hosts and with exaptations to cultivated tomato. Also, increased fitness on cultivated tomato may have occurred without affecting the low fitness of EU or CH2 on wild hosts. However, our data suggest that adaptation is the most likely mechanism involved in PepMV emergence.
Host adaptation in viruses is often conditioned by across-host trade-offs in fitness that result in evolution toward specialism (4, 51). In our analyses, a fitness trade-off is apparent both for tomato and wild host viral strains between S. lycopersicum and the wild tomato species of Solanum. Evidence of across-host fitness trade-offs for plant viruses is abundant from experimental evolution of serially passaged viruses in different hosts (see, e.g., references 52 to 55) but rarely derives from the analysis of field isolates (but see references 56 to 58). It should be underscored that the support for across-hosts fitness trade-offs reported here for PepMV isolates derives from the analysis of field isolates that were subject to only one passage in the same multiplication host and is highly coherent with experimental results with other systems. Although, given the observed trade-off, a single passage though domestic tomato may lead to fixation of mutations increasing virus fitness in this host, we followed a procedure which minimizes this effect (see Materials and Methods). More importantly, comparison of CP sequences of 10 PepMV PES isolates before and after passage showed no nucleotide changes (not shown).
In summary, the results from this work contribute to understanding the pathway to emergence of PepMV as an important pathogen of tomato crops worldwide. From an initial situation in which different PepMV strains would be found infecting wild Solanum spp. in Peru, virus spread would have occurred to tomato directly (e.g., for the CH2 strain) or perhaps through previous emergence in S. muricatum (for the EU strain). The ability of wild Solanum isolates to efficiently multiply in different Solanum species, shown for the LP and PES strains (19, 28; this work) would favor the establishment of PepMV in the new crop hosts as an epidemic pathogen. Last, adaptation to tomato, possibly traded off against adaptation to wild Solanum species, would feed back tomato epidemics and isolate tomato populations from those in other hosts.
ACKNOWLEDGMENTS
This work was supported in part by grants PEPEIRA from the European Commission 6th Framework Programme (contract no. 044189) and AGL2008-02458, Plan Nacional de I+D+i, Spain.
We thank Alberto Anculle, Universidad de San Agustín, Arequipa, for efficient help during field work.
Footnotes
Published ahead of print 3 January 2014
REFERENCES
- 1.Woolhouse MEJ, Dye C. 2001. Population biology of emerging and re-emerging pathogens. Preface. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:981–982. 10.1098/rstb.2001.0899 [DOI] [Google Scholar]
- 2.Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 19:535–544. 10.1016/j.tree.2004.07.021 [DOI] [PubMed] [Google Scholar]
- 3.Jones RAC. 2009. Plant virus emergence and evolution: origins, new encounter scenarios, factors driving emergence, effects of changing world conditions, and prospects for control. Virus Res. 141:113–130. 10.1016/j.virusres.2008.07.028 [DOI] [PubMed] [Google Scholar]
- 4.Elena SF, Bedhomme S, Carrasco P, Cuevas JM, de la Iglesia F, Lafforgue G, Lalic J, Prosper A, Tromas N, Zwart MP. 2011. The evolutionary genetics of emerging plant RNA viruses. Mol. Plant Microbe Interact. 24:287–293. 10.1094/MPMI-09-10-0214 [DOI] [PubMed] [Google Scholar]
- 5.Cooper I, Jones RAC. 2006. Wild plants and viruses: under-investigated ecosystems. Adv. Virus Res. 67:1–47. 10.1016/S0065-3527(06)67001-2 [DOI] [PubMed] [Google Scholar]
- 6.Gómez P, Sempere RN, Aranda MA. 2012. Pepino Mosaic Virus and Tomato torrado virus: emerging viruses affecting tomato crops in the Mediterranean basin. Adv. Virus Res. 84:505–532. 10.1016/B978-0-12-394314-9.00014-2 [DOI] [PubMed] [Google Scholar]
- 7.Hanssen IM, Thomma BPHJ. 2010. Pepino Mosaic Virus: a successful pathogen that rapidly evolved from emerging to endemic in tomato crops. Mol. Plant Pathol. 11:179–189. 10.1111/j.1364-3703.2009.00600.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguilar JM, Hernández-Gallardo DM, Cenis JL, Lacasa A, Aranda MA. 2002. Complete sequence of the Pepino Mosaic Virus RNA genome. Arch. Virol. 147:2009–2015. 10.1007/s00705-002-0848-9 [DOI] [PubMed] [Google Scholar]
- 9.Mumford RA, Metcalfe EJ. 2001. The partial sequencing of the genomic RNA of a UK isolate of Pepino Mosaic Virus and the comparison of the coat protein sequence with other isolates from Europe and Peru. Arch. Virol. 146:2455–2460. 10.1007/s007050170015 [DOI] [PubMed] [Google Scholar]
- 10.Pagán I, Córdoba-Selles MC, Martínez-Priego L, Fraile A, Malpica JM, Jordá C, García-Arenal F. 2006. Genetic structure of the population of Pepino Mosaic Virus infecting tomato crops in Spain. Phytopathology 96:274–279. 10.1094/PHYTO-96-0274 [DOI] [PubMed] [Google Scholar]
- 11.Hanssen IM, Mumford R, Blystad DR, Cortez I, Hasiów-Jaroszewska B, Hristova D, Pagán I, Pereira AM, Peters J, Pospieszny H, Ravnikar M, Stijger I, Tomassoli L, Varveri C, van der Vlugt R, Nielsen S. 2010. Seed transmission of Pepino Mosaic Virus in tomato. Eur. J. Plant Pathol. 126:145–152. 10.1007/s10658-009-9528-x [DOI] [Google Scholar]
- 12.Córdoba-Sellés MC, García-Rández A, Alfaro-Fernández A, Jordá C. 2007. Seed transmission of Pepino Mosaic Virus and efficacy of tomato seed disinfection treatments. Plant Dis. 91:1250–1254. 10.1094/PDIS-91-10-1250 [DOI] [PubMed] [Google Scholar]
- 13.Jones RAC, Koenig R, Lesemann DE. 1980. Pepino Mosaic Virus, a new potexvirus from pepino (Solanum muricatum). Ann. Appl. Biol. 94:61–68. 10.1111/j.1744-7348.1980.tb03896.x [DOI] [Google Scholar]
- 14.van del Vlugt RAA, Stijger CCMM, Verhoeven JThJ, Lesemann DE. 2000. First report of Pepino Mosaic Virus on tomato. Plant Dis. 84:103. 10.1094/PDIS.2000.84.1.103E [DOI] [PubMed] [Google Scholar]
- 15.Cotillon A-C, Girard M, Ducouret S. 2002. Complete nucleotide sequence of the genomic RNA of a French isolate of Pepino Mosaic Virus (PepMV). Arch. Virol. 147:2231–2238. 10.1007/s00705-002-0873-8 [DOI] [PubMed] [Google Scholar]
- 16.French CJ, Bouthillier M, Bernardy M, Ferguson G, Sabourin M, Johnson RC, Masters C, Godkin S, Mumford R. 2001. First report of Pepino Mosaic Virus in Canada and the United States. Plant Dis. 85:1121. 10.1094/PDIS.2001.85.10.1121C [DOI] [PubMed] [Google Scholar]
- 17.Ling K-S. 2007. Molecular characterization of two Pepino Mosaic Virus variants from imported tomato seed reveals high levels of sequence identity between Chilean and US isolates. Virus Genes 34:1–8. 10.1007/s11262-006-0003-x [DOI] [PubMed] [Google Scholar]
- 18.Maroon-Lango CJ, Guaragna MA, Jordan RL, Hammond J, Bandla M, Marquardt SK. 2005. Two unique isolates of Pepino Mosaic Virus from a limited source of pooled tomato tissue are distinct from a third (European-like) US isolate. Arch. Virol. 150:1187–1201. 10.1007/s00705-005-0495-z [DOI] [PubMed] [Google Scholar]
- 19.López C, Soler S, Nuez F. 2005. Comparison of the complete sequences of three different isolates of Pepino Mosaic Virus: size variability of TGBp3 protein between tomato and L. peruvianum isolates. Arch. Virol. 150:619–627. 10.1007/s00705-004-0438-0 [DOI] [PubMed] [Google Scholar]
- 20.Hanssen IM, Paeleman A, Wittemans L, Goen K, Lievens B, Bragard C, Vanachter ACRC, Thomma BPHJ. 2008. Genetic characterization of Pepino Mosaic Virus isolates from Belgian greenhouse tomatoes reveals genetic recombination. Eur. J. Plant Pathol. 121:131–146. 10.1007/s10658-007-9255-0 [DOI] [Google Scholar]
- 21.Gómez P, Sempere RN, Elena SF, Aranda MA. 2009. Mixed infections of Pepino Mosaic Virus strains modulate the evolutionary dynamics of this emergent virus. J. Virol. 83:12378–12387. 10.1128/JVI.01486-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasiow-Jaroszevska B, Kuzniar A, Peters SA, Leunissen JAM, Pospieszny H. 2010. Evidence for RNA recombination between distinct isolates of Pepino Mosaic Virus. Acta Biochim. Pol. 57:385–388 [PubMed] [Google Scholar]
- 23.Alfaro-Fernández A, Cebrián MC, Córdoba-Selles C, Herrera-Vasquez JA, Jordá C. 2008. First report of the US1 strain of Pepino Mosaic Virus in tomato in the Canary Islands, Spain. Plant Dis. 92:1590–1591. 10.1094/PDIS-92-11-1590B [DOI] [PubMed] [Google Scholar]
- 24.French CJ, Dubeau C, Bunckle A, Ferguson G, Haesevoets R, Bouthillier M, Bernardy MG. 2008. Overview of Pepino Mosaic Virus research. Can. J. Plant Pathol. 30:373–374 [Google Scholar]
- 25.Ling K-S, Wintermantel WM, Bledsoe M. 2008. Genetic composition of Pepino Mosaic Virus population in North American greenhouse tomatoes. Plant Dis. 92:1683–1688. 10.1094/PDIS-92-12-1683 [DOI] [PubMed] [Google Scholar]
- 26.Ling K-S, Li RG, Bledsoe M. 2013. Pepino Mosaic Virus genotype shift in North America and development of a loop-mediated isothermal amplification for rapid genotype identification. Virol. J. 10:117. 10.1186/1743-422X-10-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koenig R, Lesemann DE, Jones RAC. 1989. Pepino Mosaic Virus. AAB descriptions of plant viruses, no. 350. AAB, Warwick, United Kingdom [Google Scholar]
- 28.Soler S, Prohens J, Díez MJ, Nuez F. 2002. Natural occurrence of Pepino Mosaic Virus in Lycopersicon species in central and southern Peru. J. Phytopathol. 150:49–53. 10.1046/j.1439-0434.2002.00712.x [DOI] [Google Scholar]
- 29.Hasiów-Jaroszewska B, Paeleman A, Ortega-Parra N, Borodynko N, Minicka J, Czerwoniec A, Thomma BP, Hanssen IM. 2013. Ratio of mutated versus wild-type coat protein sequences in Pepino mosaic virus determines the nature and severity of yellowing symptoms on tomato plants. Mol. Plant Pathol. 10.1111/mpp.12059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rambaut A. 1996. Se-Al: sequence alignment editor. http://evolve.zoo.ox.ac.uk/
- 32.Tamura K, Peterson P, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Posada D. 2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25:1253–1256. 10.1093/molbev/msn083 [DOI] [PubMed] [Google Scholar]
- 34.Posada D. 1998. Evaluation of methods for detecting recombination from DNA sequences: empirical data. Bioinformatics 14:817–818. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- 35.Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. 10.1093/bioinformatics/btq467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guindon S, Gascuel O. 2003. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704. 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- 37.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. 10.1186/1471-2148-7-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasiow-Jaroszewska B, Borodynko N, Pospieszny H. 2009. Infectious RNA transcripts derived from cloned cDNA of a Pepino Mosaic Virus isolate. Arch. Virol. 154:853–856. 10.1007/s00705-009-0368-y [DOI] [PubMed] [Google Scholar]
- 39.Sokal RR, Rohlf FJ. 1995. Biometry, 3rd ed. W.H. Freeman and Company, New York, NY [Google Scholar]
- 40.Canto T, Aranda MA, Fereres A. 2009. Climate change effects on physiology and population processes of hosts and vectors that influence the spread of hemipteran-borne plant viruses. Glob. Change Biol. 15:1884–1894. 10.1111/j.1365-2486.2008.01820.x [DOI] [Google Scholar]
- 41.Garrett KA, Forbes GA, Savary S, Skelsey P, Sparks AH, Valdivia C, van Bruggen AHC, Willocquet L, Djurle A, Duveiller Eckersten H, Pande S, Vera Cruz C, Yuen J. 2011. Complexity in climate-change impacts: an analytical framework for effects mediated by plant disease. Plant Pathol. 60:15–30. 10.1111/j.1365-3059.2010.02409.x [DOI] [Google Scholar]
- 42.Margosian ML, Garrett KA, Hutchinson JMS, With KA. 2009. Connectivity of the American agricultural landscape: assessing the national risk of crop pest and disease spread. Bioscience 59:141–151. 10.1525/bio.2009.59.2.7 [DOI] [Google Scholar]
- 43.Woolhouse MEJ, Haydon DT, Antia R. 2005. Emerging pathogens: the epidemiology and evolution of species jumps. Trends Ecol. Evol. 20:238–244. 10.1016/j.tree.2005.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elena SF, Agudelo-Romero P, Lalic J. 2009. The evolution of viruses in multi-host fitness landscapes. Open Virol. J. 3:1–6. 10.2174/1874357900903010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holmes EC. 2009. The evolutionary genetics of emerging viruses. Annu. Rev. Ecol. Evol. Syst. 40:353–372. 10.1146/annurev.ecolsys.110308.120248 [DOI] [Google Scholar]
- 46.ISST. 2003. Chilean Seed Industry report. http://www.seedconsortium.org/PUC
- 47.Ferreyra R. 1983. Los tipos de vegetación de la costa peruana. An. Jard. Bot. Madr. 40:241–256 [Google Scholar]
- 48.Pickersgill B. 2007. Domestication of plants in the Americas: insights from mendelian and molecular genetics. Ann. Bot. 100:925–940. 10.1093/aob/mcm193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arunyawat U, Stephan W, Staedler T. 2007. Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes. Mol. Biol. Evol. 24:2310–2322. 10.1093/molbev/msm162 [DOI] [PubMed] [Google Scholar]
- 50.Lalic J, Cuevas JM, Elena SF. 2011. Effect of host species on the distribution of mutational fitness effects for an RNA virus. PLoS Genet. 7:e1002378. 10.1371/journal.pgen.1002378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elena SF, Sanjuán R. 2003. Climb every mountain? Science 302:2074–2075. 10.1126/science.1093165 [DOI] [PubMed] [Google Scholar]
- 52.Agudelo-Romero P, Carbonell P, Pérez-Amador MA, Elena SF. 2008. Virus adaptation by manipulation of host's gene expression. PLoS One 3:e2397. 10.1371/journal.pone.0002397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang X-Z, Lee BTK, Wong S-M. 2002. Covariation in the capsid protein of Hibiscus chlorotic ringspot virus induced by serial passaging in a host that restricts movement leads to avirulence in its systemic host. J. Virol. 76:12320–12324. 10.1128/JVI.76.23.12320-12324.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wallis CM, Stone AL, Sherman DJ, Damsteegt VD, Gildow FE, Schneider WL. 2007. Adaptation of Plum pox virus to an herbaceous host (Pisum sativum) following serial passages. J. Gen. Virol. 88:2839–2845. 10.1099/vir.0.82814-0 [DOI] [PubMed] [Google Scholar]
- 55.Yarwood CE. 1979. Host passage effects with plant viruses. Adv. Virus Res. 25:169–190 [DOI] [PubMed] [Google Scholar]
- 56.Cronin JP, Welsh ME, Dekkers MG, Abercrombie ST, Mitchell CE. 2010. Host physiological phenotype explains pathogen reservoir potential. Ecol. Lett. 13:1221–1232. 10.1111/j.1461-0248.2010.01513.x [DOI] [PubMed] [Google Scholar]
- 57.Power AG, Mitchell CE. 2004. Pathogen spillover in disease epidemics. Am. Nat. 164:S79–S89. 10.1086/424610 [DOI] [PubMed] [Google Scholar]
- 58.Sacristán S, Fraile A, Malpica JM, García-Arenal F. 2005. An analysis of host adaptation and its relationship with virulence in Cucumber mosaic virus. Phytopathology 95:827–833. 10.1094/PHYTO-95-0827 [DOI] [PubMed] [Google Scholar]