

Abstract
The histone demethylase JMJD2C, also known as KDM4C/GASC1, has activity against methylated H3K9 and H3K36 and is amplified and/or overexpressed in human cancers. By the generation of Jmjd2c knockout mice, we demonstrate that loss of Jmjd2c is compatible with cellular proliferation, embryonic stem cell (ESC) self-renewal, and embryonic development. Moreover, we report that JMJD2C localizes to H3K4me3-positive transcription start sites in both primary cells and in the human carcinoma KYSE150 cell line containing an amplification of the JMJD2C locus. Binding is dependent on the double Tudor domain of JMJD2C, which recognizes H3K4me3 but not H4K20me2/me3 in vitro, showing a binding specificity different from that of the double Tudor domains of JMJD2A and JMJD2B. Depletion of JMJD2C in KYSE150 cells has a modest effect on H3K9me3 and H3K36me3 levels but impairs proliferation and leads to deregulated expression of a subset of target genes involved in cell cycle progression. Taking these findings together, we show that JMJD2C is targeted to H3K4me3-positive transcription start sites, where it can contribute to transcriptional regulation, and report that the putative oncogene JMJD2C generally is not required for cellular proliferation or embryonic development.
INTRODUCTION
Posttranslational modifications of histones have been implicated in the control of gene expression programs, genomic stability, and cell fate decisions. Several chromatin-modifying enzymes, including histone methyltransferases and demethylases, are essential for normal development, and their deregulated expression are associated with pathological disorders. Importantly, chromatin modifiers implicated in human diseases, such as cancer, have gained interest as potential therapeutic targets (reviewed in references 1–4).
JMJD2C, also known as KDM4C/GASC1, is a member of the JMJD2/KDM4 subgroup of Jumonji C domain-containing proteins which catalyzes the demethylation of tri- and dimethylated lysine 9 and lysine 36 on histone H3 (H3K9me3/me2 and H3K36me3/me2) (5–8). H3K9me3 is a hallmark of pericentric heterochromatin and is linked to transcriptional repression when found at gene regulatory elements, while H3K36me3 is found in the coding regions of actively transcribed genes (reviewed in reference 9).
Several lines of evidence suggest that JMJD2C plays a role in malignant transformation. JMJD2C is located at chromosome 9p23-24 and originally was identified as a gene amplified in esophageal squamous cell carcinoma cell lines (10). Subsequent studies have shown amplifications of JMJD2C in medulloblastomas, breast cancers, primary mediastinal B cell lymphomas (PMBL), and Hodgkin lymphomas (HL) (11–14). Depletion of JMJD2C has been reported to impair the proliferation of several tumor cell types, including cell lines featuring amplifications of the JMJD2C locus (5, 12, 14, 15), and knockdown of JMJD2C in MDA-MB-435 cells has been shown to impair tumor growth and metastases after mammary fat pad injection (16). Furthermore, ectopic expression of JMJD2C was demonstrated to induce growth factor- and anchorage-independent growth of nontransformed immortalized MCF10A cells (12).
While JMJD2C is considered an interesting drug target for cancer therapy (4), this demethylase has also been reported to fulfill vital functions during normal development. Knockdown of JMJD2C in mouse embryonic stem cells (ESCs) was found to impair ESC self-renewal (17) and depletion in oocytes reported to cause a developmental arrest before the blastocyst stage (18). In addition, JMJD2C has been implicated in lineage-specific differentiation processes, as knockdown was shown to inhibit adipocyte differentiation (19).
Little is known about the genomic targets of JMJD2C. JMJD2C has been detected at a few gene promoters, where it has been implicated in transcriptional activation (15–17, 20, 21). Other JMJD2 family members have been reported to have diverse genomic targets and have been linked to both gene activation and repression, regulation of DNA replication, and/or the DNA damage response (7, 8, 22–28). In mammalian cells, JMJD2A, JMJD2B, and JMJD2C contain PHD and double Tudor domains. The double Tudor domain of JMJD2A can bind H3K4me3 and H4K20me3/me2 in vitro (25, 29–31), and recognition of methylated H4K20 has also been reported for the double Tudor domain of JMJD2B (25).
As JMJD2C is a putative oncogene, characterization of its in vivo functions and genomic targets is relevant for future studies evaluating this demethylase as a potential drug target. Here, we report the genome-wide localization of JMJD2C in primary and transformed cells and show that loss of JMJD2C expression is compatible with cellular proliferation and embryonic development.
MATERIALS AND METHODS
Animals.
C57BL6 mice with a conditional allele of Jmjd2c were obtained from the KOMP repository (http://www.komp.org/). The Jmjd2ctm1a(KOMP)Wtsi allele targets the 9th exon of the Jmjd2c gene, shifting the reading frame leading to translational termination. Jmjd2ctm1a(KOMP)Wtsi mice were crossed with Flp-recombinase-expressing mice to generate the conditional allele and remove the Neo reporter cassette. Conditional Jmjd2c mice were further crossed with Rosa26::CreERT2 mice (obtained from the Jackson Laboratory) for the generation of conditional Jmjd2c knockout ESCs and murine embryonic fibroblasts (MEFs). In addition, conditional Jmjd2c mice were crossed with Cmv-cre transgenic mice (32) to obtain heterozygous mice, which were further intercrossed to generate Jmjd2c knockouts. Jmjd2c mice were maintained on a C57BL/6 background. All mouse work was approved by the Danish Animal Ethical Committee (Dyreforsøgstilsynet).
Cell culture and derivation of Jmjd2c knockout ESCs and MEFs.
For the generation of conditional Jmjd2c ESCs, blastocysts were isolated from the uterus of superovulated pregnant female mice at 3.5 days postcoitus. Single blastocysts were cultured in serum-containing medium (Glasgow minimum essential medium [GMEM] [Sigma-Aldrich] supplemented with 15% fetal bovine serum [FBS] [HyClone], 2 mM Glutamax [Gibco], 50 μM β-mercaptoethanol [Gibco], 0.1 mM nonessential amino acids [Gibco], 1 mM sodium pyruvate [Gibco], and leukemia inhibitory factor [LIF]) and inner cell mass (ICM) outgrowths expanded. Sex and karyotype were determined as described previously (33). For all experiments shown, ESCs were cultured without feeders on 0.2% gelatin-coated plates in serum-free 2i medium (50% Dulbecco's modified Eagle medium [DMEM]–F-12 [1:1; Invitrogen], 50% neurobasal medium [Invitrogen] supplemented with N-2 supplement [Invitrogen], B-27 serum-free supplement [Invitrogen], β-mercaptoethanol [Gibco], 0.1 mM nonessential amino acids [Gibco], 1 mM sodium pyruvate [Gibco], LIF, 1 μM MEK inhibitor [CT-99021], and 3 μM glycogen synthase kinase [GSK] inhibitor [PD-035901]) unless otherwise specified. MEFs were generated from embryonic day 13.5 embryos and cultured in DMEM (Gibco) supplemented with 10% FBS (HyClone). To induce Cre recombination, MEFs and ESCs were cultured in the presence of 500 nM 4-hydroxy-tamoxifen (OHT) (Sigma-Aldrich) for at least 96 h. The esophageal squamous carcinoma cell line KYSE150 was grown in DMEM–F-12 (1:1; Invitrogen) supplemented with 10% FBS.
Cloning and expression of Jmjd2c mutant proteins.
To generate a plasmid expressing amino acids (aa) 1 to 307 of mouse Jmjd2c (NP_659036.1), we amplified Jmjd2c cDNA and inserted a stop codon after the sequence encoded by exon 8 (NM_144787.2). The resulting cDNA was cloned into an EF-1α promoter vector and transfected into U2OS cells using Fugene HD (Roche Diagnostics) according to the manufacturer's instructions.
To generate PHD and Tudor domain-deficient Jmjd2c mutants, stop codons were inserted after the codons encoding amino acids 683 (generating mutant lacking PHD and the double Tudor domain) and 873 (for mutant lacking only the double Tudor domain) in mouse Jmjd2c. Jmjd2c mutants lacking the PHD domains (aa 687 to 863) were generated by inverse PCR. The resulting cDNAs were cloned into a modified pBabe retroviral vector. Mutations of Asp923 to Arg and Tyr951 to Ala were obtained by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene). MEFs were subjected to retrovirus transduction according to standard protocols, and transduced cells were selected by incubation with puromycin (2 μg/ml).
Antibodies.
Rabbits were immunized with keyhole limpet hemocyanin (KLH)-coupled peptides corresponding to different parts of mouse and human JMJD2C under standard conditions. The sequences were mJmjd2c C (CRQRVLSSRLKNEYVDDPVYRTFLKS), mJmjd2c K (KTWLQRKKKLRKPPKSLQGNKPLC), hJMJD2C (ESHGNGLEPGEIPAVPSGE), and mJmjd2b (CFMESLLQAQGRPGAPF). The polyclonal antibodies were affinity purified from serum on the peptide antigens. In addition, the following antibodies were used for Western blotting (WB), chromatin immunoprecipitation (ChIP), or immunofluorescence (IF) analyses: antivinculin (V9131; Sigma-Aldrich), anti-Nanog (ab80892; Abcam), anti-Oct4 (ab19857; Abcam), rabbit IgG (Sigma-Aldrich), antihemagglutinin (anti-HA) probe (Y-11) (sc-805; Santa Cruz), anti-HA (clone 16B212, MMS-101P; Covance), anti-glutathione S-transferase (anti-GST) (27-4577; Amersham), anti-JMJD2A (A300-861A [Bethyl] or 5328 [Cell Signaling Technology]), anti-H3K4me3 (17-614 [Millipore] or 9751 [Cell Signaling]), anti-H3K9me3 (ab8898; Abcam), anti-H3K36me3 (ab9050; Abcam), anti-H3K9me2 (07-441; Millipore), anti-H3K36me2 (07-369 Millipore), anti-H3 (ab1791; Abcam), and anti-H4 (2592; Cell Signaling Technology).
ChIP and ChIP-seq.
ChIP experiments were performed essentially as described previously (34). Reads were mapped to the human (hg18 assembly) or mouse (mm9 assembly) genome with the Bowtie tool v0.12.7 (35) with default settings, except m = 1 (suppress all alignments if >1 exist), -S (write output in SAM format), excluding all haplotype variants and random and unknown chromosomal sequences. The number of reads obtained for each sample is listed in Table S1 in the supplemental material. Chromosomal positions were annotated according to the RefSeq database (hg18/mm9; 7 January 2013) using the UCSC refFlat tables (36). Any gene not uniquely mapped to the genome was excluded. In our annotation, a transcription start site (TSS) region is defined as ±1 kb relative to the TSS. In total, our filtered and processed hg18 RefSeq annotation contained 40,108 transcripts (29,560 for mm9), corresponding to 23,188 unique genes (22,969 for mm9). Mouse and human samples for JMJD2C and/or H3K4me3 were analyzed using MACS2 v2.0.9 (37). For the mouse Jmjd2c ChIP followed by high-throughput sequencing (ChIP-seq) data sets, the OHT-treated samples were used as negative controls (default parameters, except for MEF [−mfold = 15,30, −qvalue = 0.1 − shift-control − g mm] and E1 ES [−shift-control − g mm]). For the human JMJD2C ChIP-seq data set, the shJMJD2C sample depleted of JMJD2C was used as a negative control (default parameters, except −mfold = 15,40, −qvalue = 1e−05 −shift-control − g hs). IgG was used as a negative control for H3K4me3 mappings in KYSE150 cells (default parameters, except −mfold = 15,40, −qvalue = 1e−05 − g hs). To produce a final peak set, the MACS2-called peaks were postprocessed by filtering according to false discovery rate (FDR) employing cutoffs of qvalue = 1e−16 for KYSE150 JMJD2C and H3K4me3 ChIP-seq data sets and qvalue = 1e−02 for the MEF Jmjd2c ChIP-seq data set. H4K4me3 and control samples from Mikkelsen et al. (38) (GSE12241) and Marks et al. (39) (GSE23943) were aligned in Bowtie with the same parameters mentioned above and analyzed in MACS2 with default parameters. The typically more diffuse H3K9me3- and H3K36me3-enriched sites were predicted using SICER (window size, 200 bp; fragment size, 300 bp; gap size, 600 bp; FDR, 0.05; redundancy threshold, 1; effective genome fraction, 0.8; species, hg18) (40).
For bigwig files and density plots, we allowed only one read per chromosomal position, eliminating potential spurious spikes. Each remaining read was extended from its 5′ end to a total length of 250 bases. Each bigwig file was also scaled to TPM (tags per million) based on the number of unique read positions. For density plots, the average read densities in 100 bins across TSS were extracted from the bigwig files and plotted in R. The density heat maps were made in seqMINER (41), and a constant read number between control and JMJD2C-depleted samples was employed for comparisons of Jmjd2c, H3K36me3, and H3K9me3 data sets. Gene ontology (GO) classifications were performed using the Panther program (42), and overlaps were made using Galaxy (43). Primers for ChIP-quantitative PCR (qPCR) analyses were designed using the Primer3 software, and real-time PCR was performed on an ABI Prism 7300 sequence detection system using SYBR green qPCR master mix (Fermentas).
WB analysis.
Protein extracts for WB were made using either a high-salt lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.5% Triton X-100, 1% SDS, 1 mM EDTA, 1 mM dithiothreitol [DTT], leupeptin, aprotinin, and 1 mM phenylmethylsulfonyl fluoride phenylmethylsulfonyl fluoride [PMSF]) or urea buffer (25 mM Tris-HCl, pH 6.8, 1% SDS, 9 M urea, 1 mM EDTA, 0.7 M β-mercaptoethanol) and sonication. Immunoblotting was performed according to standard protocols.
Gene expression analyses.
RNA was purified using RNeasy (Qiagen) and reverse transcribed using TaqMan reverse transcription reagents (Applied Biosystems) according to the manufacturer's instructions. In reverse transcription-qPCR (RT-qPCR) experiments, expression values were normalized relative to the housekeeping gene RPLP0. For microarray analyses, RNA was hybridized on Affymetrix human genome U133A 2.0 arrays by the RH Microarray Center at Rigshospitalet, Copenhagen, Denmark, in accordance with Affymetrix procedures and analysis. Data processing and statistics were done in R using the affy (RMA) and limma packages. The microarray data were subjected to a two-way analysis of variance (ANOVA), and the cutoff was set at an adjusted P value of <0.05 using Bonferroni correction to correct for multiple testing. Fold change was calculated relative to the scrambled short hairpin RNA (shRNA) control sample (shCtrl).
RNA interference (RNAi).
For shRNA-mediated depletion of human JMJD2C or JMJD2A, oligonucleotides with the following target sequences were cloned into pRetrosuper: shJMJD2C, AGATAGCAGCAATGAAGAA; shJMJD2A, GGACTTAGCTTCATAACTA; and shCtrl, AGCATATAAGTCGAGTTAG. KYSE150 cells were transfected with shRNA constructs using Fugene 6 (Roche Diagnostics) according to the manufacturer's instructions and selected 24 h after transfection with 2 μg/ml puromycin (Invitrogen) for 72 h. Knockdown of Dpy-30 was obtained using a pLKO vector (NM_024428.2-302s1c1) obtained from Open Biosystems, and MEFs were transduced with lentiviral particles for 16 h and selected with 2 μg/ml puromycin 48 h after transduction. For short interfering RNA (siRNA) transfections, KYSE150 cells were reverse transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. siRNAs were purchased from Sigma-Aldrich and had the following target sequences with dTdT overhangs: siJMJD2C a, AGATAGCAGCAATGAAGAA; siJMJD2C b, TCCCAAGGGTGTGATGCATTT, and siJMJD2C c, CCTTGCATACATGGAGTCTAA. siJMJD2C a and shJMJD2C share the same target sequence. The sequence for siJMJD2C b was modified from reference 15. siUNC (Sigma-Aldrich) was used as a negative control.
GST fusion proteins.
cDNA fragments encoding the double Tudor domain of human JMJD2A (aa 895 to 1011; NP_055478.2), mouse JMJD2C (aa 875 to 989; NP_659036.1), and human JMJD2C (aa 877 to 991; NP_055876.2) were amplified by PCR and subcloned into pGEX-6p-1. The GST fusion proteins were expressed in Escherichia coli BL21 cells by induction with a final concentration of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 20°C overnight. Pellets were resuspended in a high-salt buffer (25 mM Tris-HCl, pH 7.5, 300 mM NaCl, 0.1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 100 μM ZnCl2, 1 mM DTT, leupeptin, aprotinin, and 1 mM PMSF) and broken by sonication. The resulting lysates were centrifuged and the GST fusion proteins absorbed to glutathione-Sepharose 4B beads (Amersham Pharmacia Biotech), washed 5× with high-salt buffer, and eluted from the beads with high-salt buffer containing 20 mM glutathione. All procedures were performed on ice or at 4°C. The eluted fractions were analyzed by SDS-PAGE followed by Coomassie staining (SimplyBlue safe stain; Invitrogen), flash frozen in liquid N2, and stored at −80°C.
In vitro peptide binding experiments.
For immunoprecipitations, biotinylated peptides (Millipore) were immobilized on Dynabeads M-280 streptavidin blocked with bovine serum albumin (BSA) (Invitrogen) in binding buffer (50 mM Tris-HCl, pH 7.2, 300 mM NaCl, 0.5% IGEPAL CA-630, 1 mM EDTA, pH 8.0, 1 mM DTT, leupeptin, aprotinin, and 1 mM PMSF), washed, and incubated with GST fusion protein for 4 h with rotation at 4°C. After five washes with binding buffer, beads were boiled in protein loading buffer and fractionated by SDS-polyacrylamide gel electrophoresis.
For enzyme-linked immunosorbent assay (ELISA), GST fusion proteins were incubated with histone peptides (JPT Peptides Technology GmbH) immobilized on streptavidin plates. GST fusion proteins and primary (anti-GST; G7781; Sigma-Aldrich) and secondary antibodies were resuspended in binding buffer (phosphate-buffered saline [PBS]–0.1% Tween, 5% FBS). After extensive washing with PBS containing 0.1% Tween, substrate (TMB) and stop solution (1 N H2SO4) were added and absorbance was measured.
Immunofluorescence.
For immunofluorescence analyses, U2OS cells or MEFs plated on coverslips were fixed for 10 min in 4% paraformaldehyde, permeabilized by incubation in 0.2% Triton X-100 for 5 min, and then subjected to antibody and 4′,6-diamidino-2-phenylindole (DAPI) staining. Pictures were acquired on an Axiovert 200M LSM 510 microscope (Carl Zeiss, Inc.).
Cell cycle synchronization and flow cytometry.
For synchronization assays, siRNA-transfected KYSE150 cells were subjected to a double-thymidine block starting approximately 18 h after siRNA transfection. Cells were incubated with 2 mM thymidine (Sigma-Aldrich) for 19 h, washed, released into medium containing 24 μM 2-deoxycytidine-HCl (Sigma-Aldrich), and washed 10 h later prior to incubation with 2 mM thymidine for 16 h. MEFs were synchronized by serum starvation. In brief, MEFs were grown to confluence and then starved in medium containing 0.1% FBS for 72 h prior to replating in DMEM supplemented with 20% FBS and 120 ng/ml nocodazole (Sigma-Aldrich). Cells were analyzed according to standard protocols. In brief, cells were fixed in 70% ethanol, permeabilized using 0.2% Triton X-100, and stained with anti-H3S10P (06-570; Millipore). DNA was counterstained with 0.1 mg/ml propidium iodide (PI) and treated with RNase for 30 min at 37°C. Analysis was performed on a FACSCalibur (BD Biosciences) using CellQuest software (Becton, Dickinson).
Oxygen consumption and extracellular acidification rate analyses.
Oxygen consumption and extracellular acidification rates (OCR and ECAR) were measured using an XF96 extracellular flux analyzer (Seahorse Biosciences) as described previously (44). Culture media were exchanged for XF assay medium (Seahorse Biosciences) supplemented with 1 mM sodium pyruvate and 4.5 g/liter glucose D, pH 7.4, 1 h before the assay. The OCR and ECAR values were reported in pmol/min and mpH/min, respectively, and baseline levels were normalized to cell numbers.
Data accession number.
ChIP-seq and gene expression data are available at NCBI's Gene Expression Ominbus database under accession number GSE28332.
RESULTS
Generation of Jmjd2c-deficient mouse ESCs.
To define the role of Jmjd2c in embryonic stem cells (ESCs), we established conditional targeted Jmjd2c knockout mouse ESCs in which Jmjd2c exon 9 is flanked by loxP sites (Fig. 1A) and Cre-ERT2 is expressed from the Rosa26 locus (Jmjd2cf/f; Rosa26::CreERT2 ESCs). The administration of 4-hydroxy-tamoxifen (OHT) leads to Cre-mediated deletion of the flanked exon, causing a frameshift in the open reading frame at the end of the sequence encoding the Jumonji C (JmjC) domain (Fig. 1A and B) and abrogating a zinc finger motif associating the JmjC domain with the following C-terminal domain (45, 46). The putative truncated protein product (aa 1 to 307), which could be encoded after Cre-mediated deletion, would be expected to have an altered structural conformation around the catalytic center and to lack nuclear localization signal and PHD and double Tudor domains. We cloned the sequence encoding aa 1 to 307 of Jmjd2c and found by immunofluorescence analyses that the truncated protein product lacks catalytic activity against H3K9me3 and H3K36me3 in vivo (see Fig. S1 in the supplemental material). RT-qPCR analyses with exon-specific primers confirmed the absence of transcripts containing exon 9 in OHT-treated ESCs and further demonstrated that only very low levels of residual Jmjd2c transcript could be detected (Fig. 1C). This suggests that the putative transcript that could encode the N-terminal region of Jmjd2c is degraded by nonsense-mediated RNA decay. Full-length Jmjd2c protein became undetectable by Western blotting after the addition of OHT, while expression of other Jmjd2 family members was not significantly altered in OHT-treated ESCs (Fig. 1D).
FIG 1.

Generation of Jmjd2c knockout ESCs. (A) Overview of the Jmjd2c locus with the location of loxP sites (arrows) indicated relative to exon positions (squares). The domain structure of Jmjd2c is shown below. (B) Agarose gels showing PCR amplification of Jmjd2c floxed (∼1,200 bp) and deletion (∼400 bp) alleles for ESCs homozygous or heterozygous for the conditional Jmjd2c allele grown in the absence or presence of OHT to induce Cre recombination. (C) Relative Jmjd2c mRNA levels in control (−) and OHT-treated (+OHT) Jmjd2c knockout ESCs determined by quantitative RT-PCR analyses with primers covering different exons. Transcript levels were normalized to Rplp0, and error bars show standard deviations for technical replicates. (D) Immunoblots of conditional Jmjd2c knockout ESCs probed with an antibody raised against an internal peptide of Jmjd2c (α-Jmjd2c K) or antibodies recognizing Jmjd2a, Jmjd2b, or vinculin (loading control).
Jmjd2c is dispensable for ESC self-renewal and embryonic development.
Previous studies have reported that knockdown of Jmjd2c affects embryonic stem cell self-renewal as well as preimplantation development, leading to deregulated expression of pluripotency factors such as Nanog (17, 18). Strikingly, however, we observed that conditional Jmjd2c knockout ESCs did not show decreased expression of Nanog or of the other pluripotency markers, Oct4/Pou5f1 and Sox2, upon OHT treatment, as assessed by RT-qPCR and/or WB (Fig. 2A and B; also see Fig. S2A in the supplemental material). Furthermore, the mutant Jmjd2c ESCs maintained undifferentiated ESC morphology and displayed a normal growth rate (Fig. 2C and data not shown). In addition, RT-qPCR analyses did not reveal substantially increased expression of tested differentiation markers (Fig. 2D). Similar results were obtained for two different conditional Jmjd2c knockout ESC lines grown under serum-free 2i conditions or in serum-containing medium (Fig. 2; also see Fig. S2B and C). Taken together, these data demonstrate that Jmjd2c is dispensable for ESC self-renewal.
FIG 2.

Jmjd2c is dispensable for ESC self-renewal and embryonic development. (A) Western blot of control (−) and OHT-treated (+) Jmjd2c knockout ESCs. ESCs were cultured under serum-free 2i conditions. (B) Relative transcript levels of pluripotency factors determined by RT-qPCR. (C) Growth curve of control (−) and Jmjd2c knockout (+OHT) ESCs. (D) Relative transcript levels of selected differentiation markers determined by RT-qPCR. (E) Table showing the number of live pups obtained with the indicated genotypes from Jmjd2c+/− intercrosses.
To further determine the role of Jmjd2c during embryonic development, we crossed Jmjd2cf/f mice with mice constitutively expressing Cre recombinase to obtain Jmjd2c+/− mice, which were further intercrossed to generate Jmjd2c−/− mice. Homozygous Jmjd2c knockout mice were born at almost the expected frequency (19% versus 25%) (Fig. 2E) and were viable, showing no gross phenotype. Both male and female Jmjd2c knockout mice were fertile, and normal-sized litters were obtained from knockout intercrosses. From these data we conclude that Jmjd2c is dispensable for maintaining pluripotency, and its loss is compatible with embryonic development and postnatal life.
Jmjd2c localizes to H3K4me3-positive TSS in ESCs.
To gain further insight into the cellular functions of JMJD2C, we decided to map its binding patterns genome-wide. Chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) was performed on mock- and OHT-treated ESCs grown under 2i conditions with an antibody recognizing Jmjd2c. Defined peaks could be observed in the mock-treated sample but were generally absent from the OHT-treated sample devoid of Jmjd2c (Fig. 3A). The OHT-treated sample was used as a negative control in subsequent bioinformatics analyses, which led to the prediction of 19,730 peaks representative of Jmjd2c binding sites (see Table S2 in the supplemental material). Independent ChIP experiments demonstrated that Jmjd2c binding to selected targets could be validated with the antibody used for global mapping as well as with another polyclonal antibody specific for Jmjd2c (Fig. 3B).
FIG 3.

Jmjd2c localizes to H3K4me3-positive transcription start sites in ESCs. (A) Examples of ChIP-seq tracks. (B) ChIP validations. Anti-Jmjd2c K was used for the ChIP-seq experiment. (C) Diagram illustrating the overall distribution of identified Jmjd2c peaks with regard to annotated transcription start sites and open reading frames. TES, transcription end site. (D) Density plot displaying average Jmjd2c tag distributions relative to TSSs. The y axis shows mean tags per million (TPM). (E) Genes were divided into 5 groups based on RPKM (reads per kilobase per million mapped reads) values reported for ESCs grown under 2i conditions (39), and average Jmjd2c tag counts for corresponding TSSs were plotted. Expr., expression. (F) Unsupervised k-means clustering of ChIP-seq tags within 5 kb up- or downstream of TSSs. H3K4me3 and H3K27me3 ChIP-seq data for ESCs grown under serum-free conditions were obtained from reference 39. (G) Bar graph showing the percentages of Jmjd2c peaks displaying at least a 1-bp overlap with an H3K4me3-positive region identified by MACS2 analyses of H3K4me3 ChIP-seq data from reference 39. Jmjd2c peaks were designated TSS associated (TSS) or non-TSS associated (Non-TSS) depending on whether or not they were located less than 1 kb from an annotated TSS.
Annotation of the identified peaks revealed that these were enriched within gene-rich areas. Thus, 92.1% of the identified peaks were located less than 5 kb from an annotated TSS or within a coding region (Fig. 3C). The Jmjd2c peaks were generally centered at TSSs showing a slight bimodal distribution, with 82% (16,182 peaks) located less than 1 kb from an annotated TSS (Fig. 3C and D). In total, 11,879 genes were found to have one or more Jmjd2c binding sites located within 1 kb of an annotated TSS (see Table S2 in the supplemental material). Thus, Jmjd2c localizes to a large number of transcription start sites in ESCs.
To gain further insights into the nature of the Jmjd2c-bound TSSs, we took advantage of previously published data sets. Comparisons to RNA-seq data showed that Jmjd2c binding sites are especially enriched over genes with high to intermediate expression levels (Fig. 3E) (39). Unsupervised hierarchical clustering suggested that the Jmjd2c-bound transcription start sites were generally H3K4me3 positive, and that Jmjd2c was enriched at bivalent TSSs marked by both H3K4me3 and H3K27me3 (Fig. 3F). Indeed, the majority of TSSs classified as bivalent in ESCs grown under 2i conditions were bound by Jmjd2c (951 out of 1,014 [39]). Further analyses showed that 92.8% of TSS-associated Jmjd2c peaks overlapped an H3K4me3 peak (Fig. 3G). Notably, 63.1% of the Jmjd2c peaks located more than 1 kb away from annotated TSSs also colocalized with H3K4me3-positive regions (Fig. 3G). ChIP qPCR experiments validated that randomly selected Jmjd2c binding sites were H3K4me3 positive (see Fig. S3A and B in the supplemental material). Taking these results together, we conclude that Jmjd2c localizes to a large number of transcription start sites in ESCs, and that Jmjd2c binding shows a strong correlation with H3K4 trimethylation.
JMJD2C binding patterns are conserved between cell types.
Having observed a widespread localization of JMJD2C to H3K4me3-positive TSSs in ESCs, we wondered whether this binding pattern was conserved between cell types. ChIP-seq was performed on conditional Jmjd2c knockout MEFs grown in the absence or presence of OHT (Fig. 4A and B), and selected hits were validated with the two antibodies recognizing Jmjd2c (Fig. 4C). The Jmjd2c binding patterns showed strong similarity between ESCs and MEFs. Also in MEFs, the vast majority of Jmjd2c binding sites were located within 1 kb of an annotated TSS (7,476 out of 8,228; 90.9%) (Fig. 4D and E) (see Table S3 in the supplemental material), and comparisons to published data sets showed that the majority of Jmjd2c-bound regions overlapped H3K4me3-positive sites (Fig. 4F and G) (38).
FIG 4.

Jmjd2c localizes to H3K4me3-positive transcription start sites in MEFs. (A) Western blot of control (−) and OHT-treated Jmjd2c knockout MEFs. (B) Examples of ChIP-seq tracks. (C) Independent ChIP validations. Anti-Jmjd2c C was used for the ChIP-seq experiment. (D) Diagram illustrating the overall distribution of identified Jmjd2c peaks relative to TSSs and coding regions. (E) Density plot displaying average Jmjd2c tag distributions relative to TSSs. The y axis shows mean tags per million (TPM). (F) Unsupervised clustering of ChIP-seq tags for IgG, Jmjd2c, H3K4me3, and H3K27me3 within 5 kb up- or downstream of TSSs. H3K4me3 and H3K27me3 ChIP-seq data for MEFs were obtained from reference 38. (G) Bar graph showing the percentages of Jmjd2c peaks displaying at least a 1-bp overlap with an H3K4me3-positive region as identified by MACS2 analyses of H3K4me3 ChIP-seq data from reference 38.
The JMJD2C locus is amplified in human cancers, and several reports suggest that JMJD2C plays a role in malignant transformation. Having found JMJD2C specifically at transcription start sites in two primary cell types, we wanted to investigate if this binding pattern was maintained in transformed cells with JMJD2C amplifications. We have previously reported that knockdown of JMJD2C expression results in impaired proliferation for the human esophageal carcinoma cell line KYSE150 (5) (see Fig. S4A in the supplemental material), which harbors an amplification of the JMJD2C locus (10). Of note, the conditional Jmjd2c knockout MEFs do not display significant growth defects upon induction of Jmjd2c depletion (see Fig. S4B).
We developed a polyclonal antibody which specifically recognizes human JMJD2C and used this to map binding sites in KYSE150 cells. The ChIP-seq experiment was performed on KYSE150 cells expressing an shRNA targeting JMJD2C or a control scrambled shRNA (shCtrl) (Fig. 5A). Independent ChIP experiments confirmed that JMJD2C could be found at the identified sites and that the signal obtained with the polyclonal antibody was significantly reduced in cells depleted of JMJD2C, as observed in the ChIP-seq experiment (Fig. 5B and C). Bioinformatics analyses identified 13,991 peaks in shCtrl versus shJMJD2C samples and revealed that 85.9% of these (12,018) were centered around ±1 kb of a TSS (Fig. 5D and E) corresponding to 10,982 genes having one or more JMJD2C binding sites near an annotated TSS (see Table S4 in the supplemental material). By comparing the JMJD2C binding sites to H3K4me3-marked regions in KYSE150 cells, we could demonstrate that also in this cell line, JMJD2C peaks showed a highly significant colocalization with H3K4me3. Thus, 99.7% of TSS-associated peaks and 89.7% of non-TSS-associated JMJD2C peaks overlapped an H3K4me3-positive region (Fig. 5F to G).
FIG 5.

JMJD2C localizes to H3K4me3-positive transcription start sites in the human cancer cell line KYSE150. (A) Western blot of KYSE150 cells transfected with a scrambled shRNA (shCtrl) or shRNA against JMJD2C (shJMJD2C). (B) Examples of ChIP-seq tracks. (C) Independent ChIP validations. HDAC1, GMNN, and TOP2B were identified as JMJD2C targets in the ChIP-seq analysis. (D) Diagram illustrating the overall distribution of identified JMJD2C peaks relative to TSS and coding regions. (E) Density plot showing average JMJD2C tag densities over TSSs. The y axis shows mean tags per million (TPM). (F) Heat map presenting unsupervised k-means clustering for ChIP-seq tag counts within 5 kb of TSSs. (G) Bar graph showing the percentages of JMJD2C peaks displaying at least a 1-bp overlap with an H3K4me3-positive region as identified by MACS2 analyses. (H) GO analysis of JMJD2C-bound genes in KYSE150 cells. The 10 most significantly overrepresented GO categories (biological function) are shown. Nucleic acid metabolic process is short for nucleobase, nucleoside, nucleotide, and nucleic acid metabolic process, and RNA Pol II transcription is short for transcription from the RNA polymerase II promoter.
Other JMJD2 family members have been suggested to function at heterochromatic repetitive sequences (8, 22), but these are difficult to analyze based on ChIP-seq data. However, ChIP followed by qPCR did not reveal binding of JMJD2C to tested centromeric or repetitive elements in KYSE150 cells or primary cells (Fig. 5C; also see Fig. S3A in the supplemental material).
As expected based on the localization of JMJD2C to H3K4me3-positive regions, we found that a significant fraction of the JMJD2C target genes are shared between ESCs, MEFs, and KYSE150 cells (see Fig. S5A in the supplemental material). Gene Ontology (GO) analyses showed a strong overrepresentation of genes involved in metabolic processes, transcription, and cell cycle regulation among the JMJD2C targets (Fig. 5H; also see Fig. S5B to D).
Taking these findings together, we conclude that JMJD2C has a similar binding pattern in ESCs, MEFs, and in a human cancer cell line containing amplification of the JMJD2C locus. In all cell types, JMJD2C shows widespread association with transcription start sites and a general localization to H3K4me3-positive regions.
The double Tudor domain of JMJD2C is required for binding to target genes.
As the genome-wide mapping studies had revealed a strong correlation between JMJD2C binding patterns and H3K4me3 distributions across different cell types, we wondered if JMJD2C binding could be regulated by this mark. The JMJD2 family members JMJD2A, JMJD2B, and JMJD2C all contain PHD and double Tudor domains. The double Tudor domain of JMJD2A has been demonstrated to bind H3K4me3 and H4K20me2/me3 in vitro (29–31), and recognition of H4K20me2 has also been reported for the double Tudor domain of JMJD2B (25).
The double Tudor domains of JMJD2A and JMJD2C show significant homology in amino acid sequence (see Fig. S6A in the supplemental material). Importantly, several residues demonstrated to be important for the interaction between JMJD2A and an H3K4me3 peptide are conserved between JMJD2A and JMJD2C. In contrast, an aspartate residue in JMJD2A (Asp939), which forms a salt bridge with the H4K20me3 peptide (31) and is conserved between JMJD2A and JMJD2B, is replaced by arginine in both mouse and human JMJD2C (see Fig. S6A; also data not shown). To elucidate the binding capacities of the JMJD2C double Tudor domain, we expressed the Tudor domains of JMJD2C and JMJD2A as GST fusion proteins (see Fig. S6B) and used these for in vitro binding studies with biotinylated histone peptides. Immunoprecipitations showed that the double Tudor domain of mouse Jmjd2c bound histone tails with di- or trimethylated H3K4 (Fig. 6A). However, unlike the double Tudor domain of JMJD2A, the double Tudor domain of Jmjd2c did not recognize H4K20me3 (Fig. 6A). We further tested the binding capacities of the double Tudor domain of JMJD2C using a library of histone peptides from all four major histones containing most known methylation, acetylation, and phosphorylation modifications (see Fig. S7A to C). The results confirmed that the double Tudor domains of mouse and human JMJD2C specifically recognize H3K4me3 and did not reveal significant binding to other modified histone residues (see Fig. S7A to C). In the ELISA, we did not detect binding of JMJD2C to H3K4me2, which could reflect a difference in sensitivity between the two assays.
FIG 6.

The double Tudor domain of Jmjd2c recognizes H3K4me2/me3 and is required for binding to target genes. (A) Biotin-labeled histone H3 or histone H4 peptides were used in IP experiments with the indicated GST fusion proteins. (B) Schematic representation of the domain structure of wild-type (wt) Jmjd2c and the generated mutants lacking PHD and/or double Tudor domains. (C to F) Conditional Jmjd2c knockout MEFs were transduced with an empty vector or vectors encoding wt or Jmjd2c mutants, treated with OHT or left untreated as indicated, and analyzed by WB (C and E) and ChIP (D and F). (G and H) MEFs transduced with shRNA targeting Dpy30 or a scrambled control were analyzed by WB (G) and used for ChIP experiments (H). For histone marks, ChIP signals were normalized to H3 ChIP values.
To test the importance of the JMJD2C double Tudor domain for in vivo binding, we generated deletion mutants lacking the C-terminal regions containing PHD and/or the double Tudor domain and expressed these in conditional Jmjd2c knockout MEFs (Fig. 6B and C). The mutant proteins localized to the nucleus as determined by immunofluorescence (see Fig. S8 in the supplemental material; also data not shown). As shown in Fig. 6D, exogenously expressed wild-type Jmjd2c was recruited to Jmjd2c target genes in the OHT-treated Jmjd2c knockout MEFs. Importantly, Jmjd2c mutants lacking the double Tudor domain failed to localize to the same binding sites, while deletion of the PHD domains did not abrogate binding (Fig. 6D). To further characterize the importance of H3K4me3 for binding, we took advantage of previous reports demonstrating that specific point mutations in the double Tudor domain of JMJD2A severely reduce binding to an H3K4me3 peptide (29, 31). We mutated corresponding residues in full-length Jmjd2c (D923R and Y951A) (see Fig. S6A) and tested recruitment to endogenous binding sites. ChIP experiments showed that introduction of these point mutations severely compromised Jmjd2c binding to all tested targets (Fig. 6E and F).
To further verify a role for H3K4 methylation in determining JMJD2C binding patterns, we depleted Dpy30, which is a common subunit of H3K4 methyltransferase complexes (47, 48). As shown in Fig. 6G, targeting of Dpy30 by shRNA led to a strong reduction in global H3K4me3 levels without affecting Jmjd2c protein levels in MEFs. ChIP experiments showed that reduction of H3K4me3 at individual target genes was accompanied by decreased Jmjd2c binding (Fig. 6H). Only partial reductions in Jmjd2c ChIP signals were observed, and this could reflect that the residual levels of H3K4me3 are sufficient for Jmjd2c recruitment and/or stabilization of binding. However, as JMJD2C binding does not completely mirror H3K4me3 levels (Fig. 3F; also see Fig. S3 in the supplemental material), other factors also could contribute to the regulation of JMJD2C binding patterns.
Taking these results together, we conclude that the double Tudor domain of JMJD2C recognizes H3K4me2/me3 in vitro and is important for recruitment to chromatin and/or stabilization of binding. Furthermore, our data imply that JMJD2C binding patterns are at least in part regulated by H3K4 methylation.
Depletion of JMJD2C affects expression of a subset of target genes in KYSE150 cells.
The localization of JMJD2C to transcription start sites implies a role in transcriptional regulation. The fact that Jmjd2c is dispensable for ESC and MEF proliferation suggests that Jmjd2c in itself is not a critical determinant for transcription patterns in these cell types despite the abundance of binding sites. However, more vital roles for JMJD2C might be found in other cell types during specific developmental processes or malignant transformation.
To obtain insight into the roles of JMJD2C in transformed cells, we monitored changes in gene expression profiles caused by depletion of JMJD2C and/or the closely related JMJD2A in KYSE150 cells (Fig. 7A). After JMJD2C knockdown, 480 transcripts were identified as being significantly downregulated with a log2(fold change) of <−1 in KYSE150 cells, with JMJD2C occupying the transcription start site of 431 transcripts, as determined by the ChIP-seq analysis. In comparison, 181 transcripts were upregulated after JMJD2C depletion, and JMJD2C bound to 153 of these (Fig. 7B; also see Table S5 in the supplemental material). Thus, only smaller subsets of JMJD2C-bound genes show significant transcriptional changes after JMJD2C depletion in KYSE150 cells. The fact that more bound genes are down- than upregulated suggests that JMJD2C could have a role in facilitating gene expression. RT-qPCR analyses did not reveal similar expression changes in ESCs and MEFs upon loss of Jmjd2c expression (see Fig. S9A to C). As also suggested by the different effects on cellular proliferation, this implies that Jmjd2c depletion has distinct effects on gene expression programs in different cell types.
FIG 7.
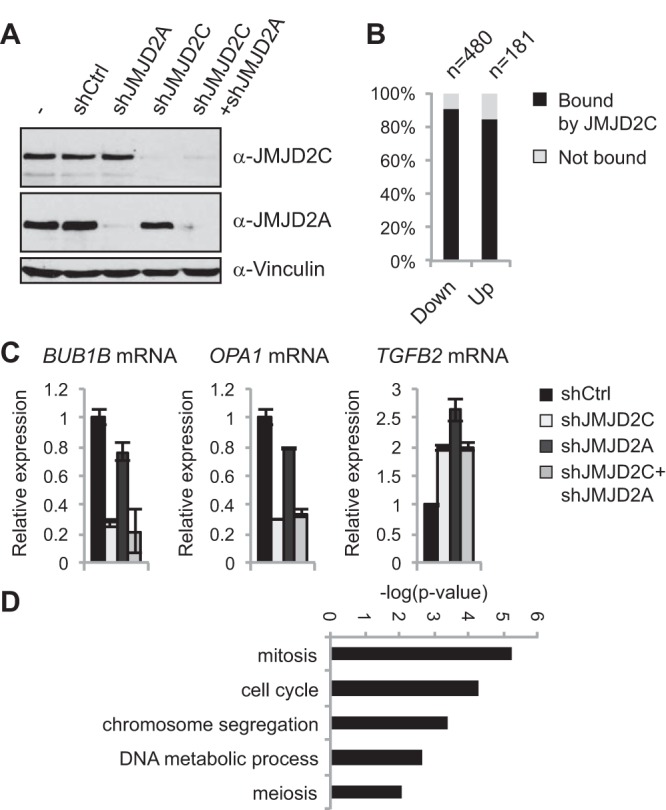
Depletion of JMJD2C in KYSE150 cells affects expression of a subset of target genes. (A) Western blot of KYSE150 cells transfected with a control scrambled shRNA (shCtrl) or shRNAs targeting JMJD2C and/or JMJD2A. (B) Bar graph showing the percentage of down- and upregulated genes, which were identified as containing a JMJD2C binding site within 1 kb of a TSS in the ChIP-seq analyses. JMJD2C binding sites were more frequently found at down- or upregulated genes compared to all genes present on the array (one-sided Fischer's exact test; P = 7.21E−55 and P = 2.34E−13, respectively). (C) RT-qPCR validation of selected targets. The gene expression analysis identified BUB1B and OPA1 as downregulated in the JMJD2C- but not JMJD2A-depleted samples, while TGFB2 was induced under both conditions. (D) GO analysis of JMJD2C-bound and downregulated genes.
JMJD2C has been reported to interact with the closely related JMJD2A (49, 50). The microarray experiment demonstrated that knockdown of JMJD2A and JMJD2C had distinct effects on gene expression, and depletion of both demethylases generally was not found to enhance the transcriptional effects (Fig. 7C; see Fig. S9D in the supplemental material; also data not shown). Thus, JMJD2C and JMJD2A do not appear to cooperate in gene regulation in KYSE150 cells.
The JMJD2C-bound genes in KYSE150 cells are enriched for functions in metabolic processes, transcription, and cell cycle regulation (Fig. 5H). Interestingly, GO analysis demonstrated that the JMJD2C-bound genes showing decreased expression upon JMJD2C depletion were generally involved in mitosis, cell cycle progression, and DNA metabolic processes, while the upregulated genes were not enriched for any GO term (Fig. 7D and data not shown). Functional assays showed that after release from a G1/S arrest, JMJD2C-depleted KYSE150 cells displayed a delayed and lower mitotic index as evaluated by staining with the mitotic marker phosphorylated H3S10. This phenotype was observed with 3 different siRNAs targeting JMJD2C (see Fig. S10A and B in the supplemental material). The transcriptional response could mostly represent secondary effects of JMJD2C knockdown; however, a likely explanation for these results is that JMJD2C depletion in KYSE150 cells affects cellular proliferation through transcriptional deregulation of certain cell cycle genes. In contrast, we did not obtain evidence that Jmjd2c knockout MEFs show an altered cell cycle progression, in agreement with their normal proliferation rate (see Fig. S10C). While a significant fraction of JMJD2C-bound genes are involved in metabolic processes, JMJD2C depletion was not found to affect the two metabolic parameters, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) (see Fig. S10D to F). OCR indicates the level of mitochondrial respiration, while ECAR correlates with glycolytic activity (44).
Taken together, our data show that JMJD2C, like many other chromatin-modifying enzymes, is not a critical transcriptional regulator of the majority of genes to which it binds but may function to facilitate gene expression and fine-tune the overall transcriptional output. We find that JMJD2C-bound genes are involved in metabolic processes and cell cycle regulation, and that JMJD2C depletion in the KYSE150 cell line is accompanied by altered expression of genes involved in cell cycle progression.
JMJD2C depletion has modest effects on the genome-wide distribution of H3K9me3 and H3K36me3.
JMJD2C is a histone demethylase specific for H3K9me3/me2 and H3K36me3/me2 with preferential activity against trimethylated H3K9 and H3K36 in vitro (5–8). As shown in Fig. 8A to C (see also Fig. S2A and B in the supplemental material), depletion of JMJD2C in ESCs, MEFs, or KYSE150 cells did not lead to significant changes in the global levels of H3K9me3 or H3K36me3. However, this did not exclude the possibility that local changes occur at JMJD2C binding sites.
FIG 8.

JMJD2C depletion has modest effects on histone methylation at binding sites. (A and B) Western blot of ESCs (A) and MEFs (B) heterozygous or homozygous for the conditional Jmjd2c allele (f) and expressing Cre-ERT2 from the Rosa26 locus. Cells were treated with OHT as indicated. (C) Western blot of KYSE150 cells transfected with the indicated shRNAs. (D and E) ChIP-seq data obtained for KYSE150 cells subjected to unsupervised k-means clustering. Regions within 5 kb of TSSs are shown. (F) shRNA-transfected KYSE150 cells were used in ChIP experiments with the indicated antibodies. For histone marks, ChIP signals were normalized to H3 ChIP values.
To systematically examine the effect of JMJD2C depletion, we generated genomic maps of H3K9me3 and H3K36me3 for both primary MEFs and KYSE150 cells. As expected, based on the localization of JMJD2C to H3K4me3-positive TSS, we found that the vast majority of H3K36me3-positive open reading frames were bound by JMJD2C at the TSS and that H3K36me3 levels increased downstream of the JMJD2C-bound regions (Fig. 8D; also see Fig. S11A in the supplemental material). In both cell types, most JMJD2C-bound transcription start sites appeared to be devoid of significant levels of H3K9me3 (Fig. 8E; also see Fig. S11B). Enrichment of H3K9me3 was observed at imprinted control regions, confirming the validity of the ChIP-seq results (data not shown).
JMJD2C depletion did not lead to gross overall alterations in H3K9me3 or H3K36me3 distributions in either cell line (Fig. 8D and E; also see Fig. S11A and B in the supplemental material). However, when examining JMJD2C-bound TSSs, slight alterations in methylation levels could be detected (see Fig. S12A and B). Bioinformatics analyses were employed to identify differentially methylated regions between the control and JMJD2C-depleted cells. For H3K9me3, 0.5% of JMJD2C target genes in KYSE150 cells were found to display a significant increase in H3K9me3 levels within a region of 1 kb of a JMJD2C-bound TSS (see Fig. S12C). Surprisingly, the observed variations in H3K9me3 did not show a general correlation with JMJD2C binding or the transcriptional changes elicited upon knockdown of JMJD2C. Thus, the modest increases in H3K9me3 levels were not restricted to JMJD2C-bound genes, nor were they more frequently observed at the TSSs of downregulated genes compared to all JMJD2C-bound genes (see Fig. S12C).
Analysis of the H3K36me3 ChIP-seq data showed that the levels increased slightly near the TSS and into the open reading frame in JMJD2C-depleted cells (see Fig. S12A and B in the supplemental material). Visual inspection of the ChIP-seq data and bioinformatics analyses suggested that the overall shifts in H3K36me3 levels represented the sum of many smaller changes, which were likely to be heterogeneous throughout the cell populations. In KYSE150 cells, 10.8% of JMJD2C target genes were found to display detectable increases in H3K36me3 extending into a region of 1 kb of a TSS. Importantly, as observed for H3K9me3, the changes in H3K36me3 were not strongly correlated with the transcriptional changes induced in KYSE150 as a result of JMJD2C depletion (see Fig. S12D).
To validate the H3K9me3 and H3K36me3 mapping results, independent ChIP-qPCR experiments were conducted. As expected, we found that the majority of tested JMJD2C binding sites displayed low levels of H3K36me3 and H3K9me3, and that depletion of JMJD2C was accompanied by minor or no effects on trimethylated as well as dimethylated H3K9 and H3K36 (Fig. 8F).
In summary, our results are consistent with a model in which the localization of JMJD2C- to H3K4me3-positive transcription start sites primarily fulfills a safeguard function with regard to removing aberrant methylation.
DISCUSSION
Several lines of evidence imply that JMJD2C is involved in malignant transformation, and this demethylase has also been suggested to fulfill important functions during development. Here, we report that JMJD2C generally is not required for proliferation or cellular homeostasis, as Jmjd2c is dispensable for mouse embryonic development and postnatal life. Furthermore, we show that JMJD2C is targeted to chromatin by its double Tudor domain and localizes to H3K4me3-positive transcription start sites in primary cells as well as in a transformed cell line with amplification of the JMJD2C locus.
Previous reports have suggested that Jmjd2c is important for ESC self-renewal and early embryonic development (17, 18). In these studies, Jmjd2c expression was depleted by shRNA transfections or double-stranded RNA injections, which could have the disadvantage of potential off-target effects as well as incomplete reduction of protein levels. To establish the role of Jmjd2c in early embryonic development, we generated conditional Jmjd2c knockout ESCs. Despite the widespread binding patterns, we find that Jmjd2c is dispensable for ESC self-renewal. The generation of viable and fertile Jmjd2c mutant mice further confirms that Jmjd2c deficiency is compatible with embryonic and postnatal development. In mammalian cells, four JMJD2 family members exist, namely, JMJD2A, JMJD2B, JMJD2C, and JMJD2D. Interestingly, Jmjd2b and Jmjd2d knockout mice were recently reported to be viable without gross abnormalities (51, 52). Evidence was provided that Jmjd2b deletion delays mammary gland development (52), while mice with a heart-specific Jmjd2a deletion demonstrate altered responses to cardiac stress (27). Most likely the four JMJD2 family members have different tissue-specific functions, but it is also plausible that they have redundant roles at some developmental stages. In Caenorhabditis elegans, loss of the only JMJD2 family member leads to germ cell apoptosis (7), and Drosophila mutants lacking both JMJD2 family members die in the second-instar larval stage, while single mutants display less severe phenotypes (53).
Another main finding of this paper is that JMJD2C shows a general localization to H3K4me3-positive transcription start sites across cell types with recruitment being at least in part dependent on the double Tudor domain. The H3K4me3 mark serves as a nucleation factor for several transcriptional regulators, including chromatin remodeling and histone acetyltransferase complexes, as well histone demethylases, such as the H3K9me2/me1 demethylase PHF8 (reviewed in reference 54). Trimethylated H3K4me3 is found at the transcription start sites of actively transcribed genes and at most CpG islands, which, through the binding capacities of CXXC domains, are also targeted by other enzymes, such as the H3K36me2/me1 demethylases FBXL10 and FBXL11 (reviewed in reference 55). Thus, numerous chromatin-modifying enzymes are broadly recruited to cis-acting sequences, where they may cooperatively contribute to the establishment of a specialized chromatin structure. It is noteworthy that several demethylases with activity against H3K9 and H3K36 methylation generally are recruited to TSSs and/or CpG islands.
Our data show that JMJD2C does not function as a key transcriptional regulator at the majority of its binding sites. Despite the widespread binding patterns, Jmjd2c is not required for embryonic development, and while depletion of JMJD2C impairs the proliferation of KYSE150 cells, it is accompanied by modest transcriptional regulation of smaller subsets of bound genes in this cell type. Likewise, several other chromatin modifiers have been found to have limited transcriptional effects, suggesting that these often contribute to the transcriptional potential of associated genes while not defining transcriptional output alone (reviewed in reference 3).
Genome-wide analyses showed that depletion of JMJD2C generally is not accompanied by gross alterations in H3K9me3 or H3K36me3 at its binding sites. It is possible that other JMJD2 family members compensate for the loss of JMJD2C function. However, it is also likely that several regulatory mechanisms ensure that H3K9me3 and H3K36me3 are not generally deposited at the JMJD2C-bound H3K4me3-positive regions. For H3K36me3, current evidence suggests that this mark is targeted specifically to transcribed regions by the methyltransferase SETD2, which can associate with the hyperphosphorylated elongating form of RNA polymerase (Pol) II (56, 57). We observed slight alterations in H3K36me3 levels in JMJD2C-depleted cells, and the effects could be consistent with a model in which JMJD2C resides at transcription start sites to remove low levels of H3K36me3 aberrantly targeted during transcription. In Saccharomyces cerevisiae and Drosophila, increased levels of H3K36me3 within open reading frames have been observed upon mutation of JMJD2 family members (24, 58). However, our data do not indicate general binding of JMJD2C downstream of the TSS, suggesting that alterations in H3K36me3 levels within open reading frames are not directly mediated by JMJD2C in mammalian cells.
Several reports have described JMJD2C-dependent modulation of H3K9me3 at selected tested promoter regions (14–17, 20), and global increases in H3K9me3 have been reported for certain cell types (17, 19). While our data do not exclude the possibility that JMJD2C, in certain contexts, functions to counteract H3K9me3, the genome-wide analyses suggest that JMJD2C rarely encounters high levels of H3K9me3 at its binding sites in MEFs or in KYSE150 cells. We speculate that the general recruitment of JMJD2C to H3K4me3-positive TSSs primarily fulfills a safeguard function with regard to ensuring the correct chromatin modification signature. At H3K4me3-positive regions, JMJD2C could remove aberrant methylation but also be in position to counteract, e.g., H3K9 methylation induced by specific stimuli.
Surprisingly, we did not find evidence that transcriptional effects were significantly correlated with changes in H3K9me3 or H3K36me3. The gene expression profiling captures both direct and indirect effects of JMJD2C depletion, making it difficult to deduce which of the bound genes are directly regulated by JMJD2C. It is possible that most transcriptional changes directly elicited by JMJD2C depletion are linked to removal of aberrant methylation, with modest increases in H3K36me3 or H3K9me3 in specific contexts influencing overall transcription levels. However, our data are also consistent with the possibility that JMJD2C has roles in transcriptional regulation that are uncoupled from H3K36me3/H3K9me3 methylation. In vitro studies suggest that JMJD2C has multiple targets and is capable of demethylating not only trimethylated but also dimethylated H3K9 and H3K36, as well as H1.4K26 and several nonhistone proteins, including Polycomb 2 protein (Pc2) (21, 59, 60). Functions independent of catalytic activity also could be expected; in line with this, JMJD2C and other JMJD2 family members were reported to interact with the SWI/SNF complex (52). Interestingly, a recent study demonstrated that while some transcriptional changes elicited by loss of the Drosophila JMJD2 family member, dKDM4A, involved its catalytic activity, most target genes did not appear to be regulated by histone demethylation (61).
The JMJD2 proteins, in particular JMJD2C, have been implicated in malignant transformation, but the underlying mechanisms are incompletely understood. Within recent years, deregulated expression of JMJD2 family members has been found to modulate gene expression but also has been reported to have effects independent of transcriptional regulation. In mammalian cells, JMJD2A was shown to regulate DNA replication by influencing chromatin accessibility and was detected at satellite repeats (22). The double Tudor domains of JMJD2A and JMJD2B recognize methylated H4K20, and this interaction has also been linked to roles in the DNA damage response (25). Interestingly, we find that the double Tudor domain of JMJD2C differs in specificity from those of JMJD2A/B, recognizing only methylated H3K4. In line with this, we find that JMJD2C localizes specifically to H3K4me3-positive transcription start sites also in a cell line containing amplification of the JMJD2C locus. This suggests that the putative roles of JMJD2C in malignant transformation and during developmental processes are linked to effects on gene expression.
Our results imply that the binding pattern of JMJD2C is, at least in part, regulated by the double Tudor domain recognizing H3K4me3. Interestingly, JMJD2B has been reported to interact with the MLL2 H3K4 methyltransferase complex, providing another link between JMJD2 proteins and H3K4 methylation (62). While we find a very strong overlap between JMJD2C binding and H3K4me3 patterns, the correlation is not absolute. It is an interesting possibility that combinations of histone marks could influence the binding of JMJD2C; however, transcription factors also could contribute to JMJD2C targeting, as has been reported for hypoxia-inducible factor 1 (16).
Our genome-wide analyses show that JMJD2C has the potential to regulate genes involved in metabolic processes, transcription, and cell cycle progression in different cell lines. Especially genes involved in cell cycle regulation and mitosis especially are downregulated upon JMJD2C knockdown in KYSE150 cells, which show impaired progression through the G2/M phases of the cell cycle. A possible explanation for these results is that JMJD2C is required for the correct expression of certain genes involved in cell cycle progression in KYSE150 cells. Our data suggest that JMJD2C depletion does not elicit a similar transcriptional response in ESCs and MEFs. It is likely that distinct subsets of genes respond to JMJD2C depletion in different cell types, which may also differ in their cellular response to the overall transcriptional effects. While several cancer cell lines have been reported to show impaired growth after JMJD2C depletion, our data demonstrate that JMJD2C generally is dispensable for cellular proliferation and homeostasis. This supports the notion that JMJD2C is an interesting target for the development of anticancer therapies.
In summary, we have shown that JMJD2C is not required for cellular proliferation, ES cell self-renewal, and mouse embryonic development. Moreover, we report that JMJD2C localizes specifically to H3K4me3-positive transcription start sites in both normal and malignant cells, and that this localization is dependent on its double Tudor domain. Based on our results, we propose that JMJD2C fulfills a safeguard function in transcriptional regulation on most of its target genes, and that JMJD2C can contribute to specific developmental processes and tumorigenesis through the regulation of H3K4me3-marked genes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Klaus Hansen for help with the generation of the antibody against human JMJD2C and Marie Sophie Isidor and Jacob Bo Højberg Hansen for help with the Seahorse XF96 extracellular flux analyzer. We are grateful to Gitte Andersen for expert technical assistance, to Itys Comet and Ida Holst-Pedersen for assistance with the histone peptide library, and to Susanne M. Kooistra for help with the generation of MEFs. We thank the EUCOMM consortia for providing the Jmjd2c loxP+ mice. We thank members of the Helin laboratory for vigorous discussions, technical advice, and support.
This work was supported by the Danish National Research Foundation, the Danish Cancer Society, the Novo Nordisk Foundation, the Danish Medical Research Council, the Lundbeck Foundation, the P. Carl Petersens Foundation, and the Excellence Programme of the University of Copenhagen.
Footnotes
Published ahead of print 6 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00864-13.
REFERENCES
- 1.Dambacher S, Hahn M, Schotta G. 2010. Epigenetic regulation of development by histone lysine methylation. Heredity 105:24–37. 10.1038/hdy.2010.49 [DOI] [PubMed] [Google Scholar]
- 2.Albert M, Helin K. 2010. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 21:209–220. 10.1016/j.semcdb.2009.10.007 [DOI] [PubMed] [Google Scholar]
- 3.Pedersen MT, Helin K. 2010. Histone demethylases in development and disease. Trends Cell Biol. 20:662–671. 10.1016/j.tcb.2010.08.011 [DOI] [PubMed] [Google Scholar]
- 4.Berry WL, Janknecht R. 2013. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 73:2936–2942. 10.1158/0008-5472.CAN-12-4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. 2006. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442:307–311. 10.1038/nature04837 [DOI] [PubMed] [Google Scholar]
- 6.Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. 2006. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature 442:312–316. 10.1038/nature04853 [DOI] [PubMed] [Google Scholar]
- 7.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. 2006. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125:467–481. 10.1016/j.cell.2006.03.028 [DOI] [PubMed] [Google Scholar]
- 8.Fodor BD, Kubicek S, Yonezawa M, O'Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. 2006. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 20:1557–1562. 10.1101/gad.388206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou VW, Goren A, Bernstein BE. 2011. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12:7–18. 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
- 10.Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. 2000. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 60:4735–4739 [PubMed] [Google Scholar]
- 11.Ehrbrecht A, Muller U, Wolter M, Hoischen A, Koch A, Radlwimmer B, Actor B, Mincheva A, Pietsch T, Lichter P, Reifenberger G, Weber RG. 2006. Comprehensive genomic analysis of desmoplastic medulloblastomas: identification of novel amplified genes and separate evaluation of the different histological components. J. Pathol. 208:554–563. 10.1002/path.1925 [DOI] [PubMed] [Google Scholar]
- 12.Liu G, Bollig-Fischer A, Kreike B, van de Vijver MJ, Abrams J, Ethier SP, Yang ZQ. 2009. Genomic amplification and oncogenic properties of the GASC1 histone demethylase gene in breast cancer. Oncogene 28:4491–4500. 10.1038/onc.2009.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, Mack S, Kongkham PN, Peacock J, Dubuc A, Ra YS, Zilberberg K, McLeod J, Scherer SW, Sunil Rao J, Eberhart CG, Grajkowska W, Gillespie Y, Lach B, Grundy R, Pollack IF, Hamilton RL, Van Meter T, Carlotti CG, Boop F, Bigner D, Gilbertson RJ, Rutka JT, Taylor MD. 2009. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 41:465–472. 10.1038/ng.336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rui L, Emre NC, Kruhlak MJ, Chung HJ, Steidl C, Slack G, Wright GW, Lenz G, Ngo VN, Shaffer AL, Xu W, Zhao H, Yang Y, Lamy L, Davis RE, Xiao W, Powell J, Maloney D, Thomas CJ, Moller P, Rosenwald A, Ott G, Muller-Hermelink HK, Savage K, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Weisenburger DD, Chan WC, Gascoyne RD, Levens D, Staudt LM. 2010. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 18:590–605. 10.1016/j.ccr.2010.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9:347–353. 10.1038/ncb1546 [DOI] [PubMed] [Google Scholar]
- 16.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. 2012. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc. Natl. Acad. Sci. U. S. A. 109:E3367–E3376. 10.1073/pnas.1217394109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh YH, Zhang W, Chen X, George J, Ng HH. 2007. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21:2545–2557. 10.1101/gad.1588207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Zhang M, Zhang Y, Kou Z, Han Z, Chen DY, Sun QY, Gao S. 2010. The histone demethylase JMJD2C is stage-specifically expressed in preimplantation mouse embryos and is required for embryonic development. Biol. Reprod. 82:105–111. 10.1095/biolreprod.109.078055 [DOI] [PubMed] [Google Scholar]
- 19.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O'Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. 2012. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483:474–478. 10.1038/nature10860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, Suzuki T. 2009. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem. Biophys. Res. Commun. 389:366–371. 10.1016/j.bbrc.2009.08.155 [DOI] [PubMed] [Google Scholar]
- 21.Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG. 2011. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147:773–788. 10.1016/j.cell.2011.08.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K, Rinehart C, Quiton J, Walsh R, Smallwood A, Dyson NJ, Whetstine JR. 2010. Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol. Cell 40:736–748. 10.1016/j.molcel.2010.11.008 [DOI] [PubMed] [Google Scholar]
- 23.Gray SG, Iglesias AH, Lizcano F, Villanueva R, Camelo S, Jingu H, Teh BT, Koibuchi N, Chin WW, Kokkotou E, Dangond F. 2005. Functional characterization of JMJD2A, a histone deacetylase- and retinoblastoma-binding protein. J. Biol. Chem. 280:28507–28518. 10.1074/jbc.M413687200 [DOI] [PubMed] [Google Scholar]
- 24.Kim T, Buratowski S. 2007. Two Saccharomyces cerevisiae JmjC domain proteins demethylate histone H3 Lys36 in transcribed regions to promote elongation. J. Biol. Chem. 282:20827–20835. 10.1074/jbc.M703034200 [DOI] [PubMed] [Google Scholar]
- 25.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. 2012. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31:1865–1878. 10.1038/emboj.2012.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Jubb AM, Pike L, Buffa FM, Turley H, Baban D, Leek R, Gatter KC, Ragoussis J, Harris AL. 2010. The histone demethylase JMJD2B is regulated by estrogen receptor alpha and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 70:6456–6466. 10.1158/0008-5472.CAN-10-0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang QJ, Chen HZ, Wang L, Liu DP, Hill JA, Liu ZP. 2011. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J. Clin. Investig. 121:2447–2456. 10.1172/JCI46277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, van Essen D, Saccani S. 2012. Cell-type-specific control of enhancer activity by H3K9 trimethylation. Mol. Cell 46:408–423. 10.1016/j.molcel.2012.05.011 [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. 2006. Recognition of histone H3 lysine-4 methylation by the double Tudor domain of JMJD2A. Science 312:748–751. 10.1126/science.1125162 [DOI] [PubMed] [Google Scholar]
- 30.Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. 2006. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 7:397–403. 10.1038/sj.embor.7400625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Thompson JR, Botuyan MV, Mer G. 2008. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 15:109–111. 10.1038/nsmb1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwenk F, Baron U, Rajewsky K. 1995. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23:5080–5081. 10.1093/nar/23.24.5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. 2007. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 27:3769–3779. 10.1128/MCB.01432-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleine-Kohlbrecher D, Christensen J, Vandamme J, Abarrategui I, Bak M, Tommerup N, Shi X, Gozani O, Rappsilber J, Salcini AE, Helin K. 2010. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell 38:165–178. 10.1016/j.molcel.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, Pohl A, Pheasant M, Meyer LR, Learned K, Hsu F, Hillman-Jackson J, Harte RA, Giardine B, Dreszer TR, Clawson H, Barber GP, Haussler D, Kent WJ. 2010. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 38:D613–D619. 10.1093/nar/gkp939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560. 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, Stunnenberg HG. 2012. The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149:590–604. 10.1016/j.cell.2012.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. 2009. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25:1952–1958. 10.1093/bioinformatics/btp340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L. 2011. seqMINER: an integrated ChIP-seq data interpretation platform. Nucleic Acids Res. 39:e35. 10.1093/nar/gkq1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13:2129–2141. 10.1101/gr.772403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. 2005. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15:1451–1455. 10.1101/gr.4086505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. 2007. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 292:C125–C136. 10.1152/ajpcell.00247.2006 [DOI] [PubMed] [Google Scholar]
- 45.Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G. 2006. Structural insights into histone demethylation by JMJD2 family members. Cell 125:691–702. 10.1016/j.cell.2006.04.024 [DOI] [PubMed] [Google Scholar]
- 46.Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U. 2007. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 448:87–91. 10.1038/nature05971 [DOI] [PubMed] [Google Scholar]
- 47.Jiang H, Shukla A, Wang X, Chen WY, Bernstein BE, Roeder RG. 2011. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 144:513–525. 10.1016/j.cell.2011.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J. Biol. Chem. 282:20395–20406. 10.1074/jbc.M701574200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shin S, Janknecht R. 2007. Diversity within the JMJD2 histone demethylase family. Biochem. Biophys. Res. Commun. 353:973–977. 10.1016/j.bbrc.2006.12.147 [DOI] [PubMed] [Google Scholar]
- 50.Verrier L, Escaffit F, Chailleux C, Trouche D, Vandromme M. 2011. A new isoform of the histone demethylase JMJD2A/KDM4A is required for skeletal muscle differentiation. PLoS Genet. 7:e1001390. 10.1371/journal.pgen.1001390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwamori N, Zhao M, Meistrich ML, Matzuk MM. 2011. The testis-enriched histone demethylase, KDM4D, regulates methylation of histone H3 lysine 9 during spermatogenesis in the mouse but is dispensable for fertility. Biol. Reprod. 84:1225–1234. 10.1095/biolreprod.110.088955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawazu M, Saso K, Tong KI, McQuire T, Goto K, Son DO, Wakeham A, Miyagishi M, Mak TW, Okada H. 2011. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS One 6:e17830. 10.1371/journal.pone.0017830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsurumi A, Dutta P, Yan SJ, Sheng R, Li WX. 2013. Drosophila Kdm4 demethylases in histone H3 lysine 9 demethylation and ecdysteroid signaling. Sci. Rep. 3:2894. 10.1038/srep02894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vermeulen M, Timmers HT. 2010. Grasping trimethylation of histone H3 at lysine 4. Epigenomics 2:395–406. 10.2217/epi.10.11 [DOI] [PubMed] [Google Scholar]
- 55.Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev. 25:1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, Zhang QH, Chen SJ, Huang QH, Chen Z. 2005. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 280:35261–35271. 10.1074/jbc.M504012200 [DOI] [PubMed] [Google Scholar]
- 57.Edmunds JW, Mahadevan LC, Clayton AL. 2008. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 27:406–420. 10.1038/sj.emboj.7601967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin CH, Paulson A, Abmayr SM, Workman JL. 2012. HP1a targets the Drosophila KDM4A demethylase to a subset of heterochromatic genes to regulate H3K36me3 levels. PLoS One 7:e39758. 10.1371/journal.pone.0039758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. 2009. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J. Biol. Chem. 284:8395–8405. 10.1074/jbc.M807818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ponnaluri VK, Vavilala DT, Putty S, Gutheil WG, Mukherji M. 2009. Identification of non-histone substrates for JMJD2A-C histone demethylases. Biochem. Biophys. Res. Commun. 390:280–284. 10.1016/j.bbrc.2009.09.107 [DOI] [PubMed] [Google Scholar]
- 61.Crona F, Dahlberg O, Lundberg LE, Larsson J, Mannervik M. 2013. Gene regulation by the lysine demethylase KDM4A in Drosophila. Dev. Biol. 373:453–463. 10.1016/j.ydbio.2012.11.011 [DOI] [PubMed] [Google Scholar]
- 62.Shi L, Sun L, Li Q, Liang J, Yu W, Yi X, Yang X, Li Y, Han X, Zhang Y, Xuan C, Yao Z, Shang Y. 2011. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. U. S. A. 108:7541–7546. 10.1073/pnas.1017374108 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.