

Abstract
11[C]Methyl iodide is the most widely used reagent for labeling radiotracers with carbon-11 (t1/2 = 20.4 min) for molecular imaging with positron emission tomography. However, some substrates for labeling, especially primary arylamines and pyrroles, are sluggishly reactive towards [11C]methyl iodide. We found that insoluble inorganic bases, especially Li3N or Li2O, are effective in promoting rapid reactions (≤ 10 min) of such substrates with no-carrier-added [11C]methyl iodide in DMF at room temperature to give 11C-methylated products in useful radiochemical yields. In particular, we discovered that some primary arylamines in Li3N-DMF were converted into their formanilides, and that these were readily N-methylated with [11C]methyl iodide, preceding easy basic hydrolysis to the desired [11C]N-methyl secondary arylamines. Use of a solid base permitted selective reaction at an arylamino group and in some cases also avoided undesirable side reaction, such as ester group hydrolysis. An ultrasound device proved useful to provide remote and constant agitation of the radioactive heterogeneous reaction mixtures, but imparted no ‘ultrasound-specific’ chemical effect.
Keywords: carbon-11, methylation, inorganic base, arylamine, ultrasound
Introduction
11C-Methylation is an important method for preparing radiotracers for application with positron emission tomography (PET).[1–3] Since carbon-11 has a very short half-life of 20.4 min, 11C-methylation procedures must be efficient over short periods, typically less than 10 min.[4,5] [11C]Methyl iodide[6–9] and [11C]methyl triflate[10] are the most accessible and most widely used 11C-methylation agents. Even so, these reagents have some limitations for the 11C-N-methylation of arylamines. [11C]Methyl iodide is often poorly effective.[11] The more reactive [11C]methyl triflate can be generated simply by passing [11C]methyl iodide over heated silver triflate,[10] and is especially useful for the 11C-methylation of arylamines, where the nucleophilicity of the amino group is reduced by conjugation to the ring.[12,13] However, if the electron density of the aryl group in a primary arylamine is further reduced by an electron-withdrawing group, even [11C]methyl triflate may fail to react.
In our program of developing radiotracers for PET imaging, we have encountered several arylamines that show only low or moderate reactivity towards either [11C]methyl iodide or [11C]methyl triflate under conventional heated basic conditions. Moreover, these relatively harsh conditions often lead to decomposition of substrate and/or product. These amines include precursors for the prospective β-amyloid plaque radioligands, [11C]1a and [11C]1b, and of a prospective PKC receptor radioligand ([11C]2) (Chart 1).
Chart 1.
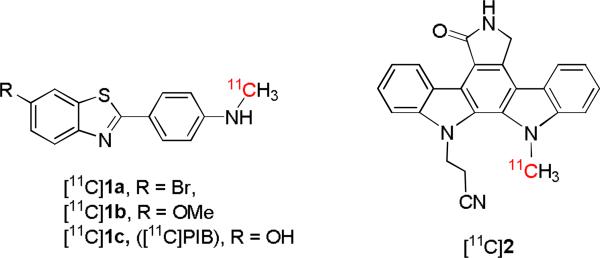
Some known[11] or prospective radiotracers having sluggishly reactive precursors in labeling reactions with [11C]methyl iodide.
In view of reports describing the successful use of solid-base-solvent combinations to achieve methylation or other alkylation reactions at anilines and pyrroles,[14,15] we decided to explore this approach for the 11C-methylation of such substrates with [11C]methyl iodide. We also aimed to explore ultrasound as a means for achieving constant agitation with potential to increase reaction yields as documented for several types of organic reaction,[16,17] including methylation reactions.[18,19] In this study, we found that the use of agitated solid inorganic bases, when paired with N,N-dimethylformamide (DMF), could overcome the sluggish reactivity of arylamines and permit rapid 11C-methylation at room temperature. For certain arylamines, this process was found to occur through unexpected N-formyl intermediates.
Results and Discussion
The aim of this study was to explore the use of solid inorganic bases for promoting rapid reactions of [11C]methyl iodide with primary arylamines and a pyrrole (Chart 1) at room temperature, using ultrasound as a means of heterogeneous reaction mixture agitation. Our major findings were: i) that the use of Li3N-DMF or Li2O-DMF provided useful (>5%) decay-corrected radiochemical yields (RCYs) of 11C-N-methylated products, including unexpected N-formyl compounds, ii) that formanilides were themselves useful reactive precursors for rapidly preparing [11C]N-methylarylamines, and iii) that ultrasound was useful for heterogeneous reaction agitation but did not impart an ultrasound-specific chemical effect.
Ultrasound is known to accelerate many types of organic chemical reaction,[16,17] especially those occurring at solid-liquid interfaces.[20,21] Micro-bubbles formed during ultrasonication of liquid-solid mixtures may collapse at the phase interface, generating a local temperature of up to 5000 °C within a fraction of a millisecond.[16,22-24] Also ultrasound between 5 and 24 kHz transfers energy to reactants without dramatically changing the temperature of the bulk medium[24] and this is an attractive feature for reactions involving compounds with thermally sensitive groups. Despite the widespread use of ultrasound as an energy source in general synthetic organic chemistry, ultrasound has not been considered previously for promoting labeling reactions with short-lived isotopes, such as carbon-11. In view of possible advantages, we decided to explore ultrasound as a means for constant heterogeneous reaction mixture agitation and for promoting 11C-methylation reactions at sluggishly reactive amino groups.
[11C]Methyl iodide was chosen for this study over the more reactive [11C]methyl triflate to avoid unwanted side-reactions. Inorganic bases were selected i) to be strong enough to deprotonate the weakly acidic amino group to be labeled; ii) to have a large lattice energy in solid form to prevent the base serving as a nucleophile; and iii) to be insoluble in the liquid phase in order to prevent other possible side-reactions, such as substrate decomposition or direct reaction with [11C]methyl iodide. For the methylation of arylamines, we focused on the use of solid Li3N, Li2O, BaO or KOH as base. For the 11C-methylation of the pyrrole 12, we used solid K2CO3.
In the choice of a supporting liquid phase for the reactions, protic solvents and those capable of reacting with strong base were excluded. Solvents with low water-solubility were avoided so that labeled products could be purified with reverse phase HPLC. THF (tetrahydrofuran) is known to participate in chemical reactions under strongly basic conditions[25] and we also found that some substrates decomposed extensively in THF within 10 min of ultrasonication (unpublished results). Hexamethylphosphoramide and pyridine were found to give only very low RCYs (< 1%) for reactions of the primary aromatic amines 7a and 7b in the presence of Li3N, and hence their use was not further explored. Therefore DMF became the liquid phase of choice. We limited all reaction times to 10 min or less to avoid excessive physical decay of carbon-11 during reaction.
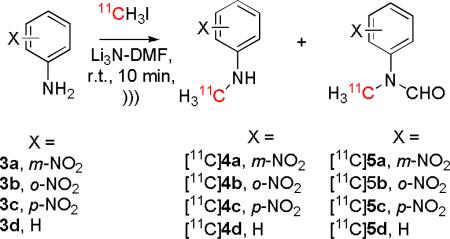
Simple anilines and in particular anilines carrying a p-nitro or another electron-withdrawing group, are well-known to be poorly reactive towards alkyl halides, even in the presence of strong base.[26-29] For example, alkylations of aniline in hydrocarbon solvents promoted by alumina or zeolites take several hours at elevated temperatures.[15] We first tested the 11C-methylation of simple anilines in Li3N-DMF. m-Nitroaniline gave a very low yield (3%) of [11C]N-methyl-m-nitroaniline ([11C]4a; Table 1, entry 1) whereas o- and p-nitroaniline gave high (62%) and moderate yields (37%) of the respective [11C]N-methyl products, [11C]4b and [11C]4c (Table 1, entries, 2 and 3). Such reactions might be expected to proceed through deprotonation of the weakly acidic amino group to create a highly nucleophilic anion.[30] However, the radiochemical yields in these reactions appear opposite to the expected rank of basicities and nucleophilicities of the anions (m-nitroaniline > p-nitro ~ o-nitro). A possible explanation is that the generated concentration of the anion decreased with increase in the pKa of the amine for proton loss, although we note some evidence for the N-alkylation of weak N-H acids in the presence of a solid base to occur by complex catalytic mechanisms, independent of amine deprotonation.[31] Aniline was unexpectedly converted in high radiochemical yield (68%) into the 11C-methylated formamide ([11C]5d), whereas no labeled formamide derivatives were produced from the nitroanilines (Table 1, entry 4). This may suggest that the anion of aniline (aniline pKa, 30.7)[32] and not any of the anions of the nitroanilines (p-nitroaniline pKa, 20.9)[32] is sufficiently nucleophilic to displace Me2NH from DMF. DMF has previously been reported to act as a source of the formyl group in a number of reactions,[33] including the N-formylation of halo and carboxyl anilines in DMF under reflux in the presence of sodium methoxide.[34]
Table 1.
RCYs of labeled products from the reactions of anilines with [11C]methyl iodide in the presence of Li3N-DMF after ultrasonication for 10 min at r.t.
| |||||
|---|---|---|---|---|---|
| Entry | Precursor | Amine product | RCY[a] (%) | Amide product | RCY (%) |
| 1 | 3a | [11C]4a | 3 | [11C]5a | 0 |
| 2 | 3b | [11C]4b | 62 ± 3 (59) | [11C]5b | 0 |
| 3 | 3c | [11C]4c | 37 ± 2 (25) | [11C]5c | 0 |
| 4 | 3d | [11C]4d | 4 | [11C]5d | 68 |
Errors are range for n = 2. Values in prentheses are for shaken ‘silent’ reactions.
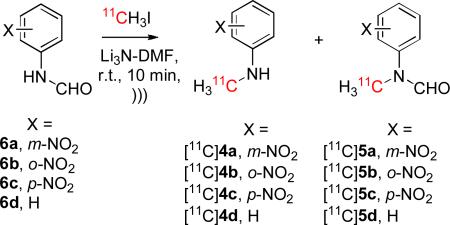
A low radiochemical yield (4%) of [11C]N-methylaniline ([11C]4d) was co-produced in the reaction of aniline (Table 1, entry 4). We wondered if this product may have been generated from aniline directly or from the major N-formyl product [11C]5d during aqueous quench of the reaction. Accordingly, we tested the influence of the N-formyl group on radiolabeling. All the tested formanilides (6a–6d) gave aggregate 11C-N-methylation (Table 2) in higher yields than the corresponding anilines (3a–3d) (Table 1). The most significant difference arose between N-formyl-m-nitroaniline (6a) and m-nitroaniline (3a) with 6a giving 45% 11C-methylation and 3a only 3% (c.f. Table 2, entry 1 with Table 1, entry 1). For p-nitroaniline, no radiolabeled formamide was present after product isolation, probably because of the high reactivity of any formamide towards the LiOH that would have been generated in situ from Li3N upon quench with water. Formanilides and N-methylformanilides are indeed known to be readily hydrolyzed in alkaline solution and both N-methylformanilide (5d) and N-methyl-m-nitroformanilide (5a) are much less prone to alkaline hydrolysis than N-methyl-pnitroformanilide (5c).[35] These facts appear remarkably consistent with our findings in Table 2.
Table 2.
RCYs of labeled products from the reactions of simple formanilides with [11C]methyl iodide in the presence of Li3N-DMF after ultrasonication for 10 min at r.t.
| |||||
|---|---|---|---|---|---|
| Entry | Precursor | Amine product | RCY (%) | Amide product | RCY (%) |
| 1 | 6a | [11C]4a | 0 | [11C]5a | 45 |
| 2 | 6b | [11C]4b | 20 | [11C]5b | 61 |
| 3 | 6c | [11C]4c | 76 | [11C]5c | 0 |
| 4 | 6d | [11C]4d | 0 | [11C]5d | 77 |
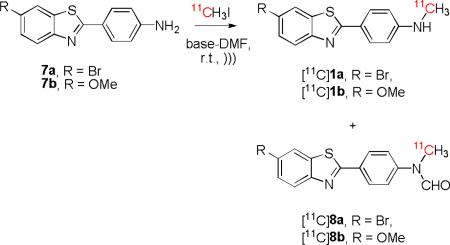
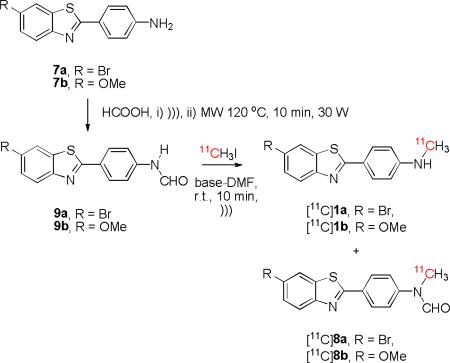
We are developing prospective radiotracers for imaging brain β-amyloid with PET, such as [11C]1a and [11C]1b, which are structurally-related to the prototypical radiotracer [11C]PIB ([11C]1c)[11] (Chart 1). In view of our findings on the 11C-methylation of the simple nitroanilines (Table 1), we were interested to test the same procedure for preparing the structurally more complex targets, [11C]1a and [11C]1b. For the reaction of the primary arylamine 7a with [11C]methyl iodide under ultrasonication for 5 min, Li3N in DMF gave the highest aggregate yield of labeled products (14%), in this case the target 11C-labeled secondary amine [11C]1a as minor product plus the N-formyl derivative [11C]8a as major product (Table 3, entry 1). Increase in ultrasonication time from 5 to 10 min had negligible effect on yield for [11C]1a (entry 2). The closely related arylamine 7b gave similar yields of the corresponding [11C]N-formyl and [11C]N-methyl products ([11C]8b and [11C]1b, respectively) under these conditons (entry 3). Since the major products were [11C]8a from 7a and [11C]8b from 7b, DMF had clearly acted as a source of the formyl group, as in the reaction of aniline.
Table 3.
RCYs of labeled products from the reaction of the primary arylamines 7a and 7b with [11C]methyl iodide in the presence of solid inorganic base-DMF under ultrasonication at r.t.
| |||||||
|---|---|---|---|---|---|---|---|
| Entry | Precursor | Solid base | Time (min) | Amine product | RCY[a] (%) | Amide product | RCY[a] (%) |
| 1 | 7a | Li3N | 5 | [11C]1a | 6 | [11C]8a | 8 |
| 2 | 7a | Li3N | 10 | [11C]1a | 6 ± 0.1 | [11C]8a | 9 ± 0.1[b] |
| 3 | 7b | Li3N | 10 | [11C]1b | 4 ± 2 (9) | [11C]8b | 9 ± 10[c] (2) |
| 4[d] | 7a | Li3N | 10 | [11C]1a | 3 | [11C]8a | 25 |
| 5[d] | 7a | Li2O | 10 | [11C]1a | 2 | [11C]8a | 22 |
| 6[d] | 7b | Li2O | 10 | [11C]1b | 2 | [11C]8b | 19 |
Values in parentheses are for shaken reactions.
Errors are range for n = 2.
Mean ± SD for n = 4.
The precursor and Li3N-DMF were sonicated for 30 min before [11C]methyl iodide was introduced.
Interestingly, for 7a ultrasonication of a reaction mixture for 30 min before the addition of [11C]methyl iodide gave a much higher yield of radiolabeled products (28%), with the [11C]N-formyl compound much more prevalent than the [11C]N-methyl compound (Table 3, entry 4). Replacement of Li3N with Li2O resulted in similar radioactive product distribution and yields (entry 5). Arylamine 7b gave similar results to 7a under these conditions, again with the N-formyl compound ([11C]8b) as major radioactive product (entry 6). Our interpretation of these results is that presonication in DMF likely generated substantial concentrations of the formanilides and that these reacted more readily than the parent amines with [11C]methyl iodide under basic conditions. Again reactions likely proceeded through the generation of nucleophilic anions. Consideration of reported pKa values suggests that anions of formanilide intermeditaes would indeed be formed much more easily than anions of the parent anilines. Thus, the pKa of formanilide (19.4)[35] is much lower than that of aniline (30.7)[32], and that of p-nitroformanilide (12.5)[36] is much lower than that of p-nitroaniline (20.9)[32].
We obtained evidence that the N-formyl compounds [11C]8a and [11C]8b were progenitors of the labeled amines, [11C]1a and [11C]1b, respectively. Thus, after exposure of the amines 7a and 7b in DMF to [11C]methyl iodide and ultrasound, we isolated the respective N-formyl compounds [11C]8a and [11C]8b and showed that each may be readily hydrolyzed with 1M KOH at room temperature for 5 min to give the target 11C-N-methyl compounds, [11C]1a and [11C]1b.
Our results plus literature precedent[37] describing the ready alkylation of lithium salts of formanilides prompted us to prepare the formanilides 9a and 9b (Table 4) to test their susceptibilities to 11C-N-methylation. We found that 7a and 7b could be converted into their respective non-radioactive formanilides, 9a and 9b, with formic acid under microwave irradiation.[38] Ultrasonication of 9a or 9b with [11C]methyl iodide in the presence of Li3N-DMF gave high aggregate yields of labeled products (68 and 50%, respectively), in each case composed of a mixture of the [11C]N-methyl derivative with the desformyl analog (Table 4, entries 1 and 2,). Presumably, the [11C]N-methyl derivatives [11C]1a and [11C]1b were produced by hydrolyses of the major products, the respective N-formyl-N-methyl compounds, [11C]8a or [11C]8b. Use of the base-solvent pair KOH-DMF with 9a or 9b cleanly gave useful radiochemical yields (66 and 32%) of the respective secondary amines [11C]1a or [11C]1b, (entries 3 and 4). These results further emphasize the strong utility of the N-formyl group for activating primary arylamines for 11C-N-methylation. Importantly, from these results, the 11C-N-methylation of formanilides followed by alkaline hydrolysis emerged clearly as a new rapid method for preparing [11C]N-methylanilines under mild conditions.
Table 4.
RCYs of labeled products from the reaction of formanilides 9a and 9b with [11C]methyl iodide in the presence of solid inorganic base-DMF with ultrasonication for 10 min at r.t.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Precursor | Solid base | Amine product | RCY[a] (%) | Amide product | RCY[a] (%) |
| 1 | 9a | Li3N | [11C]1a | 25 (21) | [11C]8a | 43 (45) |
| 2 | 9b | Li3N | [11C]1b | 9 (23) | [11C]8b | 41 (50) |
| 3 | 9a | KOH | [11C]1a | 66 (53) | [11C]8a | 0 (0) |
| 4 | 9b | KOH | [11C]1b | 32 (35) | [11C]8b | 0 (0) |
Values in parentheses are for shaken ‘silent’ reactions.
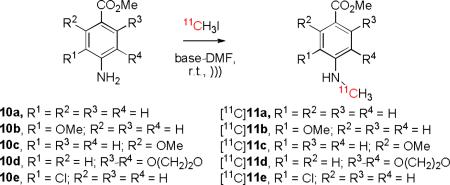
We have explored p-aminobenzoic acid esters as prospective radioligands for imaging serotonin subtype 4 receptors with PET.[39] A method to label such compounds with carbon-11 in the N-methyl group would be useful. We therefore next attempted the 11C-N-methylation of p-aminobenzoic acid methyl esters 10a–e in DMF under ultrasonic conditions to assess whether methylation could be achieved without accompanying ester hydrolysis. Li3N gave no labeled product from p-aminobenzoic acid methyl ester (10a) (Table 5, entry 1), while Li2O gave a modest radiochemical yield (15%) of the desired [11C]11a (entry 2). Li3N also gave extremely low or negligible yields from the aryl ring methoxy-substituted substrates 10b (entry 3) and 10c (entries 4), whereas Li2O gave a small yield from 10c (entries 5 and 6). The amino-benzodioxan 10d gave only a somewhat higher yield (14%) of [11C]N-methylated product [11C]11d with Li3N (entry 7) and no yield with Li2O (entry 8). For anilines 10a–d radiochemical yields therefore depended on the steric and electronic features of aryl ring substituents and their effects on amino group acidity, which may be expected to determine the effective concentration of nucleophilic anion. A methoxy substituent located ortho to the ester group, likely prevented co-planar arrangement of the ester group with the aryl ring, in turn reducing the electron-withdrawing power of the ester group on the amino group. An electron-donating methoxy group ortho to the amino group would also have reduced anion availability. In 10d both effects operated. By contrast, if a chloro substituent was ortho to the amino group, as in 10e, a useful radiochemical yield (up to 31%) of the [11C]N-methyl compound ([11C]11e) could be obtained (entries 9–11). In Li2O-DMF the yield of [11C]11e was maximal after 6 min of sonication (entry 10). The specific radioactivity of [11C]11e was found to be 6.1 Ci/μmol at the end of radiosynthesis, an acceptably high value for PET applications. BaO and Li3N gave much lower radiochemical yields (Table 5, entries 12 and 13, respectively).
Table 5.
RCYs of labeled products from the reaction of variously substituted 4-aminobenzoic acid methyl esters with [11C]methyl iodide in the presence of solid inorganic base-DMF with ultrasonication at r.t.
| ||||||
|---|---|---|---|---|---|---|
| Entry | Precursor | Ring substituent | Solid base | Time (min) | Product | RCY (%) |
| 1 | 10a | None | Li3N | 10 | [11C]11a | 0 |
| 2 | 10a | None | Li2O | 10 | [11C]11a | 15 ± 1[a] |
| 3 | 10b | 3-MeO | Li3N | 10 | [11C]11b | 4 ± 3 |
| 4 | 10c | 2-MeO | Li3N | 10 | [11C]11c | 0 |
| 5 | 10c | 2-MeO | Li2O | 6 | [11C]11c | 6 ± 1[a] |
| 6 | 10c | 2-MeO | Li2O | 10 | [11C]11c | 7 |
| 7 | 10d | O(CH2)2O | Li3N | 10 | [11C]11d | 14 |
| 8 | 10d | O(CH2)2O | Li2O | 6–10 | [11C]11d | 0[a] |
| 9 | 10e | 3-Cl | Li2O | 2 | [11C]11e | 21 ± 10[a] |
| 10 | 10e | 3-Cl | Li2O | 6 | [11C]11e | 31 ± 18[b] |
| 11 | 10e | 3-Cl | Li2O | 10 | [11C]11e | 16 ± 13[b] |
| 12 | 10e | 3-Cl | BaO | 10 | [11C]11e | 8 |
| 13 | 10e | 3-Cl | Li3N | 12 | [11C]11e | 11 |
Errors are range for n = 2.
Mean ± SD for n >5.
Remarkably, no ester hydrolysis was observed for substrates 10a–e under the labeling conditions. The chosen solid inorganic bases showed selectivity for imparting amino group deprotonation versus ester hydrolysis. We attribute the lack of nucleophilicity of the bases to their high lattice energies and to their insolubility in the reaction solvents.
Pyrrole nitrogens are weakly acidic (e.g., pKa for 1H-dibenzo[a,i]carbazole, 17.7[30]) and recognized to be poorly reactive towards methyl iodide. We have been interested to produce the [11C]N-methyl-pyrrole, [11C]2, as a prospective radiotracer for imaging the enzyme PKC by 11C-N-methylation of the pyrrole 12.[40] We have previously observed that strong bases eliminate acrylonitrile from 12 (reverse Michael addition) (Scheme 1). Even with the weak base, K2CO3, in the absence of ultrasound, but at elevated temperature (80 °C), elimination was complete within 10 min. If an N-methyl group was also present, as in the target 2, the steric crowding further favored elimination under thermal conditions. However, we found that reaction of 12 with [11C]methyl iodide in DMF-K2CO3 during ultrasound at room temperature reduced elimination significantly (Scheme 1). In fact only about 50% of the labeled product underwent elimination, and the target [11C]N-methyl-pyrrole [11C]2 was obtained in appreciable radiochemical yield (25%). This product was obtained with a specific radioactivity of 2.1 Ci/μmol at end of radiosynthesis.
Scheme 1.

Labeling of prospective PKC ligand 2 with carbon-11 by treatment of pyrrole 12 with [11C]methyl iodide in K2CO3-DMF for 10 min at r.t. with ultrasonication.
Clearly, moderate but useful radiochemical yields of [11C]N-methyl compounds were obtained from quite unreactive substrates by brief treatment with 11C-methyl iodide in solid base-DMF at room temperature under the influence of ultrasound. However, we wished to establish whether the ultrasound imparted a specific effect in these reactions, other than achieving desired reaction mixture agitation. For this reason, we conducted control reactions in the absence of ultrasound but with occasional shaking to promote uniformity in the heterogeneous reaction mixture. We found that the yields of labeled products in nearly all cases were comparable to those acheived under ultrasonication (Table 1, entries 2 and 3; Table 3, entry 3; Table 4, entries 1–4). This strongly indicates that in this study the major role imparted by ultrasound was mainly limited to constant agitation and promotion of reaction mixture uniformity. Nonetheless, the apparatus employed here proved attractive for this purpose, since it can be operated remotely at room temperature and with full radiation safety in a lead shielded ‘hot-cell‘.
Conclusions
The use of inorganic base-DMF proved especially effective for the rapid 11C-N-methylation of sluggishly reactive arylamines and of a pyrrole at room temperature with no-carrier-added [11C]methyl iodide. The bases appear effective at removing protons at weakly acidic nitrogen but inactive towards promoting the hydrolysis of sensitive benzoic acid esters. Furthermore, this study revealed formanilides to be useful precursors to secondary [11C]N-methylarylamines. The described ultrasound device was found to be a convenient means for heterogeneous radioactive reaction mixture agitation.
Experimental Section
Materials
The following solid inorganic bases were purchased from Aldrich (St. Louis, MO): lithium nitride (Li3N; 80 mesh), lithium oxide (Li2O; 97%, 60 mesh), barium oxide (BaO, technical grade, 90%, powder), potassium hydroxide (KOH, 85% pastilles, crushed in glove-box) and potassium carbonate (K2CO3, anhydrous powder, crushed in glove-box). N,N-Dimethylformamide (DMF) was purchased from Alfa Aesar (Ward Hill, MA). m-Nitroaniline (3a), o-nitroaniline (3b), p-nitroaniline (3c), aniline (3d), N-methyl-3-nitroaniline (4a), N-methyl-2-nitroaniline (4b), N-(2-nitrophenyl)formamide (6b), N-(4-nitrophenyl)formamide (6c), methyl 4-aminobenzoate (10a), methyl 4-amino-2-methoxybenzoate (10c), methyl 4-amino-3-chlorobenzoate (10e) and methyl 4-(methylamino)-3-chlorobenzoate (11e) were purchased from Aldrich (St. Louis, MO). N-Methyl-4-nitroaniline (4c), N-(3-nitrophenyl)formamide (6a), and methyl 4-(methylamino)benzoate (11a) were obtained from Alfa Aesar (Ward Hill, MA). N-Methyl-aniline (4d) was obtained from TCI (Portland, OR). N-Methyl-N-phenylformamide (5d), and N-phenylformamide (6d) were purchased from Acros (Pittsburgh, PA). Methyl 4-amino-3-methoxybenzoate (10b) was purchased from Ryan Scientific (Mt. Pleasant, SC).
General Methods and Instruments
Synthesized non-radioactive compounds were analyzed with HPLC on an X-bridge C18 column (5 μm; 10 × 250 mm; Waters Corp., Milford, MA) eluted with aq. NH4OH (0.025% v/v)-MeCN at 6.2 mL/min. Eluates were monitored for absorbance at 254 nm (System Gold 168 detector; Beckman, Fullerton, CA). The purity of each compound was expressed as its area percentage of all chromatogram peak areas.
1H-NMR (400 MHz) and proton-decoupled 13C-NMR (100 MHz) spectra were acquired on an Avance 400 instrument (Bruker, Billerica, MA) using the chemical shifts of residual deuterated solvent as internal standard. Chemical shift (δ) data for the proton and carbon resonances are reported in parts per million (ppm) downfield relative to TMS. s, d, dd, brs and dm denote singlet, doublet, double doublet, broad singlet and double multiplet, respectively.
LC-MS spectra were acquired with an LCQDECA instrument (Thermo Fisher Scientific, Waltham, MA) fitted with a Luna C18 column (5 μm; 2.0 × 150 mm; Phenomenex, Torrance, CA) eluted at 150 μL/min with a MeOH-H2O mixture.
High-resolution mass spectra (HRMS) were acquired with either electron or electron spray ionizations at the Mass Spectrometry Laboratory, University of Illinois at Urbana-Champaign (Urbana, IL).
An industrial ultrasonic processor instrument (UIS250L; 250 W; 24 kHz; described at http://downloads.german-pavilion.com/downloads/pdf/exhibitor_19738.pdf), was used for agitating heterogeneous radioactive reaction mixtures in closed vials. These vials (1-mL volume) and their caps were custom-made from inert fluoropolymers. The reaction vial was designed to fit snugly into a port of the instrument so that ultrasound was efficiently transmitted to the reaction mixture. In order to allow reagents to be added and reaction mixtures to be sampled, the septum liner of the vial cap was designed to withstand multiple needle punctures without leaking.
Chemical syntheses
The following compounds were synthesized as published: 4-(6-bromobenzo[d]thiazol-2-yl)-N-methylaniline (1a),[11] 4-(6-methoxybenzo[d]thiazol-2-yl)-N-methylaniline (1b),[11] 3-(13-methyl-7-oxo-6,7-dihydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-12(13H)-yl)propanenitrile (2),[41,42] N-methyl-4-nitroaniline (4c),[43] N-methyl-N-(3-nitrophenyl)formamide (5a),[44] N-methyl-N-(2-nitrophenyl)formamide (5b),[45] N-methyl-N-(4-nitrophenyl)formamide (5c),[44] 4-(6-bromobenzo[d]thiazol-2-yl)aniline (7a),[11] 4-(6-methoxybenzo[d]thiazol-2-yl)aniline (7b),[11] methyl 8-amino-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (10d),[45] methyl 3-methoxy-4-(methylamino)benzoate (11b),[46-48] methyl 2-methoxy-4-(methylamino)benzoate (11c),[46,47] methyl 8-(methylamino)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate (11d),[44,45] and 3-(7-oxo-6,7-dihydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-12(13H)-yl)propanenitrile (12).[41,42]
N-(4-(6-Bromobenzo[d]thiazol-2-yl)phenyl)-N-methylformamide (8a)
N-Formyl-arylamine 9a (20 mg, 0.06 mmol), Li3N (5 mg, 0.14 mmol) and CH3I (5 μL, 0.08 mmol) were suspended in DMF (0.5 mL). The suspension was placed in the ultrasonic device (described above) which was then set at full power level for half of a 30 min period. The reaction mixture was then quenched with aq. HCOONH4 (5M; 0.5 mL). Solvent was evaporated off and the residue was dissolved in DMF. The product 8a was separated with HPLC on an X-bridge C18 column (5 μm; 19 × 150 mm; Waters Corp.) eluted with aq. NH4OH (0.025% v/v)-MeCN (49: 51 v/v). Purity >98%. Yield (4.2 mg; 20%). 1H-NMR (CDCl3): δ 8.65 (s, 1H, CHO), 8.12 (dm, 3JHH = 8.6 Hz, 2H, Ar-H), 8.05 (d, 4JHH = 1.8 Hz, 1H, Ar-H), 7.92 (d, 3JHH = 8.6 Hz, 1H, Ar-H), 7.61 (dd, 3JHH = 8.7 Hz, 4JHH = 1.9 Hz, 1H, Ar-H), 7.30 (dm, 3JHH = 8.6 Hz, 2H, Ar-H), 3.41 (s, 3H, NCH3). 13C-NMR (CDCl3): δ 167.1 (CHO), 161.9, 153.0, 144.6, 136.7, 130.9, 130.1, 128.8, 124.3, 124.2, 121.7, 119.0, 31.7 (NCH3). MS, TOF, m/z: 163.0 (5%), 350.9 (5%), 165.0 (7%), 381.2 (13%), 349.9 (21%), 346.9 (98%, [M+H]+). HRMS: calc‘d for [M+H]+ (C15H12BrN2OS): 346.9854, found: 346.9850. Error (ppm): –1.2.
N-(4-(6-Methoxybenzo[d]thiazol-2-yl)phenyl)-N-methylformamide (8b)
The method used to synthesize 8a was used to prepare 8b from 9b (20 mg, 0.07 mmol). The crude product was dissolved in DMF (2.0 mL) and purified with HPLC on an X-bridge C18 column (5 μm; 19 × 150 mm; Waters Corp.) eluted with aq. NH3 (0.025% v/v)-MeCN (73: 27 v/v). Purity >99%. Yield (4.8 mg; 23%). 1H-NMR (CDCl3): δ 8.62 (s, 1H, CHO), 8.09 (dm, 3JHH = 8.8 Hz, 2H, Ar-H), 7.95 (d, 3JHH = 9.0 Hz, 1H, Ar-H), 7.37 (d, 4JHH = 2.5 Hz, 1H, Ar-H), 7.29 (dm, 3JHH = 8.8 Hz, 2H, Ar-H), 7.11 (dd, 3JHH = 9.0 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 3.90 (s, 3H, OCH3), 3.37 (s, 3H, NCH3). 13C-NMR (CDCl3): δ 163.8 (CHO), 161.7, 157.7, 148.4, 143.6, 136.2, 131.4, 128.2, 123.5, 121.5, 115.6, 104.0, 55.6 (OCH3), 31.5 (NCH3). MS, TOF, m/z: 301.1 (8%), 300.1 (23%), 299.0 (98%, [M+H]+). HRMS: calc‘d for [M+H]+ (C16H15N2O2S): 299.0854, found: 299.0850. Error (ppm): –1.3.
N-(4-(6-Bromobenzo[d]thiazol-2-yl)phenyl)formamide (9a)
4-(6-Bromobenzo[d]thiazol-2-yl)aniline (7a, 20 mg, 0.06 mmol) was dissolved with the aid of ultrasonication in formic acid (1.0 mL). The solution was then placed in a microwave apparatus (Discover CEM, Matthews, NC) at 120 °C for 10 min using 30 W power and a pressure limit of 200 psi. After reaction was complete, the solvent was evaporated off. The residue was washed with anhydrous diethyl ether to afford crude 9a, which was then dissolved in DMF (2.0 mL) and purified with HPLC on an X-bridge C18 column (5 μm; 19 × 150 mm; Waters Corp.) eluted with aq. NH3 (0.025% v/v)-MeCN (17: 83 v/v). Purity >99%. Yield (5.5 mg; 25%). 1H-NMR (CDCl3), δ 10.6 (brs, 1H, NH), 8.44 (d, 3JHH = 1.9 Hz, 1H, CHO), 8.37 (d, 3JHH = 1.6 Hz, 1H, Ar-H), 8.07 (d, 3JHH = 8.7 Hz, 2H, Ar-H), 7.96 (d, 3JHH = 8.6 Hz, 1H, Ar-H), 7.79 (d, 3JHH = 8.7 Hz, 2H, Ar-H), 7.68 (dd, 3JHH = 8.7 Hz, 4JHH = 2.0 Hz, 1H). 13C-NMR (CDCl3), δ 167.8 (CHO), 160.1, 152.6, 141.2, 136.3, 129.7, 128.3, 124.8, 124.1, 119.5, 117.8, 117.3. MS, TOF, m/z: 336.9 (5%), 335.9 (21%), 334.9 (98%), 332.9 (93%, [M+H]+). HRMS: calc'd for [M+H]+ (C14H10BrN2OS): 332.9697, found: 332.9697. Error (ppm): 2.4.
N-(4-(6-Methoxybenzo[d]thiazol-2-yl)phenyl)formamide (9b)
The method used to synthesize 9a was used with 4-(6-methoxybenzo[d]thiazol-2-yl)aniline (7b, 20 mg, 0.07 mmol) to give crude 9b which was dissolved in DMF (2.0 mL) and purified by HPLC on an X-bridge C18 column (5 μm; 19 × 150 mm; Waters Corp.) eluted with aq. NH3 (0.025% v/v)-MeCN (13: 87 v/v). Purity >99%. Yield (5.3 mg; 24%). 1H-NMR (MeOD): δ 8.66 (s, 1H, CHO), 8.12 (dm, 3JHH = 8.7 Hz, 2H, Ar-H), 7.91 (d, 3JHH = 9.0 Hz, 1H, Ar-H), 7.56 (d, 4JHH = 2.5 Hz, 1H, Ar-H), 7.49 (dm, 3JHH = 8.7 Hz, 2H, Ar-H), 7.15 (dd, 3JHH = 9.0 Hz, 4JHH = 2.6 Hz, 1H, Ar-H), 3.92 (s, 3H, OCH3). 13C-NMR (CD3OD): δ 166.1 (CHO), 164.2, 159.8, 149.5, 145.6, 138.6, 132.6, 129.4, 124.3, 123.2, 117.3, 105.3, 56.4 (OCH3). MS, TOF, m/z: 362.1 (25%), 360.1, 324.1 (100%), 285.1 (900%, [M+H]+). HRMS: calc‘d for C15H13N2O2S: 285.0698, found: 285.0696. Error (ppm): – 0.7.
General radiochemistry procedure
No-carrier-added (NCA) [11C]carbon dioxide was prepared via the 14N(p,α)11C nuclear reaction by irradiation of nitrogen gas (164 psi) containing oxygen (1%) with protons (16.5 MeV, 45 μA) generated with a cyclotron (PETtrace 200; GE Healthcare, Milwaukeee, WI). Radiochemistry was performed in lead-shielded hot-cells for radiation protection to personnel. The [11C]carbon dioxide was converted in an automated apparatus (PETrace MeI MicroLab; GE) into [11C]methyl iodide by reduction to [11C]methane followed by gas-phase reaction with iodine. Within an inert glovebox, equipped with both oxygen and moisture sensors (showing oxygen < 10 ppm and moisture < 0.5 ppm), solid inorganic base (~ 5 mg), substrate (1.0 mg) and solvent (0.3 mL) were loaded into a fluoropolymer reaction vial (volume, 1 mL) and the vial crimp-sealed with a fluoropolymer septum. NCA [11C]methyl iodide (~ 100 mCi) in nitrogen carrier gas was then bubbled into the reaction mixture. The sealed vial was then placed in the port of the ultrasound apparatus (Ultrasonic Processor, UIS250L) and irradiated at full power for 50% of the time, unless otherwise indicated, for a set period (≤ 10 min). The reaction mixture was filtered through a celite plug to obtain a clear solution (syntheses of [11C]11a–11e only) or quenched by addition of water (0.5 mL). The radioactive product was then separated with reverse phase HPLC eluted with 0.1M HCOONH4 (A)-MeCN (B), with eluate monitored for radioactivity and absorbance, as detailed below. The identities of labeled products were confirmed chromatographically by observation of co-elution with non-radioactive reference compound in HPLC, and/or by LC-MS of associated carrier. Decay-corrected radiochemical yields (RCYs) of measured isolated radioactive product were calculated from the amount of [11C]methyl iodide added to the reaction vial. The specific radioactivities (Ci/μmol) of some products were calculated by measurement of the mass of carrier with calibrated analytical HPLC, through absorbance detection and measurement of the associated radioactivity. Some reactions were performed with ultrasound irradiation before addition of [11C]methyl iodide. Some control reactions were performed without ultrasound irradiation, but with shaking at 0, 5 and 10 min. Thus, were prepared:
[11C]4-(6-Bromobenzo[d]thiazol-2-yl)-N-methylaniline ([11C]1a)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:7 v/v) at 6.2 mL/min (λ = 350 nm) (tR = 13.0 min).
[11C]4-(6-Methoxybenzo[d]thiazol-2-yl)-N-methylaniline ([11C]1b)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (2:3 v/v) at 6.2 mL/min (λ = 350 nm) (tR = 10.0 min).
[11C]3-(13-Methyl-7-oxo-6,7-dihydro-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazol-12(13H)-yl)propanenitrile ([11C]2)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (9:11 v/v) at 6.2 mL/min (λ = 292 nm) (tR = 7.5 min); elimination side product (tR = 8.3 min).
[11C]N-Methyl-3-nitroaniline ([11C]4a)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:2 v/v) at 6.2 mL/min (λ = 254 nm) (tR = 13.2 min).
[11C]N-Methyl-2-nitroaniline ([11C]4b)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (1:3 v/v) at 6.2 mL/min (λ = 254 nm) (tR = 6.1 min) or on the same column eluted at 6.2 mL/min with a gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min) (λ = 254 nm) (tR = 21.4 min).
[11C]N-Methyl-4-nitroaniline ([11C]4c)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:2 v/v) at 6.2 mL/min (λ = 254 nm) (tR = 9.4 min) or on the same column eleuted at 6.2 mL/min with a gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min) (λ = 254 nm) (tR = 12.6 min).
[11C]N-Methylaniline ([11C]4d)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted at 6.2 mL/min with a gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min) (λ = 254 nm) (tR = 13.3 min).
[11C]N-methyl-N-(3-nitrophenyl)formamide ([11C]5a)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted at 6.2 mL/min with a gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min) (λ = 254 nm) (tR = 7.3 min).
[11C]N-methyl-N-(2-nitrophenyl)formamide ([11C]5b)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted at 6.2 mL/min with a gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min) (λ = 254 nm) (tR = 8.1 min).
[11C]N-methyl-N-phenylformamide ([11C]5d)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with gradient of A:B (30% B for 1 min increased linearly to 50% B at 30 min, v/v) at 6.2 mL/min (λ = 254 nm) (tR = 7.8 min).
[11C]N-(4-(6-Bromobenzo[d]thiazol-2-yl)phenyl)-N-methylformamide ([11C]8a)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:7 v/v) at 6.2 mL/min (λ = 350 nm) (tR = 8.7 min).
[11C]N-(4-(6-Methoxybenzo[d]thiazol-2-yl)phenyl)-N-methylformamide ([11C]8b)
HPLC isolation: Luna C18 column (5 μm, 10 × 250 mm) eluted with A:B (2:3 v/v) at 6.2 mL/min (λ = 350 nm) (tR = 7.0 min).
[11C]Methyl 4-(methylamino)benzoate ([11C]11a)
HPLC isolation: Gemini-NX C18 column (5 μm, 10 × 250 mm) eluted with A:B (1:1 v/v) at 6 mL/min (λ = 254 nm) (tR = 5.4 min).
[11C]Methyl 3-methoxy-4-(methylamino)benzoate ([11C]11b)
HPLC isolation: Gemini-NX C18 column (5 μm, 10 × 250 mm) eluted with A:B (1:1 v/v) at 6 mL/min (λ = 254 nm) (tR = 6.7 min).
[11C]Methyl 2-methoxy-4-(methylamino)benzoate ([11C]11c)
HPLC isolation: Gemini-NX C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:2 v/v) at 4 mL/min (λ = 254 nm) (tR = 7.4 min).
[11C]Methyl 8-(methylamino)-2,3-dihydrobenzo[b][1,4]dioxine-5-carboxylate ([11C]11d)
HPLC isolation: Gemini-NX C18 column (5 μm, 10 × 250 mm) eluted with A:B (3:2 v/v) at 5 mL/min (λ = 254 nm) (tR = 7.6 min).
[11C]Methyl 3-chloro-4-(methylamino)benzoate ([11C]11e)
HPLC isolation: Gemini-NX C18 column (5 μm, 10 × 250 mm) eluted with A:B (1:1 v/v) at 6 mL/min (λ = 254 nm) (tR = 9.5 min).
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (National Institute of Mental Health). We are grateful to the NIH Clinical Center PET Department (Chief: Dr. P. Herscovitch) for the production of carbon-11.
References
- 1.Bolton R. J. Label. Compd. Radiopharm. 2001;44:701–736. [Google Scholar]
- 2.Elsinga PH. Methods. 2002;27:208–217. doi: 10.1016/s1046-2023(02)00076-2. [DOI] [PubMed] [Google Scholar]
- 3.Miller PW, Long NJ, Vilar R, Gee AD. Angew. Chem., Int. Ed. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 4.Fowler JS, Wolf AP. Acc. Chem. Res. 1997;30:181–188. [Google Scholar]
- 5.Allard M, Fouquet E, James D, Szlosek-Pinaud M. Curr. Med. Chem. 2008;15:235–277. doi: 10.2174/092986708783497292. [DOI] [PubMed] [Google Scholar]
- 6.Långström B, Lundqvist H. Int. J. Appl. Radiat. Isot. 1976;27:357–363. doi: 10.1016/0020-708x(76)90088-0. [DOI] [PubMed] [Google Scholar]
- 7.Marazano C, Mazière M, Berger G, Comar D. Int. J. Appl. Radiat. Isot. 1977;28:49–52. doi: 10.1016/0020-708x(77)90159-4. [DOI] [PubMed] [Google Scholar]
- 8.Crouzel C, Långström B, Pike VW, Coenen HH. Appl. Radiat. Isot. 1987;38:601–603. [Google Scholar]
- 9.Larsen P, Ulin J, Dahlström K, Jensen M. Appl. Radiat. Isot. 1997;48:153–157. [Google Scholar]
- 10.Jewett DM. Appl. Radiat. Isot. 1992;43:1383–1385. doi: 10.1016/0883-2889(92)90012-4. [DOI] [PubMed] [Google Scholar]
- 11.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. J. Med. Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 12.Solbach C, Uebele M, Reischl G, Machulla H-J. Appl. Radiat. Isot. 2005;62:591–595. doi: 10.1016/j.apradiso.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Philippe C, Haeusler D, Mitterhauser M, Ungersboeck J, Viernstein H, Dudczak R, Wadsak W. Appl. Radiat. Isot. 2011;69:1212–1217. doi: 10.1016/j.apradiso.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Hayat S, Rahman A, Choudhary MI, Khan KM, Schumann W, Bayer E. Tetrahedron. 2001;57:9951–9957. [Google Scholar]
- 15.Onaka M, Umezono A, Kawai M, Izumi Y. J. Chem. Soc., Chem. Commun. 1985:1202–1203. [Google Scholar]
- 16.Einhorn C, Einhorn J, Luche J-L. Synthesis. 1989:787–813. [Google Scholar]
- 17.Li J-T, Wang S-X, Chen G-F, Li T-S. Curr. Org. Synthesis. 2005;2:415–436. [Google Scholar]
- 18.Davidson RS, Patel AM, Safdar A, Thornthwaite D. Tetrahedron Lett. 1983;24:5907–5910. [Google Scholar]
- 19.Jurczak J, Ostaszewski R. Tetrahedron Lett. 1988;29:959–960. [Google Scholar]
- 20.Bokatzian-Johnson SSB, Maier ML, Bell RH, Alston KE, Le BY, Cioffi EA. J. Label. Compd. Radiopharm. 2007;50:380–383. [Google Scholar]
- 21.Suslick KS, Didenko Y, Fang MM, Hyeon T, Kolbeck KJ, McNamara WB, Mdleleni MM, Wong M. Phil. Trans. R. Soc. Lond. A. 1999;357:335–353. [Google Scholar]
- 22.Doktycz SJ, Suslick KS. Science. 1990;247:1067–1069. doi: 10.1126/science.2309118. [DOI] [PubMed] [Google Scholar]
- 23.Suslick KS, Skrabalak SE. In: Sonocatalysis. In Handbook of Heterogeneous Catalysis. Ertl G, Knözinger H, Schüth F, Weitkamp J, editors. Wiley-VCH; Weinheim: 2008. pp. 2006–2017. [Google Scholar]
- 24.Mason TJ. Chem. Soc. Rev. 1997;26:443–451. [Google Scholar]
- 25.Imai T, Muramoto T, Tsuji T. Chem. Lett. 1995:355–356. [Google Scholar]
- 26.Salvatore RN, Yoon CH, Jung KW. Tetrahedron. 2001;57:7785–7811. [Google Scholar]
- 27.De K, Legros J, Crousse B, Bonnet-Delpon D. J. Org. Chem. 2009;74:6260–6255. doi: 10.1021/jo9012699. [DOI] [PubMed] [Google Scholar]
- 28.Lee I, Sung DD. In: Chemistry of Anilines. Rappoport Z, editor. Wiley; Chichester: 2007. pp. 537–581. [Google Scholar]
- 29.Brotzel F, Chu YC, Mayr H. J. Org. Chem. 2007;72:3679–3688. doi: 10.1021/jo062586z. [DOI] [PubMed] [Google Scholar]
- 30.Vlasov VM, Os'kina IA. Russ. J. Org. Chem. 2002;38:1705–1718. [Google Scholar]
- 31.Dehmlow EV, Thieser R, Zahalka HA, Sasson Y. Tetrahedron Lett. 1985;26:297–300. [Google Scholar]
- 32.Bordwell FG, Algrim D, Vanier NR. J. Org. Chem. 1977;42:1817–1819. [Google Scholar]
- 33.Muzart J. Tetrahedron. 2009;65:8313–8323. [Google Scholar]
- 34.Pettit GR, Thomas EG. J. Org. Chem. 1959;24:895–896. [Google Scholar]
- 35.Maran F, Celadon D, Severin MG, Wanello E. J. Am. Chem. Soc. 1991;113:9320–9329. [Google Scholar]
- 36.DeWolfe RH, Newcomb RC. J. Org. Chem. 1971;36:3870–3878. [Google Scholar]
- 37.Stein AR, Tan S-H. Can. J. Chem. 1974;52:4050–4061. [Google Scholar]
- 38.Bose AK, Ganguly SN, Manhas MS, Guha A, Pombo-Villars E. Tetrahedron Lett. 2006;47:4605–4607. [Google Scholar]
- 39.Xu R, Hong J, Morse CL, Pike VW. J. Med. Chem. 2010;53:7035–7047. doi: 10.1021/jm100668r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cai L, Ozaki H, Fujita M, Hong JS, Bukhari M, Innis RB, Pike VW. J. Label. Compd. Radiopharm. 2009;52:S337. [Google Scholar]
- 41.Kleinschroth J, Hartenstein J, Rudolph C, Schachtele C. Bioorg. Med. Chem. Lett. 1993;3:1959–1964. [Google Scholar]
- 42.Bergman J, Pelcman B. J. Org. Chem. 1989;54:824–828. [Google Scholar]
- 43.Ge Y, Hu L. Tetrahedron Lett. 2007;48:4585–4588. [Google Scholar]
- 44.Du P. CN. 101580473:2009. [Google Scholar]
- 45.Bosmans JRM, Gijsen HJM, Mevellec LAJ. 2005 BE; EP/2004/0739785; WO/ 2005/000838 (A1) [Google Scholar]
- 46.Högberg T, Bjurling AE, Receveur J, Little PB, Elling CE, Nørregaard PK, Ulven T. 2003 PCT/DK2003/000231; WO/2003/087045. [Google Scholar]
- 47.Sellarajah S, Lekishvili T, Bowring C, Thompsett AR, Rudyk H, Birkett CR, Brown DR, Gilbert IH. J. Med. Chem. 2004;47:5515–5534. doi: 10.1021/jm049922t. [DOI] [PubMed] [Google Scholar]
- 48.Akada Y, Matsura K. PCT/JP2003/015005; WO/2004/048326. [Google Scholar]