

Abstract
Tuberous sclerosis complex (TSC) is characterized by the formation of tumors in multiple organs and is caused by germline mutation in one of two tumor suppressor genes, TSC1 and TSC2. As for other tumor suppressor gene syndromes, the mechanism of somatic second-hit events in TSC tumors is unknown. We grew fibroblast-like cells from 29 TSC skin tumors from 22 TSC subjects and identified germline and second-hit mutations in TSC1/TSC2 using next-generation sequencing. Eighteen of 22 (82%) subjects had a mutation identified, and 8 of the 18 (44%) subjects were mosaic with mutant allele frequencies of 0 to 19% in normal tissue DNA. Multiple tumors were available from four patients, and in each case, second-hit mutations in TSC2 were distinct indicating they arose independently. Most remarkably, 7 (50%) of the 14 somatic point mutations were CC>TT ultraviolet ‘signature’ mutations, never seen as a TSC germline mutation. These occurred exclusively in facial angiofibroma tumors from sun-exposed sites. These results implicate UV-induced DNA damage as a cause of second-hit mutations and development of TSC facial angiofibromas and suggest that measures to limit UV exposure in TSC children and adults should reduce the frequency and severity of these lesions.
INTRODUCTION
Tuberous sclerosis complex (TSC) is a member of a large family of autosomal dominant syndromes in which a germline mutation in one allele of a tumor suppressor gene (TSG) predisposes patients to developing multiple tumors, often in a characteristic pattern, that exhibit somatic mutation of the second allele. A wide variety of genetic mechanisms contribute to somatic second hits in TSGs, although the specific cause of these somatic events is unknown, and they are thought to be stochastic in nature (1).
TSC is caused by inactivating mutations in either of two tumor suppressor genes TSC1 or TSC2 leading to aberrant activation of mTORC1 (mammalian Target Of Rapamycin Complex 1) and tumor development (2). Distinctive tumors are seen in the skin, brain, heart, kidney and other sites. Among TSC tumors, kidney angiomyolipomas have been studied most extensively and typically lose the second allele of TSC1 or TSC2 through large genomic deletion, mitotic exchange or gene conversion, all detectible by loss of heterozygosity (LOH) analyses (3). Here we report the first detailed analysis of genetic events in TSC skin lesions. We find strong evidence for a UV cause for second-hit mutations in TSC2 in facial angiofibromas in TSC patients.
RESULTS
Dermal fibroblast-like cells from TSC skin tumors and fibroblasts from normal-appearing skin were grown from explants (4,5) of >50 skin samples from 22 subjects with TSC (Table 1) (6). Immunoblot analysis revealed that most tumor cell lines showed reduction in the expression of TSC2 with elevated phospho-S6 (Ser235/236) levels, consistent with two hit loss/inactivation of TSC2 and activation of mTORC1 (Fig. 1), as is seen in TSC tumors from other organs (7,8). DNA preparations from 29 tumor cell lines (17 facial angiofibromas, 7 periungual fibromas, 2 shagreen patches, 2 fibrous plaques and 1 oral fibroma) were analyzed by next-generation sequencing (NGS) using an approach that enables high read-depth (>5000x) across all of the coding exons, adjacent introns, promoter regions and most of the genomic extent of TSC1 and TSC2 (9). Multiplex ligation-dependent probe amplification (MLPA) and RT-PCR sequencing of RNA were also performed to search for genomic deletions and splicing defects, respectively. All findings were confirmed by secondary studies using Sanger bidirectional sequencing in the case of variants seen at >20% allele ratio and SNaPshot analysis for those seen at <20% allele ratio and compared with analysis of DNA isolated from subject normal skin fibroblasts, blood, or saliva. Eighteen of 22 (82%) subjects had a mutation in TSC1/TSC2 identified in one or more skin samples (Table 2). Eight of the 18 (44%) were mosaic, with allele frequencies in the tumor cell lines ranging from 2 to 50%. In these 8 subjects, mosaic mutations were seen at frequencies from 0 to 19% in the analysis of normal tissue samples. In one mosaic subject's tumor culture (P4T), we identified deletion of portions of TSC2 by MLPA that was not detected in the normal skin sample (Table 2). The deletion of TSC2 was homozygous (94% copy number loss) in the tumor, suggesting somatic gene conversion as the mechanism of the second hit. In two other cases (P5 and P11), the mosaic mutation was not seen (NGS read frequency <0.01%) in normal skin fibroblasts, suggesting extreme low-level mosaicism or selective loss of the germline mutation-bearing clone in these cultures. Mosaicism is well-known in TSC but is uncommon (2). The high frequency seen here likely reflects enrichment of these subjects for those with adult diagnosis or presentation of TSC (Table 3) (6).
Table 1.
Summary of clinical features of the TSC subjects
| Age | 39 ± 11 years, range 20–64 | |
| Sex | 22 female; 0 male | |
| Family Hx | 8/21 (1 of 22 adopted, 5 subjects with an affected child) | |
| Major features | Number (%) | |
| Angiofibromas or forehead plaque | 22 (100) | |
| Hypomelanotic macules | 18 (82) | |
| Ungual fibromas | 18 (82) | |
| Shagreen patch | 15 (68) | |
| Retinal hamartomas | 4/15 (27) | |
| Cortical tuber | 21 (95) | |
| Subependymal nodules | 7 (32) | |
| Subependymal giant cell astrocytoma | 0 (0) | |
| Cardiac rhabdomyoma | 0/14 (0) | |
| Lymphangioleiomyomatosis | 20 (91) | |
| Renal angiomyolipomas | 19 (86) | |
| Minor features | ||
| Dental pitting | 16/20 (80) | |
| Oral fibromas | 15/20 (75) | |
| Confetti lesions | 8 (36) | |
| Non-renal hamartoma | 6/11 (55) | |
| Retinal achromic patch | 0/15 (0) | |
| Multiple renal cysts | 8 (36) | |
Figure 1.

Immunoblot analysis of skin tumor and control fibroblasts from TSC subjects. Subjects are indicated as P#. C, control fibroblasts; T, skin tumor fibroblasts and multiple skin tumors from one subject are indicated as T#. Immunoblotting is shown for TSC1, TSC2, ribosomal protein S6, phospho-S6 (Ser-235/236) (pS6) and β-actin.
Table 2.
Mutations and allele frequencies in TSC skin tumors
| Patient | TSC group | Tumor sample | Lesion and location | Mutations identified by NGS | Allele frequency | Mutation frequency in control skin (C)/ blood (B) | MLPA | cDNA sequencing |
|---|---|---|---|---|---|---|---|---|
| P1 | 2 | P1T | Angiofibroma R nose | TSC2 c.4830G>A, p.W1610* | 0.5 | P1C: 0.5 | ND | ND |
| P2 | 2 | P2T | Periungual fibroma, 5th toe | 0.5 | P2C: 0.5 | TSC2 del ex. 1–41 | TSC2 normal | |
| P3 | 1 | P3T1 | Periungual fibroma, 3rd toe | TSC2 c.4620C>G, p.Y1540* | 0.54 | P3C: 0.02 | ND | ND |
| TSC2 c.3691_3707del | 0.5 | P3C: 0 SS | ||||||
| P3T2 | Periungual fibroma, 5th toe | TSC2 c.4620C>G, p.Y1540* | 0.12 | P3C: 0.02 | TSC2 normal | TSC2 normal | ||
| TSC1 c.1888_1891del | 0.02 | P3C: 0 SS | ||||||
| P4 | 1 | P4T | Angiofibroma, R nose | 0.94 | P4C: not detected | TSC2 del ex.6–11, ex.21 | ND | |
| P5 | 1 | P5T1# | Angiofibroma, L nose | TSC2 c.1074G>A, p.W358* | 0.47 | P5B: 0.07; not seen in P5C | ND | ND |
| TSC2 c.3441_3442CC>TT, p.Q1148* | 0.47 | P5C: 0 SS | ||||||
| P5T2# | Angiofibroma, R nose | TSC2 c.1074G>A, p.W358* | 0.1 | P5B: 0.07 | ND | ND | ||
| TSC2 c.5228_5229GG>AA, p.R1743Q | 0.06 | P5C: 0 SS | ||||||
| P5T3 | Oral fibroma, mouth | TSC2 c.1074G>A, p.W358* | 0.49 | P5B: 0.07 | ND | |||
| 0.4 | P5C: 0 SS | TSC2 del ex.1–25, ex.39–41 | ||||||
| P6 | 1 | P6T1 | Angiofibroma, L nose | TSC2 c.4375C>T, p.R1459* | 0.56 | P6C: 0.5 | TSC2 normal | TSC2 normal |
| P6T2# | Angiofibroma, R nose | TSC2 c.4375C>T, p.R1459* | 0.5 | P6C: 0.5 | ND | ND | ||
| TSC2 c.2250_2251CC>TT, p.R751* | 0.48 | |||||||
| TSC2 c.245G>A, p.W82* | 0.15 | |||||||
| P7 | 2 | P7T1# | Angiofibroma, R nose | TSC2 c.1803C>G | 0.48 | P7C: 0.5 | ND | c.1803 C>G leads to aberrant splice site |
| TSC2 c.1830_1831CC>TT, p.R611W | 0.53 | P7C: 0 SS | ||||||
| P7T2# | Angiofibroma, L nose | TSC2 c.1803C>G | 0.51 | P7C: 0.5 | ND | c.1803 C>G leads to aberrant splice site | ||
| TSC2 c.1830_1831CC>TT, p.R611W | 0.32 | P7C: 0 SS | ||||||
| TSC2 c.1444-2A>G | 0.18 | P7C: 0 SS | ||||||
| P8 | 1 | P8T | Fibrous plaque, supraclavicular | TSC2 c.3750C>G, p.Y1250* | 0.52 | P8C: 0.5 | TSC2 normal | TSC2 normal |
| P9 | 1 | P9T | Fibrous plaque, forehead | TSC2 c.5197insC | 0.5 | P9C: 0.5 | ND | ND |
| TSC2 c.3412C>T, p.R1138* | 0.24 | P9C: 0 SS | ||||||
| P10 | 2 | P10T | Angiofibroma, L nose | TSC2 c.4502T>C, p.F1501S | 0.18 | P10C and P10B: 0.06 | ND | ND |
| TSC2 c.336delG | 0.08 | P10C: 0 SS | ||||||
| P11 | 1 | P11T | Angiofibroma, R nose | TSC2 c.1832G>A, p.R611Q | 0.02 | Not seen in P11C and P11B | TSC2 normal | TSC2 normal |
| P12 | 2 | P12T | Angiofibroma, R chin | TSC2 c.944insG | 0.39 | P12C: 0.003 | ND | ND |
| TSC2 c.2747T>G, p.L916R | 0.34 | P12C: 0 SS | ||||||
| P13 | 2 | P13T1 | Shagreen patch, back | TSC2 c.1513C>T p.R505* | 0.5 | P13C: 0.5 | TSC2 normal | TSC2 normal |
| P13T2 | Angiofibroma, L nose | TSC2 c.1513C>T p.R505* | 0.5 | P13C: 0.5 | TSC2 normal | TSC2 normal | ||
| P13T3 | Periungual fibroma, 1st-toe | TSC2 c.1513C>T p.R505* | 0.5 | P13C: 0.5 | TSC2 normal | TSC2 normal | ||
| TSC2 c.2647C>T p.Q883* | 0.01 | P13C: 0 SS | ||||||
| P14 | 2 | P14T | Angiofibroma, L nose | No mutation found | TSC1 and TSC2 normal | TSC1 and TSC2 normal | ||
| P15 | 2 | P15T | Angiofibroma, L nose | TSC2 c.139-1G>A | 0.47 | P15C: 0.5 | TSC2 normal | Splice variant results in aberrant splicing |
| P16 | 3 | P16T | Periungual fibroma, 5th toe | No mutation found | TSC1 and TSC2 normal | TSC1 and TSC2 normal | ||
| P17 | 2 | P17T | Periungual fibroma, 5th toe | TSC1 c.1027C>T p.Q343* | 0.49 | P17C: 0.5 | TSC1 normal | TSC1 normal |
| P18 | 3 | P18T | Periungual fibroma, 2nd toe | No mutation found | TSC1 and TSC2 normal | TSC1 and TSC2 normal | ||
| P19 | 2 | P19T | Shagreen patch, back | TSC2 c.4490C>T p.P1497L | 0.03 | P19C: 0.09, P19B: 0.19 | ND | ND |
| TSC2 c.759C>A p.C253* | 0.01 | P19C: 0 SS | ||||||
| P20 | 3 | P20T | Angiofibroma, L nose | No mutation found | TSC1 and TSC2 normal | TSC1 and TSC2 normal | ||
| P21 | 1 | P21T# | Angiofibroma, L nose | TSC2 c.1488delC | 0.35 | P21B: 0.1 | ND | ND |
| TSC2 c.1116_1117CC>TT p.Q373* | 0.03 | P21B: 0 SS | ||||||
| TSC2 c.2250_2251CC>TT p.R751* | 0.1 | P21B: 0 SS | ||||||
| P22 | 1 | P22T | Angiofibroma, tip of nose | TSC2 c.1461_1462delAG | 0.47 | P22C: 0.5 | ND | ND |
| TSC2 c.4318C>T p.Q1440* | 0.16 | P22C: 0 SS |
First column is patient sample number. Second column refers to age of onset of TSC; with 1 denoting childhood onset and diagnosis; 2, childhood onset and adult diagnosis and 3, adult presentation. Germline mutations are highlighted in bold. # denotes angiofibromas with one or two UV-induced mutations. SS, Sanger sequencing; MLPA of TSC1 and TSC2;
ND, not done.
Table 3.
Age (years) of patients with TSC mutation identified or NMI
| Mutation identified (n = 18) | NMI (n = 4) | P-valuea | |
|---|---|---|---|
| Mean age at first criterion (S.D.) | 2.2 (4.3) | 15 (11) | 0.003 |
| Mean age at penetrance (S.D.) | 6.2 (4.6) | 29 (16) | 0.013 |
| Mean age at diagnosis (S.D.) | 19 (15) | 37 (12) | N.S. |
| Mean age at enrollment (S.D.) | 38 (12) | 43.8 (3.9) | N.S. |
Age (years) at first criterion = age at which first major criterion manifested by patient history; age at penetrance = age at which patient manifested at least two major criteria by patient history; S.D. = standard deviation; N.S. = not significant (P ≥ 0.05); NMI = no mutation identified.
aMann–Whitney U-test.
Sixteen of 25 (64%) tumor cell lines with at least one mutation in TSC1/TSC2 had two or three mutations identified. These additional, second-hit mutations were not seen in any control DNA sample from the same patient and were often present at low allele frequency likely due to normal cell contamination of the cultures. Three of these 16 cell lines had two TSC2 somatic mutations, consistent with subclonality, possibly due to coalescence of closely spaced tumors. Interestingly, we identified distinct second-hit mutations in TSC2 in multiple tumors that were available from four patients (P3, P5, P6 and P7) indicating that they developed from different progenitor cells, despite being derived from similar regions on the body. In one sample from one of these patients (P3T2), who had a mosaic mutation in TSC2, we detected an unexpected somatic mutation in TSC1 at 2% mutant allele frequency. It seems unlikely that a mono-allelic mutation in TSC1 would complement a mutation in TSC2 and lead to mTOR activation. While this mutation may be chance observation, its presence in only one tumor, combined with the absence of the second-hit TSC2 mutation identified in P3T1, supports the divergent origins of the two tumors in this patient.
Strikingly, 17 (89%) of the 19 second-hit mutations that were identified were small indels or point mutations and not large genomic deletions or recombination events that would be detected as LOH, in contrast to TSC kidney angiomyolipoma where LOH is seen in ∼70% (3). In one culture, the germline and somatic mutations in TSC2 occurred within 50 nt of each other, and Integrative Genomics Viewer (IGV) (10) examination of NGS sequencing reads confirmed that the two mutations had occurred in trans, affecting both alleles (Fig. 2).
Figure 2.

IGV screenshot demonstrating that TSC2 c.1803C>G (splice) and TSC2 c.1830_1831CC>TT, p.R611W mutations do not occur in the same amplified DNA molecules generated from cultured skin tumor P7T1 and thus arose on opposite alleles of TSC2.
Most remarkably, 7 (50%) of the 14 somatic point mutations were CC>TT, a mutation type that has never been seen in TSC germline analysis, with >1500 germline mutations identified in TSC1/TSC2 (2) (http://chromium.liacs.nl/LOVD2/TSC/home.php, last accessed on 30 November 2013.) (Figs 2 and 3; Table 2). CC>TT mutations are well known to be a product of sunlight-induced DNA damage caused by the formation of cyclobutane pyrimidine dimers at adjacent pyrimidines in DNA in response to ultraviolet A or ultraviolet B radiation (11). Furthermore, CC>TT mutations in TSC1/TSC2 were seen exclusively in cultured cells from facial angiofibromas, 7 (54%) of 13, and not in other cutaneous tumors (fibrous plaque, shagreen patch, ungual fibroma and oral fibroma), 0 of 6 (P = 0.0436, Fisher exact test).
Figure 3.
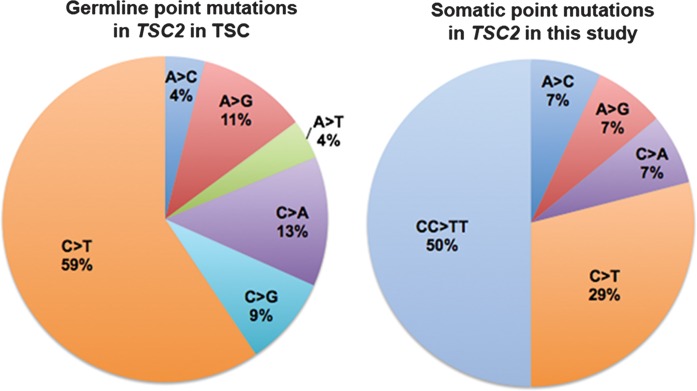
Distribution of types of point mutations seen in the germline in TSC2 (2, http://chromium.liacs.nl/LOVD2/TSC/home.php), left; and seen in this study as somatic mutations in TSC2, right.
DISCUSSION
Each type of TSC tumor has a distinctive natural history and time course, for which the mechanism is largely unknown (12). TSC facial angiofibromas are rare in infants, typically begin to appear at about age 3 to 5 years, and attain greatest severity during the teenage years and later (13). Our observations that somatic second-hit mutations of the form CC>TT occur in TSC facial angiofibromas at high frequency, indicative of sunlight-induced DNA damage, have several important implications. First, this is the first instance of a tumor suppressor gene syndrome in which the mechanism of somatic second-hit mutations has been identified, providing a deeper understanding of the pathogenesis of this disease. Sunlight-induced mutations are well known to occur in TP53 in both sporadic and Gorlin syndrome-associated basal cell carcinoma of the skin, but second-hit inactivation of PTCH in Gorlin basal cell carcinoma is typically due to LOH (14,15). Second, these observations indicate that somatic mutations critical for the pathogenesis of many TSC facial angiofibromas occur after birth in response to environmental sunlight exposure. The cell of origin and the timing of second-hit events critical for the development of TSC tumors are uncertain for non-brain tumors, and our data suggest that it is a resident skin cell in the facial dermis that sustains a second-hit event in TSC1/TSC2, and possibly other events, and begins to develop into a facial angiofibroma in many cases. This sequence and timing of events fits the clinical time course of the development of facial angiofibromas. Finally, our observations suggest that simple sun protective measures, such as avoidance of sun exposure and use of hats or sunscreen, may reduce the number and potentially the severity of these tumors, which are a common (seen in ∼80%) and major problem in TSC.
MATERIALS AND METHODS
Subjects
All subjects were patients with TSC seen at the National Heart, Lung, and Blood Institute at the National Institutes of Health Clinical Center where there has been a clinical program focused on lymphangioleiomyomatosis under the direction of JM for many years. Diagnosis of TSC was based on the presence of at least two major or one major and two or more minor diagnostic features (16). Inclusion criteria were age 18 years or older and being capable of providing written informed consent, which was provided by all patients. All patients were female because this cohort was enriched with patients known to have lymphangioleiomyomatosis, a disorder that occurs predominantly in women (17). Samples of blood, saliva and skin were obtained from the patients. In addition, they were evaluated by clinical history, physical examination, pulmonary function testing, chest radiography, abdominal computed tomography, high-resolution computed tomography of the chest and brain magnetic resonance imaging.
Cell culture
Samples of angiofibromas, periungual fibromas, shagreen patches, forehead plaques, oral fibromas and normal-appearing skin were obtained for routine pathology and cell culture. To isolate fibroblast-like cells, the skin samples were cut into small pieces and placed in DMEM supplemented with 10% FBS (Gibco), 100 units/ml penicillin and 100 μg/ml streptomycin in culture dishes. Medium was changed twice weekly until the fibroblasts migrated out to cover the dishes. Cells were harvested for serial passage and cryopreservation (5).
Western blot
Cells were seeded overnight into 60-mm dishes at 5 × 105 cells in DMEM with 10% FBS and switched to serum-free DMEM for 24 h. Cells were lysed in protein extraction buffer [20 mm Tris (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 20 mm NaF, 2.5 mm Na4P2O7, 1 mm β-glycerophosphate, 1 mm benzamidine, 10 mm p-nitrophenyl phosphate, 0.1 mm phenylmethylsulfonyl fluoride]. Total cellular protein was measured using BCA reagent (Pierce Chemical Co.). Equivalent samples of total proteins were separated using 10% (wt/vol) polyacrylamide gels and transferred to 0.45-μm Invitrolon PVDF membranes (Invitrogen) before immunoblotting with anti-Tuberin/TSC2 (D93F12), anti-Hamartin/TSC1, anti-phospho-S6 ribosomal protein (Ser-235/236), anti-S6 ribosomal protein antibodies (Cell Signaling) and β-actin (Sigma–Aldrich), horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) and Immobilon Western Chemiluminescent HRP Substrate (Millipore).
DNA and RNA analysis
Genomic DNA was isolated from TSC patient cells using DNeasy Blood & Tissue Kit (QIAGEN). Total RNA was extracted from the cells using the RNeasy Mini Kit (QIAGEN).
Next-generation sequencing and validation
Next-generation sequencing of most of the genomic extent of TSC1 and TSC2 was performed on the HiSeq 2000 (Illumina, San Diego, CA, USA) as described previously (9). Briefly, long-range PCR was performed on DNA prepared from the tumor cell cultures to amplify all of the coding exons and most of the intronic sequence of each of TSC1 and TSC2. Amplicons were purified and used to prepare a small fragment library for Illumina sequencing (9). Libraries from different samples were generated with different indices and then mixed at an equimolar ratio and sequenced on an Illumina GAIIx or HiSeq 2000 sequencer for 50–75 nt reads.
Sequencing data output was analyzed using a combination of standard tools and custom software to enable detection of sequence variants at ≥1% frequency. The primary data were deconvoluted using the index sequences to individual sample files and converted to FASTQ format, aligned to the human genome using bwa-0.5.8c (Burrows-Wheeler Alignment) (18), filtered to eliminate reads of low quality and to reduce redundancy to a uniform 50 reads starting at each nucleotide position of interest in each direction. The data were then analyzed for sequence variants using tools from the Genome Analysis Toolkit (GATK) (19), including IndelGenotyperV2 and Unified Genotyper, to identify both indels and single-nucleotide variants. A second approach was used in parallel to analyze the sequence data, with capture of read calls at all positions using Pileup (SAMtools), followed by custom processing in Python and Matlab to determine nucleotide call frequency at each position in each read orientation. The output from these analyses was reviewed in comparison with those from other samples, including controls, to exclude artifacts derived from the sequencing process. All variants seen at a frequency of ≥1% more than that seen in other samples were directly reviewed using the IGV (10) (IGV; www.broadinstitute.org/software/igv/) to help confirm bona fide variant calls and to exclude sequencing artifacts. A median read-depth for each exon of TSC1 and TSC2 of >5000 was achieved by this procedure.
Single-nucleotide variants and indels that were identified as novel and/or of possible significance were confirmed by secondary analysis of the DNA sample, using Sanger bidirectional sequencing in the case of variants seen at >20% allele ratio and SNaPshot analysis for those seen at <20% allele ratio. SNaPshot extension products were analyzed on an ABI 3100 sequencer (Applied Biosystems, Carlsbad, CA, USA), and the proportion of alleles was quantified using GeneMapper version 3.0 (Applied Biosystems) (20). In this analysis, small peaks were seen for variant nucleotides in some cases because of spontaneous base misincorporation. However, comparison with control samples permitted discrimination of bona fide variants at allele frequencies as low as 2%. The allele frequency of mutant alleles was determined as M/(M + W), where M and W are the peak areas of the mutant and wild-type allele products, respectively, after subtraction of the signal seen in control samples. All SNaPshot experiments were replicated at least once. For variants seen at allele frequency of <2%, confirmation was performed by repeat NGS analysis of individual amplicons. Several apparent germline variants were not seen in control skin cultures. For these samples as well, targeted NGS was performed, enabling analysis of >10 000 reads at the nucleotide position of interest to search for very low frequency alleles.
Multiplex ligation-dependent probe amplification (MLPA) and RT-PCR sequencing
MLPA was performed using commercially available probe sets for every exon of TSC1 and TSC2 and flanking exons, available from MRC-Holland, as described previously (21). RT-PCR sequencing was performed by standard methods using unfractionated RNA, and cDNA probe sets that cover the entire coding region of each of TSC1 and TSC2 in six and eight overlapping amplicons, respectively, as described previously (21).
Statistics
Statistical analyses were performed using the Fisher exact test and the Mann–Whitney U (Wilcoxon rank-sum) test in Prism version 1.0.
Study approval
Patients with TSC were enrolled between 2002 and 2012 in protocols 00-H-0051, 95-H-0186 and/or 96-H-0100 approved by the National Heart, Lung, and Blood Institute/National Institutes of Health Institutional Review Board. All subjects provided written informed consent.
FUNDING
This work was supported by NIH 2R37NS031535 and R01AR062080, the Sulzberger Dermatological Research and Education Endowment, and the Tuberous Sclerosis Alliance, and the Intramural Research Program of the National Institutes of Health, National Heart, Lung, and Blood Institute.
ACKNOWLEDGEMENTS
The authors thank the LAM Foundation and the Tuberous Sclerosis Alliance for assistance with patient recruitment, as well as the patients participating in this research. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health, the Department of Defense or the Uniformed Services University of the Health Sciences.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Foulkes W.D. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008;359:2143–2153. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 2.Kwiatkowski D.J. Genetics of tuberous sclerosis complex. In: Kwiatkowski D.J., Whittemore V.H., Thiele E.A., editors. Tuberous Sclerosis Complex. Wiley-VCH, Weinheim, Germany; 2010. pp. 29–60. [Google Scholar]
- 3.Henske E.P., Scheithauer B.W., Short M.P., Wollmann R., Nahmias J., Hornigold N., van Slegtenhorst M., Welsh C.T., Kwiatkowski D.J. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet. 1996;59:400–406. [PMC free article] [PubMed] [Google Scholar]
- 4.Li S., Thangapazham R.L., Wang J.A., Rajesh S., Kao T.C., Sperling L., Moss J., Darling T.N. Human TSC2-null fibroblast-like cells induce hair follicle neogenesis and hamartoma morphogenesis. Nat. Commun. 2011;2:235. doi: 10.1038/ncomms1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S., Takeuchi F., Wang J.A., Fuller C., Pacheco-Rodriguez G., Moss J., Darling T.N. MCP-1 overexpressed in tuberous sclerosis lesions acts as a paracrine factor for tumor development. J. Exp. Med. 2000;202:617–624. doi: 10.1084/jem.20042469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seibert D., Hong C.H., Takeuchi F., Olsen C., Hathaway O., Moss J., Darling T.N. Recognition of tuberous sclerosis in adult women: delayed presentation with life-threatening consequences. Ann. Intern. Med. 2011;154:806–813. doi: 10.1059/0003-4819-154-12-201106210-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goncharova E.A., Goncharov D.A., Eszterhas A., Hunter D.S., Glassberg M.K., Yeung R.S., Walker C.L., Noonan D., Kwiatkowski D.J., Chou M.M., et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM) J. Biol. Chem. 2002;277:30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- 8.El-Hashemite N., Zhang H., Henske E.P., Kwiatkowski D.J. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 9.Badri K.R., Gao L., Hyjek E., Schuger N., Schuger L., Qin W., Chekaluk Y., Kwiatkowski D.J., Zhe X. Exonic mutations of TSC2/TSC1 are common but not seen in all sporadic pulmonary lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2013;187:663–665. doi: 10.1164/ajrccm.187.6.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikehata H., Ono T. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011;52:115–125. doi: 10.1269/jrr.10175. [DOI] [PubMed] [Google Scholar]
- 12.Thiele E.A., Jozwiak S. Natural history of tuberous sclerosis complex and overview of manifestations. In: Kwiatkowski D.J., Whittemore V.H., Thiele E.A., editors. Tuberous Sclerosis Complex. Wiley-VCH, Weinheim, Germany; 2010. pp. 11–20. [Google Scholar]
- 13.Darling T.N., Moss J., Mauser M. Dermatologic manifestations of tuberous sclerosis complex (TSC) In: Kwiatkowski D.J., Whittemore V.H., Thiele E.A., editors. Tuberous Sclerosis Complex. Wiley-VCH, Weinheim, Germany; 2010. pp. 285–310. [Google Scholar]
- 14.Kim M.Y., Park H.J., Baek S.C., Byun D.G., Houh D.J. Mutations of the p53 and PTCH gene in basal cell carcinomas: UV mutation signature and strand bias. J. Dermatol. Sci. 2002;29:1–9. doi: 10.1016/s0923-1811(01)00170-0. [DOI] [PubMed] [Google Scholar]
- 15.Ling G., Ahmadian A., Persson A., Undén A.B., Afink G., Williams C., Uhlén M., Toftgård R., Lundeberg J., Pontén F. PATCHED and p53 gene alterations in sporadic and hereditary basal cell cancer. Oncogene. 2001;20:7770–7778. doi: 10.1038/sj.onc.1204946. [DOI] [PubMed] [Google Scholar]
- 16.Roach E.S., Gomez M.R., Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J. Child. Neurol. 1998;13:624–628. doi: 10.1177/088307389801301206. [DOI] [PubMed] [Google Scholar]
- 17.Glasgow C.G., Steagall W.K., Taveira-Dasilva A., Pacheco-Rodriguez G., Cai X., El-Chemaly S., Moses M., Darling T., Moss J. Lymphangioleiomyomatosis (LAM): molecular insights lead to targeted therapies. Respir. Med. 2010;104:S45–S58. doi: 10.1016/j.rmed.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H., Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qin W., Kozlowski P., Taillon B.E., Bouffard P., Holmes A.J., Janne P., Camposano S., Thiele E., Franz D., Kwiatkowski D.J. Ultra deep sequencing detects a low rate of mosaic mutations in tuberous sclerosis complex. Hum. Genet. 2010;127:573–582. doi: 10.1007/s00439-010-0801-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin W., Bajaj V., Malinowska I., Lu X., MacConaill L., Wu C.L., Kwiatkowski D.J. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS One. 2011;6:e24919. doi: 10.1371/journal.pone.0024919. [DOI] [PMC free article] [PubMed] [Google Scholar]