

Abstract
Two original chiral diphosphines, SYNPHOS and DIFLUORPHOS, have been synthesized on multigram scales. Their steric and electronic profiles have been established in comparison with the commonly used 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl and 6,6′-dimethoxy-2,2′-bis(diphenylphosphino)-1,1′-biphenyl ligands. A screening study of the four ligands in RuII-catalyzed asymmetric hydrogenation of prochiral ketones and olefins has been performed. It revealed that the stereoelectronic features of the ligand and the substrate deeply influenced the enantioselectivities obtained in asymmetric hydrogenation, SYNPHOS and DIFLUORPHOS being fully complementary in terms of enantioselectivity for this reaction.
Homogeneous asymmetric catalysis is undoubtedly a powerful synthetic tool of the organic chemist on both a laboratory and a production scale (1). In recent years, progress in this field has been spectacular and ultimately recognized by the 2001 Nobel Prize awarded to Knowles (2) and Noyori (3) for enantioselective hydrogenation and to Sharpless (4) for enantioselective oxidation catalysis. In particular, enantioselective hydrogenation (5–9) using molecular hydrogen to reduce prochiral C X (X = N,O) or CC double bonds is one of the most efficient and versatile methods to build chiral compounds and valuable synthetic intermediates. Effective homogeneous hydrogenation catalysts are organometallic complexes that consist of one or more (chiral) ligands coordinated to an ionic metal center (Ru, Rh, Ir,...). The choice of the chiral ligand is decisive both for catalytic activity and for achieving a high level of chiral induction. The most common ligands have a chiral backbone with two coordinating heteroatoms. The pioneering work by Kagan, who reported the first chiral biphosphine ligand DIOP (10) for rhodium-catalyzed asymmetric hydrogenation in the 1970s, resulted in several significant breakthroughs in ligand design. C2 symmetry is an important structural feature of diphosphines, as confirmed by the discovery of 1,2-bis(o-anisylphenyl-phosphino)-ethane by Knowles in 1975 (11). Noyori discovered the 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) ligand in 1980 (12), which resulted in an extraordinary expansion of the scope of asymmetric hydrogenation in the academic field. The extensive use of BINAP-based catalysts on a production scale (13) by Takasago International (Tokyo) encouraged research groups to devote their efforts toward the design and synthesis of new atropisomeric biaryl diphosphines (for recent reviews, see refs. 14 and 15). The elaboration of new ligand families, such as the biphenyl series by Hoffmann-LaRoche as described in ref. 16 or the [(4,4′-bi-1,3-benzodioxole)-5,5′-diyl]bis(diphenylphosphine) (SEGPHOS) series by Takasago International as described in ref. 17, are significant in the current challenges to chemists in the field of asymmetric catalysis (Fig. 1). Following our continued interest in ligand design (18, 19), we report the synthesis of two atropisomeric diphosphines SYNPHOS and DIFLUORPHOS (20, 21), with stereoelectronically complementary backbones, based on bi(benzodioxane) (see also ref. 22) and bi(difluorobenzodioxole) moieties (Fig. 1), respectively. We also propose detailed structural profiling of these ligands and catalytic evaluation in asymmetric Ru-mediated hydrogenation compared with other leading atropisomeric diphosphines BINAP and MeO-BIPHEP.
X (X = N,O) or CC double bonds is one of the most efficient and versatile methods to build chiral compounds and valuable synthetic intermediates. Effective homogeneous hydrogenation catalysts are organometallic complexes that consist of one or more (chiral) ligands coordinated to an ionic metal center (Ru, Rh, Ir,...). The choice of the chiral ligand is decisive both for catalytic activity and for achieving a high level of chiral induction. The most common ligands have a chiral backbone with two coordinating heteroatoms. The pioneering work by Kagan, who reported the first chiral biphosphine ligand DIOP (10) for rhodium-catalyzed asymmetric hydrogenation in the 1970s, resulted in several significant breakthroughs in ligand design. C2 symmetry is an important structural feature of diphosphines, as confirmed by the discovery of 1,2-bis(o-anisylphenyl-phosphino)-ethane by Knowles in 1975 (11). Noyori discovered the 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) ligand in 1980 (12), which resulted in an extraordinary expansion of the scope of asymmetric hydrogenation in the academic field. The extensive use of BINAP-based catalysts on a production scale (13) by Takasago International (Tokyo) encouraged research groups to devote their efforts toward the design and synthesis of new atropisomeric biaryl diphosphines (for recent reviews, see refs. 14 and 15). The elaboration of new ligand families, such as the biphenyl series by Hoffmann-LaRoche as described in ref. 16 or the [(4,4′-bi-1,3-benzodioxole)-5,5′-diyl]bis(diphenylphosphine) (SEGPHOS) series by Takasago International as described in ref. 17, are significant in the current challenges to chemists in the field of asymmetric catalysis (Fig. 1). Following our continued interest in ligand design (18, 19), we report the synthesis of two atropisomeric diphosphines SYNPHOS and DIFLUORPHOS (20, 21), with stereoelectronically complementary backbones, based on bi(benzodioxane) (see also ref. 22) and bi(difluorobenzodioxole) moieties (Fig. 1), respectively. We also propose detailed structural profiling of these ligands and catalytic evaluation in asymmetric Ru-mediated hydrogenation compared with other leading atropisomeric diphosphines BINAP and MeO-BIPHEP.
Fig. 1.

Atropisomeric diphosphine ligands.
Materials and Methods
Materials. Starting materials 6-bromo-2,3-dihydro-1,4-benzodioxine and 5-bromo-2,2-difluoro-1,3-benzodioxole were purchased from Acros Organics (Noisy-Le-Grand, France) and Avocado (La Tour du Pin, France), respectively, and purified by distillation before use. Hydrogenation substrates 5a–5k were commercially available and used as received. Metallic precursors RuCl3·nH2O, [Ru(η6-benzene)Cl2]2, and [Rh(CO)2Cl]2 were purchased from Strem Chemicals (Bischheim, France) or Aldrich. [Ru(1,5-cyclooctadiene)(η3-(CH2)2CCH3)2] was synthesized from RuCl3·nH2O as reported (23).
Instrumentation. 1H NMR spectra were recorded on a Bruker AC 200 at 200 MHz. IR spectra were obtained on a Brücker FT IFS 48 instrument. GC analyses were performed on a Hewlett–Packard 5890 series II instrument connected to a Merck D-2500 or D-2000 integrator with a flame-ionization detector. HPLC analyses were conducted by using a Waters 600 system with Daicel chiral stationary-phase columns.
Typical Procedure for Asymmetric Hydrogenation with in Situ [Ru-(Chiral Diphosphine [P*P])Br2] Catalysts. Diphosphine (0.011 mmol) and [Ru(1,5-cyclooctadiene)(η3-(CH2)2CCH3)2] (3.2 mg, 0.01 mmol) were placed in a 10-ml flask, and 1 ml of degassed anhydrous acetone was added under argon. A methanolic solution of HBr (122 μl, 0.18 M, 0.022 mmol) was added dropwise to the suspension. The reaction mixture was stirred at room temperature for 30 min, and a resulting orange suspension was observed. The solvent was removed under vacuum. The orange solid residue was used without further purification as a catalyst for the hydrogenation reaction of the desired substrate (1 mmol) in 2 ml of MeOH or EtOH. The reaction vessel was placed in a 500-ml stainless steel autoclave, which was adjusted at the desired pressure and temperature for 24 h. The solvent was evaporated and the crude product was filtered on a short pad of silica gel. Conversion and enantiomeric excess (ee) were determined by 1H NMR and chiral GC or HPLC, respectively.
Computational Methods. All molecular geometries incorporating diphosphine ligands (free diphosphine, β1 and β2 structures) were optimized with the same molecular mechanics program (cache worksystem pro 5.0), using the same parameter set: force field parameters, MM2; calculation type, structure optimization; optimization method, conjugate gradient for no more than 3,000 updates or until convergence to 10–4 kcal·mol–1 (1 cal = 4.184 J).
Results
Scale-up Development Synthesis of SYNPHOS (24) and DIFLUORPHOS (25). Both ligands were synthesized on multigram scales according to similar synthetic strategies (Fig. 2). Phosphorylation of 6-bromo-1,4-benzodioxane 1 and 5-bromo-2,2-difluoro-1,3-benzodioxole 1′ was realized in both syntheses by the Grignard reagent, which was reacted with chlorodiphenylphosphine (X = C2H4) and further oxidized by hydrogen peroxide to obtain phosphine oxide 2, or with chlorodiphenylphosphine oxide (X = CF2) directly to obtain compound 2′. Dimerization of phosphine oxides 2 and 2′ required long and meticulous adjustments, in particular, to adapt the laboratory-scale synthesis to the kilogram scale for compound 2. A two-step sequence consisting of lithiation/iodination followed by Ullmann coupling was preferred to direct oxidative coupling of the lithiated intermediate in both syntheses. Compounds 2 and 2′ were subjected to regioselective ortholithiation by using lithium diisopropylamide in tetrahydrofuran (THF) at –78°C. In the fluorinated series (X = CF2), direct addition of iodide in THF at –78°C to the resulting solution gave satisfactory yields (88%) in iodinated compound 3′. On the contrary, if X = C2H4, the inverted addition of the solution containing the lithiated intermediate to the iodine solution at –10°C was necessary to avoid the formation of degradation byproducts (mainly resulting from benzodioxane ring opening) and to obtain compound 3 in good yield (85%). Homocoupling of iodinated intermediates 3 and 3′ was successfully performed under Ullmann-type conditions (Cu, dimethylformamide, 120°C) and provided racemic diphosphine oxides 4 and 4′ in 80% and 69% yield, respectively. A resolution step was then necessary to obtain enantiopure ligands. This key step required intensive optimization in the nonfluorinated series and was performed on a multikilogram scale in 78–79% yield based on racemic 4 by using O,O-dibenzoyltartaric acid [(+)- or (–)-DBTA] as described (24). Because the use of DBTA proved unsuccessful to resolve racemic 4′, separation of the two atropisomers was performed by using chiral preparative HPLC in 90% yield based on racemic 4′. Enantiomeric purities of both enantiomers of 4 and 4′ were checked by using chiral HPLC (24, 25) and turned out to be >99%. The final reduction step of enantiopure 4 and 4′ was accomplished by treatment with trichlorosilane and tributylamine in refluxing xylene and afforded (R)- and (S)-SYNPHOS and (R)- and (S)-DIFLUORPHOS in 90% and 91% yield, respectively.
Fig. 2.

Scale-up development synthesis of SYNPHOS and DIFLUORPHOS ligands (38% and 33% overall yield, respectively). Reagents and conditions: (i) Mg, THF, then ClPPh2, then H2O2, MeOH; (ii) Mg, THF, then ClP(O)Ph2; (iii) lithium diisopropylamide, –78°C, THF, 3.5h; then I2, –10°C (inverted addition); (iv) lithium diisopropylamide, –78°C, THF, 2 h; then I2, –78°C (direct addition); (v) Cu, dimethylformamide, 120°C; (vi) obtainment of (+)-(R)-4: (–)-DBTA, CHCl3/AcOEt, filtration, then CH2Cl2, filtration, then KOHaq (1 N); obtainment of (–)-(S)-4: (+)-DBTA, CHCl3/AcOEt, filtration, then CH2Cl2, filtration, then KOH aq (1 N); (vii) chiral preparative HPLC, Chirose C3 column; (viii) HSiCl3, Bu3N, xylene, 140°C.
Steric Profiling of SYNPHOS and DIFLUORPHOS. As already pointed out, the choice of the ligand is often decisive for the catalytic performance of a metal complex. As far as Ru-catalyzed asymmetric hydrogenation is concerned, its structure influences how the metal reacts with H2 and the substrate. Thus, we were eager to evaluate the similarities and differences of these two ligands with other diphosphines of the same structural family, especially BINAP and MeO-BIPHEP (Fig. 1). The most commonly used mechanism (8, 26) for (diphosphine)Ru-catalyzed hydrogenation of functionalized ketones is depicted in Fig. 3. The hydrogenation proceeds by a RuII monohydride species A (27, 28), which is expected to react reversibly with the substrate to afford the adduct B (28). Further intramolecular hydride transfer provides the ruthenium alkoxide C (29). The β-hydroxy ester is then released by the action of solvent to afford the ruthenium species D, which reacts with H2 to complete the catalytic cycle. The intramolecular hydride transfer B → C is considered as the stereodetermining step of the catalytic cycle. Two chelation modes of the bidentate substrate (re-or si-face) can occur, affording two diastereomers of complex B. The sense of enantioselection is assumed to depend on the relative stability of the two diastereomeric transition states of the B → C step. The asymmetric spatial environment displayed by the diphosphine ligand directly influences this stability (Fig. 4). For atropisomeric diphosphines, such a chiral environment is defined by the general rule of the “quadrants”: the steric hindrance around the ruthenium is created by the phosphorus substituents in pseudoequatorial positions (bottom left and upper right quadrants for an (S)-atropisomeric diphosphine, as shown in Fig. 4). The dihedral angle θ of the biaryl backbone (Fig. 4) is geometrically related to the bite angle β (30) and is known to determine the proximity of the pseudoequatorial aryl groups and the chelating substrate around the ruthenium (31). Based on steric considerations only, the smaller θ is, the higher the ligand–substrate interaction should be, providing a better chiral discrimination of the catalyst (17, 32, 33). Steric evaluation of SYNPHOS and DIFLUORPHOS was achieved similar to BINAP and MeO-BIPHEP by measuring the dihedral angle of the biaryl skeleton in free and chelated diphosphines. Molecular modeling proved to be the tool of choice for rapid and easy quantification of θ (34). The β1 and β2 structures {RuHCl[(S)-P*P][substrate]} were chosen as representative models for the two diastereomeric transition states of step B → C, for the asymmetric hydrogenation of the simple substrate methyl acetoacetate (Fig. 5). Energy minimization using molecular mechanics calculations was performed on both β1 and β2 structures for each of the four ligands BINAP, MeO-BIPHEP, SYNPHOS, and DIFLUORPHOS. As reported in Table 1, for each ligand, the β1 structure displayed a lower relative energy than its diastereomer β2 (ΔE ≈ 2–4 kcal·mol–1). In the β1 structure, the substrate is chelated to ruthenium through its re-face, whereas the β2 represents the si-face chelation mode. Because of steric interactions between the bottom left “quadrant” of the (S)-ligand and the methyl group of the ketone, the β2 transition state is not favored (see Fig. 5). This difference in relative energy of the two transition states of step B → C explains the major obtainment of methyl (3S)-hydroxybutanoate at the experimental level by using (S)-atropisomeric diphosphines.
Fig. 3.

Commonly used mechanism of diphosphine/Ru-catalyzed asymmetric hydrogenation of functionalized ketones.
Fig. 4.

Quadrants rule: schematic representations of the steric hindrance created by phosphorus substituents (ax, pseudoaxial position; eq, pseudoequatorial position). (Left) Front view of (S)-SYNPHOS ligand. (Right) General top view of atropisomeric diphosphines. Pseudoaxial phenyl groups are omitted for clarity.
Fig. 5.

Structures minimized by molecular mechanics calculations. *, Biaryl atropisomeric backbone is not represented. ax, pseudo-axial position; eq, pseudo-equatorial position. †, CAChe MM2 representation. H omitted for clarity; purple = P, red = O, green = F, blue = Cl, gray = C, and white = Ru. ‡, Sterically favored according to the rule of the quadrants. §, Sterically unfavored according to the rule of the quadrants.
Table 1. Dihedral angles of the biaryl backbone in minimized structures of free diphosphines, β1 and β2, and corresponding relative energies (CAChe MM2 calculations).
| Dihedral angle θ, °
|
Relative energy, kcal·mol-1
|
||||
|---|---|---|---|---|---|
| Diphosphine ligand | Free p*p | β1 | β2 | β1* | β2† |
| BINAP | 86.2 | 79.5 | 78.1 | -51.87 | -49.40 |
| BIPHEMP | 74.5 | 77.8 | 76.5 | -32.03 | -29.22 |
| MeO-BIPHEP | 72.3 | 75.7 | 75.3 | -29.27 | -27.20 |
| SYNPHOS | 70.7 | 75.4 | 73.2 | -27.55 | -23.12 |
| SEGPHOS | 67.2 | 73.3 | 72.6 | -7.22 | -3.09 |
| DIFLUORPHOS | 67.6 | 73.3 | 72.7 | -9.31 | -4.89 |
Sterically favored according to quadrants rule.
Sterically unfavored according to quadrants rule.
As depicted in Table 1, whatever the structure modeled by this method (P*P, β1, and β2), the relative order in dihedral angles remained unchanged: θ(BINAP) > θ(BIPHEMP) > θ(MeO-BIPHEP) > θ(SYNPHOS) > θ(DIFLUORPHOS) = θ(SEGPHOS), which assured us of the ability of molecular modeling to exhibit correct trends in dihedral angles as long as the structures and the calculation methods remain the same for all of the ligands studied. This relative order seemed independent of the type of complex containing the diphosphine. This result was in good agreement with our previous independent comparative studies of SYNPHOS (33) and DIFLUORPHOS (25) with BINAP and MeO-BIPHEP, and the results found by Saito and coworkers (17) using the same program to model BINAP (73.49°), BIPHEMP (72.07°), MeO-BIPHEP (68.56°), and SEGPHOS (64.99°) ligands in complexes of type [NH2Me2][{RuCl[(R)-P*P]}2(μ-Cl)3]. Therefore, SYNPHOS and DIFLUORPHOS displayed the narrowest dihedral angles of the series, which conferred on them apriori ideal steric properties for asymmetric hydrogenation. However, we expected their electronic properties to differ radically from each other, which is the point of the next section.
Electronic Profiling of SYNPHOS and DIFLUORPHOS (25). The electronic coordination mode of phosphine ligands to a transition metal involves σ-donation from the ligand to the metal and π-retrodonation from the metal to the ligand. Resulting electronic donor–acceptor properties of diphosphines can be conveniently evaluated by measuring the carbonyl stretching frequency of [RhCl(P*P)(CO)] complexes by IR spectroscopy. The higher the carbonyl stretching frequency [ν(CO)], the lower the electronic density on the rhodium center and the higher the π-acidic character of the ligand. Takaya used these complexes to show that p-F-BINAP and p-Cl-BINAP, two halogenated analogues of BINAP, had more π-acidic character than BINAP (35). [RhCl(P*P)(CO)] complexes E (25) were obtained by reaction of the dimeric precursor [Rh(CO)2Cl]2 with the diphosphine ligand. We noticed that [RhCl(P*P)(CO)] complexes were highly sensitive to oxidation by dioxygen. Therefore, preparation of complexes E were strictly performed under an inert atmosphere of argon to avoid the formation of [RhCl(P(O)*P)(CO)] (36). The four diphosphines of this comparative study were ordered on an decreasing π-acidity as follows: DIFLUORPHOS [ν(CO) = 2,023 cm–1] > BINAP [ν(CO) = 2,017 cm–1] > MeO-BIPHEP [ν(CO) = 2,014 cm–1] > SYNPHOS [ν(CO) = 2,012 cm–1]. SYNPHOS and DIFLUORPHOS were placed at both ends of this electronic scale, displaying, respectively, the highest and the lowest electronic availability. With stereoelectronic data concerning four atropisomeric ligands, we performed screening tests in asymmetric hydrogenation of a series of prochiral substrates.
Screening Tests in Asymmetric RuII-Mediated Hydrogenation Reactions. According to our convenient and versatile procedure (23), the catalysts [Ru(P*P)Br2] were prepared as shown in Fig. 6. SYNPHOS and DIFLUORPHOS were directly compared with their commercially available analogues BINAP and MeO-BIPHEP in terms of enantioselectivity in the hydrogenation of substrates 5a–5k. We conducted hydrogenation reactions at least twice for each substrate and used strictly the same conditions for the four ligands: temperature, pressure, reaction time, substrate concentration (0.5 M), and substrate/catalyst ratio (= 100). The main goal of this comparative study was to observe the direct influence of the ligand on enantioselectivities rather than to optimize each diphosphine/substrate system. Results are described in Tables 2 and 3. Whatever the diphosphine used, β-keto esters bearing linear (5a, entry 1) or branched (5b, entry 2) alkyl functionalities in the γ-position were hydrogenated with ee ≥98% by using mild hydrogenation conditions [4 bar (1 bar = 100 kPa), 50°C]. On the contrary, significant differences in selectivity between the four ligands were perceptible in the hydrogenation reactions of substrates 5c–5g (Table 2). Under 10 bar and 80°C, the hydrogenation of ethyl benzoylacetate (5c, entry 3) afforded the corresponding β-hydroxy ester 6c with ee ranging from 88% (BINAP) to 97% (SYNPHOS). α-Keto esters, such as 5d and 5e, are known to be sensitive substrates that often require accurate optimization of the hydrogenation conditions (37); acidic additives may be necessary to increase both activity and selectivity (35). We were pleased to observe that, even without acidic additives, the results showed a marked superiority in selectivity for MeO-BIPHEP and SYNPHOS ligands: ee = 94% for 6d (entry 4) and ee = 90% and 92%, respectively, for 6e (entry 5). In the hydrogenation of hydroxyacetone 5f, oxygenated diphosphines MeO-BIPHEP, SYNPHOS, and DIFLUORPHOS were more selective than BINAP and afforded 1,2-propandiol 6f in 96–97% ee (entry 6). Dimethyl itaconate 5g (entry 7) was also reduced efficiently by using SYNPHOS ligand (92% ee; 20 bar and 50°C). Other diphosphines displayed ee ranging from 85% (DIFLUORPHOS) to 90% (MeO-BIPHEP) in the same hydrogenation conditions. For the seven substrates 5a–5g (series 1, Table 2), SYNPHOS ligand turned out to be the ligand of choice to obtain the best enantioselectivities (92–99%).
Fig. 6.

General procedure for screening tests in asymmetric hydrogenation reactions with in situ [Ru(diphosphine)Br2] as catalysts.
Table 2. Screening of ligands in the asymmetric hydrogenation of substrates 5a–5g (series 1).
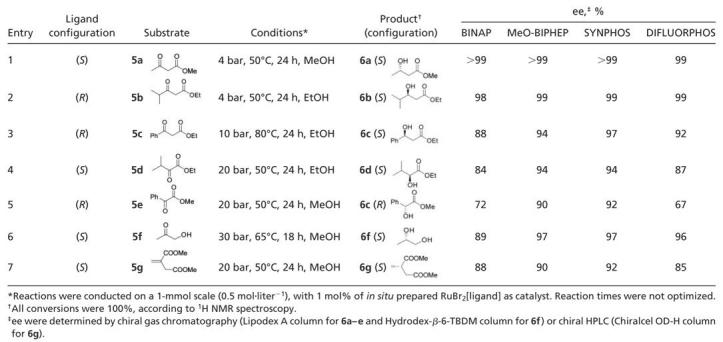 |
Table 3. Screening of ligands in the asymmetric hydrogenation of substrates 5h–5k (series 2).
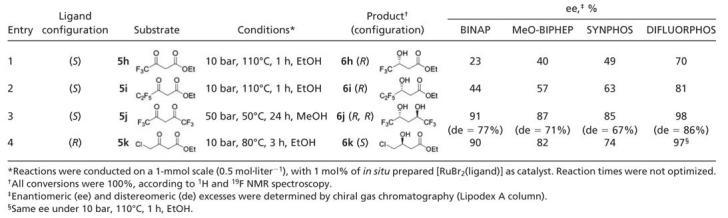 |
On the contrary, relative order in ee was different for substrates 5h–5k (Table 3). The β-hydroxy esters bearing perfluorinated alkyl groups in γ-position, 6h and 6i, are valuable building blocks for the synthesis of fluorinated drugs (38), but their corresponding β-keto esters are known to give poor ee in asymmetric hydrogenation with Ru/BINAP (39) or Ru/MeO-BIPHEP (40) catalysts. Indeed, even at high temperature (110°C), 6h and 6i were obtained in only moderate 23–57% ee with these two diphosphines (entries 1 and 2). Significant improvement in selectivity was achieved by using SYNPHOS (49% and 63% ee for 6h and 6i, respectively) and, in particular, DIFLUORPHOS (70% and 81% ee for 6h and 6i, respectively). The asymmetric hydrogenation of fluorinated diketone 5j has rarely been studied and has furnished only moderate ee for corresponding anti-1,1,1,5,5,5-hexafluoro-2,4-pentandiol 6j [diastereomeric excess = 70% and ee = 70% for the (R,R)-enantiomer with Ru{(S)-(–)-p-Tol-BINAP}(PF6)2 as catalyst; ref. 41]. Once again (S)-DIFLUORPHOS displayed the best results among the four ligands of this comparative study, affording (R,R)-6j with good diastereomeric (86%) and excellent enantiomeric (98%) excesses (entry 3). The same trend was observed for the chlorinated substrate 5k (entry 4). For this substrate, high temperature is necessary to achieve high ee, presumably avoiding a competitive chelation of chloride to ruthenium (42). Indeed results were highly dependent on the temperature for rich ligands, such as MeO-BIPHEP (ee = 82% at 80°C) or SYNPHOS (only 74% ee at 80°C). The use of BINAP afforded improved ee of 90% at 80°C. In addition, DIFLUORPHOS results on this substrate were not only excellent, but independent of the temperature: ee = 97%, under 10 bar, at either 80 or 110°C. For the four substrates of series 2 (5h–5k), which all have electron-withdrawing and/or chelating groups in the γ-position, DIFLUORPHOS exhibited the best selectivity in the ligand-screening tests.
In summary, this preliminary comparative study provided us important clues to estimate the matching pairs (giving the best ee) between atropisomeric diphosphines and classical hydrogenation substrates. In series 1, the matching substrate/ligand couples were systematically obtained with SYNPHOS, whereas, in series 2, they were successfully realized with DIFLUORPHOS. SYNPHOS and DIFLUORPHOS were indeed fully complementary regarding their behavior in Ru-mediated asymmetric hydrogenation, insofar as the highest enantioselectivity could be achieved by choosing the appropriate ligand.
Discussion
Steric and electronic profiling studies performed on BINAP, MeO-BIPHEP, SYNPHOS and DIFLUORPHOS ligands helped us to get a deeper insight into the intimate role of diphosphine ligands in ruthenium-mediated asymmetric hydrogenation. Fig. 7 describes the superimposition of steric and electronic scales established for the four ligands, arbitrarily lined up on BINAP. It reveals parallel stereoelectronic tendencies for BINAP, MeO-BIPHEP, and SYNPHOS (the smaller the dihedral angle, the higher the electronic density on phosphorus), whereas DIFLUORPHOS does not follow the same trend and is placed at opposite ends of steric and electronic scales, with the narrowest dihedral angle and the highest π-acidity. This result confers on DIFLUORPHOS a totally atypical structural profile compared with other diphosphines.
Fig. 7.

Comparative steric (Left) and electronic (Right) scales of BINAP, MeO-BIPHEP, SYNPHOS, and DIFLUORPHOS ligands (arbitrarily lined up on BINAP).
At this point, it was of interest to correlate these stereoelectronic characteristics with the ee obtained in asymmetric hydrogenation. Fig. 8 displays the respective influence of dihedral angle θ (Fig. 8 a and b) and π-acidity (Fig. 8 c and d) of ligands on the ee for each substrate, in series 1 and 2 separately. As shown in the preceding section, substrates 5a and 5b (Table 2) did not provide any discrimination between the four ligands and were not represented on these graphics. Correlations between θ and ee had been described previously for Ru-mediated asymmetric hydrogenation, but concerned only electronically close ligands (33) and/or were performed on few (17) or nondiscriminating (31) substrates. Actually, none of these studies had taken into account the simultaneous impact of the steric and electronic properties of the ligand and the influence of the substrate. The narrowest dihedral angle did not always provide the best ee (Fig. 8a) for substrates 5c–5g (series 1). As long as the steric and electronic scales of the diphosphines remained parallel (BINAP, MeO-BIPHEP, and SYNPHOS), the ligand with the narrowest dihedral angle (which is also the most electron-rich) displayed the best ee for the substrates of series 1; however, the DIFLUORPHOS ligand, despite its narrowest dihedral angle, induced a marked decrease in ee with this series, which revealed the overwhelming impact of the electronic properties of the ligand for these substrates. As shown in Fig. 8c, for this series of substrates (especially for substrates 5e and 5g), the ee values were almost lineally related to the basicity of the ligand; the less π-acidic the ligand, the better the enantioselectivity. In summary, SYNPHOS was the best compromise in terms of stereoelectronic features (a narrow dihedral angle combined with a higher electronic density on phosphorus) to fit the requirements of substrates in series 1. The influence of dihedral angle parameter seemed more evident for fluorinated substrates 5h and 5i (Fig. 8b); the ee obtained for the corresponding β-keto esters were directly dependent on θ and reached their maximum for DIFLUORPHOS ligand, which has the smallest dihedral angle. In this case, electronic features of the ligand seem to match perfectly the requirement of the substrates. Fluorinated diketone 5j and ethyl 4-chloroacetoacetate 5k were more sensitive to the π-acidic character of the diphosphine. As shown in Fig. 8d, enantiomeric excesses obtained in the hydrogenation reactions of these substrates were almost linearly related to π-acidity and consequently reached their maximum with DIFLUORPHOS.
Fig. 8.

Respective influence of steric (Upper) and electronic (Lower) parameters of diphosphines on the enantioselectivities (ee) measured in Ru-mediated asymmetric hydrogenation of substrates 5c–5k.
Conclusion
In summary, we have described effective scaled-up development syntheses of two atropisomeric diphosphines, SYNPHOS and DIFLUORPHOS. Practical steric and electronic profiling, applicable to a wide range of other diphosphines, has been performed to quantify the stereoelectronic parameters of these ligands. SYNPHOS (with a narrow dihedral angle and a stronger basicity) is fully complementary to DIFLUORPHOS (with a narrow dihedral angle and a higher π-acidity) in terms of structural features and selectivity in RuII-catalyzed asymmetric hydrogenation of prochiral insaturations. Substrate families corresponding to preferred ligand/substrate pairs were identified. Studies concerning the systematic impact of steric and electronic properties of diphosphine ligands in other asymmetric catalytic reactions are needed.
Acknowledgments
We thank Dr. A. Solladié-Cavallo (Department of Fine Organic Chemistry, Ecole Européenne de Chimie Polyméres et Matériaux, Strasbourg, France) for permission to use molecular modeling facilities and Dr. R. Schmid (Hoffmann-LaRoche) for the gift of (R)- and (S)-MeO-BIPHEP. This work was supported by the Centre National de la Recherche Scientifique and Synkem S.A.S. (S.J. and S.D.P.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ee, enantiomeric excess; P*P, chiral diphosphine; THF, tetrahydrofuran; DBTA, O,O-dibenzoyltartaric acid; BINAP, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; BIPHEMP, 6,6′-dimethyl-2,2′-bis(diphenylphosphino)-1,1′-biphenyl; MeO-BIPHEP, 6,6′-dimethoxy-2,2′bis(diphenylphosphino)-1,1′-biphenyl; SEGPHOS, [(4,4′-bi-1,3-benzodioxole)-5,5′-diyl]bis(diphenylphosphine).
References
- 1.Blaser, H.-U., Spindler, F. & Studer, M. (2001) Appl. Catal. A 221, 119–143. [Google Scholar]
- 2.Knowles, W. S. (2002) Angew. Chem. Int. Ed. Engl. 41, 1998–2007. [Google Scholar]
- 3.Noyori, R. (2002) Angew. Chem. Int. Ed. Engl. 41, 2008–2022. [PubMed] [Google Scholar]
- 4.Sharpless, K. B. (2002) Angew. Chem. Int. Ed. Engl. 41, 2024–2032. [PubMed] [Google Scholar]
- 5.Noyori, R. (1994) in Asymmetric Catalysis in Organic Synthesis (Wiley, New York), pp. 1–93.
- 6.Genet, J.-P. (1996) in Reductions in Organic Synthesis, ACS Symposium Series 641, ed. Magid, A. (American Chemical Society, Washington, DC), pp. 31–51.
- 7.Ohkuma, T., Kitamura, M. & Noyori, R. (2000) in Catalytic Asymmetric Synthesis, ed. Ojima, I. (Wiley-VCH, New York), pp. 1–110.
- 8.Noyori, R. & Ohkuma, T. (2001) Angew. Chem. Int. Ed. Engl. 40, 40–73. [PubMed] [Google Scholar]
- 9.Genet, J.-P. (2003) Acc. Chem. Res. 36, 908–918. [DOI] [PubMed] [Google Scholar]
- 10.Kagan, H. B. & Dang, T. P. (1971) Chem. Commun. 481.
- 11.Knowles, W. S., Sabacky, M. J., Vineyard, B. D. & Weinkauff, D. J. (1975) J. Am. Chem. Soc. 97, 2567–2568. [Google Scholar]
- 12.Miyashita, A., Yasuda, A., Takaya, H., Toriumi, K., Ito, T., Souchi, T. & Noyori, R. (1980) J. Am. Chem. Soc. 102, 7935–7936. [Google Scholar]
- 13.Akutagawa, S. (1995) Appl. Catal. A 128, 171–207. [Google Scholar]
- 14.McCarthy, M. & Guiry, P. J. (2001) Tetrahedron 57, 3809–3844. [Google Scholar]
- 15.Tang, W. & Zhang, X. (2003) Chem. Rev. 103, 3029–3070. [DOI] [PubMed] [Google Scholar]
- 16.Schmid, R., Broger, E. A., Cereghetti, M., Crameri, Y., Foricher, J., Lalonde, M., Müller, R. K., Scalone, M., Schoettel, G. & Zutter, U. (1996) Pure Appl. Chem. 68, 131–138. [Google Scholar]
- 17.Saito, T., Yokozawa, T., Ishizaki, T., Moroi, T., Sayo, N., Miura, T. & Kumobayashi, H. (2001) Adv. Synth. Catal. 343, 264–267. [Google Scholar]
- 18.Guerreiro, P., Ratovelomanana-Vidal, V., Genet, J.-P. & Dellis, P. (2001) Tetrahedron Lett. 42, 3423–3426. [Google Scholar]
- 19.Michaud, G., Bulliard, M., Ricard, L., Genet, J.-P. & Marinetti, A. (2002) Chem. Eur. J. 8, 3327–3330. [DOI] [PubMed] [Google Scholar]
- 20.Duprat de Paule, S., Champion, N., Vidal, V., Genet, J.-P. & Dellis, P. (2001) French Patent 2,830,254.
- 21.Duprat de Paule, S., Champion, N., Vidal, V., Genet, J.-P. & Dellis, P. (2003) World Patent 03,029,259.
- 22.Pai, C.-C., Li, Y.-M., Zhou, Z.-Y. & Chan, A. S. C. (2002) Tetrahedron Lett. 43, 2789–2792. [Google Scholar]
- 23.Genet, J.-P., Pinel, C., Ratovelomanana-Vidal, V., Mallart, S., Pfister, X., Caño de Andrade, M. C. & Laffitte, J. A. (1994) Tetrahedron: Asymmetry 5, 665–674. [Google Scholar]
- 24.Duprat de Paule, S., Jeulin, S., Ratovelomanana-Vidal, V., Genet, J.-P., Champion, N., Deschaux, G. & Dellis, P. (2003) Org. Process Res. Dev. 7, 399–406. [Google Scholar]
- 25.Duprat de Paule, S., Jeulin, S., Ratovelomanana-Vidal, V., Genet, J.-P., Champion, N. & Dellis, P. (2004) Angew. Chem. Int. Ed. 43, 320–325. [DOI] [PubMed] [Google Scholar]
- 26.Kitamura, M., Yoshimura, M., Kanda, N. & Noyori, R. (1999) Tetrahedron 55, 8769–8785. [Google Scholar]
- 27.Ashby, M. T. & Halpern, J. (1991) J. Am. Chem. Soc. 113, 589–594. [Google Scholar]
- 28.Wiles, J. A. & Bergens, S. H. (1999) Organometallics 18, 3709–3714. [Google Scholar]
- 29.Daley, C. J. A. & Bergens, S. H. (2002) J. Am. Chem. Soc. 124, 3680–3691. [DOI] [PubMed] [Google Scholar]
- 30.Dierkes, P. & van Leeuwen, P. W. N. M. (1999) J. Chem. Soc. Dalton Trans. 1519–1529.
- 31.Zhang, Z., Qian, H., Longmire, J. & Zhang, X. (2000) J. Org. Chem. 65, 6223–6226. [DOI] [PubMed] [Google Scholar]
- 32.Duprat de Paule, S., Jeulin, S., Ratovelomanana-Vidal, V., Genet, J.-P., Champion, N. & Dellis, P. (2003) Tetrahedron Lett. 44, 823–826. [Google Scholar]
- 33.Duprat de Paule, S., Jeulin, S., Ratovelomanana-Vidal, V., Genet, J.-P., Champion, N. & Dellis, P. (2003) Eur. J. Org. Chem. 1931–1941.
- 34.van Leeuwen, P. W. N. M., Kamer, P. C. J., Reek, J. N. H. & Dierkes, P. (2000) Chem. Rev. 100, 2741–2769. [DOI] [PubMed] [Google Scholar]
- 35.Mashima, K., Kusano, K., Sato, N., Matsumura, Y., Nozaki, K., Kumobayashi, H., Sayo, N., Hori, Y., Ishizaki, T., Akutagawa, S. & Takaya, H. (1994) J. Org. Chem. 59, 3064–3076. [Google Scholar]
- 36.Bunten, K. A., Farrar, D. H., Poë, A. J. & Lough, A. (2002) Organometallics 21, 3344–3350. [Google Scholar]
- 37.Genet, J.-P., Pinel, C., Ratovelomanana-Vidal, V., Mallart, S., Pfister, X., Bischoff, L., Caño de Andrade, M. C., Darses, S., Galopin, C. & Laffitte, J. A. (1994) Tetrahedron: Asymmetry 5, 675–690. [Google Scholar]
- 38.Antolini, L., Forni, A., Davoli, P., Moretti, I. & Prati, F. (1998) Tetrahedron: Asymmetry 9, 285–292. [Google Scholar]
- 39.Sayo, N., Akutagawa, S., Saito, T., Noyori, R., Kumobayashi, H. & Takaya, H. (1988) Eur. Patent 295,109.
- 40.Blanc, D., Ratovelomanana-Vidal, V., Gillet, J.-P. & Genet, J.-P. (2000) J. Organomet. Chem 603, 128–130. [Google Scholar]
- 41.Sayo, N., Saito, T., Kumobayashi, H., Akutagawa, S., Noyori, R. & Takaya, H. (1989) European Patent (Takasago International Corp.) EP297,752.
- 42.Kitamura, M., Ohkuma, T., Takaya, H. & Noyori, R. (1988) Tetrahedron Lett. 29, 1555–1556. [Google Scholar]