

Abstract
A series of pateamine A (1) derivatives were synthesized for structure/activity relationship (SAR) studies and a selection of previous generation analogs were re-evaluated based on current information regarding the mechanism of action of these translation inhibitors. Structural modifications in the new generation of derivatives focused on alternations to the C19-C22 Z, E-diene and the trienyl side chain of the previously described simplified, des-methyl, des-amino pateamine A (DMDAPatA, 2). Derivatives were tested for anti-proliferative activity in cell culture and for inhibition of mammalian cap-dependent translation in vitro. Activity was highly dependent on the rigidity and conformation of the macrolide and the functionality of the side chain. The only well tolerated substitutions were replacement of the N,N-dimethyl amino group found on the side chain of 2 with other tertiary amine groups. SAR reported here suggests that this site may be modified in future studies to improve serum stability, cell-type specificity, and/or specificity towards rapidly proliferating cells.
Keywords: Pateamine A, DMDAPatA, Stille coupling, Translation initiation, eIF4A
1. Introduction
Natural products continue to be a significant source of novel and potential therapeutics because of their structural diversity and evolutionarily optimized structure and function in the environment of a living system. Therapeutic natural products or their semi-synthetic derivatives have been discovered from a wide range of living organisms such as fungi, bacteria, and plants.1–3 Furthermore, significant biological inquiry has been accomplished using natural products as chemical probes to interrogate disease-related phenomena.4 We have previously reported the total synthesis of the marine natural product pateamine A (PatA, 1, Figure 1).5 PatA initially isolated from the marine sponge Mycale sp. demonstrated potent in vitro antifungal and cytotoxic activity.6, 7 We and others have identified that the mammalian cellular protein target of PatA is the translation initiation factor eIF4A (eukaryotic initiation factor 4A),8, 9 which is found in three forms (I, II and III) in human cells and are RNA-dependent ATPases and ATP-dependent RNA helicases.10–13 The very similar eIF4AI and II isotypes (approximately 90% identical at the protein level) are necessary for translation initiation of mRNAs by cap-dependent translation initiation,10–15 while eIF4AIII (approximately 60% identical to I and II at the protein level) is a member of the exon-junction complex16 of proteins that are deposited onto mRNA after splicing. By directly binding to eIF4AI/II, PatA inhibits eIF4AI/II function and prevents cap-dependent translation initiation leading to the induction of apoptosis.6–9, 17, 18 Treatment of cells in culture with PatA or DMDAPatA (2) (see below) induced stress granule formation9, 19 and inhibited nonsense-mediated mRNA decay.20
Figure 1.
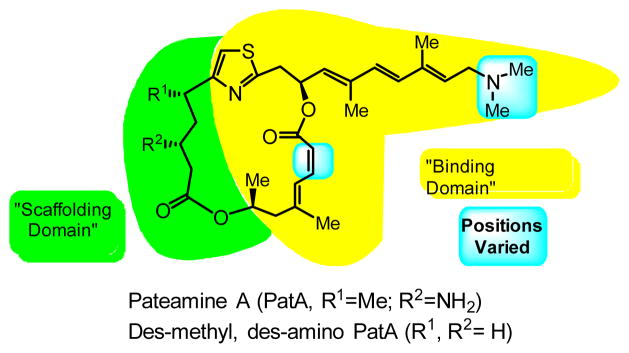
Structures of pateamine A (PatA, 1) and C5-des-methyl, C3-des-amino pateamine A (DMDAPatA, 2) highlighting proposed ‘binding’ and ‘scaffolding’ domains.
In addition, DMDAPatA has been found to inhibit S-phase DNA synthesis in some cell lines with direct in vitro inhibition of DNA polymerases α and β at much higher concentrations (IC50 of 3–19 μM).21 By these mechanisms, PatA has proven to be an extremely potent anti-proliferative agent in cell culture with IC50 values in the sub-nanomolar range in a variety of cancer cell lines.6, 9 The search for novel anti-neoplastic agents is an active area of research, and small-molecule inhibition of translation initiation is being increasingly recognized as a viable target for therapy as deregulated translational control has become more evident in various diseases including cancer.22–28
In addition to PatA, we have previously reported a simplified, nearly equipotent derivative of PatA, C5-des-methyl, C3-des-amino pateamine A (DMDAPatA, 2 in Figure 1), which exhibited high potency as an anti-proliferative agent in cell culture with approximately single-digit nanomolar IC50 values across a number of cancer lines.9, 21, 29 This derivative, which is simpler to synthesize than the natural product (i.e. 14 vs. 24 steps for the longest linear sequence), led us to identify and propose ‘binding’ and ‘scaffolding’ domains in PatA29 (see Figure 1) with respect to its interaction with eIF4A. Furthermore, DMDAPatA also showed potent anticancer activity in several xenograft mouse models with especially significant regressions in melanoma models,21 and more recently PatA demonstrated promise at low doses in preventing cachexia.30 Thus, PatA and its simplified derivate DMDAPatA warrant further structure-activity relationship studies as novel anticancer agents.
Initial reports on the activity of PatA in mixed lymphocyte reaction assays suggested that PatA may have immunosuppressive activity.5, 29 Our initial structure/activity relationship (SAR) studies focused on the activity of several structural analogs of PatA in an IL-2 (interleukin-2) reporter assay system.29 As this was a cell-based assay, these results may have been reporting on the anti-proliferative activity. To take into account more recent findings of the anti-proliferative activity of PatA, we have sought to re-evaluate the previously reported derivatives in cell proliferation assays. In addition, we have synthesized and investigated a second-generation of DMDAPatA derivatives for study. From our previous SAR29, the scaffolding domain consists of the flexible region on the western half of the macrolide ring (C1–C5), while the binding domain is comprised of the more rigid eastern half of the macrolide (C18–25) and rigid side chain (C10–C17) terminating in an N,N-dimethyl tertiary amine. In this report, we have sought to clarify our previous results based on IL-2 reporter assays, and to extend our SAR analysis with the synthesis of novel derivatives of DMDAPatA to further clarify the roles of the various structural features of PatA and DMDAPatA required for activity in the binding domain.
2. Materials and Methods
General information for synthetic experiments
All reactions were carried out under a nitrogen atmosphere in flame-dried glassware. Acetonitrile, dichloromethane, methanol were purified by passage through activated molecular sieves or alumina (solvent system). Tetrahydrofuran was freshly distilled over sodium and benzophenone. DMF was dried over molecular sieves before use. All commercial reagents were used as received. 1H and 13C NMR spectrum were recorded on INOVA-500. 1H NMR chemical shifts are reported as δ values in ppm relative to CDCl3 (7.26 ppm), coupling constants (J) are reported in Hertz (Hz), and multipilicity follows convention. Unless indicated otherwise, deuterochloroform (CDCl3) served as an internal standard (77.2 ppm) for all 13C spectra. Flash column chromatography was performed using 60Å Silica Gel (Silicycle, 230–400 mesh, cat. no. R10030B) as a stationary phase using a gradient solvent system (EtOAc/hexanes as eluent unless specified otherwise). Mass spectra were obtained at the center for Chemical Characterization and Analysis (Texas A&M University). Thin layer chromatography (TLC) was performed using glass-backed silica gel 60F254. For detailed synthesis of compounds 2, 4, 8, 9a–g, and 10a–g see Supplemental Material.
Synthesis of dienes 5 and 6
The macrocyclic enyne 4 (372 mg, 0.78 mmol) was placed in a 100 mL round-bottomed flask and dissolved in MeOH (30 mL). Lindlar catalyst (180 mg) was added under N2. The flask was evacuated and purged several times with H2, and the reaction mixture was stirred under H2 atmosphere (1 atm) for 15 h until TLC analysis confirmed the completion of the reaction. The mixture was filtered through a short Celite pad, rinsed with MeOH (5 × 2 mL). The combined filtrate was concentrated and the residue was purified by flash chromatography on silica gel (10 → 40% EtOAc/hexanes) to give Z-isomer 5 (274 mg, 73%) as a white foam. The E-isomer 6 (52 mg) was also isolated in 14% yield.
General procedure for Stille coupling reaction
A stock solution of Pd(0) catalyst was prepared by mixing Pd2dba3•CHCl3 (17 mg, 0.016 mmol) and PPh3 (35 mg, 0.133 mmol, 8.3 equiv) in 1 mL of degassed THF and stirring for 5 min to give a clear yellow solution. The final concentration of Pd(0) stock solution was ~0.032 M. The macrocycle (0.025 mmol, 1. 0 equiv) and vinylstannane reagent (0.028~0.050 mmol, 1.1~2.0 equiv) were combined in a 5 mL round-bottom flask and purged with N2. Degassed THF (0.8 mL) was added and to this was transferred by microliter syringe a solution of the freshly prepared Pd(0) catalyst stock solution (0.1 mL, 0.0032 mmol, ~13 mol %) and the mixture was stirred at ambient temperature (23 °C) under N2 for 2 h. An additional 0.2 mL of Pd(0) stock solution (~26 mol %) was added and stirring was continued at ambient temperature for 20 h or until the reaction was judged complete by TLC analysis. The crude reaction mixture was concentrated to dryness and the residue was purified by flash chromatography on silica gel to afford the product.
Cell culture
All cells were cultured in appropriate media, DMEM (Thermoscientific) for HeLa and SK-MEL2 cells, RPMI 1640 (Cellgro) for Jurkat T cells, that were supplemented with penicillin-streptomycin (Amresco) and 10% FBS (Atlanta Biologicals) Cells were incubated at 37 °C in a humidified Symphony 5.3A air-jacketed humidified incubator (VWR, Radnor, PA) with 5% CO2.
Cell proliferation assays
First generation analogs were assayed for anti-proliferative activity as previously described9 with minor modifications. Adherent cell lines were aliquoted into 96-well plates at 2000 cells/well in 200 μl of growth media and allowed to recover overnight before addition of compound at concentrations spanning 0.001 nM to 100 nM across 10 wells. Compounds were dissolved in carrier solvent DMSO with final concentration of DMSO at 0.1% and 0.1% DMSO only was used for control. After 24 h, DNA synthesis levels were assayed as previously described.9 For non-adherent Jurkat T cells, cells were aliquoted at 5000 cells/well without a recovery period before addition of compounds. Each compound was tested in a minimum of 3 independent assays with mean IC50 values reported.
Second-generation analogs and DMDAPatA were assayed for anti-proliferative activity using MTT (Amresco). Adherent cells were aliquoted at 2000 cells/well in 96-well plates and allowed to recover overnight prior to addition of compounds. Treatment of cells was performed as described above. Cells were then incubated for 72 h followed by assay of proliferation using MTT. For non-adherent Jurkat T cells, cells were aliquoted at 5000 cells/well without a recovery period before addition of compounds, followed by 72 h incubation and assay for cell proliferation by MTT assay. In each independent assay data points were collected in quadruplicate and IC50 determination was performed by curve fitting using Prism® 5 (GraphPad Software, Inc., La Jolla, CA). Each compound was tested in a minimum of two independent assays and IC50 values (Table 2) are reported (where applicable) with 95% confidence intervals.
Table 2.
SAR study of cell proliferation inhibition activity for second-generation DMDAPatA derivatives.
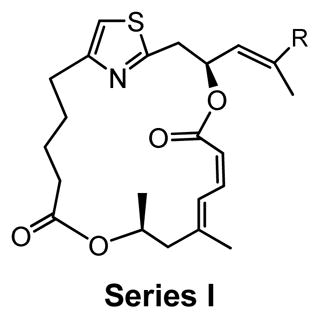
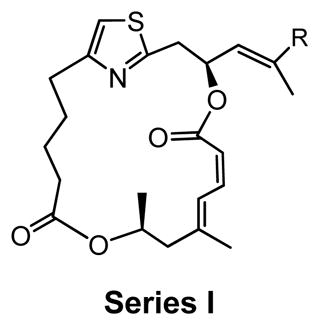 Series I |
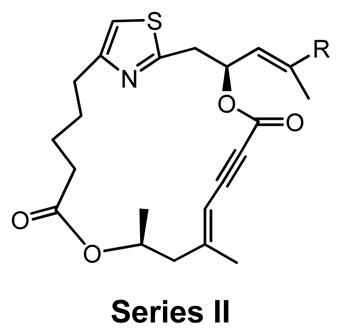 Series II |
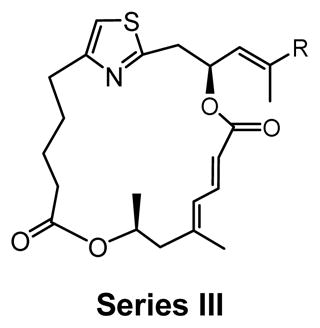 Series III |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R | Compd | IC50 (nM) | Compd | IC50 (nM) | Compd. | IC50 (nM) | ||||||
| HeLa | SK-MEL2 | Jurkat T | HeLa | SK MEL2 | Jurkat T | HeLa | SK MEL2 | Jurkat T | ||||
| Br | 5 | * N.E. (500) | †>104 | †>104 | 4 | * N.E. (500) | * N.E. (104) | * N.E. (104) | 6 | * N.E. (500) | * N.E. (104) | * N.E. (104) |
|
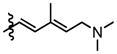
2 | 9.8 a(8.2–10.2) | 13 a(10.8–16.4) | 11 a(7.5–17.6) | 10a | * N.E. (500) | * N.E. (104) | * N.E. (104) | 8 | 860 a(790–870) | 950 a(898–991) | †>104 |
|
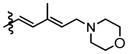
9b | 430 a(354–490) | 320 a(236–444) | * N.E. (104) | 10b | * N.E. (500) | †>5×103 | * N.E. (104) | ||||
|
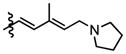
9c | 23 a(19.4–25) | 27 a(20–28.2) | 25 a(19.9–27) | 10c | * N.E. (500) | * N.E. (104) | * N.E. (104) | ||||
|
|
9d | * N.E. (500) | †>5×103 | * N.E. (104) | 10d | * N.E. (500) | * N.E. (104) | * N.E. (104) | ||||
|
|
9e | †>5×103 | †>5×103 | †>104 | 10e | * N.E. (500) | * N.E. (104) | * N.E. (104) | ||||
|
|
9f | * N.E. (500) | * N.E. (104) | * N.E. (104) | 10f | * N.E. (500) | * N.E. (104) | †>104 | ||||
|
|
9g | * N.E. (500) | †>5×103 | †>5×103 | 10g | * N.E. (500) | * N.E. (104) | * N.E. (104) | ||||
N.E.: no effect was observed up to concentration tested indicated in parentheses.
activity was observed, but IC50 values were not determined due to incomplete data sets for curve fitting.
95% confidence interval for IC50 value.
In vitro translation assay
pSP/(CAG)33/FF/HCV/Ren.pA518, 31 was linearized using BamHI. In vitro transcription was carried out using RiboMAX™ SP6 Large Scale RNA Production System (Promega). Briefly, 100 μL reactions containing 5 μg linearized DNA, 20 μl SP6 Transcription buffer, 20 μl rNTPs, 10 μl SP6 Transcription enzyme and nuclease-free water were mixed at room temperature and incubated at 37°C for 4 h. 10 μl of RQ-1DNase (Promega) was added followed by extraction using Trizol (Life Technologies, Grand Island, NY). RNA was purified as per instructions for E.Z.N.A Mag-Bind mRNA Enrichment Kit (Omega Biotek). Translation was performed using the Flexi Rabbit Reticulocyte Lysate System (Promega). 200 ng of RNA was combined in 20 μl reactions containing 10 μl RRL, 0.2 μl each of –Met and –Leu amino acid mixtures, 70 mM KCl, 2 mM DTT, 10 U RNasin and 1 μl of DMSO or compound dissolved in DMSO. Reactions were incubated at 30 °C for 1.5 h, and then 5 μL were assayed for luciferase activity as per instructions of Dual Luciferase reporter Assay System (Promega). Each independent assay was performed in duplicate, and each compound was tested in 4 independent assays. IC50 determination was determined by curve fitting using Prism® 5 (GraphPad Software, Inc.) and reported IC50 values (Table 3) are means of all independent assays.
Table 3.
SAR study of cap-dependent translation inhibition activity for second-generation DMDAPatA derivatives.
 Series I |
 Series II |
 Series III |
||||
|---|---|---|---|---|---|---|
| R | Compd | IC50 (nM) | Compd | IC50 (nM) | Compd | IC50 (nM) |
| Br | 5 | * N.E. (500) | 4 | ND | 6 | ND |
|
2 | 21.5 ± 2 | 10a | * N.E. (500) | 8 | †>500 |
|
9b | 72.2 ± 1 | 10b | ND | ||
|
9c | 54.8 ± 2 | 10c | ND | ||
|
|
9d | * N.E.(500) | 10d | ND | ||
|
|
9e | †>500 | 10e | N.D. | ||
|
|
9f | * N.E.(500) | 10f | ND | ||
|
|
9g | * N.E.(500) | 10g | ND | ||
ND, not determined, compounds were not assayed due to lack of activity in cell proliferation assay (see Table 2)
IC50 values reported with ± 1 standard deviation of the mean
N.E., no effect was observed up to concentration tested indicated in parentheses.
activity was observed, but IC50 values were not determined due to incomplete data sets for curve fitting.
Stability assay
A stock solution of 2, 9b, or 9c in DMSO was added to the pre-heated mixture of serum (0.2 mL) (Human blood serum from male AB plasma, HBAB, Sigma or Fetal Bovine Serum, FBS, GIBCO) and RPMI 1640 (1.8 mL) to reach the final concentration of 10 μM or 1 μM. The mixture was heated at 37 °C. At regular time intervals 200 μL was removed and mixed with 400 μL of CH3CN. The mixture was vortexed to precipitate out the proteins. Then 400μL of the clear solution was mixed with 5 μL of internal standard (a solution of naphthalene in CH3CN, 1mg/mL) followed by HPLC (Phenomenex® Gemini-NX 3μ C18 110A. Size: 150 × 4.60 mm) using a mobile phase A of HCOOH-NH3·H2O in CH3CN/water (9:1 v/v) at pH 9.5; the mobile phase B was a buffer solution of HCOOH-NH3·H2O in CH3CN at pH 9.5. The conversion % of the compounds was calculated based on the integration ratio of the substrate peak and the internal standard peak. All compounds were assayed in triplicate. For stability in RPMI without serum, the stock solutions of 2, 9b, or 9c were added to 2 mL of RPMI to reach the final concentration of 10 μM or 1 μM. The remaining procedure was the same.
3. Results and Discussion
First generation PatA derivatives
Given the new insights into the mechanism of PatA since our initial SAR studies, we have sought to re-evaluate a selection of the derivatives 2 and 3a–g previously presented by Romo et al.29 The structures are shown in Figure 2 and Table 1. For comparison purposes, the new cell proliferation data for these analogs is presented alongside the previously reported activity of the first generation PatA derivatives in the IL-2 assays (Table 1). In all human cell lines tested (HeLa, cervical carcinoma, RKO, colon carcinoma, and Jurkat immortalized T cells), PatA demonstrated sub-nanomolar IC50 values. Similar IC50 values for proliferation inhibition were also observed in the immortalized human keratinocyte cell line HaCaT cells and in primary proliferating BAEC (bovine aortic endothelial) cells. These sub-nanomolar IC50 values for anti-proliferative activity were in good agreement with the single digit nanomolar IC 50 (4 nM) for the IL-2 reporter assay performed in Jurkat T cells. The anti-proliferative IC50 values for the highly active derivative DMDA-PatA (2) also demonstrated good agreement with previously reported IC50 values for IL-2 reporter assay (single-digit nanomolar for proliferation versus sub-nanomolar for IL-2 reporter assay).
Figure 2.
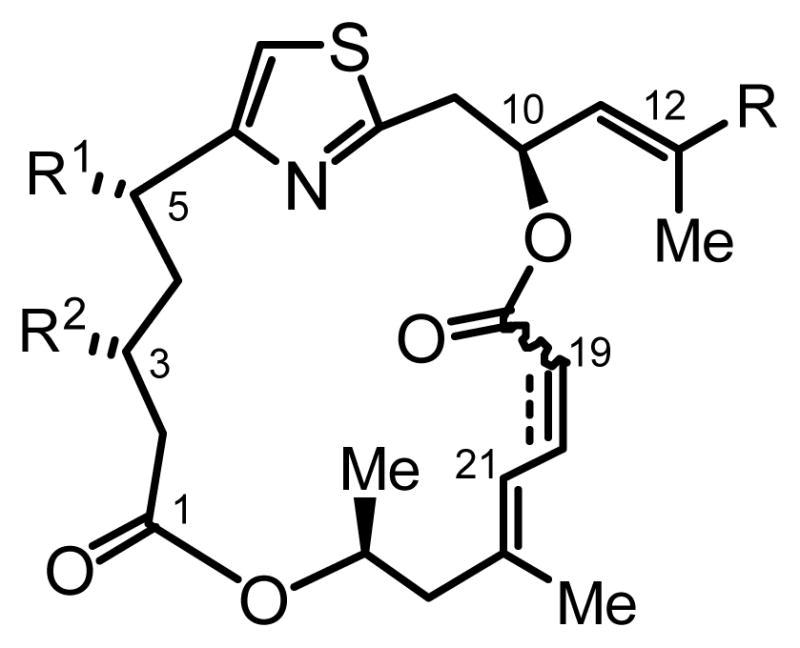
General structures of PatA derivatives 2 and 3a–g. For first generation PatA analogs see Table 1 for modifications at R1, R2 and R. The fourth site of derivatization was level of unsaturation at C19–C20.
Table 1.
SAR study of cell proliferation inhibition activity of first-generation Pateamine A derivatives
| Cmpd | R | R1 | R2 | C19–C20 | IC50 (nM) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| HeLa | RKO | HaCaT | Jurkat T | BAEC | IL-2 reporter gene assay* | |||||
| PatA (1) |
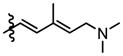
|
Me | NH2 |
|
0.72 ± 0.08 | 0.38 ± 0.02 | 0.42 ± 0.03 | 0.60 ± 0.02 | 0.5 ± 0.04 | 4.0 ± 0.9 |
| DMDA PatA (2) |
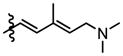
|
H | H |
|
4.5 ± 0.4 | 1.2 ± 0.05 | 0.9 ± 0.1 | 3.0 ± 0.1 | 1.97 ± 0.1 | 0.81 ± 0.3 |
| 3a |
|
Me | NHBoc |
|
ND | ND | >100 | ND | >20 | 330 ± 120 |
| 3b |
|
Me | NHBoc |
|
ND | ND | ND | ND | ND | >1000 |
| 3c |
|
Me | NHBoc |
|
ND | ND | ND | ND | ND | >1000 |
| 3d |
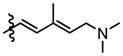
|
Me | NHC(O)CF3 |
|
>100 | >100 | >100 | ND | ND | 300 ± 93 |
| 3e |
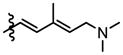
|
Me | NHC(O)Ph |
|
16.1 ± 0.7 | 4.2 ± 0.5 | 3.4 ± 0.4 | 11.6 ± 1.4 | 12.8 ± 0.3 | 15 ± 6.1 |
| 3f |
|
Me | NHBoc |
|
>100 | >100 | >100 | ND | >20 | 340 ± 180 |
| 3g |
|
Me | NHBoc |
|
>100 | >100 | >100 | ND | >20 | 55 ± 16 |
data previously reported in Romo et al.29
ND, not determined
IC50 values reported with ± 1 standard deviation of the mean
Given limited availability of the first generation derivatives, proliferation assays were only performed to concentrations of 100–200 nM leading to an inability to determine precise IC50 value estimates for the majority of derivatives. In addition, not all of the compounds in Table 1 were tested in all cell lines. Nevertheless, as can be seen in Table 1, all the derivatives that demonstrated poor activity or no activity in the IL-2 reporter assay also showed poor activity in the cell proliferation assays. The only derivative aside from 2 with reasonable activity, was the benzoyl protected β-amino derivative 3e (R2 = PhC(O)) which demonstrated anti-proliferative activity on par with the previously reported IL-2 reporter assay inhibition activity (IC50 values ranging from 3.4 ± 0.4 to 16.1 ± 0.7 nM (cell proliferation) versus 15 ± 6.1 nM (IL-2)).
Given the literature published since our initial SAR and the results presented here, we propose that the general trends of our previously reported IC50 values for IL-2 inhibition by PatA and its derivatives may be a result of the anti-proliferative effects of the compound(s) as the assay for determining these values was a cell-based assay. A complication of utilizing the cell-based assay for assessing inhibition of IL-2 promoter activity is that PatA/DMDAPatA have well documented cytotoxic effects on cells in culture, including Jurkat T cells, and induce apoptosis. Thus, read-out of IL-2 transcription under these conditions may also be impacted by other signaling events. Furthermore, IL-2 reporter gene read-out (i.e., luciferase activity) would also rely on translation of the reporter gene mRNA. It is our contention that as the IL-2 reporter assay was performed in a cellular environment, namely in cultured Jurkat T cells, the conclusions of the previous SAR are not invalidated with respect to anti-proliferative activity and targeting of eukaryotic translation initiation. Due to the good agreement between the cell proliferation assays and the IL-2 reporter assay, we believe our previous conclusions are still valid: (i) the methyl at C3 and the primary amino group at C5 are not essential for high potency, and (ii) the overall structure of PatA consists of a flexible scaffold domain (C1–C5) and a rigid binding domain (C6–C25) consisting of the eastern half of the macrolide ring and the side chain.
Synthesis of second-generation PatA derivatives
In light of the new cellular mechanism of action studies since our initial SAR study, we sought to further evaluate the PatA family with a new series of derivatives specifically targeting the previously identified rigid binding domain (Figure 3). This second-generation of analogs was generated to examine more closely the side chain (R in Figure 2) and the olefin geometry of C19–C20. Given the fact that the substitutions at C3 and C5 provided only marginal improvement, i.e., the simplified derivative DMDAPatA (2) is only slightly less active than PatA (1), and that DMDAPatA is more easily accessible by chemical synthesis (the synthesis of DMDAPatA is 10 fewer steps than that of PatA for the longest linear sequence), the second-generation of derivatives were designed based on DMDAPatA as a starting point with a focus on modifying the side chain and the diene system, while leaving C3 and C5 unsubstituted.
Figure 3.
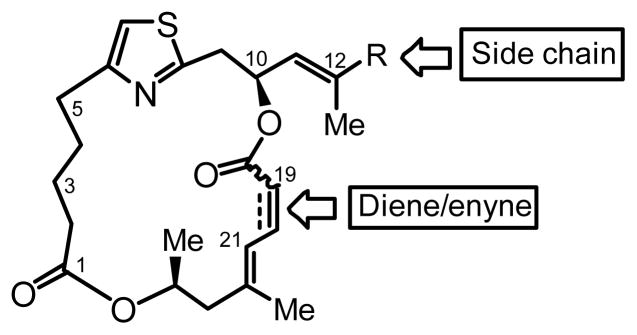
Sites of derivatization for second-generation PatA analogs. Diversification of: (i) the side chain R with different olefins with amino or oxygen terminus, and (ii) the C19–C20 bond as alkyne, cis-alkene or trans-alkene. Second-generation derivatives were unsubstituted at C3 and C5 and were based on DMDAPatA (2).
The synthesis of this new series of second-generation derivatives (5, 6, 8, 9b–g, 10a–g) is shown in Scheme 1. The key intermediate, enyne macrocycle 4, was synthesized based on our previously reported procedure shown in abbreviated form in Scheme 1.5, 29 Hydrogenation of 4 with Lindlar’s catalyst under particular conditions gave a mixture of Z-and E-dienes (5 and 6) that were separable on silica gel. A series of vinylstannane reagents (Bu3SnR, 7a–g) were prepared following known procedures enabling preparation of several side-chain variants.5, 29 Application of the Stille coupling allowed access to enyne macrocycle derivatives 10a–g from enyne macrocycle 4 and organostannanes 7a–g using Pd(PPh3)4 or Pd(AsPh3)4. Alternatively, use of diene macrocycles 5 and 6 led to E-diene derivative 8 and Z-dienes 9b–g including DMDAPatA (2). Using the described routes, a total of 15 new DMDAPatA derivatives were synthesized in addition to further quantities of DMDAPatA for comparative biological assays.
Scheme 1.

Synthesis of second-generation of PatA analogs. Only the final synthetic steps are shown in detail, including (i) reduction of the enyne core 4 to dienes 5 and 6; (ii) Stille couplings between the brominated macroclycles (4, 5, and 6) and organo tin reagents (7a–g) to form 9a–g and 10a–g in moderate yields. All other synthetic steps leading to 4 were based on our previous reports5, 29 and are not shown in detail.
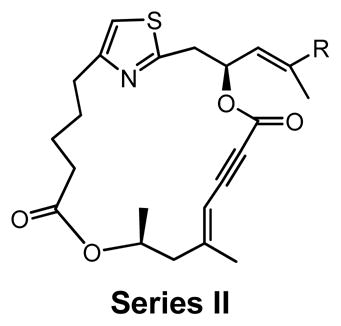
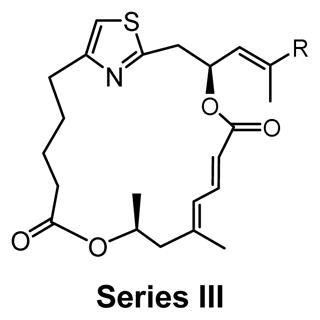
Biological activity of second-generation DMDAPatA derivatives
As shown in Table 2, the second-generation derivatives were segregated into 3 new series (series I, II and III) based on the structural characteristics of the macrolide ring. Series I derivatives (5, 2, 9b–g) contained the same macrolide as DMDAPatA (2), series II derivatives (4, 10a–g) possessed an enyne moiety (C19–C23), and series III derivatives (6 and 8) consisted of the diene with the trans-configuration of the C19–C20 olefin. These analogs were tested for cell proliferation inhibition activity in three cell lines, HeLa, Jurkat T cells, and SK-MEL2. HeLa and Jurkat T cells were included for comparison with the first generation analogs, and SK-MEL2 was included given the in vivo activity previously observed against melanoma animal models.21 Full data sets for SK-MEL2 cells are presented in Figure 4 because of this in vivo data. Although a slightly different protocol was utilized for the second-generation analogs (see Materials and Methods, MTT assay versus tritiated thymidine incorporation, second-generation versus first generation respectively), the results were comparable between both methods for DMDAPatA (4.5 nM and 3 nM for tritiated thymidine incorporation for HeLa and Jurkat T cells respectively versus 9.8 nM and 11 nM for MTT assay). To obtain a more accurate evaluation, compounds in the second-generation were also tested with much higher concentrations.
Figure 4.
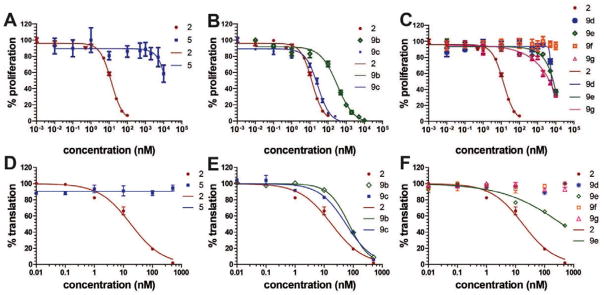
Anti-proliferative activity against SK-MEL2 melanoma cells and cap-dependent translation inhibition activity of second- generation DMDAPatA derivatives. (A–C) The SK-MEL2 melanoma cells were cultured in the presence of indicated compounds at indicated concentrations followed by determination of cellular proliferation. DMSO was present at final concentration of 0.1%. Data points were collected in quadruplicate, and two independent assays were performed for each compound, with each independent assay producing similar results (only one independent assay set is shown). Data points are the mean of the quadruplicate readings with ± 1 S.E.M. represented by the error bars. (D–F) In vitro inhibition of cap-dependent translation in rabbit reticulocyte lysate using a luciferase reporter gene. Assays included indicated concentrations of compounds dissolved in DMSO at 5% (v/v) in the final reaction. Four independent assays with duplicate readings were performed for all compounds, and one representative data set is shown. Data points are the mean of the two replicates and error bars represent ± 1 S.E.M. In all panels, compounds were compared with DMDAPatA (2). Sigmoidal dose-response curves were generated by curve-fitting to data points and legends indicate curves associated with each compound. See main text for description of compound structures and Tables 2 and 3 for IC50 values.
In addition to cell proliferation assays, the second-generation analogs were also tested in a rabbit reticulocyte lysate (RRL) in vitro translation assay using a bicistronic vector where translation at the first cistron was initiated by cap-dependent means (eIF4AI/II-dependent) and the second cistron was translated using the hepatitis C virus (HCV) internal ribosome entry site (IRES) (eIF4AI/II-independent). This dual-luciferase system was previously used in the target/mechanism identification of PatA function.8, 9, 31 Only a subset of second-generation analogs were tested in the translation assay and were chosen for evaluation based on the cellular activities reported in Table 2. All analogs reported did not show any inhibitory activity towards cap-independent HCV-IRES (eIF4AI/II-independent) initiated translation at the highest concentrations tested (data not shown), thus exhibiting similarities to the parent compounds PatA and DMDAPatA. All further discussion of translation inhibition is of cap-dependent (eIF4AI/II-dependent) translation initiation.
Series I derivatives: investigating the trienyl amine side chain of DMDAPatA
The brominated derivative 5, prior to Stille coupling, showed a significantly reduced potency, resulting in at least a 700-fold loss in activity for SK-MEL2 and Jurkat T cells (Table 2 and Figure 4). For HeLa cells, no activity was observed for derivative 5 up to the highest concentration tested (500 nM, data not shown). Similarly, no activity was observed for 5 in the translation assay (Table 3 and Figure 4). These results indicate that the trienyl amine side chain is likely an absolute requirement for activity.
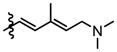
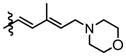
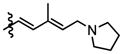
Compounds 9b and 9c were utilized to investigate the structural variation possible with the tertiary amine found in the trienyl side chain. Furthermore, as a means to increase the metabolic stability of DMDAPatA by precluding the N-demethylation metabolic process32, the N,N-dimethyl tertiary amine of 2 was replaced with a morpholine ring (9b) or a pyrollidine ring (9c), with both derivatives still possessing tertiary amines. Substitution with the morpholine ring resulted in a greater than 400-fold loss of activity in HeLa cells, at least 700-fold loss in Jurkat T cells and an approximately 25-fold loss in SK-MEL2 cells (Table 2 and Figure 4). Interestingly, substitution with a pyrrolidine ring (9c) resulted in a comparably active compound with only an approximate 2-fold loss of activity in all cell lines. However, in in vitro translation assay, the morpholine substitution (9b) showed only a roughly 3-fold loss of activity in cell assay which was comparable to the pyrollidine substitution that demonstrated again a 2-fold loss in activity (Table 3).
These results suggest that there may be a larger pocket available near the N,N-dimethyl tertiary amine binding site in the target protein (assuming that each analog still properly targets the eIF4A proteins within cells). The dramatically decreased activity of the morpholine moiety may be due to the larger 6-membered ring and/or electrostatic repulsion, i.e., due to the presence of the electron pairs on oxygen, prohibiting strong target binding. However, this seems unlikely as only a modest loss of activity was observed in in vitro translation assay. It is also interesting to note that the morpholine ring was slightly better tolerated in terms of activity in the SK-MEL2 cell line compared to other cell lines considering the in vivo activity of 2 in the melanoma animal model noted above. The dramatic loss in activity for 9b compared to 9c in the cellular assay, but comparable activities (less than 2-fold difference) in in vitro translation assays (Figure 4, panel B vs. panel E) may suggest cell penetration effects or other target accessibility issues such as sequestration by other cellular targets were responsible for the loss in activity. The N,N-dimethyl and pyrolidine substituents (2 and 9c respectively) have similar pKa values of approximately 11 while the morpholine (9b) has a significantly less basic pKa value of 8.36, thus the base characteristics of the terminal functional group of the side chain arm may be an important feature for cell permeability. Furthermore, the differential degrees of loss of activity for 9b in cell assays may suggest that future substitutions of the N,N-dimethyl tertiary amine could generate increased cell specificity for anti-proliferative activity. Other possibilities include differential metabolism of the compounds and further studies are warranted as the potential causes presented here are speculative and are not exhaustive.
The remainder of series I consisted of more dramatic structural changes to the trienyl amine side chain involving replacement of the N,N-dimethyl tertiary amine with ether (9d, 9f) and alcohol (9e, g) functionalities and decreased rigidity in the side chain by removal of a vinyl methyl substituent at C15 and one unsaturation at C15–C16. In all instances, a significant loss (approximately 500-fold) of potency was observed in cell proliferation assays. These losses in activity were also observed in in vitro translation assays, with only 9e demonstrating a degree of activity at high concentration (Figure 4F). A comparison of 9e activity in translation assay with 9g, where the only structural difference was the loss of unsaturation at C15–C16, demonstrates again the importance of the rigidity of the side chain. The comparable pair of analogs, 9d and 9f did not show this trend, however, for these analogs, the side chain terminated in an ether functionality in place of the alcohol found in 9e and 9g, suggesting that identity of the terminal functional group may supersede side chain rigidity in determination of translation inhibition activity in vitro. Also of note is that 9f, the analog possessing the same macrolide as 2, but with the most flexible side chain terminating in an ether demonstrated the lowest levels of activity across all cell lines and in the translation assay. However, these analogs (9d–g) all demonstrated significant loss of activity in both cell proliferation assay and in translation assay compared to 2, 9b and 9c (trienyl amine side chains).
Although the ether and alcohol substituted derivatives, 9d and 9e lacked a methyl moiety at C15, the unsaturation of the side chain arm would still confer rigidity. Under the assumption that a loss of the methyl substituent at C15 does not significantly impact target protein binding, the dramatic loss of activity for 9d and 9e in comparison to the tertiary amine terminated derivatives (2, 9b and 9c) in the translation assay in conjunction with the lower activity observed for 9b may also suggest a basic requirement at this location for protein target binding as well as for cell permeability as discussed above. Due to the fact that at least two structural changes from DMDAPatA were made for 9d-g, it is difficult to assess the individual contributions to activity for each of the altered structural features. Nevertheless, the results support the requirement of a rigid side chain with a tertiary amine at the terminus of the chain for highest potency.
Series II derivatives: Probing diene versus enyne macrocycles
In series II derivatives, the diene structure (C19–C22) was replaced with an enyne structure and all derivatives demonstrated a significant loss of activity in cell proliferation assay (Table 2). This result was not surprising as the enyne imparts a major change in the expected preferred conformations of the macrocycle, a feature previously found to be important for activity (Table 1).29 A direct evaluation of the enyne replacement can be made by comparing DMDAPatA (2) to enyne 10a, where this is the only structural difference. For all cell lines tested, no effect was observed with enyne 10a up to 50 times the IC50 values for DMDAPatA (2) and no effect was observed on cap-dependent translation for 10a up to the highest concentration tested (Table 3).
Series III derivatives: comparing Z versus E configuration of the C19-C20 olefin
Two E,E-diene derivatives 6 and 8 were synthesized in series III to again investigate the effects of altering the macrocycle conformation, which was previously proposed to be part of the binding domain of the macrolide ring. Bromo derivative 6, the E-isomer of compound 5, also lacks the side chain. As expected from the results of 5, derivative 6 was also inactive in cell proliferation assay. Compound 8, however, contained the same side chain as 2 with the only structural difference being the E,E-diene. In all three cell lines tested, E,E-diene 8 demonstrated a 50- to 100-fold loss in activity, again suggesting that the degree of unsaturation and, therefore the macrocycle conformation, has a dramatic impact on potency. In translation assay, 8 demonstrated only minor activity (~80% translation at 500 nM, data not shown) in agreement with cell proliferation data.
Comparing activity trends between the first and second- generation derivatives
The first- and second-generation derivatives were designed to interrogate different regions of the starting compounds, PatA (1) and DMDAPatA (2). The first generation derivatives focused on simplifications of the more flexible western half of the macrolide in addition to changes to the more rigid eastern region, the presumed binding domain. The second-generation series focused more specifically on the latter regions of these eIF4A inhibitors. When the diene was replaced by the enyne, the trend of compounds in both Tables 1 and 2 was toward loss of activity. One discrepancy between the data for the two series was that 3f and 3g (C19–C22 enyne structure) still retained a degree of activity in the IL-2 reporter assay (340 nM and 55 nM respectively), but the second-generation derivatives with the C19–C22 enyne structure and identical side chain arms, 10g and 10f respectively, did not demonstrate any significant cell proliferation inhibition activity. For the comparisons of 3f and 3g to 10g and 10f, two items should be noted, i) the observed activity for 3f and 3g was in the IL-2 assay and ii) the structures were not equivalent outside of the side chain as 3f and 3g possessed substitution at C3 (methyl moiety) and C5 (NHBoc).
Thus, even though it is not possible to make a direct comparison, it is conceivable that the other structural differences resulted in a compensatory effect not found for 10f and 10g. Another possibility is that PatA and its derivatives do possess IL-2 transcriptional inhibition at higher concentrations that are masked by the more potent anti-proliferative activity. By reducing or eliminating the anti-proliferative activity in 3f and 3g, the anti-IL-2 transcriptional activation activity may have been revealed. This intriguing possibility could be further explored as our original interest in PatA lay in the development of a novel immunosuppressive agent.29 Furthermore, the more potent IL-2 inhibitor derivative 3g possessed the least rigid side chain, and the ether terminal functional group, which appears to contribute to loss of inhibition of eIF4A-dependent translation initiation as demonstrated by 9f. In the context of this line of analysis, it is also interesting to note that in our initial target identification9 we found a second protein, serine-threonine kinase receptor-associated protein (STRAP), also known as UNR-interacting protein (unrip), that was also capable of specific interaction with our biotin-conjugated PatA derivative. STRAP/unrip is a component of the SMN (survival motor neuron) complex33, 34 which is necessary for assembly and cellular localization of spliceosomal small nuclear ribonucleoproteins (snRNPs) and named for its interaction with the TGF-β receptor.35 Although the biochemical mechanisms of action of STRAP/unrip are not worked out to the detail of the eIF4As, it is possible that the IL-2 inhibiting activity may act through this alternate protein target. More detailed follow-up studies would be necessary to investigate these possibilities, which is beyond the scope of this report and it should also be noted that studies by others did not identify STRAP/unrip as a protein target of PatA.8 Furthermore, potential alternative targets such as DNA polymerases α and γ21 have not been investigated here. Activity against alternative targets unrelated to eukaryotic cap-dependent translation initiation cannot be assessed at this time.
The general trend from both generations of derivatives was that loss of side chain rigidity in the binding domain resulted in loss of biological activity. However, derivative 3a, which lacked one unsaturation (C15–C16) and the methyl group at C15, did not have a total loss of activity in the IL-2 assay. In this case, the retained activity observed in the IL-2 assay may again be due to other compensatory effects imparted by the presence of the N,N-dimethyl tertiary amine in the side chain and the presence of substitution at C3 and C5. We again cannot exclude the possibility that this analog also possessed IL-2 inhibitory activity independent of cell proliferation inhibition activity.
Selectivity towards rapidly proliferating cells
PatA has demonstrated selectivity towards rapidly proliferating cells over slower growing cells.7, 21 To confirm that this selectivity was retained in the active analogs, DMDAPatA (2), 9b and 9c were tested in proliferation assays performed on SK-MEL2 cultured with reduced serum, 5% FBS, to reduce cell proliferation (see Supplemental Material, Figure S1, panels A–C). 2 demonstrated roughly 2-fold reduced anti-proliferative activity with an IC50 value of 30.0 nM (95% C.I. of 22.6–38.7 nM) under these conditions. 9c also demonstrated reduced activity of nearly 6-fold with an IC50 value of 154.0 nM (95 % C.I. of 95.7–248 nM). Interestingly, compound 9b demonstrated an approximately 2-fold increase of activity with an IC50 value of 157 nM (95% C.I. of 118–210 nM). It should be noted that there was reduced confidence in the precision of the IC50 values for 5% FBS conditions as 100% inhibition was not observed for the highest concentrations tested. In addition, examination of the data for 9b (see Supplementary Material Figure S1B) may suggest less of an activity difference under 5% FBS compared to 10% FBS when the complete data sets are examined instead of comparing only the IC50 values. Nevertheless, given the fact that 9b consistently demonstrated higher, and more variable IC50 values for cell proliferation inhibition under 10% FBS (see Table 2), the results for 9b may indicate that the proposed cell permeability or target accessibility issues associated with this compound may be independent of the state of cell proliferation.
Compounds 2, 9b and 9c were also assayed for anti-proliferative activity against slower growing immortalized mouse embryonic fibroblasts (MEFs). All three compounds did not demonstrate 100% inhibition at the highest concentrations tested (see Supplemental Material Figure S1D). For 2, the IC50 value was estimated to be >50 nM, giving a roughly 5-fold decrease in activity compared to cancer cell lines. 9b also demonstrated an estimated 5-fold loss in activity compared with cancer cell lines with an estimated IC50 value of >2 μM. It again should be noted that 9b demonstrated the greatest variability amongst the three compounds between different cell lines. 9c demonstrated an estimated 4- to 5-fold loss in activity with an estimated IC50 value >100 nM. These results along with the observations under 5% FBS for SK-MEL2 strongly suggest that selectivity towards rapidly proliferating cells was retained within the active analog 9c and also for 9b against certain cell-types. Accordingly, further modifications at the terminus of the triene side chain may be possible to further increase selectivity.
Compound stability
In order to assess the potential to develop inhibitors 2, 9b and 9c as anticancer agents, a series of solution stability studies were conducted in RPMI-1640 formulation alone (control) and in the presence of plasma/serum on these three compounds (HBAB: Human blood Serum from male AB plasma and FBS: Fetal Bovine Serum, Table 4). Several trends were observed in the analysis. First, all three compounds were more stable in the presence of serum/plasma than in RPMI alone. Second, in RPMI and FBS, the morpholine analog 9b was less stable than the other two derivatives. However, in the human plasma, 9b was significantly more stable at a concentration of 10 μM than 2 and 9c. Finally, 2 was more stable at the lower concentration of 1 μM in all three mediums. The greater stability of all three compounds in serum/plasma was likely due to binding to plasma proteins however, other serum components cannot be ruled out. Binding to plasma proteins almost certainly protects the compounds from degradation in the slightly basic (pH 7.5) RPMI medium. The decomposition of the compounds in RPMI medium may be due to base catalyzed hydrolysis of the macrolide C18-ester to form an acyclic compound. Future assessment includes serum protein-binding assay, identification of the product(s) of the decomposition and identification of formulations with improved stability of the derivatives.
Table 4.
Half-lives of tertiary amine side chain terminated derivatives of PatA in cell culture media
| compd conc. | 2 1 μM |
2 10 μM |
9b 10 μM |
9c 10 μM |
|
|---|---|---|---|---|---|
| t1/2 (h) | HBAB | 24.0 ± 2.08 | 13.0 ± 1.01 | 17.8 ± 1.63 | 11.7 ± 0.15 |
| FBS | 19.9 ± 1.57 | 12.5 ± 0.81 | 9.23 ± 0.28 | 14.8 ± 0.82 | |
| RPMI | 5.11 ± 0.12 | 3.25 ± 0.28 | 0.89 ± 0.043 | 3.87 ± 0.58 |
HBAB: human blood serum and RPMI media
FBS: fetal bovine serum and RPMI media
RPMI: (control) RPMI media only
Half-lives are reported with ± 1 standard deviation
4. Conclusions and Future Directions
The results presented here further re-affirm our previous SAR findings for the eukaryotic initiation inhibitors in the PatA family. Given the fact that we have reassessed a selection of our previous compounds (first generation analogs) and have presented a second-generation of analogs, we conclude that our previous findings of a flexible scaffolding region in the western half of the macrolide ring, and a more rigid binding domain consisting of the eastern half of the macrolide and a rigid side chain arm are still valid despite the fact that our previous assay was for IL-2 transcriptional inhibition.
One assumption being made in this study is that each of the second-generation analogs that demonstrated cellular activity still functions in a mechanistically similar manner as DMDAPatA (2). The in vitro translation assay data strongly suggests that the active second-generation analogs do target eIF4A as only eIF4A-dependent translation was inhibited. In addition, we have demonstrated that replacement of the diene with an enyne structure at C19–C22 significantly abrogated potency, and that the E,Z-diene stereochemistry was absolutely critical for activity. Our investigation of the terminal N,N-dimethyl tertiary amine on the rigid side chain suggests that this region of the molecule may present further opportunities for derivatization to address any solubility or metabolism liabilities as DMDAPatA progresses into preclinical studies. In particular, the greater stability of a pyrrolidine ring over a dimethyl amino group in the presence of human serum is an important finding that can be used for further preclinical studies of this family of anticancer agents.
The terminal position of the triene may also serve as a site of modification for increasing cell-type specificity as the morpholine ring substitution (9b) resulted in drastic loss of cellular activity, but to different degrees for the different cell lines tested, and only a more moderate loss in cap-dependent translation inhibition was observed. The potential importance of the terminal position of the triene for cell-type specificity and selectivity against rapidly proliferating cells is also supported by the results observed under slower growth conditions. Side chain substituents alone (R-groups) should also be assayed in biological and biochemical assays. This would help in identifying potential metabolic breakdown products that may be responsible for activity. It is acknowledged that further substitutions at C3 and C5 should also be explored such as re-installation of the C3-amino and C4-methyl given the comparative potencies observed between 3a, 3f, and 3g with the second-generation compounds. This is also reflected in the fact that DMDAPatA, while highly potent, has consistently demonstrated roughly 10-fold lower activity than PatA in cell proliferation assays yet DMDAPatA demonstrated greater inhibition in IL-2 reporter gene assay, again taking into account the limitations of this assay noted above (see Table 1).
Supplementary Material
Acknowledgments
We thank the NIH (GM052964, D.R), St. John’s University (W.L.) and FAMRI (J.O.L.) for support of this work. The Office of the Vice President for Research, the College of Science, and the Department of Chemistry at Texas A&M provided seed funding for the TAMU Natural Products LINCHPIN Laboratory. We thank Dr. Jerry Pelletier (McGill University) for providing the in vitro bicistronic reporter vector and Dr. Andre Nussenzweig (NIH) for provision of immortalized MEFs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2012;75:311. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li JW, Vederas JC. Science. 2009;325:161. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 3.Zhang MQ, Wilkinson B. Curr Opin Biotechnol. 2007;18:478. doi: 10.1016/j.copbio.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Carlson EE. ACS Chem Biol. 2010;5:639. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rzasa RM, Shea HA, Romo D. Journal of the American Chemical Society. 1998;120:591. [Google Scholar]
- 6.Hood KA, West LM, Northcote PT, Berridge MV, Miller JH. Apoptosis. 2001;6:207. doi: 10.1023/a:1011340827558. [DOI] [PubMed] [Google Scholar]
- 7.Northcote PT, Blunt JW, Munro MHG. Tetrahedron Letters. 1991;32:6411. [Google Scholar]
- 8.Bordeleau ME, Matthews J, Wojnar JM, Lindqvist L, Novac O, Jankowsky E, Sonenberg N, Northcote P, Teesdale-Spittle P, Pelletier J. Proc Natl Acad Sci U S A. 2005;102:10460. doi: 10.1073/pnas.0504249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Low WK, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, Liu JO. Mol Cell. 2005;20:709. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Parsyan A, Svitkin Y, Shahbazian D, Gkogkas C, Lasko P, Merrick WC, Sonenberg N. Nat Rev Mol Cell Biol. 2011;12:235. doi: 10.1038/nrm3083. [DOI] [PubMed] [Google Scholar]
- 11.Rocak S, Linder P. Nat Rev Mol Cell Biol. 2004;5:232. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 12.Rogers GW, Jr, Komar AA, Merrick WC. Prog Nucleic Acid Res Mol Biol. 2002;72:307. doi: 10.1016/s0079-6603(02)72073-4. [DOI] [PubMed] [Google Scholar]
- 13.Andreou AZ, Klostermeier D. RNA Biol. 2013;10:19. doi: 10.4161/rna.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinnebusch AG, Lorsch JR. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hershey JWB, Sonenberg N, Mathews MB. In: Protein Synthesis and Translational Control; Hershey JWB, Sonenberg N, Mathews MB, editors. Cold Spring Harbor Laboratory Press; New York: 2012. p. 1. [Google Scholar]
- 16.Bono F, Gehring NH. RNA Biology. 2011;8:24. doi: 10.4161/rna.8.1.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bordeleau ME, Cencic R, Lindqvist L, Oberer M, Northcote P, Wagner G, Pelletier J. Chem Biol. 2006;13:1287. doi: 10.1016/j.chembiol.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Low WK, Dang Y, Bhat S, Romo D, Liu JO. Chem Biol. 2007;14:715. doi: 10.1016/j.chembiol.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 19.Dang Y, Kedersha N, Low WK, Romo D, Gorospe M, Kaufman R, Anderson P, Liu JO. J Biol Chem. 2006;281:32870. doi: 10.1074/jbc.M606149200. [DOI] [PubMed] [Google Scholar]
- 20.Dang Y, Low WK, Xu J, Gehring NH, Dietz HC, Romo D, Liu JO. J Biol Chem. 2009;284:23613. doi: 10.1074/jbc.M109.009985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuznetsov G, Xu Q, Rudolph-Owen L, Tendyke K, Liu J, Towle M, Zhao N, Marsh J, Agoulnik S, Twine N, Parent L, Chen Z, Shie JL, Jiang Y, Zhang H, Du H, Boivin R, Wang Y, Romo D, Littlefield BA. Mol Cancer Ther. 2009;8:1250. doi: 10.1158/1535-7163.MCT-08-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blagden SP, Willis AE. Nat Rev Clin Oncol. 2011;8:280. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]
- 23.Le Quesne JP, Spriggs KA, Bushell M, Willis AE. J Pathol. 2010;220:140. doi: 10.1002/path.2627. [DOI] [PubMed] [Google Scholar]
- 24.Lee T, Pelletier J. Future Med Chem. 2012;4:19. doi: 10.4155/fmc.11.150. [DOI] [PubMed] [Google Scholar]
- 25.Lindqvist L, Pelletier J. Future Med Chem. 2009;1:1709. doi: 10.4155/fmc.09.122. [DOI] [PubMed] [Google Scholar]
- 26.Malina A, Mills JR, Pelletier J. Cold Spring Harb Perspect Biol. 2012;4:a012377. doi: 10.1101/cshperspect.a012377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polunovsky VA, Bitterman PB. RNA Biol. 2006;3:10. doi: 10.4161/rna.3.1.2718. [DOI] [PubMed] [Google Scholar]
- 28.Sonenberg N, Hinnebusch AG. Mol Cell. 2007;28:721. doi: 10.1016/j.molcel.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 29.Romo D, Choi NS, Li S, Buchler I, Shi Z, Liu JO. J Am Chem Soc. 2004;126:10582. doi: 10.1021/ja040065s. [DOI] [PubMed] [Google Scholar]
- 30.Di Marco S, Cammas A, Lian XJ, Kovacs EN, Ma JF, Hall DT, Mazroui R, Richardson J, Pelletier J, Gallouzi IE. Nat Commun. 2012;3:896. doi: 10.1038/ncomms1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novac O, Guenier AS, Pelletier J. Nucleic acids research. 2004;32:902. doi: 10.1093/nar/gkh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meegan MJ, Lloyd DG. Frontiers in Medicinal Chemistry - Online. 2005;2:183. [Google Scholar]
- 33.Carissimi C, Baccon J, Straccia M, Chiarella P, Maiolica A, Sawyer A, Rappsilber J, Pellizzoni L. FEBS Lett. 2005;579:2348. doi: 10.1016/j.febslet.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 34.Grimmler M, Otter S, Peter C, Muller F, Chari A, Fischer U. Hum Mol Genet. 2005;14:3099. doi: 10.1093/hmg/ddi343. [DOI] [PubMed] [Google Scholar]
- 35.Datta PK, Chytil A, Gorska AE, Moses HL. J Biol Chem. 1998;273:34671. doi: 10.1074/jbc.273.52.34671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.