

Key Points
Adhesion of sRBCs is synergistically regulated by hypoxia and low NO bioavailability.
P-selectin and p38 kinase pathways play a role in the synergistic adhesion of sRBCs.
Abstract
The molecular mechanisms by which nitric oxide (NO) bioavailability modulates the clinical expression of sickle cell disease (SCD) remain elusive. We investigated the effect of hypoxia and NO bioavailability on sickle red blood cell (sRBC) adhesion using mice deficient for endothelial NO synthase (eNOS) because their NO metabolite levels are similar to those of SCD mice but without hypoxemia. Whereas sRBC adhesion to endothelial cells in eNOS-deficient mice was synergistically upregulated at the onset of hypoxia, leukocyte adhesion was unaffected. Restoring NO metabolite levels to physiological levels markedly reduced sRBC adhesion to levels seen under normoxia. These results indicate that sRBC adherence to endothelial cells increases in response to hypoxia prior to leukocyte adherence, and that low NO bioavailability synergistically upregulates sRBC adhesion under hypoxia. Although multiple adhesion molecules mediate sRBC adhesion, we found a central role for P-selectin in sRBC adhesion. Hypoxia and low NO bioavailability upregulated P-selectin expression in endothelial cells in an additive manner through p38 kinase pathways. These results demonstrate novel cellular and signaling mechanisms that regulate sRBC adhesion under hypoxia and low NO bioavailability. Importantly, these findings point us toward new molecular targets to inhibit cell adhesion in SCD.
Introduction
Although sickle cell disease (SCD) arises from a single mutation of β-globin, clinical severity varies significantly.1 A well-known clinical modifier is fetal hemoglobin, which is expressed at variable levels in patients and inhibits the polymerization of sickle hemoglobin.2 The frequency of vaso-occlusive crisis was also used to evaluate disease severity of SCD patients.3 Vaso-occlusive crisis is likely triggered by multiple physiological insults such as infection and cytokine-mediated inflammation that may cause tissue hypoxia, and mediated by multistep cell adhesion mechanisms involving sickle red blood cells (sRBCs).4 The degree of sRBC adhesion to endothelial cells was shown to correlate with SCD severity.1
Nitric oxide (NO) bioavailability is another physiological factor capable of modulating clinical severity.5 NO bioavailability decreases during vaso-occlusive crisis, primarily because of NO scavenging by cell-free hemoglobin6 as well as other mechanisms including arginase,7 reactive oxygen species,8 and NO synthases.9 NO metabolite levels vary among patients even at steady state and may inversely correlate with the frequency of vaso-occlusive crisis.10 We showed that inhalation of low-dose NO permitted SCD mice to survive hypoxic stress.11 The gender difference in the clinical severity of SCD may result in part from distinct levels of NO bioavailability.12 Thus, heterogeneity of the clinical expression in SCD appears to be mediated by multiple hematologic and physiological parameters.
We previously reported that NO increases the oxygen affinity of sickle hemoglobin in SCD patients.13 NO was recently found to physically disrupt sickle hemoglobin polymers by altering the electric charge.14 Although NO donors reduce sRBC adhesion to endothelial cells in vitro,15 the molecular and signaling mechanisms by which NO bioavailability modulates cell adhesion remain unclear. In this study, we investigated the mechanisms by which NO bioavailability regulates the adhesion of sRBCs and leukocytes to endothelial cells under hypoxia. Although SCD mice16 provide an excellent in vivo tool to study cell adhesion, blood cells in such mice are likely exposed to both low oxygen tension and low NO bioavailability because a number of sRBCs are circulating in peripheral blood and NO metabolite levels are diminished. In contrast, endothelial NO synthase (eNOS)-deficient mice17 have NO metabolite levels comparable to those of SCD model mice but without hypoxemia. To differentiate the effect of oxygen tension on cell adhesion from that of NO bioavailability, we used eNOS-deficient mice in this study. We demonstrate that adhesion of sRBCs, but not leukocytes, to endothelial cells in eNOS-deficient mice markedly increases in as few as 15 minutes of mild hypoxia, suggesting that sRBC adhesion precedes leukocyte adhesion and is synergistically regulated by hypoxia and low NO bioavailability. The synergistic adhesion of sRBCs to endothelial cells is found to be mediated by P-selectin whose expression is upregulated by p38 kinase pathways.
Materials and methods
Mice
eNOS-deficient mice17 were obtained from the Jackson Laboratory. SCD mice were provided by Dr T. M. Ryan.16 Animal studies were approved by the Institutional Animal Care and Use Committee of the Georgia Regents University.
Plasma NO metabolites and arterial blood gas analysis in mice
Plasma NO metabolite levels in mouse blood were determined using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical). Partial pressure of oxygen in arterial blood (Pao2) was measured by a VetScan i-STAT cartridge (Abaxis) and i-STAT Portable Clinical Analyzer (Heska). The detailed methods are described in the supplemental Methods (available on the Blood Web site).
Hypoxic treatment procedures and intravital microscopy for cell adhesion in mice
Control (C57BL/6J) or eNOS-deficient mice were placed under normoxia (fraction of inspired oxygen [Fio2] = 21%) or hypoxia (Fio2 = 12%) for 15 minutes (Figure 1, procedures I and II). In procedures III and IV, mice were rendered to breathe NO gas of 4 or 20 parts per million (ppm) under normoxia or hypoxia. Cell adhesion to endothelial cells in the skull bone marrow microvascular network in mice was analyzed by intravital microscopy as described previously.18 The detailed methods for treatment of mice under normoxia or hypoxia, animal surgery procedure, staining of sRBCs and leukocytes, antibody infusion, and intravital microscopic analysis for cell adhesion are described in the supplemental Methods.
Figure 1.

Treatment protocol of mice to study the effects of hypoxia and NO bioavailability on sRBC adhesion. Control and eNOS-deficient mice were treated with 4 different procedures (I-IV). The numbers of mice used for the treatments were as follows: I, 5 control mice and 5 eNOS-deficient mice; II, 5 control mice and 7 eNOS-deficient mice; III, 7 eNOS-deficient mice; and IV, 4 eNOS-deficient mice. A bolus of 2,7-bis-(carboxyethyl)-5-(and-6) carboxyfluorescein (BCECF)-labeled RBCs was injected into the mice, and 3 minutes later, the recording of microcirculation images was initiated and completed by analyzing 4 1-minute video segments. Fio2 indicates fraction of inspired oxygen in a gas mixture.
Culture of human umbilical vein endothelial cells
Human umbilical vein endothelial cells (HUVECs) (CRL#1730; ATCC) were grown in vascular cell basal media (ATCC) supplemented with endothelial cell growth supplements according to the manufacturer’s protocol. Prior to all experiments, HUVEC monolayers were serum starved for 18 hours. To mimic hypoxic stress, cells were exposed (1 hour, 37°C) to a gas mixture (5% CO2-balanced N2) to obtain 12% O2 in a PROOX 110-sealed hypoxia chamber provided with oxygen sensor (BioSperix Limited, Lacona, NY) in the presence or absence of 100 μM NG-nitro-l-arginine methyl ester (l-NAME) (Sigma-Aldrich). To study effects of p38 kinases on P-selectin expression, HUVECs were treated with anisomycin (100 nM) and SB203580 (5 μM).
Immunofluorescence microscopy
HUVECs were rinsed with phosphate-buffered saline (PBS) and fixed in ice-cold methanol. Cells were blocked with PBS-containing 2% bovine serum albumin and incubated with anti–P-selectin or anti–VCAM-1 antibody (Santa Cruz Biotechnology). Cells were then incubated with appropriate secondary Rhodamine or Texas Red conjugated antibody (Santa Cruz Biotechnology). Cells were analyzed using a ×40 oil objective on the Axiovert 200 microscope (Carl Zeiss, Thornwood, NY). Images were captured by OImaging Retiga 1300 CCD digital camera (OImaging, Burnaby, BC, Canada). Mean fluorescence intensity (MFI) of images was analyzed using ImageJ (National Institutes of Health [NIH]).
Membrane proteins and total cell lysate preparation
For the isolation of membrane proteins, HUVECs were washed once in PBS, centrifuged for 5 minutes at 350g, and resuspended in 10 mL of an ice-cold cell lysis buffer.19 The cells were sonicated and then centrifuged for 60 minutes at 35 000 rpm at 4°C in an ultracentrifuge (SW45; Beckman Coulter, Brea, CA). Plasma membrane fractions were dissolved in radio immunoprecipitation assay (RIPA) buffer (Santa Cruz Biotechnology) containing protein phosphatase inhibitors. For total cell lysates, HUVECs were resuspended in RIPA buffer containing protein phosphatase inhibitors.
Western blot analysis and screening of intracellular signaling pathways regulating P-selectin expression in HUVECs
Membrane proteins or total cell lysates were separated by sodium dodecyl sulfate–polyacrylamide gels, and western blotting was performed as described previously.19 Briefly, membranes were blocked with a high-salt buffer solution containing 2× PBS, 3% bovine serum albumin, 1% polyethylene glycol, and 1% polyvinylpyrrolidone (pH 7.4) or Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk. Antibodies were rabbit polyclonal anti–P-selectin (Santa Cruz Biotechnology) and phospho-p38 mitogen-activated protein kinase (MAPK) (Thr180/Tyr182; R&D Systems). Screening for intracellular signaling pathways involved in P-selectin expression in HUVECs was performed using Proteome Profiler Human Phospho-MAPK Array (R&D Systems) according the manufacturer’s instruction.
RNA interference knockdown for p38 kinases in HUVECs
HUVECs were plated at 6.0 × 104 cells per well of a 24-well plate. The p38α small interfering RNA (siRNA) construct (siRNA# SI00300769; Qiagen) or negative control siRNA was transfected at a final concentration of 5 nM into HUVECs using HiPerFect Reagent (Qiagen). Expression levels of p38α were confirmed by western blotting 72 hours after transfection. P-selectin expression in HUVECs transfected with p38α siRNA and negative control siRNA constructs was evaluated by immunofluorescence microscopy and quantitated as described previously.
Statistical analysis
All data were expressed as means ± standard errors of means (SEMs). Student t test or Mann-Whitney U test was used to determine the difference of cell adhesion scores between animal groups. Percent reductions of mean arterial blood pressure (MABP) in response to hypoxia were analyzed using unpaired Student t test. P values of < .05 were considered statistically significant.
Results
Physiological comparison of eNOS-deficient mice with SCD mice
To determine the physiological conditions to which blood cells were exposed, we first compared plasma NO metabolite levels and Pao2 in the arterial blood of control mice, SCD mice, and eNOS-deficient mice. The NO metabolite level of eNOS-deficient mice was much lower than that of control mice but comparable to that of SCD mice (Figure 2A) (P = .46), suggesting the possibility that SCD and eNOS-deficient mice have similar levels of NO bioavailability in the peripheral circulation. In contrast, there was no significant difference in Pao2 between control mice and eNOS-deficient mice; however, the Pao2 levels of SCD mice were lower than those of control mice (Figure 2B) (P < .01). These results demonstrate that blood cells in SCD mice even at normoxia are likely exposed to low NO bioavailability and low oxygen tension, whereas those in eNOS-deficient mice are under a low NO metabolite condition but without low oxygen tension.
Figure 2.

NO metabolite levels and Pao2 in control and eNOS-deficient mice. (A) Steady-state plasma nitrate/nitrite levels in control, SCD, and eNOS-deficient mice. Plasma nitrate/nitrite levels were determined using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical). NS, not significant. (B) Pao2 levels in control, SCD, and eNOS-deficient mice. Arterial blood was drawn at baseline conditions for blood gas analysis. Values were mean ± SEM obtained from 3 to 4 mice in each group. P values are shown at the top of the figure.
Hypoxic stress under low NO metabolites leads to synergistic sRBC adhesion to endothelial cells
To study the effects of hypoxia and NO bioavailability on cell adhesion, we first examined sRBC adhesion to endothelial cells in control mice and eNOS-deficient mice under normoxia as well as in an early phase of hypoxia (Figure 1, procedures I and II). RBCs prepared from control mice showed no adherence to endothelial cells in control mice or eNOS-deficient mice (Figure 3A, lanes 1 and 4), indicating that our in vivo system could eliminate nonspecific RBC interactions with endothelial cells. Under normoxia (Fio2 = 21%), eNOS-deficient mice injected with sRBCs exhibited much higher adhesion scores (2.5 ± 0.56, lane 5) than control mice (0.6 ± 0.23, lane 2) (Figure 3A). Of note, under low oxygen tension (Fio2 = 12%), the adhesion scores of control mice increased from 0.6 ± 0.23 (lane 2) to 2.7 ± 0.74 (lane 3) (Figure 3A); whereas eNOS-deficient mice showed much higher increases, from 2.5 ± 0.56 (lane 5) to 13.7 ± 1.69 (lane 6) (Figure 3A), demonstrating that sRBC adhesion in mice with low NO bioavailability is enhanced synergistically at the onset of hypoxia. Representative video images for control and eNOS-deficient mice under normoxia and hypoxia are shown in Figure 3B.
Figure 3.

Adhesion of sRBC and leukocytes in control and eNOS-deficient mice under normoxia (procedure I) or hypoxia (procedure II). (A) Analysis of RBC adhesion to endothelial cells by intravital microscopy. Labeled RBCs were prepared from either control or SCD mice and injected into control or eNOS-deficient mice. Note that RBCs prepared from control mice did not adhere to endothelial cells in either control or SCD mice, suggesting that the intravital microscopy analysis eliminates false-positive cell–cell interactions. Lanes are shown at the top of figure, and the origins of injected RBCs and oxygen tensions are shown at the bottom of figure. Values were mean ± SEM obtained from 3 to 7 mice in each group. P values are shown at the top of the figure. (B) Frame-captured images from videotaped intravital microscopy of bone marrow venules in control (left panels) and eNOS-deficient mice (right panels) after injection of BCECF-labeled sRBCs under normoxia (upper panels) and hypoxia (low panels). Arrows indicate adhered sRBCs. (C) Percent reduction in MABP in response to hypoxia (Fio2 = 12%) in control and eNOS-deficient mice. Values were mean ± SEM from 5 to 7 mice in each group. (D-E) Leukocyte rolling (D) and leukocyte adhesion (E) to endothelial cells in eNOS-deficient mice at normoxia (procedure I) or at hypoxia (procedure II). PE rat anti-CD45 antibody was injected into eNOS-deficient mice. Leukocyte adhesion was quantified by counting the number of adherent cells (stationary for >30 seconds) in a 100-μM length of the vessel. Note that only leukocyte rolling is increased at the onset of hypoxia. Values were mean ± SEM from 5 to 7 mice in each group. P values are shown in the figure.
Cell adhesion to endothelial cells could be affected by the level of wall shear rates (WSRs).20 We next examined whether synergistic enhancement of sRBC adhesion in eNOS-deficient mice by hypoxia is associated with such physiological changes as venular WSRs and MABP levels in venules. Hypoxia did not cause significant changes in the diameters of venules in either eNOS-deficient mice (P = .33) or control mice (P = .07) (Table 1). Although hypoxia significantly reduced RBC velocities (P < .001) and WSRs (Table 1), there was no difference between control and eNOS-deficient mice. Further, there was no statistically significant difference in the reduction in MABP levels in eNOS-deficient mice under hypoxia compared with control mice (Figure 3C). These results indicate that WSRs and MABP levels have no role in increasing sRBC adhesions in eNOS-deficient mice under hypoxia.
Table 1.
Hemodynamic parameters in the skull bone marrow venules of control and eNOS-deficient mice infused with sRBCs
| Venular diameter (μm) | Mean RBC velocity (μm/s) | Calculated WSR (s−1) | |
|---|---|---|---|
| Control mice | |||
| Normoxia | 29.7 ± 1.9 | 236.8 ± 40.4 | 63.7 ± 8.8 |
| Hypoxia | 26.1 ± 1.0 | 109.9 ± 12.4*** | 33.7 ± 3.5** |
| eNOS-deficient mice | |||
| Normoxia | 28.5 ± 2.6 | 217.5 ± 23.0 | 70.0 ± 11.6 |
| Hypoxia | 33.7 ± 3.3 | 117.3 ± 13.9*** | 29.4 ± 3.4*** |
| Hypoxia with 4 ppm NO | 27.5 ± 1.7 | 190.7 ± 13.0* | 56.8 ± 4.1* |
| Hypoxia with 20 ppm NO | 30.5 ± 2.2 | 167.2 ± 16.9* | 50.6 ± 6.6* |
All data were expressed as means ± SEM. Values calculated from 6 mice for each group. ***P < .001 compared with corresponding normoxia; **P < .01 compared with corresponding normoxia; *P < .05 compared with corresponding hypoxia without NO.
We then examined leukocyte adhesion to endothelial cells. In eNOS-deficient mice, leukocyte rolling flux increased in response to hypoxia from 4.1 ± 0.71 to 11.0 ± 1.02 cells per minutes (P < .001; Figure 3D), whereas leukocyte adhesion remained unaffected (1.5 ± 0.20 vs 1.4 ± 0.19 cells per 100 μm, P = .82; Figure 3E).
Restoration of NO metabolites to physiological levels in eNOS-deficient mice is sufficient for inhibiting sRBC adhesion
To verify that low NO bioavailability indeed plays a role in a synergistic increase of sRBC adhesion under hypoxia, we examined sRBC adhesion in eNOS-deficient mice whose NO metabolite levels were raised by inhaling various concentrations of NO. Mice that breathed 4 or 20 ppm of NO gas had increased levels of NO metabolites at low oxygen tension of 12% O2 (Figure 4A). Although the NO metabolite levels of mice inhaling 4 ppm NO were statistically comparable to those of control mice, 20 ppm NO gas raised NO metabolite levels to more than 3 times physiological levels (Figure 4A). We next assessed sRBC adhesion to endothelial cells in eNOS-deficient mice (Figure 4B). Compared with hypoxic eNOS-deficient mice (Figure 4B, lane 4), the sRBC adhesion scores in eNOS-deficient mice breathing 4 ppm NO under hypoxia decreased by ∼75% and were statistically similar to those of the mice under normoxia (lanes 1 and 5). Inhalation of 20 ppm NO increased the NO metabolite levels approximately threefold, but no further reduction was seen with the sRBC adhesion scores (lane 6), indicating that restoring NO metabolites to physiological levels is sufficient to inhibit the synergistic sRBC adhesion; representative video images for eNOS-deficient mice are shown in Figure 4C. The sRBC adhesion scores of eNOS-deficient mice breathing 4 ppm NO at normoxia were also diminished to levels comparable to those in control mice injected with sRBCs under normoxia (0.9 ± 0.23 vs 0.6 ± 0.23; Figure 4B, lane 2 vs Figure 3A, lane 2). Although we observed some changes in hemodynamic parameters including WSRs and MABP levels (Table 1), there were no significant correlations between such parameters and sRBC adhesion scores.
Figure 4.
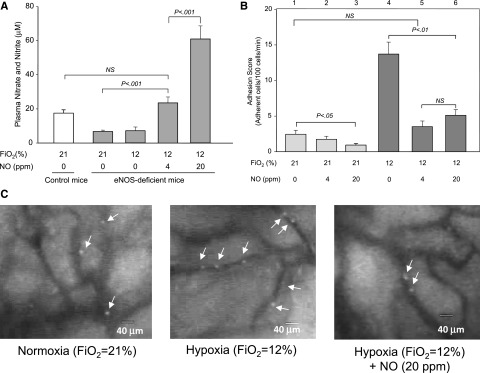
Effects of NO inhalation on NO metabolite levels and sRBC adhesion in eNOS-deficient mice. (A) Plasma nitrate/nitrite levels in eNOS-deficient mice that inhaled NO gas under normoxia or hypoxia. Plasma nitrate/nitrite levels were determined using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical). Values were mean ± SEM from 3 to 4 mice in each group. P values are shown in the figure. (B) Modulation by NO inhalation of sRBC adhesion in postcapillary venules of eNOS-deficient mice. eNOS-deficient mice were treated with 4 or 20 ppm NO gas at normoxia (procedure III) or hypoxia (procedure IV). Values were mean ± SEM from 5 to 7 mice in each group. P values are shown in the figure. (C) Frame-captured images from videotaped intravital microscopy of bone marrow postcapillary venules in eNOS-deficient mice after injection of BCECF-labeled sRBCs under normoxia, hypoxia, and hypoxia with 20 ppm NO. Arrows indicate adhered sRBCs.
Adhesion molecules mediating synergistic adhesion of sRBCs to endothelial cells
To identify adhesion molecules that mediated synergistic sRBC adhesion to endothelial cells at the onset of hypoxia under low NO bioavailability, we first examined expression levels of vascular cell adhesion molecule 1 (VCAM-1) on the cell surface of HUVECs because VCAM-1 expression is induced by sRBCs and required for hypoxia-induced sRBC adhesion.21 However, the MFIs of VCAM-1 on HUVECs did not increase after 1-hour exposure to hypoxia (Fio2 = 12%) in the absence or presence of the NOS inhibitor l-NAME (100 μM) (supplemental Figure 1), suggesting that VCAM-1 is unlikely to be involved in sRBC adhesion at the onset of hypoxia under low NO bioavailability. This is probably because VCAM-1 expression in HUVECs is regulated at the level of transcription, and it may take at least a few hours to upregulate VCAM-1 messenger RNA expression.21 In contrast, P-selectin is rapidly translocated from Weibel-Palade bodies to the plasma membrane upon hypoxic stress.22 We then examined the MFIs of P-selectin on HUVECs that were exposed to hypoxia with or without treatment with 100 μM l-NAME (Figure 5A). Hypoxic stress and l-NAME increased the MFIs for P-selectin by 43% and 33%, respectively, whereas a combination of hypoxia and NOS inhibition by l-NAME resulted in an 87% increase over the control levels, suggesting an additive effect of hypoxia and low NO bioavailability on P-selectin expression in HUVECs. Because P-selectin expressed on the cell surface is glycosylated,23 we next isolated membrane preparations from HUVECs and examined whether the expression levels of the glycosylated form of P-selectin in the membrane preparations increased in response to hypoxic stress and/or l-NAME treatment. The band intensities of both faster- and slower-mobility forms of P-selectin were significantly enhanced following hypoxic stress as well as low NO conditions generated by l-NAME treatment (Figure 5B). This is consistent with our finding that P-selectin expression on HUVECs is increased additively in response to hypoxia and low NO bioavailability. We further confirmed the involvement of P-selectin in the sRBC adhesion in eNOS-deficient mice. Anti–P-selectin antibody was infused into eNOS-deficient mice, and the adhesion scores of sRBCs to endothelial cells were measured using intravital microscopy (Figure 5C). Administration of anti–P-selectin antibody decreased the sRBC adhesion score by more than 70%. In contrast, antibodies against VCAM-1 and E-selectin as well as αvβ3-integrin blocking peptide did not have any significant effects on the adhesion scores (Figure 5C).
Figure 5.

Expression of P-selectin on HUVECs is regulated by hypoxia and NO bioavailability and its role in sRBC adhesion. (A) Effects of hypoxia and the eNOS inhibitor l-NAME on P-selectin expression on HUVECs. P-selectin expression on the cell surface of HUVECs was evaluated by immunofluorescence microscopy. Cells were treated with room air (Fio2 = 21%) or hypoxia of 12% O2 for 1 hour with or without l-NAME (100 μM). Images were taken at ×400 (upper panel). MFIs were calculated by scanning fluorescence images with ImageJ v1.43 (NIH). Results with MFI for each experiment are shown in the lower panel. Values were mean ± SEM from 3 to 4 images taken for each treatment with at least 5 cells analyzed per image. (B) Analysis of P-selectin expression in HUVECs by western blotting. Cells were treated as described previously, and the cell membrane fractions were isolated and subjected to western blot analysis. Note that the intensity of a slower-mobility protein band at 140 kDa is increased in response to hypoxia and l-NAME treatment. (C) Effect of blocking antibodies on sRBC adhesion in eNOS-deficient mice under hypoxia. eNOS-deficient mice were infused with antibody against P-selectin, VCAM-1, and E-selectin or αvβ3-integrin blocking peptide before intravital experiments, and the adhesion scores of sRBCs were measured. Values were mean ± SEM from 5 to 7 mice in each group. P values are shown at the top of the figure.
Intracellular signaling pathways involved in synergistic sRBC adhesion
To identify new molecular targets to inhibit sRBC adhesion under hypoxia, we next investigated the intracellular signaling pathways that mediated P-selectin expression under hypoxia and low NO bioavailability. Recent studies showing activation of p38 kinases by hypoxia24,25 led us to focus on MAPK pathways. First, phosphorylation levels of MAPKs in HUVECs exposed to hypoxic stress, treated with 100 μM l-NAME, or both were assessed using Proteome Profiler Human Phospho-MAPK Array. The phosphorylation levels of p38α increased in an additive manner in response to hypoxia of 12% O2 and l-NAME treatment (supplemental Figures 2 and 3). Next, we performed western blotting to confirm the results. Again, the phosphorylation level of the p38α kinase was markedly increased in response to hypoxia and l-NAME treatment (Figure 6A). To determine the role for p38 kinases in the induction of P-selectin expression, HUVECs were next treated with the p38 activator anisomycin (100 nM) and the selective p38 inhibitor SB203580 (5 μM). P-selectin expression was increased and decreased by anisomycin and SB203580, respectively, demonstrating a role for p38 kinases in P-selectin expression in HUVECs (Figure 6B). To verify the role of p38 kinases in P-selectin expression under hypoxia and low NO bioavailability, the expression of p38α in HUVECs was downregulated by transfecting with 2 siRNA constructs targeting p38α, and the knock-down efficacy was determined by western blotting. Expression of the p38α kinase was suppressed by 50% to 66%, whereas negative control siRNA had no effect (Figure 6C). Next, we examined whether expression of P-selectin was induced by hypoxia or l-NAME on the cell surface of HUVECs transfected with siRNA constructs for p38α kinase. Compared with HUVECs transfected with control negative siRNA, in which P-selectin expression was induced by hypoxia and l-NAME (black columns), inhibition of p38α by a p38 siRNA construct resulted in marked downregulation of P-selectin expression by hypoxia and l-NAME (gray columns, Figure 6D).
Figure 6.

P-selectin expression in HUVECs is regulated by p38 kinase pathways. (A) Phosphorylation of p38 kinases increases in HUVECs treated with hypoxia for 1 hour with or without the NOS inhibitor l-NAME (100 μM). A representative blot was shown from 3 independent experiments. (B) P-selectin expression on the cell surface of HUVECs is regulated by p38 kinases. HUVECs were treated with anisomycin (100 ng/mL) (upper panel) or SB203580 (5 μM) (lower panel) for 1 hour, and P-selectin expression on the cell surface was evaluated by immunofluorescence microscopy as described in “Materials and methods.” MFIs were calculated by scanning fluorescence images with ImageJ v1.43 (NIH). (C) Knockdown of p38 kinase expression in HUVECs by siRNA constructs. Western blot showing that p38 siRNA, but not control negative siRNA, decreased total p38 protein by 50% to 66% in HUVECs at 48 hours. (D) P-selectin expression in HUVECs transfected with siRNA constructs for p38α kinase (gray columns) and negative control siRNA (black columns). Expression of P-selectin on the cell surface of HUVECs was quantitated by immunofluorescence microscopy as described in “Materials and methods,” and MFIs were calculated by scanning fluorescence images with ImageJ v1.43 (NIH).
Discussion
The significant variability of NO bioavailability among SCD patients26 is assumed to contribute to the clinical heterogeneity.5,10,12 Despite studies of the physiological effects of NO,12,26 the mechanisms by which NO bioavailability modulates the pathophysiology remain elusive. To determine the role for NO bioavailability under various oxygen tensions in sRBC adhesion, we used eNOS-deficient mice, which have an NO bioavailability comparable to that of SCD mice.16 We found that hypoxic stress increases sRBC adhesion modestly in mice with normal NO metabolite levels but synergistically enhances it in eNOS-deficient mice that had reduced NO metabolite levels. Conversely, restoration of NO metabolite levels to physiological levels in eNOS-deficient mice drastically reduced sRBC adhesion to the levels found at normoxia. These results may suggest a role for NO bioavailability in sRBC adhesion at the onset of hypoxia. Our result showing a marked reduction in sRBC adhesion following NO breathing may be relevant to a physiological basis for the clinical improvements SCD patients experienced after NO inhalation during vaso-occlusive crisis27 as well as to pain reduction.28,29 Also, given that SCD patients with low NO metabolite levels experience higher levels of pain,10 our results suggest that lower NO bioavailability increases the risk of vaso-occlusive crisis under hypoxia.26 Thus, the differences in the risk for cell adhesion might contribute to the clinical heterogeneity among SCD patients because sRBC adhesion is a crucial determinant for clinical severity.1 In contrast to the synergistic mechanisms for sRBC adhesion, leukocyte rolling influx was elevated, but leukocyte adhesion to endothelial cells was unaffected at the onset of hypoxia, which is consistent with studies by Frenette and coworkers.4
Identifying the adhesion molecules and signaling pathways involved in the synergistic sRBC adhesion mediated by hypoxia and low NO bioavailability will allow us to develop novel therapeutics to inhibit cell adhesion. Although VCAM-1 is involved in the adhesion of sRBCs to endothelial cells and its expression is increased in response to hypoxia,26 our in vitro studies were unable to detect an increase in VCAM-1 expression on HUVECs. Instead, we found that P-selectin expression, which plays a critical role in sRBC adhesion,30 is induced in an additive manner by hypoxia and low NO bioavailability. Inhibition by a specific antibody of P-selectin, but not VCAM-1 or E-selectin, reduced the in vivo adhesion score of sRBC by more than 70%. These results demonstrate a major role for P-selectin in sRBC adhesion. In this regard, it is interesting to note that the adhesion scores of eNOS-deficient mice at normoxia (Figure 3A, lane 5) are comparable to those of control mice under hypoxia (Figure 3A, lane 3). Because P-selectin expression is upregulated by low oxygen tension31 as well as by low NO bioavailability32 in multiple types of cells, it is reasonable to expect such results. We also observed an increase in leukocyte rolling influx, which is mediated by P-selectin.33 These also provide further support to our conclusion that P-selectin plays a critical role in sRBC adhesion at hypoxia as well as low NO bioavailability. Furthermore, our in vitro studies demonstrated that P-selectin expression in HUVECs is upregulated through the activation of p38 kinase pathways; anisomycin increased P-selectin expression on HUVECs, whereas its expression was decreased by SB203580. The role for p38 kinases in P-selectin expression was further confirmed by studies using siRNAs (Figure 6C-D). It is possible that selective inhibitors of p38 kinases can be developed as sRBC adhesion inhibitors to treat SCD.
NO inhalation for SCD patients under vaso-occlusive crisis reduced pain scores28,29; however, another clinical trial was unable to reproduce the beneficial effects of NO inhalation.34 Results from this study may suggest possible clues to the mechanisms underlying these conflicting results. Our intravital microscopic studies demonstrated that increasing NO metabolite levels beyond physiological levels by NO inhalation did not further reduce sRBC adhesion, suggesting that restoring NO metabolites to physiological levels may be critical for the efficient inhibition of sRBC adhesion. In a clinical trial by Gladwin et al,34 however, the baseline NO metabolite levels of the NO inhaled group, even while under vaso-occlusive crisis, did not show substantial reductions, but rather were comparable to those at steady state in other studies.26 Importantly, the results shown in Figure 4B suggest that NO inhalation may not reduce sRBC adhesion among SCD patients with normal NO bioavailability. Heterogeneity of NO metabolite levels among SCD patients may obfuscate clinical trials and thereby lead to differences in clinical outcomes from NO inhalation. It may be helpful to compare the clinical effects of NO inhalation among patient groups with distinct NO metabolite levels. Further studies are necessary to more accurately determine the physiological effects of NO inhalation.
Results obtained in this study also convey important implications for pharmacologic treatments of SCD. SCD patients treated by hydroxyurea experience improved clinical symptoms including less frequent vaso-occlusive crisis, before fetal hemoglobin levels rise.35 We and others demonstrated activation of the soluble guanylate cyclase-cyclic guanosine monophosphate pathway by NO generated by the metabolism of hydroxyurea.36 NO generated from hydroxyurea may also function to reduce cellular adhesion in the microvasculature, which may in turn contribute to improved clinical manifestations. Our results in this study also suggest that increased levels of NO metabolites after hydroxyurea therapy may decrease sRBC adhesion to endothelial cells, resulting in reduced pain scores. NO derived from hydroxyurea may directly inhibit the polymerization of sickle hemoglobin by altering its electric charge, as reported recently.14 Second, pharmacologic induction of fetal hemoglobin is a potential therapy for SCD.37 Previous studies showed that p38 kinases play a role in fetal hemoglobin induction by sodium butyrate38 and thalidomide.39 In this study, however, we demonstrated that p38 kinases are involved in the upregulation of P-selectin expression under hypoxia and low NO bioavailability, which presumably leads to the initiation of vaso-occlusive crisis. Recently, extracellular signal-regulated kinases were shown to participate in sRBC adhesion to endothelial cells.40 Thus, the activation of MAPK pathways likely enhances sRBC adhesion to endothelial cells, which in turn may lead to the initiation of vaso-occlusion. Caution should be taken when deciding whether the benefits of fetal hemoglobin induction through the activation of MAPK pathways outweigh the risks for enhanced sRBC adhesion that may elicit vaso-occlusion. In contrast, the induction of fetal hemoglobin by stimulating cyclic guanosine monophosphate–dependent or cyclic adenosine monophosphate–dependent pathways36,41 may preclude such possible adverse effects, but rather may be associated with the reduction of cell adhesion between multiple types of cells.42
Despite our findings in the molecular and signaling mechanisms regulating sRBC adhesion, a few caveats come with this study. First, we did not investigate whether the synergistic sRBC adhesion data observed in eNOS-deficient mice are reproducible in SCD mice. In our previous study with SCD mice,18 hypoxic stress (12% O2) increased sRBC adhesion by more than threefold. A lower level of induction of sRBC adhesion by hypoxia might be attributable in part to low NO bioavailability and hypoxemia of SCD mouse blood (Figure 2A-B). Second, a recent study by Frenette and coworkers demonstrated a role of E-selectin in the development of vaso-occlusion.43 In their study, tumor necrosis factor α was injected into their SCD mice to induce vaso-occlusions. However, tumor necrosis factor α selectively upregulates the expression of E-selectin rather than P-selectin,44,45 which might complicate their conclusions. Third, although we demonstrated that P-selectin expressed on HUVECs plays a role in the synergistic adhesion of sRBCs by hypoxia under low NO bioavailability, other molecules expressed on sRBCs or endothelial cells might play a role in sRBC adhesion, as sRBCs also adhered to endothelial cells in control mice.46
In conclusion, our in vivo and in vitro studies demonstrated that under the physiological condition of low NO bioavailability, sRBCs adhere to endothelial cells in a synergistic manner at the onset of hypoxia, whereas leukocyte adhesion to endothelial cells remains unaffected. Regarding the means of efficiently inhibiting sRBC adhesion in SCD, our results suggest the interruption of synergistic sRBC adhesion by hypoxia and low NO bioavailability, for instance, the restoration of NO bioavailability by NO inhalation,29 administration of sodium nitrite47 or sodium nitrate,48 or increasing oxygen tension by oxygen supply. However, continued oxygen supply could suppress erythropoiesis by reducing the secretion of erythropoietin.49 Thus, our findings in this study may help us develop novel treatment modalities for SCD.
Supplementary Material
Acknowledgments
We thank INO Therapeutics LLC for supplying NO gas; James L. Smith, Carson Strickland, and Kelly M. Robbins for their contribution; and Nadine Odo for critical reading of the manuscript.
This study was supported in part by NIH Exploratory Grant (P20MD003383) (T.I.).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.R.G. performed the research, analyzed the data, and wrote the manuscript; P.M.-H., J.B.P., and S.D.Y. performed research; T.I. designed the research, analyzed the data, and wrote the manuscript; and C.A.H. conceived the study, designed the research, analyzed the data, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for P.M.-H. is 56 G St, Boston, MA 02127.
Correspondence: Tohru Ikuta, Department of Anesthesiology and Perioperative Medicine, Medical College of Georgia, Georgia Regents University, 1120 15th St, BIW-2144, Augusta, GA 30912-2700; e-mail: tikuta@gru.edu; and C. Alvin Head, Department of Anesthesiology and Perioperative Medicine, Medical College of Georgia, Georgia Regents University, 1120 15th St, BIW-2144, Augusta, GA 30912-2700; e-mail: ahead@gru.edu.
References
- 1.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302(18):992–995. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 2.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin polymerization. I. Studies using temperature-jump and laser photolysis techniques. J Mol Biol. 1985;183(4):591–610. doi: 10.1016/0022-2836(85)90174-3. [DOI] [PubMed] [Google Scholar]
- 3.Ataga KI, Smith WR, De Castro LM, et al. ICA-17043-05 Investigators. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111(8):3991–3997. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
- 4.Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9(2):101–106. doi: 10.1097/00062752-200203000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Morris CR, Kuypers FA, Larkin S, Vichinsky EP, Styles LA. Patterns of arginine and nitric oxide in patients with sickle cell disease with vaso-occlusive crisis and acute chest syndrome. J Pediatr Hematol Oncol. 2000;22(6):515–520. doi: 10.1097/00043426-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med. 2002;8(12):1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 7.Kato GJ, Wang Z, Machado RF, Blackwelder WC, Taylor JG, VI, Hazen SL. Endogenous nitric oxide synthase inhibitors in sickle cell disease: abnormal levels and correlations with pulmonary hypertension, desaturation, haemolysis, organ dysfunction and death. Br J Haematol. 2009;145(4):506–513. doi: 10.1111/j.1365-2141.2009.07658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aslan M, Thornley-Brown D, Freeman BA. Reactive species in sickle cell disease. Ann N Y Acad Sci. 2000;899:375–391. doi: 10.1111/j.1749-6632.2000.tb06201.x. [DOI] [PubMed] [Google Scholar]
- 9.Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest. 1995;96(6):2661–2666. doi: 10.1172/JCI118332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez BL, Barnett J, Ballas SK, Christopher TA, Davis-Moon L, Ma X. Nitric oxide metabolite levels in acute vaso-occlusive sickle-cell crisis. Acad Emerg Med. 1996;3(12):1098–1103. doi: 10.1111/j.1553-2712.1996.tb03367.x. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Ruiz R, Montero-Huerta P, Hromi J, Head CA. Inhaled nitric oxide improves survival rates during hypoxia in a sickle cell (SAD) mouse model. Anesthesiology. 2001;94(6):1113–1118. doi: 10.1097/00000542-200106000-00028. [DOI] [PubMed] [Google Scholar]
- 12.Gladwin MT, Schechter AN, Ognibene FP, et al. Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation. 2003;107(2):271–278. doi: 10.1161/01.cir.0000044943.12533.a8. [DOI] [PubMed] [Google Scholar]
- 13.Head CA, Brugnara C, Martinez-Ruiz R, et al. Low concentrations of nitric oxide increase oxygen affinity of sickle erythrocytes in vitro and in vivo. J Clin Invest. 1997;100(5):1193–1198. doi: 10.1172/JCI119631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikuta T, Thatte HS, Tang JX, et al. Nitric oxide reduces sickle hemoglobin polymerization: potential role of nitric oxide-induced charge alteration in depolymerization. Arch Biochem Biophys. 2011;510(1):53–61. doi: 10.1016/j.abb.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Space SL, Lane PA, Pickett CK, Weil JV. Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am J Hematol. 2000;63(4):200–204. doi: 10.1002/(sici)1096-8652(200004)63:4<200::aid-ajh7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 16.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278(5339):873–876. doi: 10.1126/science.278.5339.873. [DOI] [PubMed] [Google Scholar]
- 17.Shesely EG, Maeda N, Kim HS, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1996;93(23):13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutsaeva DR, Parkerson JB, Yerigenahally SD, et al. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117(2):727–735. doi: 10.1182/blood-2010-05-285718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue A, Kuroyanagi Y, Terui K, Moi P, Ikuta T. Negative regulation of γ-globin gene expression by cyclic AMP-dependent pathway in erythroid cells. Exp Hematol. 2004;32(3):244–253. doi: 10.1016/j.exphem.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Russell J, Cooper D, Tailor A, Stokes KY, Granger DN. Low venular shear rates promote leukocyte-dependent recruitment of adherent platelets. Am J Physiol Gastrointest Liver Physiol. 2003;284(1):G123–G129. doi: 10.1152/ajpgi.00303.2002. [DOI] [PubMed] [Google Scholar]
- 21.Shiu YT, Udden MM, McIntire LV. Perfusion with sickle erythrocytes up-regulates ICAM-1 and VCAM-1 gene expression in cultured human endothelial cells. Blood. 2000;95(10):3232–3241. [PubMed] [Google Scholar]
- 22.Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111(11):5271–5281. doi: 10.1182/blood-2008-01-078204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stenberg PE, McEver RP, Shuman MA, Jacques YV, Bainton DF. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101(3):880–886. doi: 10.1083/jcb.101.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D. Selective activation of p38α and p38γ by hypoxia. Role in regulation of cyclin D1 by hypoxia in PC12 cells. J Biol Chem. 1999;274(33):23570–23576. doi: 10.1074/jbc.274.33.23570. [DOI] [PubMed] [Google Scholar]
- 25.Gao N, Jiang B-H, Leonard SS, et al. p38 Signaling-mediated hypoxia-inducible factor 1alpha and vascular endothelial growth factor induction by Cr(VI) in DU145 human prostate carcinoma cells. J Biol Chem. 2002;277(47):45041–45048. doi: 10.1074/jbc.M202775200. [DOI] [PubMed] [Google Scholar]
- 26.Stuart MJ, Setty BNY. Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood. 1999;94(5):1555–1560. [PubMed] [Google Scholar]
- 27.Sullivan KJ, Goodwin SR, Evangelist J, Moore RD, Mehta P. Nitric oxide successfully used to treat acute chest syndrome of sickle cell disease in a young adolescent. Crit Care Med. 1999;27(11):2563–2568. doi: 10.1097/00003246-199911000-00039. [DOI] [PubMed] [Google Scholar]
- 28.Weiner DL, Hibberd PL, Betit P, Cooper AB, Botelho CA, Brugnara C. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive crisis in pediatric patients with sickle cell disease [published correction appears in JAMA. 2004;292(8):925]. JAMA. 2003;289(9):1136–1142. doi: 10.1001/jama.289.9.1136. [DOI] [PubMed] [Google Scholar]
- 29.Head CA, Swerdlow P, McDade WA, et al. Beneficial effects of nitric oxide breathing in adult patients with sickle cell crisis. Am J Hematol. 2010;85(10):800–802. doi: 10.1002/ajh.21832. [DOI] [PubMed] [Google Scholar]
- 30.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98(6):1955–1962. doi: 10.1182/blood.v98.6.1955. [DOI] [PubMed] [Google Scholar]
- 31.Closse C, Seigneur M, Renard M, et al. Influence of hypoxia and hypoxia-reoxygenation on endothelial P-selectin expression. Thromb Res. 1997;85(2):159–164. doi: 10.1016/s0049-3848(96)00233-2. [DOI] [PubMed] [Google Scholar]
- 32.Armstead VE, Minchenko AG, Schuhl RA, Hayward R, Nossuli TO, Lefer AM. Regulation of P-selectin expression in human endothelial cells by nitric oxide. Am J Physiol. 1997;273(2, pt 2):H740–H746. doi: 10.1152/ajpheart.1997.273.2.H740. [DOI] [PubMed] [Google Scholar]
- 33.Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106(7):2334–2339. doi: 10.1182/blood-2005-04-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gladwin MT, Kato GJ, Weiner D, et al. DeNOVO Investigators. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305(9):893–902. doi: 10.1001/jama.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charache S, Terrin ML, Moore RD, et al. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 36.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci USA. 2001;98(4):1847–1852. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mabaera R, West RJ, Conine SJ, et al. A cell stress signaling model of fetal hemoglobin induction: what doesn’t kill red blood cells may make them stronger. Exp Hematol. 2008;36(9):1057–1072. doi: 10.1016/j.exphem.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Pace BS, Qian XH, Sangerman J, et al. p38 MAP kinase activation mediates gamma-globin gene induction in erythroid progenitors. Exp Hematol. 2003;31(11):1089–1096. doi: 10.1016/s0301-472x(03)00235-2. [DOI] [PubMed] [Google Scholar]
- 39.Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;110(8):2864–2871. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zennadi R, Whalen EJ, Soderblom EJ, et al. Erythrocyte plasma membrane-bound ERK1/2 activation promotes ICAM-4-mediated sickle red cell adhesion to endothelium. Blood. 2012;119(5):1217–1227. doi: 10.1182/blood-2011-03-344440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuroyanagi Y, Kaneko Y, Muta K, et al. cAMP differentially regulates γ-globin gene expression in erythroleukemic cells and primary erythroblasts through c-Myb expression. Biochem Biophys Res Commun. 2006;344(3):1038–1047. doi: 10.1016/j.bbrc.2006.03.203. [DOI] [PubMed] [Google Scholar]
- 42.Clancy RM, Abramson SB. Acetylcholine prevents intercellular adhesion molecule 1 (CD54)-induced focal adhesion complex assembly in endothelial cells via a nitric oxide-cGMP-dependent pathway. Arthritis Rheum. 2000;43(10):2260–2264. doi: 10.1002/1529-0131(200010)43:10<2260::AID-ANR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 43.Chang J, Patton JT, Sarkar A, Ernst B, Magnani JL, Frenette PS. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116(10):1779–1786. doi: 10.1182/blood-2009-12-260513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gedeit RG. Tumor necrosis factor-induced E-selectin expression on vascular endothelial cells. Crit Care Med. 1996;24(9):1543–1546. doi: 10.1097/00003246-199609000-00019. [DOI] [PubMed] [Google Scholar]
- 45.Bennett BL, Cruz R, Lacson RG, Manning AM. Interleukin-4 suppression of tumor necrosis factor alpha-stimulated E-selectin gene transcription is mediated by STAT6 antagonism of NF-kappaB. J Biol Chem. 1997;272(15):10212–10219. doi: 10.1074/jbc.272.15.10212. [DOI] [PubMed] [Google Scholar]
- 46.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11, suppl):S83–S86. [PubMed] [Google Scholar]
- 47.Mack AK, McGowan Ii VR, Tremonti CK, et al. Sodium nitrite promotes regional blood flow in patients with sickle cell disease: a phase I/II study. Br J Haematol. 2008;142(6):971–978. doi: 10.1111/j.1365-2141.2008.07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carlström M, Larsen FJ, Nyström T, et al. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc Natl Acad Sci USA. 2010;107(41):17716–17720. doi: 10.1073/pnas.1008872107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Embury SH, Garcia JF, Mohandas N, Pennathur-Das R, Clark MR. Effects of oxygen inhalation on endogenous erythropoietin kinetics, erythropoiesis, and properties of blood cells in sickle-cell anemia. N Engl J Med. 1984;311(5):291–295. doi: 10.1056/NEJM198408023110504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.