

Abstract
Adrenocortical carcinoma (ACC) is a rare endocrine malignancy, often with an unfavorable prognosis. Here we summarize the knowledge about diagnosis, epidemiology, pathophysiology, and therapy of ACC. Over recent years, multidisciplinary clinics have formed and the first international treatment trials have been conducted. This review focuses on evidence gained from recent basic science and clinical research and provides perspectives from the experience of a large multidisciplinary clinic dedicated to the care of patients with ACC.
Introduction
Epidemiology
Genetic Predisposition
Patient Presentation/Clinical Characteristics
-
Diagnosis
Biochemistry
Imaging
Differential diagnosis
Pathology
-
Molecular Pathology
Molecular genetics
Pathophysiology of cellular signaling pathways
Prognostic Factors
-
Therapy
Surgical therapy
Adjuvant therapy
Medical therapy
Radiation therapy
Other therapies
Surveillance
Future Perspectives
I. Introduction
In recent years, it has become evident that patients with malignant disease are best cared for by multidisciplinary teams of physicians and associated healthcare providers. This is particularly true for rare disorders such as adrenocortical carcinoma (ACC). The care for patients with rare diseases by the nonexpert is often based on extrapolation from other more common diseases or from the scarce evidence available through the medical literature. The formation of dedicated multidisciplinary clinics providing care for a larger referral community (eg, state- or nationwide) is a crucial step in gathering, preserving, and enhancing knowledge about these uncommon disorders. These multidisciplinary clinics have become essential for the exchange of scientific and clinical knowledge, the coordination of international multicenter trials, and the ultimate enrichment of evidence-based care for patients with rare disorders.
A multidisciplinary team that can provide high-level care for ACC patients ideally consists of endocrinologists, endocrine surgeons, medical and radiation oncologists, pathologists, radiologists, nuclear medicine physicians, and genetic counselors as well as clinical research coordinators. At the University of Michigan Health Systems, a multidisciplinary endocrine oncology program, mainly caring for patients with ACC has been in place for over 10 years. This current review is a result of discussions and experiences in our clinic. It aims to gather and present the evidence in diagnosis and therapy of ACC and to provide expert opinions where evidence is lacking. Figure 1 serves as a summary of the diagnosis of and therapy for ACC.
Figure 1.
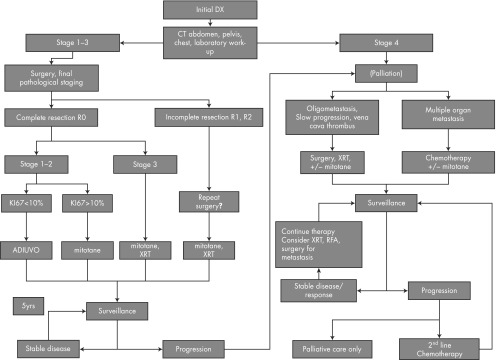
Flow chart for ACC therapy. Abbreviations: Dx, diagnosis; XRT, radiation therapy.
II. Epidemiology
Adrenal tumors are very common, affecting 3% to 10% of the human population, and the majority are small benign nonfunctional adrenocortical adenomas (ACA) (1). ACC, in contrast, is a very rare disease. The National Institutes of Health Office of Rare Diseases Research defines rare diseases by a prevalence of fewer than 200 000 affected patients in the United States (2). According to this definition, ACC might be regarded as an ultrarare disease. The incidence is believed to be 1 to 2 per million per year, but valid data are lacking (3). The Surveillance, Epidemiology, and End Results (SEER) database provides an estimation of incidence of approximately 0.72 per million cases per year leading to 0.2% of all cancer deaths in the United States (4). In Southern Brazil, the incidence during childhood is 2.9 to 4.2 per million per year compared with an estimated incidence of 0.2 to 0.3 per million children per year worldwide (5). This is mainly attributed to the high prevalence of the p.R337H low-penetrance allele of TP53 (6–8).
The median age of diagnosis is in the fifth to sixth decade, with the German ACC Registry reporting a median age at diagnosis of 46 years (9). This is in accordance with a median age of 46 years in a large single center series in France (10). Analysis of the SEER database gives a slightly older mean age of 55 years (11). Whether a second peak of increased incidence during childhood can be detected seems to be dependent on the prevalence of regional predisposing factors and biases (12, 13). A bimodal distribution was definitely observed in a concise review of case series reported and in the SEER data (13, 14). The fact that 1.3% of all childhood cancers are ACCs as opposed to 0.02% to 0.2% of adult cancers confirms a higher relative incidence early in life (12, 15–17). In the adult as well as in the pediatric population, there is a predilection for the female gender (the ratio of female to male ranges from 1.5–2.5:1) (10, 17). Aside from genetic predisposition (see below), no risk factors have been firmly established. A review of data from the 1986 National Mortality Followback Survey identified smoking in men and contraceptive use in women, especially before age 25 as risk factors (18). A role of estrogens has also been suggested by the observation of a probable relative increase of diagnosis of ACC during pregnancy (10, 19). Interestingly, recent in vitro studies confirm growth-promoting effects of estrogen on the ACC cell line NCI-H295 (20).
III. Genetic Predisposition
Epidemiological data on ACC from larger cancer registries is sparse, and they are often grouped with other endocrine malignancies, which makes analysis challenging (21, 22). In addition, detailed analyses of ACC patients' family histories have not been systematically conducted. However, there are certain clinical features supporting genetic predisposition. ACC appears to be relatively more common in children (6, 17). There are several descriptions of coexistence of childhood ACC and other tumors (23). In the adult population, the proportion of second malignancies is about 10% to 20% (24–26). However, no association or specific tumor pattern has been cataloged in previous studies. In roughly 2% to 10% of ACC patients, a contralateral tumor is present, in some cases probably presenting a synchronous and in other cases a metachronous ACC. It is of course difficult to determine whether the contralateral tumor is an independent primary tumor vs a metastasis to the contralateral gland. Clonal analyses supporting either of these theories are currently lacking, and there are only occasional reports supporting the diagnosis of 2 different primary ACCs (27).
The relative increase in incidence in childhood is mainly explained by germline TP53 mutations, which are the underlying genetic cause of ACC in ∼50% to 80% of children with ACC (Table 1)(28–30). Childhood ACC is a core malignancy of Li Fraumeni syndrome (LFS). Other core cancers are choroid plexus tumors, sarcomas, early-onset breast cancers, brain cancers, and leukemias. Approximately 3% to 10% of LFS-associated cancers are ACCs, suggesting that germline TP53 mutations infer a significant relative risk increase (31, 32). Therefore, according to the Chompret testing criteria, TP53 germline testing is recommended for any patient with a diagnosis of ACC (33, 34). However, the contribution of germline TP53 mutations to ACC development in adults had been not well researched until 2 recent studies determined the prevalence of TP53 mutations between 3% and 7% in the adult population (35, 36). Most importantly, TP53 germline testing should not be dismissed because of the absence of a family history. Up to 25% of TP53 mutations occur de novo, and these patients lack a significant family history (34). Because of the impact of a diagnosis of LFS for the patient and at-risk relatives, TP53 germline testing should be considered in all ACC patients. Adjuvant radiotherapy should be considered with caution for mutation-positive patients because of the increased risk of secondary malignancies in the radiation field. Most TP53 mutations affect the DNA binding and tetramerization domains (14). One particular hot spot mutation has been described to date, which is the low-penetrance tetramerization domain p.R337H mutation in Southern Brazil (14, 37–39). Although it was initially believed that this mutation specifically predisposes to ACC development in childhood, it is now well recognized that this mutation causes other LFS-associated tumors as well as a Li Fraumeni-like syndrome in affected families (37). The p.R337H mutation was initially considered not to be a result of common ancestry; however, recent analyses suggest a founder effect in most cases, although in some cases, de novo mutations may still exist (7, 39). The high frequency of germline mutations in the Southern Brazilian population has also recently been confirmed in a population-based screening study (40).
Table 1.
Hereditary Syndromes in Patients with ACC
| Syndrome | Prevalence in ACC Patients | Prevalence in General Population | Gene Mutation | Other Phenotype |
|---|---|---|---|---|
| LFS | Common (3%–7% of adults, 50%–80% children) | 1:20 000 to 1:1 000 000 (358) | TP53 | Sarcoma, choroid plexus tumor, brain cancer, early breast cancer, leukemia, lymphoma |
| MEN1 | Rare (1%–2% of adults) | 1:30 000 (359) | MENIN | Foregut neuroendocrine tumors, pituitary tumors, parathyroid hyperplasia, collagenoma, angiofibroma, adrenal adenoma/hyperplasia |
| Lynch syndrome | 3% of adults | 1:440 | MSH2, MSH6, MLH1, PMS2 | Colorectal cancer, endometrial cancer, sebaceous neoplasms, ovarian cancer, pancreatic cancer, brain cancer |
| BWS | Very rare, only children | 1:13 000 (360–362) | IGF2, CDKN1C, H19 locus changes on 11p15 | Wilms' tumor, hepatoblastoma, macrosomia, adrenocortical cytomegaly, adrenal adenoma, adrenal cyst, hemihypertrophy, macroglossia, omphalocele, ear pits |
| FAP | Very rare (<1%) | 1:30 000 (363–365) | APC | Intestinal polyps, colon cancer, duodenal carcinoma, thyroid cancer, desmoid tumor, adrenal adenoma, supernumerary teeth, congenital hypertrophy of the retina, osteoma, epidermoid cysts |
| Neurofibromatosis type 1 | Very rare (<1%) | 1:3000 (366, 367) | NF1 | Malignant peripheral nerve sheet tumor, pheochromocytoma, café au lait spots, neurofibroma, optic glioma, Lisch nodule, skeletal abnormalities |
| Carney complex | Very rare (case reports) | ∼700 patients worldwide (368) | PRKAR1A | Primary pigmented nodular adrenal disease, large-cell calcifying Sertoli cell tumors, thyroid adenoma, myxoma, somatotroph pituitary adenoma, lentigines |
Beckwith-Wiedemann syndrome (BWS) spectrum disorders, such as classical BWS and idiopathic isolated hemihypertrophy, also increase the risk for ACC (Table 1). The underlying genetics of these syndromes are complex. A hallmark is alterations of DNA methylation of the 11p15 locus, which harbors the coding regions for IGF2, the cell cycle regulator CDKN1C, and the nontranslated RNA, H19 (41). The common sequelae of all these changes are an upregulation of IGF2 expression and a downregulation of the other two transcripts (41). The main adrenal phenotype as initially described by Beckwith is adrenocortical cytomegaly (42). Several benign and malignant tumors are classically associated with BWS. Specifically, the risk for Wilms' tumor and hepatoblastoma is increased, and regular screening for these cancers is recommended during childhood. The most frequent macroscopic adrenal pathologies described are adrenal cysts and ACAs (43). Although data vary significantly, ACC comprises 5% to 15% of malignancies in BWS (43, 44). Due to the low overall incidence (<1% of children with BWS will develop an ACC), no specific screening recommendations for ACC exist. As observed with other embyronal tumors that exhibit a developmental window of presentation, the cancer risk of children with BWS decreases through adolescence and then remains at the level of the general population.
Multiple endocrine neoplasia type 1 (MEN1) is caused by mutations in the MENIN gene on chromosome 11q13. Its classical manifestations are hyperparathyroidism, caused by 4-gland hyperplasia, foregut neuroendocrine tumors (most commonly in the pancreas and duodenum, but also thymus and lung), and pituitary adenomas (prolactinomas are most common). Associated adrenal lesions, mainly ACAs and uni- or bilateral hyperplasia, occur in 20% to 55% of MEN1 cases (45–48). Although hormone production has been well-described for adrenal tumors in MEN1, most of the tumors are nonfunctional. A small fraction of patients with MEN1 will develop ACC (46–51). Recent analysis of a French multicenter registry determined that ∼10% of MEN1 patients have distinct adrenal tumors, and of these, up to 14% are malignant (48). These are usually characterized by relatively fast growth, but no other predictive factors have been established. The current guidelines do not recommend regular monitoring of the adrenal glands in this setting. However, because development of ACC from preexisting adrenal lesions has been well described in MEN1, special attention should be given to these organs during annual or biennial imaging of the pancreas for neuroendocrine tumors (52).
ACC has also been reported in patients with Lynch syndrome (53–56). Lynch syndrome is caused by mutations in genes involved in DNA mismatch repair genes MSH2, MSH6, PMS2, MLH1, and TACSD1/EPCAM. Patients with Lynch syndrome have a significant increase in lifetime risk of cancer, specifically for the core malignancies, colorectal and endometrial cancer (57). Screening for Lynch syndrome is recommended in all patients with colorectal cancer (58). This includes immunohistochemistry for the 4 gene products as well as microsatellite instability analysis. The vast majority of Lynch syndrome-associated colorectal cancers show loss of immunostaining for at least 1 of the gene products and are microsatellite unstable (59). Screening has been proven cost-effective, and surveillance for colon cancer with regular colonoscopies significantly decreases morbidity and mortality in affected patients (60, 61). Recently, a systematic analysis has defined the prevalence of Lynch syndrome in patients with ACC to be ∼3%, comparable to the prevalence in colorectal and endometrial cancer, estimated at 2% to 5% (62). Immunohistochenistry was informative in most cases; however, all tested ACCs were microsatellite stable at the usual microsatellite markers. Routine screening for Lynch syndrome in ACC tumors by immunohistochemistry may be warranted regardless of family history.
There are several reports of ACCs in patients with familial adenomatous polyposis (FAP), neurofibromatosis type 1, and Werner syndrome (63–72). Most recently, ACC has also been reported in 2 cases of patients with Carney complex (73, 74). Some cases of ACC in conjunction with congenital adrenal hyperplasia (CAH) have been described (75). However, the co-occurrence of a rare tumor and a fairly common genetic syndrome make this association unconvincing at this point (76). Furthermore, there is currently no support for this association from large CAH registries. However, it has become clear over the last decade that patients with CAH commonly develop adrenal myelolipomas (77).
Understanding the relationship between ACC and hereditary cancer syndromes has been valuable in revealing mechanisms of tumorigenesis and identifying new targets for therapy. For example, the relation of adrenal tumors with FAP led to the discovery of the role of β-catenin signaling in adrenal tumors. The relation of ACC to BWS together with findings from gene expression arrays led to the hypothesis that the IGF-1 receptor may be a target for ACC therapy. This hypothesis has now been tested in several phase 1 to phase 3 clinical trials (78).
In terms of clinical recommendations, it is the authors' opinion that every ACC patient should receive a basic physical examination aimed at finding clues for hereditary diseases. A minimum of a 3-generation family history should be obtained with focused extension on second- and third-degree relatives with malignancies. Every ACC patient should be offered TP53 mutation screening, ideally in the context of an evaluation by a professional genetic counselor or clinical geneticist (36). Furthermore, any adrenal lesion observed in a patient with LFS, MEN1, Lynch syndrome, BWS, or FAP should deserve a clinical and hormonal work-up as well as close follow-up imaging with an increased suspicion for malignancy.
IV. Patient Presentation/Clinical Characteristics
There are 3 main clinical scenarios in which ACC patients present. For 40% to 60% of patients, the major presenting complaints are symptoms and signs of hormone excess (3, 9, 10). Another third present with nonspecific symptoms due to local tumor growth, such as abdominal or flank pain, abdominal fullness, or early satiety (9, 10). Roughly 20% to 30% of ACCs are incidentally diagnosed by imaging procedures for unrelated medical issues (25). Patients with ACC only rarely present with classical tumor symptoms, such as cachexia or night sweats (3, 10). Paraneoplastic syndromes are uncommon. However, tumor-associated hypoglycemia is a well-described phenomenon, historically termed Anderson's syndrome, which may be attributed to IGF-2–mediated hypoglycemia (79–81). However, it is unclear why this symptom is less prevalent in the modern medical era. Other rare paraneoplastic syndromes are hyperreninemic hyperaldosteronism, erythropoietin-associated polycythemia, and leukocytosis (caused by chemokine release from the tumor) (82–84).
Biochemically or clinically apparent adrenocortical hormone production is evident in up to 45% to 70% (9, 10, 85). In these patients, symptoms related to the hormone excess are the major cause for presentation, leading to imaging and clinical investigation. However, syndromes of hormone excess are often not readily recognized by physicians, leading to delay in diagnosis and subsequent surgical and/or medical therapy.
Hypercortisolism is the most common presentation of patients presenting with hormone excess (50%–80% of hormone-secreting ACCs), causing classic symptoms including plethora, diabetes mellitus, muscle weakness/atrophy, and osteoporosis. Frequently, very high cortisol levels in ACC saturate the renal HSD11B2 system, resulting in glucocorticoid-mediated mineralocorticoid receptor activation. Therefore, hypokalemia and hypertension are commonly observed in ACC patients with hypercortisolism. Together with pronounced muscle weakness, these symptoms of rapidly progressive Cushing's syndrome are generally indicative of a malignant adrenal tumor. The second most commonly produced hormones in patients with ACC are adrenal androgens (40%–60% of hormone-secreting ACCs), causing rapid-onset male pattern baldness, hirsutism, virilization, and menstrual irregularities in women. Concurrent androgen and cortisol production is evident in roughly half of all ACC patients with hormone excess. However, isolated hyperandrogenism in male patients is often unrecognized due to the paucity of significant symptoms. Instead, it is the peripheral conversion of androgens to estrogens and/or the cosecretion of estrogen from the ACC that induces significant symptoms. Estrogen production occurs in 1% to 3% of male ACC patients, causing gynecomastia and testicular atrophy (through suppression of the gonadal axis). In the evaluation of adrenal tumors, regardless of size, androgen or estrogen production should always raise the suspicion of a malignant tumor. Autonomous aldosterone secretion (which classically leads to hypertension and hypokalemia) is rare in ACC (85, 86). More commonly, mineralocorticoid effects are mediated by high cortisol levels or possibly steroid precursors with mineralocorticoid activity, such as 11-deoxycorticosterone (87, 88).
At the time of presentation, ACCs are generally large tumors, measuring on average 10 to 13 cm (9, 85, 89). Only a minority of tumors are <6 cm (9%–14%), with only 3% presenting as lesions <4 cm (89, 90). In 2004, the World Health Organization and Union for International Cancer Control introduced a staging system for ACC based on the traditional McFarlane classification, modified by Sullivan (Table 2). This classification system has been recently challenged due to several shortcomings and the newly introduced European Network for the Study of Adrenal Tumors (ENSAT) system became widely adopted by the ACC community due to the better reflection of ENSAT stage to patient outcome (91). The ENSAT staging system defines 4 stages. Stage 1 (≤5 cm) and stage 2 (>5 cm) tumors are confined to the adrenal gland. Stage 3 tumors extend into surrounding tissue (eg, para-adrenal adipose tissue or adjacent organs) or involve locoregional lymph nodes. Stage 4 is reserved for patients with distant metastasis. Most ACCs are diagnosed at an advanced stage, although this might be predicted to change in the near future due to the persistently increasing use of abdominal imaging procedures. Although earlier studies found 49% of patients with metastatic disease (stage 4) at presentation, currently only 25% to 30% of patients present with metastatic disease (9, 13, 25). In the Michigan Endocrine Oncology Repository that contains data from >400 patients with a diagnosis of ACC, the mean stage at diagnosis is as follows: stage 1, 14%; stage 2, 45%; stage 3, 27%; and stage 4, 24% (T.E., unpublished results). The most common metastatic sites are lung (40%–80%), liver (40%–90%), and bone (5%–20%) (92). A contralateral adrenal tumor can be found in ∼5% of patients, although it is difficult to differentiate this from metachronous or synchronous tumors. Other sites, such as brain and skin, are much less affected by tumor spread (<5%). After initial resection, locoregional recurrence becomes a challenge with pelvic, peritoneal, or retroperitoneal metastases.
Table 2.
Staging Systems for ACC (91)
| Staging System |
||
|---|---|---|
| UICC/WHO | ENSAT | |
| Stage 1 | T1, N0, M0 | T1, N0, M0 |
| Stage 2 | T2, N0, M0 | T2, N0, M0 |
| Stage 3 | T1–2, N1, M0 | T1–2, N1, M0 |
| T3, N0, M0 | T3–4, N0, M0 | |
| Stage 4 | T1–4, N0–1, M1 | T1–4, N0–1, M1 |
| T3–4, N1, M0 | ||
| T4, N0, M0 | ||
Abbreviations: UICC, International Union Against Cancer; WHO, World Health Organization.
Tumors are classified as follows: T1, ≤5-cm tumor; T2, >5-cm tumor; T3, tumor infiltration into surrounding tissue; T4, tumor invasion into adjacent organs; N0, no positive lymph nodes; N1, positive lymph node(s); M0, no distant metastases; M1, presence of distant metastasis.
At the time of diagnosis, the initial evaluation should include a thorough physical examination and patient history with particular respect to symptoms and signs of hormone excess. Patients should undergo basic biochemical evaluation including creatinine, liver function tests, and a complete blood count. These values will guide further therapy and disease management. An initial hormonal evaluation is crucial (Table 3). Staging should at the minimum include a computed tomography (CT) scan or magnetic resonance imaging (MRI) of the abdomen/pelvis and a CT of the chest. Other imaging should be guided by clinical suspicion (eg, bone scan for skeletal metastasis). A focus on family history is essential to identify possible hereditary contributions.
Table 3.
Initial Staging and Laboratory Work-up
| Mandatory | Optional/Depending on Suspicion | |
|---|---|---|
| Cross-sectional imaging | MRI or CT (abdomen/pelvis/chest) | [18F]FDG-PET scan, bone scan |
| Hormonal work-up | Blood: DST, 8:00 am cortisol and ACTH, DHEAS, testosterone (total or bioavailable), aldosterone and renin, metanephrine and normetanephrinea (to exclude pheochromocytoma), 24-h urine: free cortisolb | Blood: 17-OH-progesterone, 17-OH-pregnenolone, 11-deoxycorticosterone, progesterone, androstenedione, estradiol, FSH, LH |
| Other laboratory work-up | Discuss testing for TP53 mutations; AST, ALT, creatinine, lipid profile, TSH, free T4, CBC | Alkaline phosphatase, GGT, other laboratories |
Abbreviation: CBC, complete blood count.
Either 24-hour urine or plasma free metanephrines.
If 1 mg DST suggests hypercortisolism.
V. Diagnosis
A. Biochemistry
Biochemical evaluation fulfills several purposes: 1) to establish or exclude the diagnosis of hormone excess; 2) to establish the adrenocortical origin of a tumor, making further invasive work-up, such as biopsies, unnecessary; 3) to further increase the suspicion of a malignant lesion (eg, androgen or estrogen production); 4) to use steroid hormones as tumor markers for future follow-up and surveillance; and 5) to assess for the necessity of postsurgical hydrocortisone replacement therapy.
The hallmark of a biochemical evaluation is the measurement of steroid hormones produced by the tumor (Table 3 and Figure 2). Initial evaluation is in part guided by clinical symptoms (eg, cushingoid features, hirsutism, and/or new hypertension with or without hypokalemia).
Figure 2.
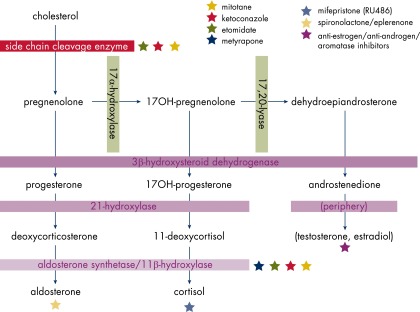
Steroidogenesis and inhibitors.
Most patients with cortisol-secreting tumors will have suppressed ACTH (<10 pg/mL) and increased cortisol on a spontaneous 8:00 am blood draw. The diagnosis of hypercortisolism is usually established by a 1 mg dexamethasone suppression test (DST), midnight salivary cortisol, or elevated 24-hour urine free cortisol. The latter will also give an estimate of the extent of hypercortisolism (93).
Screening for aldosterone production includes measurement of plasma renin activity and serum aldosterone levels. An isolated suppression of renin will often be encountered, which, in the absence of elevated levels of aldosterone, is caused by simple volume repletion or by the pathological mineralocorticoid action of cortisol or steroid precursors with mineralocorticoid activity.
Dehydroepiandrosterone sulfate (DHEAS) and total or bioavailable testosterone should be measured in every patient. Whether the measurement of other steroid metabolites, such as 17-OH-progesterone, androstenedione, and estrogen should be generally recommended is a matter of debate. However, elevated levels can certainly be useful as tumor markers and allow specific treatment with hormonal antagonists to alleviate symptoms.
Despite the presence of a large tumor, signs or symptoms of steroid hormone excess and blood levels of hormones in ACC can be absent or minimal. In comparison with the normal adrenal cortex, steroid hormone synthesis in ACC is relatively inefficient, resulting in elevated levels of a variety of steroid hormone precursors and, even in the presence of a large lesion, only modestly elevated hormone levels. Although most of these metabolites are not routinely measured clinically, they can be detected by gas chromatography/mass spectrometry analysis. Indeed, urine steroid analysis is predicted to be a sensitive method to diagnose ACCs and to follow individual steroid metabolite profiles for recurrence, progression, and/or treatment response. Several decades ago, it had been shown that urine androgens and androgen precursors can be followed as tumor markers (94). Metabolites of 11-deoxycortisol and DHEA seem to be most useful for this purpose. A recent study has shown significant differences in steroid hormone precursor and metabolite profiles in urine of patients with ACC compared with patients with benign adrenal tumors (95). This study defined the 11-deoxycortisol metabolite tetrahydro-11-deoxycortisol as the most discriminative marker, although the overall profile of several metabolites provided more information.
In addition to steroid hormone measurements, biochemical exclusion of a pheochromocytoma is warranted, especially when no steroid hormone production is evident. This is accomplished by measuring levels of metanephrine and normetanephrine in plasma or 24-hour urine and mainly serves to prevent unexpected complications during surgery or treatment (96).
B. Imaging
ACCs are typically large tumors upon clinical presentation, often measuring more than 6 cm in diameter (Figure 3 and Table 4) (97). Due to the presence of internal hemorrhage, necrosis, and calcifications, these tumors tend to vary in appearance with frequent heterogeneous enhancement. They are bilateral in 2% to 10% of cases (98, 99). Metastases to the liver, lungs, or lymph nodes can be seen, and invasion of adjacent organs or venous extension into the renal vein and/or inferior vena cava may be present. Contrast-enhanced CT or MRI is the diagnostic imaging modality of choice for initial imaging and staging as well as for follow-up. Both modalities are well suited for detecting local recurrence and metastatic disease (98). Functional imaging by positron emission tomography (PET) with [18F]fluorodeoxyglucose (FDG) and [11C]metomidate (MTO) or [123I]MTO (where available) may be used to confirm diagnosis of a malignant lesion or establish the adrenocortical origin of a tumor. NP59 ([131I]-iodocholesterol) scans are no longer available.
Figure 3.

ACC. A, Precontrast fairly homogeneous with calcification (30 HU). B, Early-phase contrast with heterogeneous enhancement. C, Delayed phase (15 minutes).
Table 4.
Imaging Characteristics of ACC
| Lesion Characteristics | ACC | ACA |
|---|---|---|
| Size | >4 cm | <4 cm |
| Necrosis | + | − |
| Hemorrhage | + | − |
| Calcification | +/− | − |
| CT density | Heterogeneous, >10 HU | Homogeneous, <10 HU |
| Chemical-shift MRI | Heterogeneous signal drop +/− | Homogeneous signal drop |
| Contrast enhancement | Heterogeneous, absolute % washout <60% | Homogeneous, absolute % washout >60% |
| SUV on [18F]FDG-PET/CT | Adrenal to liver SUV ratio >1.45 | Adrenal to liver SUV ratio <1.45 |
ACC can present as an adrenal incidentaloma, defined as an unsuspected adrenal mass discovered on a cross-sectional imaging performed for another reason (100). An incidentally discovered adrenal mass with heterogeneous appearance and a size greater than 4 cm or other imaging characteristics of malignancy should be evaluated with complete imaging for staging and will usually be treated surgically (101). The risk for malignant adrenal tumors increases with tumor size, with the index of suspicion increasing for tumors >4 cm (sensitivity, 97%; specificity, 52%) and >6 cm (sensitivity, 91%; specificity, 80%) (89). Masses 1 to 4 cm in diameter without definite benign imaging features, such as a homogenous, low-density (≤10 Hounsfield units [HU]) mass with smooth margins, need to be further assessed with a dedicated adrenal imaging protocol. If absolute percent washout is less than 60%, relative percent washout is less than 40%, or the mass has suspicious imaging features, further evaluation is warranted.
1. CT and MRI
ACCs can be distinguished from lipid-rich ACAs, which tend to be small, homogeneous masses that measure ≤10 HU on unenhanced CT or demonstrate loss of signal on chemical-shift MRI (102). Homogeneous adrenal tumors can also be further characterized using a dedicated adrenal protocol CT (see Figure 5). ACAs demonstrate a greater contrast washout than adrenal nonadenomas (103, 104). On CT imaging, ACCs are large, heterogeneous enhancing masses of soft tissue attenuation. On MRI, ACCs appear isointense to hypointense relative to liver parenchyma on T1-weighted images and hyperintense relative to liver parenchyma on T2-weighted images (98). Contrast-enhanced imaging often demonstrates heterogeneous, predominantly irregular peripheral enhancement with central nonenhancing areas secondary to hemorrhage or necrosis. Internal hemorrhage is seen as ill-defined areas of increased attenuation on non–contrast-enhanced CT and as areas of high signal intensity on T1-weighted images. Areas of necrosis have low attenuation on non–contrast-enhanced CT, high signal intensity on T2-weighted images and do not enhance after administration of iv contrast (105). Calcifications, which are best detected on CT imaging as high attenuation foci, can be present in approximately 30% of cases. These are either coarse calcifications or microcalcifications and usually centrally located (Figures 3 and 4). Calcification is also present in other adrenal pathologies such as myelolipoma (Figure 5) and 10% of pheochromocytomas and hence is not a distinguishing feature (106). Some ACCs may contain areas of intracellular lipid and rarely macroscopic fat resulting in CT density measurements of <10 HU in portions of the tumor (107). On chemical-shift MRI, the presence of intracellular lipid can cause regions of signal loss (<30% of lesion) on out-of-phase images relative to in-phase images (98). Contrast-enhanced CT scan is a reliable method of disease staging, identifying common metastatic sites such as regional and para-aortic lymph nodes, lungs, liver, and bones (98). Inferior vena cava invasion has been reported in 9% to 19% of cases at presentation (98). Due to the multiplanar capability of MRI, direct invasion of adjacent organs may be better depicted.
Figure 5.

A, Small right adrenal adenoma with <10 HU on unenhanced CT scan. B, Left adrenal adenoma with <10 HU on unenhanced CT scan. C, Myelolipoma with fat attenuation and small calcification (asterisk). D–F, Dedicated adrenal CT scan without contrast, 16 HU (D); immediately after contrast, 99 HU (E); and delayed image, 44 HU (F), identifying this lesion as a non–lipid-rich adenoma.
Figure 4.
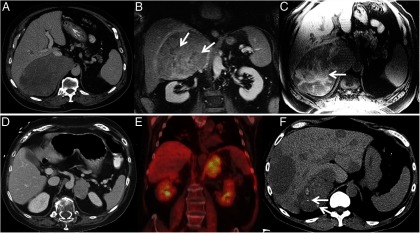
A, CT of ACC showing large heterogeneous right adrenal tumor, B, Contrast-enhanced coronal MRI in the same patient showing heterogeneous enhancement with nonenhancing areas of necrosis (arrows). C, Non–contrast-enhanced T1-weighted MRI in the same patient showing T1-weighted hyperintense areas of hemorrhage (arrows). D, CT of ACC showing left adrenal tumor. E, Intensely FDG avid left adrenal mass in the same patient. F, Metastasized ACC, calcifications in primary tumor (arrows).
2. [18F]FDG PET/CT imaging
ACC typically presents as a large, heterogeneous mass with intense FDG uptake greater than liver background (Figure 4). In a study of 77 patients with surgically proven diagnosis of ACA or ACC, [18F]FDG PET/CT had a sensitivity of 100% and specificity of 88% in distinguishing benign from malignant lesions by using cutoff value above 1.45 for adrenal to liver maximum standardized uptake value (SUV). In the same study using a cutoff value of 3.4 for adrenal maximum SUV, the sensitivity was 100% and specificity 70% (108). Assessment of morphological characteristics such as tumor size, heterogeneity, and irregular margins as well as attenuation value and metabolic activity is likely to improve accuracy. [18F]FDG PET/CT, however, cannot distinguish ACC from metastases, lymphoma, or pheochromocytoma, which also exhibit high metabolic activity (109). In a meta-analysis of published data to determine the diagnostic utility of [18F]FDG PET/CT for distinguishing benign from malignant adrenal tumors, [18F]FDG PET/CT had sensitivity of 97% and specificity of 91% (109). No significant difference in accuracy was found between visual analysis, SUV analysis, and standardized uptake ratio (defined as ratio of adrenal SUV activity to liver SUV activity) analysis.
[18F]FDG PET/CT is a useful modality for staging ACC and evaluating local recurrence. In a study on 22 patients with ACC, sensitivity of [18F]FDG PET/CT was 90% for diagnosis of metastases as compared with 88% for diagnostic CT. However, they should be considered complementary imaging modalities because 12% and 10% of lesions were seen only by [18F]FDG PET/CT or CT, respectively (110). [18F]FDG PET/CT has low sensitivity for characterization of smaller lesions, particularly for those lesions less than 10 mm in diameter (111). Intensity of FDG uptake was found to be related to survival in patients with ACC, with a maximum SUV of >10 indicating poor prognosis (111). In a study of 12 patients with previously resected ACC, [18F]FDG PET/CT correctly identified local tumor recurrence in all patients (112). [18F]FDG is not a tumor-specific tracer, and increased uptake may be seen in benign conditions including postoperative changes.
3. Experimental imaging modalities
Proton MR spectroscopy may be helpful in differentiating ACAs and pheochromocytomas from ACC and metastases using choline to creatine ratios of greater than 1.2 (92% sensitivity and 96% specificity) and choline to lipid ratios greater than 0.38 (92% sensitivity and 90% specificity) (98). However, more research data and prospective clinical evaluation are needed to substantiate this approach.
Metomidate, an inhibitor of 11β-hydroxylase (cytochrome P450 family 11 subfamily B1 [CYP11B1]) and aldosterone synthetase (CYP11B2), has high affinity and specificity for these enzymes. [11C]MTO PET can distinguish tumors of adrenocortical origin from noncortical lesions (113). It cannot, however, distinguish benign from malignant adrenocortical lesions. In a study of 11 patients with ACC, [11C]MTO PET/CT visualized all viable tumors with high tracer uptake as compared with normal adrenal gland and liver. False-negative results occurred due to tumor necrosis (113). [123I]Iodometomidate (IMTO) is a highly specific tracer for imaging of adrenocortical tissue as shown in a pilot study of 4 patients with known adrenal tumors (2 metastatic ACCs, 1 bilateral ACA, and 1 metastatic melanoma) (110).
C. Differential diagnosis
The diagnosis of ACC is often evident in the setting of a large adrenal mass with concomitant hormone excess. However, there are 2 main situations in which differential diagnoses need to be addressed: 1) an incidental large adrenal mass is discovered or 2) hormone excess is established, but imaging has not been conducted. The evaluation of incidentally discovered adrenal masses is well established by the current National Institutes of Health guidelines and guidelines by other professional organizations and has been discussed in this journal recently (1, 114). In the hormonal evaluation of such lesions, a 1 mg DST is preferred because it has a greater specificity in diagnosing subclinical Cushing's syndrome (93). Mineralocorticoid excess should be evaluated following The Endocrine Society guidelines with initial screening for increased aldosterone and suppressed renin levels (115). Mineralocorticoid excess can be caused by either bilateral hyperplasia or ACA, and these lesions are usually small (<2 cm) (115). Measurement of other steroid hormones, specifically estradiol, DHEAS, and testosterone, is not routinely recommended but should be performed in cases where lesions show imaging characteristics consistent with malignancy or where signs or symptoms suggest sex steroid excess.
Elevated adrenal steroid hormone levels can be caused by other endocrine diseases. Hypercortisolism is diagnosed according to the current guidelines for diagnosis of Cushing's syndrome by The Endocrine Society, an 8:00 am cortisol value after DST of less than 1.8 mg excludes hypercortisolism. Other suitable screening tests for hypercortisolism are midnight salivary cortisol and 24-hour urine cortisol measurement. Hypercortisolism in connection with ACC is due to autonomous cortisol secretion. The resultant ACTH-independent Cushing's syndrome is accompanied by a low ACTH level (<10 pg/mL). The main differential diagnoses in this category are ACTH-independent macronodular hyperplasia and cortisol-producing adenomas, which most often can be differentiated by imaging. Symptoms and signs of hyperandrogenemia, such as hirsutism, are also present in polycystic ovarian syndrome, ovarian hyperthecosis, and CAH or can be constitutional. However, the levels of DHEAS and testosterone are usually markedly higher in ACC, the onset of hormone excess symptoms is more pronounced, and most symptoms develop over a relatively short period of time (months).
As discussed above, the imaging characteristics of an adrenal mass weigh heavily in the diagnostic evaluation of potential ACC. Although an initial study found up to 8% ACCs among incidental adrenal tumors, another large-scale single-center study of 1049 incidental adrenal masses found only a single adrenocortical tumor of unknown malignant potential and no ACC (116, 117). In general, homogeneous lesions less than 4 cm with <10 HU or a relative washout >40% are not suspicious for ACC. Several recent studies have focused on ACCs smaller than 4 cm, and these were almost invariably suspicious for malignancy by imaging criteria. In the University of Michigan Endocrine Oncology Repository, less than 1% of ACCs were less than 4 cm on initial imaging. For an adrenal lesion greater than 4 cm, the main differential diagnoses include large ACA, myelolipoma, adrenal metastasis of another cancer, pheochromocytoma, adrenal cyst, ganglioneuroma, or other rare tumors of the adrenal gland, such as sarcomas or lymphomas. Myelolipomas have a very typical imaging appearance and can usually be readily identified. Adrenal cysts can present a challenge, because the differential diagnoses include cystic ACC, cystic pheochromocytoma, and benign cysts (eg, bronchogenic or retroperitoneal cyst). The evaluation of a large adrenal mass suspicious for malignancy should include full body imaging for cancer staging, in which a primary (nonadrenal) tumor often becomes evident. Adrenal pheochromocytomas usually produce catecholamines and can be diagnosed biochemically. Further diagnostic procedures such as biopsy are rarely indicated. The primary treatment for all large isolated adrenal tumors is surgical resection. The only exceptions to this rule are primary adrenal lymphomas. These are extremely rare, are often bilateral, may be differentiated by CT and MRI, and are treated with systemic chemotherapy (118). In case a pheochromocytoma cannot be excluded by imaging characteristics, initiating α-blockade before surgery should be considered, even if biochemical work-up is negative.
VI. Pathology
The pathological assessment of adrenocortical tumors has advanced substantially over the last 4 decades. Tumor size was initially thought to be the primary factor for determining which tumors possessed malignant potential and, accordingly, could be classified as ACC. Although tumor size still possesses diagnostic significance, current diagnostic algorithms have evolved to incorporate a variety of clinical, histological, and immunohistochemical parameters.
Work by 3 independent groups advanced the field by systematically applying histological and nonhistological parameters to clinically benign and malignant tumors (119–121). By using this approach, it was possible to define a set of diagnostic criteria that could be used to identify those tumors that possess, but did not yet manifest, malignant potential. Of these 3 overlapping diagnostic systems, the Weiss system and its modifications have gained the most acceptance in clinical practice (122). Despite the formality of these scoring systems, the criteria embedded within them represent bread-and-butter surgical pathology, ie, standard histological parameters that include invasion by tumor into capsule and adjacent vessels, changes in growth patterns, presence of tumor necrosis, increased mitotic rates, and the presence of atypical mitotic figures. Tumors with an abundance of these features (3 or more, as in the Weiss system) most often behave in a malignant fashion and can be classified as ACC, whereas tumors without these features (0–2 in the Weiss system) do not metastasize and can be classified as ACA.
Adrenocortical neoplasms, similar to other solid endocrine tumors, grow predominantly via expansion without a desmoplastic response, in contrast to other solid tumors that show infiltration of desmoplastic stroma (eg, ductal adenocarcinoma of the pancreas). As a consequence, adrenocortical tumors are usually well-delineated masses whose colors range from brown to orange to yellow, usually a function of lipid content. Benign and malignant tumors induce the creation of a fibrous capsule. The scenario of an expansile mass surrounded by a fibrous capsule is analogous to that seen in follicular tumors of the thyroid. Eventually, a tumor may acquire malignant potential, ie, the ability to invade normal tissues and metastasize distantly. The key feature that distinguishes ACC from ACA, short of the presence of metastatic disease, is the presence of invasion. Invasion can take several forms: direct invasion of the tumor capsule, invasion through the tumor capsule into extra-adrenal soft tissue, or direct invasion of lymphatic channels in and around the capsule and direct invasion of nearby blood vessels, usually veins. In some cases, the venous invasion is so advanced that the tumor invades the vena cava and extends to involve the right side of the heart. Metastatic deposits, when removed, are largely similar to the primary tumor, both in terms of cellular histology and the absence of desmoplasia. For example, ACC deposits in the liver are often intimately intertwined with hepatocytes without surrounding stroma. The key histological features of ACC are shown in Figure 6.
Figure 6.
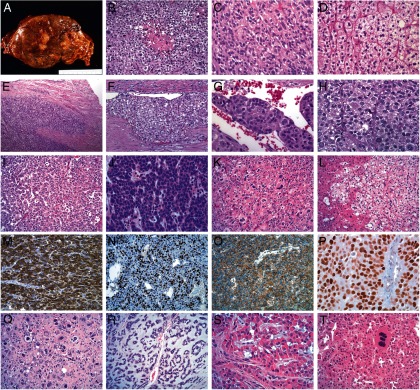
A, ACC gross. Adrenocortical tumors tend to be relatively large masses (>5 cm in largest diameter) that grow by expansion. Their cut surface ranges from brown to orange to yellow depending on the lipid content of their cells. Necrosis is almost always present. B, Typical ACC with a hypercellular population of cells with the earliest form of tumor necrosis. C, A typical ACC with a solid growth pattern and abundant eosinophilic cytoplasm with focal clear areas, consistent with lipid. Mitotic figures are present. D, A lipid-rich ACA with nested growth pattern and clear cytoplasm is shown for comparison with ACC. E, Direct invasion of the tumor capsule, representing the earliest manifestation of malignant behavior. F, Relatively differentiated ACC that has invaded a vessel within the tumor capsule. G, ACC tumor thrombus covered with endothelial cells. H, High-grade ACC with high nuclear grade, diffuse growth pattern, eosinophilic cytoplasm, and 3 visible mitotic figures. I, Low-grade ACC with abundant cytoplasm and low mitotic rate. J, High-grade ACC with minimal cytoplasm, resembling small cell carcinoma. K, Low-grade ACC with isolated nuclear pleomorphism. L, ACC metastatic to liver. Notice the lipid-rich nature of the tumor and the lack of stromal response. M, α-Inhibin immunohistochemistry showing diffuse immunoreactivity in ACC. N, Ki67 immunohistochemistry showing a high labeling index in a high-grade ACC. O, β-Catenin immunohistochemistry showing pure membranous staining in ACC, indicating a wild-type CTNNB1 gene. P, p53 immunohistochemistry showing diffuse immunoreactivity in a high-grade ACC, indicating a likely somatic TP53 mutation. Q, Postchemotherapy effect in ACC, showing large cells with bizarre nuclear forms. R, Myxoid variant of ACC with abundant extracellular myxoid material. S, Rare oncocytic variant of ACC that also has focal myxoid stroma. T, Adrenocortical oncocytoma with isolated multinucleated cells.
Despite the best efforts at the development and application of systematic classification algorithms, there are occasional adrenocortical tumors that defy classification into diagnostic categories. For instance, rare tumors that do not qualify as ACC by Weiss criteria sometimes behave in a malignant manner (for example, see Ref. 123). Conversely, some tumors diagnosed as ACC do not behave as predicted; although this issue is much more difficult to sort out because surgery can be a very effective treatment for early-stage ACCs. In these cases, pathologists have used a variety of diagnostic terms, such as atypical adenoma, adrenocortical neoplasm, and adrenocortical neoplasm of uncertain malignant potential or uncertain biological behavior.
Because of these diagnostically challenging cases, many pathologists have tried to develop ancillary techniques to refine the approach to these tumors. One such histochemical technique employs reticulin staining to highlight disruption of the reticulin network that is observed in ACC (124, 125). This observation is related to the altered growth pattern observed in ACCs and reflects one of the Weiss criteria (diffuse growth pattern greater than 25%). This simple approach is intriguing and awaits further validation.
In addition to histochemical approaches, the literature contains many studies of immunohistochemical methods designed to separate ACA and ACC. Most of these studies focus on tumor cell proliferation (126–129). Using accepted proliferation immunomarkers, such as Ki67, a general consensus has emerged that ACCs have a Ki67 labeling index >5%. Conversely, ACAs generally show a much lower index, although there is some overlap observed depending on the particular study. Although the diagnosis of ACC should not rest on any single immunomarker, proliferation markers generally correlate with mitotic accounts and do have a role to play in the evaluation of these tumors (130).
It is sometimes difficult to be certain that a particular tumor of the retroperitoneum represents ACC, usually due to spread beyond the adrenal gland and/or loss of adrenocortical differentiation. In these instances, a battery of immunostains can provide evidence of adrenocortical differentiation (131), including but not limited to the following proteins that are expressed in most ACCs: α-inhibin (132, 133), calretinin (134), synaptophysin (135), melanA (Mart1) (136), and steroidogenic factor 1 (SF1) (137, 138). In general, ACC does not express the common cytokeratins most often used in practice. Chromogranin A expression is universally not present, and if it is present, an adrenomedullary tumor should be strongly considered. In practice, most adrenocortical tumors are readily apparent on routine hematoxylin and eosin stains and do not require supplemental immunostains to document adrenocortical differentiation.
ACCs can be graded into low- and high-grade carcinoma groups based on their mitotic rates (≤20 mitoses per 50 high-power fields [HPFs] vs >20 mitoses per 50 HPFs), an observation first made by examining the individual components of the Weiss score for prognostic significance (139). Of all the criteria, mitotic rate was most closely associated with patient outcome. This observation has been essentially validated by other clinicopathological studies and extended by gene expression studies that highlighted how dominant proliferation-related genes are in these tumors (130). High-grade ACCs are enriched for mutations of TP53 (14, 140–142) and/or CTNNB1 (143–149), and these mutations tend to be mutually exclusive (reviewed in Ref. 150).
ACCs exhibit a large degree of intratumor heterogeneity, an unsurprising finding given their large size and the evolutionary nature of cancer progression. With thorough sampling, it is becoming more and more common to see tumors consisting of numerous areas and nodules with different histological phenotypes. For example, high-grade ACCs often have minority areas of low-grade ACC. Likewise, some low-grade ACCs contain areas that resemble ACA. Similarly, it is possible to find tumor nodules within a given tumor with different immunohistochemical phenotypes, ie, different Ki67 labeling indices and TP53 (tumor protein 53) and β-catenin immunoreactivities. Taken together, these observations provide support for a clonal model in which ACC can exhibit step-wise progression from low- to high-grade carcinoma. This notion is supported by some recent studies with mouse models (143).
Adrenocortical tumors do occur in the pediatric population (151). For reasons that are not entirely clear, these tumors generally behave in a more indolent fashion compared with adult ACCs (152), leading some to wonder why there are so many pediatric ACCs yet so few pediatric deaths (153). The study by Wieneke et al (154) examined a wide variety of histological features similar to the Weiss score and found that tumor weight >400 g, tumor size >10.5 cm, invasion, extension into extra-adrenal soft tissue, necrosis, severe atypia, >15 mitoses per 20 HPFs, and atypical mitotic figures were associated with malignant clinical behavior. This system was recently validated in an Italian cohort (155). Not surprising, pediatric ACCs display different molecular attributes (156, 157).
Relatively few histological variants of ACC have been described. The most common is called the oncocytic variant because the predominant cell type in this variant is an oncocyte, which is defined as a cell with abundant, granular cytoplasm related to accumulation of mitochondria and endoplasmic reticulum (158–163). Because these tumors, whether benign or malignant, display a solid growth pattern with eosinophilic cells and focal nuclear atypia, traditional Weiss scoring tends to overdiagnose these tumors as oncocytic ACC. For this reason, modified and simpler scoring methods have been devised and work well in most cases (164, 165), although challenging cases are still presented. The other significant ACC variant is called the myxoid variety due to the production of abundant extracellular myxoid substances (166–173). These cases are rare, and the point of their distinction is to recognize them diagnostically. One such myxoid ACC displayed a distinct gene expression profile compared with conventional ACC (174). Finally, sarcomotoid ACCs (carcinosarcomas) have also been described as they have for most other carcinoma types. The development of a sarcomatoid histology, although rare, generally portends aggressive tumor behavior (175).
In practical terms, a standard evaluation of an adrenocortical tumor should include thorough examination of the tumor capsule looking for capsular and vascular invasion and thorough sampling of the tumor to ensure capture of a high-grade component. An immunohistochemical panel of a primary adrenal tumor that is presumed to be adrenocortical could be limited to Ki67, with the possible addition of TP53 and β-catenin. Primary or metastatic tumors of unknown origin would involve a larger panel of the adrenocortical and adrenomedullary markers discussed above in this section as well as other nonadrenal markers (eg, thyroid transcription factor 1 in the setting of a lung nodule). The most common tumors metastasizing to the adrenal gland are lung carcinoma, melanoma, renal cell carcinoma, and breast carcinoma. With the exception of renal cell carcinoma, these tumors generally possess a distinct morphology that will immediately suggest metastatic disease. Bilateral adrenal masses strongly suggest metastatic carcinoma or lymphoma.
Finally, much work is proceeding on how the molecular pathobiology of adrenocortical tumors can be translated into practical tools that will enhance the routine pathological evaluation of these tumors beyond standard histopathology, immunohistopathology, mitotic grading, and tumor staging. Gene expression studies of adrenocortical tumors have led to a refined tumor taxonomy and provide ample opportunities for the discovery of novel ACC biomarkers that should advance the care of these patients in the coming years (150, 176). Looking forward, molecular tools should also facilitate the selection of the most appropriate therapies as they become increasing available.
VII. Molecular Pathology
A. Molecular genetics
Successive and specific genetic alterations within a cell are the principal events underlying carcinogenesis. With the use of classical genetic tools (ie, DNA content assessment, metaphase spreads, and comparative genomic hybridization [CGH]) and the advent of modern, high-resolution analytic methods (ie, tiled arrays and whole-genome sequencing), the genetic dissection of ACC has revealed genomic aberrations that are predicted to contribute to neoplastic transformation of adrenocortical cells.
1. Clonality and DNA content
Most ACAs and all ACCs initiate from monoclonal cell populations, suggesting that mutation events lead to clonal expansion and ultimate progression to cancer (177, 178). Over 30 years ago, cytogenetic and flow cytometry techniques began to be applied to study ACC (179). One of the first genetic assessments with flow cytometry revealed aneuploidy (a genomic aberration consistently observed in most cancers) in 4 of 4 ACCs, yet only diploidy or tetraploidy in normal adrenal cortices and benign adrenal tumors (180). These results were validated in 2 larger studies. In the examination of 22 adrenal neoplasms, aneuploidy was observed in 5 of 6 ACCs, whereas diploidy was observed in all 16 ACAs (181). In a separate study of 39 adrenal tumors, aneuploidy was observed in 75% of ACCs (6 of 8 samples), whereas only 10% of benign lesions (3 of 31 samples) displayed hypotriploid nuclei. The assessment of aneuploidy with histopathological criteria in 7 of 9 adrenal tumors revealed a high correlation with Weiss score >3 (indicative of malignancy) (182). Despite these data, one study revealed aneuploidy in 20% of ACAs (6 of 30 samples), albeit compared with aneuploidy in 69% of ACCs (9 of 13 samples). Moreover, no significant difference in overall survival was observed in patients with ACC exhibiting aneuploidy vs patients with ACC exhibiting diploid neoplasms (126). Although the high prevalence of aneuploidy in ACC suggests chromosomal instability, further investigation to determine aneuploidy and hyperploidy as etiological factors that drive tumorigenesis or as an epiphenomenon is required.
2. Chromosomal aberrations
CGH can identify structural chromosomal abnormalities within ACCs at a higher resolution. A number of studies have found that whereas ACAs have few regions of chromosomal losses and gains, ACCs exhibited complex chromosomal alterations. The first CGH study examined 22 adrenal tumors, 14 ACAs, and 8 ACCs, categorized by histopathological features, size, urinary steroid profile, and clinical data (183). Only 2 of 14 ACAs exhibited a maximum of 2 genetic alterations, whereas 7 of 8 ACCs contained multiple chromosomal gains or losses with a mean of 10 events. In ACCs, chromosomal gains were frequently observed in regions 4q, 4p16, 5p15, 5q12–13, 5q32-qter, 9q34, 12q13, 12q24, and 19p, and chromosomal losses were observed at 1p, 2q, 11q 17p, 22p, and 22q. Microsatellite studies identified frequent allelic losses in regions 17p13, 11q15, and 2p16 (85%, 92%, and 90% of samples, respectively) (184, 185). A follow-up study examining 35 adrenal tumors and 6 adrenocortical hyperplasias identified unique events within 12 of 12 ACCs compared with 15 of 23 ACAs. Specific events in ACC were gains at 5q12–13, 5q22-ter, 9q32-qter, 12q13–14, 12q24, and 20q and losses at 1p21–31, 3p, 2q, 3q, 6q, 9p, and 11q14-qter. Events in ACA consisted of gains at 17q11.2–21 and 17q24–25, 17p, and 9q32 (186). A confirmatory study of 25 adrenocortical tumor samples, including 14 ACCs and 8 ACAs as well as NCI-H295 and SW13 cell lines revealed similar gains in chromosomes 5 and 12 with additional gains in chromosomes 7 and 16 in ACC (187). Moreover, this study identified multiple loci of high-level, multiple amplifications specifically at 19p13.3 and 19q13.4 and revealed a positive correlation between the number of aberrations and the size of tumors.
Most recently, a study using higher-resolution CGH arrays revisited this phenomenon through examination of 138 adrenal neoplasms encompassing 86 ACAs and 52 ACCs to assess the diagnostic and prognostic value of chromosomal abnormalities (188). The study confirmed increased alterations in ACCs (44%) compared with ACAs (10%). In ACCs, the frequently observed chromosomal gains at 5, 7, 12, 16, 19, and 20 and losses at 13 and 22 were confirmed. The group identified genes within these regions with potential tumorigenic potential including fibroblast growth factor 4 (FGF4), cyclin-dependent kinase 4 (CDK4), and cyclin E1 (CCNE1). Moreover, in an independent cohort, the study confirmed the diagnostic utility of 6 loci (5q, 7p, 11p, 13q, 16q, and 22q) in the differentiation of ACA and ACC (sensitivity, 100%; specificity, 83%) (188). Although survival prediction using these data could not be established, a separate CGH study that identified a similar increase in copy number in chromosomes 5, 6q, 7, 8q, 12, 16q, and 20 and allelic losses in 1, 2q, 3, 6p, 7p, 8p, 9, 10, 11, 13q, 14q, 15q, 16, 17, 19q, and 22q determined that some of these alterations (gains in 6q, 7q, and 12q and losses in chromosomes 3, 8 10p, 16q, 17q, and 19q) were associated with decreased overall survival (189).
Although these studies together indicate genetic diversity and heterogeneity of chromosomal gains and losses in ACC, genomic aberration at chromosomes 5, 12, and 17 are predicted to harbor genes that initiate or maintain neoplastic transformation. Chromosome 17, specifically at 17p13, contains the well-known tumor suppressor gene TP53.
3. Epigenetic changes
DNA methylation involves the addition of a methyl group to the cytosine pyrimidine ring or adenine purine ring, occurring typically at CpG dinucleotides. In a normal cell, it acts as a regulatory mechanism for proper gene expression. However, in cancer, frequent dysregulation in this process is observed. A recent study of 51 ACCs and 84 ACAs revealed hypermethylation of promoters in ACCs with correlation to poor survival and identified H19, PLAGL1, G0S2, and NDRG2 as silenced genes (188). This observational study also provided insight into the possible role of methylation in ACC tumorigenesis, particularly in the 11p15 locus containing IGF2 and H19.
4. Gene expression arrays
Global gene expression studies aim to identify biomarkers that could provide diagnostic and prognostic utility in addition to the classic histological analyses and hold the promise of new potential targets for therapy. ACAs and ACCs have distinct expression profiles (174, 190–192). An initial study identified elevated expression of genes involved in cell proliferation in ACCs, such as IGF2, compared with increased expression of steroidogenic genes in ACAs (steroidogenic cluster) (190). Giordano et al (192) identified unique transcriptionally activated (12q and 5q) and repressed (11q, 1p, and 17p) chromosomal regions in 33 ACCs vs 22 ACAs in a microarray study, which confirmed the early chromosomal studies. More recently, 2 large studies have correlated expression profiles in ACC with clinical outcome. Specifically, Giordano et al (192) determined that ACCs with high histological grade exhibited marked overexpression of cell cycle and functional aneuploidy genes, which correlated with decreased overall survival. In another study, cluster analysis of ACCs again revealed 2 distinct groups with different genetic signatures and concomitant distinct clinical outcomes. ACCs with poor outcome were enriched for genes involved in cell cycle and proliferation, whereas ACCs in the better outcome group exhibited overexpression of genes involved in differentiation, metabolism, and intracellular transport. Expression levels of BUB1B and PINK1 alone identified subgroups of ACCs with different overall survival, regardless of tumor stage. Similarly, the expression levels of DLG7 and PINK1 identified subgroups of ACCs with distinct disease-free survival, regardless of tumor grade (191). These findings were later validated in a separate cohort of adult patients (193).
5. MicroRNAs
MicroRNAs (miRNAs) are evolutionarily conserved, small, noncoding, 18- to 25-nucleotide RNAs that are important in posttranscriptional regulation of gene expression. Mature miRNAs in association with the RNA-induced silencing complex are loaded onto the 3′-untranslated region of the targeted mRNA to inhibit translation or to cause degradation (194). Numerous miRNAs have been identified and implicated in the regulation of various cellular processes such as proliferation, apoptosis, and differentiation. In addition, dysregulation of miRNAs, such as overexpression or deletion, plays an important role in diseases, including various cancers (195, 196). Mistargeting of the miRNAs, resulting in inhibition or activation of various oncogenes, tumor suppressors, and/or other factors important in tumor angiogenesis, epithelial-mesenchymal transition, and metastasis, have been identified (196). The examination of 36 adrenocortical samples (10 normal tissues, 10 nonfunctional ACAs, 9 cortisol-secreting adenomas, and 7 ACCs) revealed differential expression of 22 miRNAs, with 14 miRNAs preferentially expressed in ACCs. Upregulated miRNAs in ACCs included miR-184, miR-210, and miR-503. Downregulated miRNAs included miR-214, miR-375, and miR-511 (197). Levels of miR-184, miR-503, and miR-511 alone were able to distinguish benign from malignant adrenal tumors (specificity, 80%–97%; sensitivity, 100%) (197). A recent study of 55 adrenal samples (6 normal tissues, 22 ACAs, and 27 ACCs) similarly determined an miRNA expression signature unique to ACC (198). The investigation identified 14 upregulated miRNAs and 9 downregulated miRNAs unique to ACC. In addition to validating the upregulation of miR-503 in ACC, the study identified a significant upregulation of miR-483 (diagnostic sensitivity of 80% and specificity of 100%) and downregulation of miR-195 and miR-335 in ACC (198). Lastly, miRNA expression in 25 pediatric adrenal neoplasms (18 ACCs, 6 ACAs, and 1 unknown) was compared with 5 normal adrenals (199). Unsupervised clustering of the samples according to miRNA expression resulted in clear differentiation of the tumors from the normal controls. Further differentiation between ACA and ACC could not be achieved. In this study, similar to the adult ACC study, miR-483 was found to be significantly upregulated in pediatric ACCs. However, a majority of the differentially expressed miRNAs were downregulated in ACCs, most notably miR-99a and miR-100. MiR-99a and miR-100 are bioinformatically predicted to target the 3′-untranslated regions of IGF1R, RPTOR, and FRAP1 (mTOR) and were experimentally confirmed to target several components of the IGF-1 signaling pathway (199). Moreover, miR-483 is located in an intron of IGF2. It is hypothesized that dysregulation of the IGF2 locus perturbs the expression of miR-483 (198, 200). In the hepatocarcinoma cell line HepG2, observational studies revealed the oncogenic potential of miR-483 through inhibition of apoptotic regulatory genes PUMA/BBC3 (201).
6. Gene mutations
Targeted genetic analyses, such as sequencing and single-strand confirmation analyses have identified somatic genetic changes in TP53, MEN1, IGF2, IGF2R, and p16/INK4A (CDKN2A). TP53 located on 17p13 is the most commonly mutated gene in ACC, present in at least one-third of ACCs (140, 142, 202). Examination of TP53 in 89 adrenal tumors revealed loss of heterozygosity (LOH) at the 17p13 in 11 of 13 ACCs and 23 of 76 ACAs (203). Of note, there was no overlap of LOH and mutations in the same tumors. The reason for ACC not following the canonical LOH model remains unclear. LOH in the gene encoding p16ink/p14arf, CDKN2A is observed in a subset of ACCs. The tumor suppressor function of this gene has been established in multiple cancers (204). A small-scale study revealed 3 of 7 ACCs with LOH at this locus (205). MEN1 (located on 11q13) somatic mutations are unusual in sporadic ACC. This is in contrast with LOH of 11q13, which has been identified in ∼83% of samples (185). It is unclear whether this region harbors an additional unrecognized tumor suppressor gene involved in adrenocortical tumorigenesis. Furthermore, as detailed below in Section VII.B.2., in addition to aberrant activation of critical signaling pathways such as the IGF and wingless-type (WNT) pathways, mutational analysis of the effector of the canonical Wnt pathway, the β-catenin gene, CTNNB1, has identified activating point mutations in over 25% of both ACAs and ACCs in children and adults (149, 206–208).
B. Pathophysiology of cellular signaling pathways
1. IGF pathway
The IGF signaling pathway consists of ligands (IGF-1 and IGF-2), receptors (IGF-1 receptor [IGF-1R], IGF-2R, and insulin receptor), IGF binding proteins 1–6, and IGF binding protein proteases. The binding of the mitogenic polypeptides to their receptors activates the downstream AKT/PI3K and MAPK pathways to regulate cellular processes of metabolism, differentiation, proliferation, and apoptosis. The IGF pathway mediates ACTH-induced prenatal adrenal growth, fetal and adult steroidogenesis, and organ maintenance (209–212). In the developing fetal organ, IGF1 expression is restricted to the capsule, whereas IGF2 expression is enriched in the cortex (213). In the adult adrenal cortex, both IGF-1 and IGF-2 stimulate basal and ACTH-induced steroidogenesis (210, 214). Overall, the main role of IGF-2 lies in fetal development and growth, whereas IGF-1 acts mainly postnatally. Prominent overexpression of IGF2 and alterations of the IGF2/H19 locus have been identified in sporadic ACC (174, 190, 215). The IGF2 gene is located on 11p15, which also includes a noncoding H19 gene and a cyclin-dependent kinase inhibitor, CDKN1C (p57KIP2) (216, 217), and 80% to 90% of all ACCs show very high IGF2 expression (∼100-fold over normal and ACA) (174, 218–220). Interestingly, relative expression of Igf2 is much higher than in tissues from mice resembling human BWS, in which genetic changes result in an ∼2-fold upregulation. High IGF2 expression levels in adrenal tumors, when analyzing malignant and benign tumors, are associated with a 5-fold increased risk for recurrence and a shorter disease-free survival (184, 191). Pediatric ACCs reveal an ∼20-fold overexpression of IGF2. Various cell culture studies using ACC cell lines suggest a paracrine or autocrine effect of IGF-2 and mitogenic activity through IGF-1R (156, 221–223). PEPCK-IGF2 transgenic mice that overexpress IGF2 have adrenocortical hyperplasia and enhanced steroidogenesis (224). Similar phenotypes are observed in indirect IGF2 overexpression in PEPCK-GH transgenic mice that overexpress GH (225). However, simple overexpression of IGF2 was insufficient to initiate adrenocortical tumorigenesis.
Perturbation of the IGF2 locus, with upregulation of maternally imprinted genes (IGF2), and downregulation of paternally imprinted genes (H19 and CDKN1C), is frequently observed in ACCs (226). However, 11p15 LOH has been shown to be a stronger predictor for shorter disease-free survival than simple levels of IGF2 overexpression (184). Based on this observation, it is hypothesized that additional genetic changes, such as loss of maternally expressed CDKN1C and H19, may contribute to adrenal tumorigenesis (184).
The findings of high IGF2 expression levels and the knowledge of an increased incidence of ACC in BWS led to the investigation of IGF-1R as a therapeutic target. In an NCI-H295 xenograft mouse model, IGF pathway inhibition by the small-molecule inhibitor NVP-AEW541 and the monoclonal IGF-1R antibody IMCA12 showed an antitumor effect. Furthermore, the combined treatment of NCI-H295 cells with IGF-1R antagonists and mitotane resulted in a synergistic antiproliferative effect in vitro and in vivo in tumor xenografts (223, 227).
2. WNT signaling pathway
The WNT/β-catenin signaling pathway is a major developmental pathway in multiple organ systems, including the adrenal gland. The pathway is differentiated into 3 diverging signaling cascades dependent on signal conduction through β-catenin (canonical pathway), ras homolog gene family small GTPase (planar cell polarity pathway), or phospholipase C (Wnt/calcium pathway). β-Catenin is normally sequestered in a destruction complex with adenomatous polyposis coli (APC), glycogen synthase kinase 3, and axin. In the canonical pathway, binding of the WNT ligand to its respective frizzled receptors results in release of β-catenin from the complex and translocation to the nucleus where it serves as a transcriptional cofactor with T-cell factor/lymphoid enhancer factor.
In the normal adrenal gland, the WNT/β-catenin signaling pathway plays a crucial role in both embryonic development and maintenance of the adrenal cortex (228). Temporal and spatial expression of β-catenin is limited to a subset of developing fetal adrenocortical cells and to the subcapsular cells of the adult cortex (228). Conditional knockout of β-catenin in a transgenic mouse resulted in the absence of the adrenal gland at embryonic day 18.5. In mice harboring incomplete β-catenin knockout (knockout of β-catenin in a subset of adrenocortical cells), although normal adrenal development occurred, at age 45 weeks postpartum, these mice exhibited thinned and disorganized adrenal cortex in the setting of increased apoptosis (228).
Initial alterations of the WNT/β-catenin system/pathway were identified in FAP (229, 230). The molecular bases of FAP are inactivating mutations in the tumor suppressor gene APC, resulting in constitutive activation of β-catenin with subsequent increased target gene expression (229, 230). Recent examinations of adrenocortical tumors suggest that the WNT/β-catenin signaling pathway plays an important role in sporadic adrenocortical tumorigenesis. Immunohistochemical analysis of 39 adrenal tumors revealed accumulation of β-catenin in 10 of 26 ACAs and in 11 of 13 ACCs, consistent with stabilized and hence activated β-catenin (149). Furthermore, mutational analysis of the β-catenin gene CTNNB1 identified activating point mutations in both ACAs and ACCs (149, 206–208). An activating Ser45 β-catenin mutation as well as activated β-catenin signaling was also identified in the NCI-H295 ACC cell line (149). Moreover, gene expression profiling studies revealed overexpression of β-catenin target genes such as ENC1, suggesting a role of active β-catenin signaling in ACCs (174). Moreover, inactivating mutations of AXIN2 (a component of the β-catenin destruction complex) have also been described in some adrenocortical tumors (231). Furthermore, activation of β-catenin as well as TP53 inactivation predicted poor outcome in one study of 51 samples (232).
The fact that both nuclear β-catenin accumulation and activating CTNNB1 mutations are present in ACAs as well as in ACCs suggests that WNT activation may be an early step in adrenocortical tumorigenesis, which precedes malignant transformation. A recent study on mouse models corroborates this hypothesis (143). Mice with constitutive activation of the Wnt signaling pathway obtained by adrenal-specific Apc knockout develop adrenal hyperplasia and adenomas by 30 weeks of life. On the other hand, no adrenal phenotype is observed in the adrenal-specific Igf2 overexpression mouse model. However, when the Apc-knockout mice were crossed with adrenal-specific Igf2 overexpression mice, early-onset adrenal nodular hyperplasia evolving to large tumors later in life (including an invasive cortical tumor similar to an ACC) was observed, suggesting that both pathways may have synergistic effects on adrenocortical tumorigenesis (143). This study was further validated in a similar model of adrenocortical-specific β-catenin stability and Igf2 overexpression (233).
3. Vascular endothelial growth factor
Sustained angiogenesis is a sine qua non feature of cancer. Anomalous blood vessels are a characteristic of virtually all types of cancer (234). The vascular endothelial growth factor (VEGF) is a chief regulator of cancer angiogenesis. Its effects are mediated through its receptors (VEGFRs) (235). The pharmacological inhibition of VEGFRs are considered an attractive option for cancer treatment (236). Elevated VEGF levels were identified in blood samples from ACC patients (237, 238). In addition, overexpression of VEGFR type 2 in ACC samples was observed by immunohistochemistry (239). The increased expression of VEGF correlates with the expression of IGF2 (192). Recently, several groups used targeted therapeutic methods of VEGF signaling inhibition in xenograft mouse models with relative success. Mariniello et al (240) reported marked growth inhibition using sorafenib and everolimus, for VEGFR1–2 and mammalian target of rapamycin inhibition, respectively, suggesting potential antiangiogenic and antitumor effects. However, an earlier clinical trial using bevacizumab, an anti-VEGF monoclonal antibody, proved to be ineffective (239).
VIII. Prognostic Factors
Despite the generally unfavorable prognosis of ACC, there is a marked individual variation in disease progression, recurrence, and overall survival. Even in patients with stage 4 disease, survival ranges from a few months to several years. Exceptional cases of long-term survival with the diagnosis of ACC have been reported (242). In the Michigan Endocrine Oncology Repository, roughly 5% of all patients diagnosed with ACC will have a disease course of >10 years (T.E., unpublished results). Although this may be caused by a referral bias, there is an emerging notion of an ACC population with exceptionally long survival.
Despite these variations in survival, prognostic factors have not been definitively researched. Naturally, age at diagnosis is correlated with decreased overall survival (243). However, whether this is true for tumor-free survival remains unclear. Tumor characteristics of malignancy and velocity of tumor growth are usually related to a decreased survival. Tumor extent (eg, stage), specifically the presence of distant metastasis and number of organs involved in metastatic disease, confers a worse prognosis (243, 244). High tumor grade (>20 mitoses per HPF) is also an unfavorable prognostic indicator (245). Although older studies did not show any differences in prognoses for patients harboring different hormone secretion subtypes of ACC, some recent studies identified cortisol production as an adverse prognostic factor (12, 85, 244).
IX. Therapy
Currently, the only curative approach to ACC is complete tumor resection. Adjuvant therapies aim to decrease the chance of recurrence. All therapy of unresectable or metastatic ACC must be considered palliative, a fact that needs to be discussed with the patient so that reasonable expectations are set. Although this review does not provide a detailed focus on palliative care, general principles of palliative care need to be considered at any point during the disease course. This includes improvement and sustainability of quality of life (QOL) through necessary interventions (eg, adequate control of hormonal symptoms, pain control, and prevention of fractures caused by bony metastasis) as well as minimizing side effects from antineoplastic therapies.
A. Surgical therapy
Appropriate preoperative evaluation and operative planning by a surgeon experienced in the resection of malignant adrenal tumors is of the utmost importance to assure optimal outcome. Consideration of surgical anatomy, the potential complications of surgical intervention, expected outcomes including the tempo of recovery and the various options for intervention, are all important with regard to the beneficial application of surgical management strategies for patients with ACC. However, in the United States, 45% of adrenalectomies for ACC are performed in community hospitals, 30% in academic centers, and only 15% in National Cancer Institute-designated Cancer Centers, suboptimal treatment of a rare and aggressive disease requiring specialized knowledge of surgical technique (11).
Operative planning hinges on the presumed preoperative diagnosis. Surgery should be conducted only after appropriate preoperative diagnostic tests, including biochemical evaluation and imaging. In the setting of adrenal imaging characteristics not clearly excluding malignancy, surgeons are obligated to approach the resection as a cancer operation. Failure to do so often leads to dismal outcomes, because an oncological resection for ACC is quite different from one performed for a benign adrenal mass.
Preoperative imaging should be obtained to evaluate the extent of tumor, possible invasion of surrounding anatomic structures, and technical ability of the tumor to be completely resected. Imaging studies also help to guide the surgeon as to the expected extent of resection required. Careful attention should be paid to adjacent organs, the adrenal and renal veins, the inferior vena cava, and the aorta, including the takeoff of the celiac and superior mesenteric arteries. Despite preoperative diagnostics, approximately 25% of stage 3 cases are initially suspected to be stage 2 ACC but ultimately found to have microscopic extension through the adrenal capsule. These cases go unrecognized in the preoperative and intraoperative settings and hence highlight the importance of careful surgical technique including resection of all surrounding soft tissue and adjacent organs if necessary (246). Imaging should be obtained as close as possible to the anticipated date of surgery, because many aggressive ACCs grow quickly and involvement of adjacent structures may change, thereby altering the operative plan. Intravascular ultrasound or venography may complement other imaging studies to estimate extent of tumor involvement. Other preoperative considerations include management and optimization of those patients with hormone excess, especially those with Cushing's syndrome due to the numerous deleterious effects of elevated cortisol (poor wound healing, infection, and metabolic derangements). Aggressive control of cortisol excess should be attempted in the short period between identification of the tumor and surgery, but surgery should not be unnecessarily delayed solely to tightly control hypercortisolism.
Although surgery is the treatment of choice for nonmetastastatic ACC, the decision for resection of the primary tumor in stage 4 disease needs to be individually addressed. In general, those with widespread distant metastatic disease in multiple organs or those with multiple metastatic deposits in one organ system unable to be completely resected should not undergo adrenalectomy. The primary tumor can instead be treated with external beam radiation for palliation along with other adjuncts to improve local symptoms and better control hormone excess, if present (247). Some groups attempt to assess the tempo of disease progression, waiting for several months to restage the patient by imaging and may treat with chemotherapy and/or mitotane in the interval. If tumor burden remains stable or decreases, then surgical treatment is pursued and vice versa. Adrenalectomy in the setting of tumor thrombus within the vena cava (if the tumor is otherwise technically resectable) is reasonable. Obstruction or occlusion of the vena cava by tumor thrombus can lead to significant lower body and gastrointestinal (GI) tract edema, which leads to significant patient suffering. Lack of resection in the setting of vena cava thrombus can quickly lead to death. If tumor resection is not technically feasible for other reasons, vena cava stents can be placed, leading to temporary prevention of occlusion.
Debulking for control of hormone excess in the setting of known metastatic disease is also performed in some situations. The long-term durability of hormone control is usually limited as the metastatic disease progresses. The benefits of debulking must outweigh the risks of surgery in these patients who have poor wound healing and lengthy recovery periods due to preexisting debilitation. Postoperative QOL should be carefully considered in this setting with respect to estimated length of survival.
1. Surgical approach
The first operation is the best chance for long-term local control of malignancy. Poor initial surgical treatment can rarely be corrected, whether by reoperation, radiotherapy, or chemotherapy. Lack of attention to oncological principles when resecting ACC may explain why no differences are observed in some series comparing laparoscopic and open resections. Based on the University of Michigan experience, an operation likely to be most effective for treatment of ACC includes the steps outlined in Table 5.
Table 5.
Surgery
| Surgical Steps | |
|---|---|
| 1. Incision and exploration of the peritoneal cavity. | Make an ample incision. Subcostal incisions allow for better access than midline incisions. Thoracoabdominal incisions are indicated in select situations. |
| Perform a complete and systematic evaluation of the peritoneal cavity. | |
| 2. Evaluation of liver for metastasis | Perform intraoperative ultrasound for metastatic deposits not evident on preoperative imaging. |
| 3. Containment | A self-retaining retractor system with towels or laparotomy pads should be placed in such a way as to exclude the rest of the peritoneal cavity from the area of the tumor and other organs requiring resection. |
| 4. Mobilization of organs adjacent to tumor | Fully mobilize adjacent organs overlying the tumor (if not adherent to the tumor). |
| 5. En bloc resection. | Preserve any tissue overlying the tumor. Use a no-direct-touch technique. Extreme caution should be used mobilizing the soft tissue and tumor together as the capsule is easily ruptured or tumor cells are abraded from the surface. The tumor should not be shelled out, but instead removed with the entire retroperitoneal fat pad. Adjacent organs should be included with the en bloc resection if adherent to the tumor rather than creating a plane between the tumor and organ. |
| 6. Regional lymphadenectomy | Perform when feasible for staging purposes and clearance of in-transit tumor. Few lymph nodes directly surround the adrenal. Representative draining lymph node basins can be found near the renal hilum, celiac axis, and superior mesenteric arteries. |
| 7. Provide intact en bloc specimen for pathologic review | Mark the specimen with sutures to correctly orient the pathologist. Communicate any areas of particular concern or clarify potential misconceptions regarding the resection. Do not morcellate the specimen to facilitate removal through a smaller incision as this makes portions of accurate pathological review impossible. |
| 8. Mark field to facilitate postsurgical external beam radiation therapy | Clips should be placed around the periphery of the field of resection to facilitate planning for possible external beam radiation therapy. |
| 9. Dictate operative report | Include specific details of the operation, findings, areas of metastatic disease, any areas of tumor left behind (known or possible), and acts of commission or omission to facilitate communication with the multidisciplinary team. |
2. Lymph node dissection
The role of lymph node sampling or formal regional lymph node dissection in the treatment of ACC remains unknown, and consensus within the field is needed (248). There is also no formal agreement on the extent of lymph node dissection. In general, the lymphadenectomy is performed based on following the arterial supply. In the case of adrenal tumors, the main lymphatic areas are the renal hilum and the origin of the celiac and mesenteric artery. Because lymph nodes ideally should be removed as part of the en bloc resection, surgeons need to individually balance the increased risk due to extended surgery (eg, bleeding) with the presumed benefit of radical lymph node dissection. The impact of regional lymph node metastasis upon overall survival provides impetus for earlier or more aggressive use of additional therapies when disease is present in the lymphatic system (249). In one retrospective study, locoregional lymph node dissection improved tumor staging ability and led to a more favorable oncological outcome in patients with otherwise localized ACC. Some of the improved outcome can be attributed to the upstaging of ACC patients with lymph node metastasis and subsequent more aggressive treatment. Similarly, more radical surgery in these patients can lead to increased clearance of disease as opposed to a higher rate of positive margins. Further validation in independent ACC cohorts is warranted.
3. Open vs laparoscopic surgery
Controversy surrounds the appropriateness of laparoscopic adrenalectomy (LA) for patients with ACC. LA has become the gold standard for resection of benign adrenal masses, and it has been shown to result in significantly lower morbidity, less pain, shorter hospital stays, and decreased overall time to recovery when compared with open adrenalectomy (OA). Because effective adjuncts to surgery for the treatment of ACC are extremely limited, ensuring a complete, margin-negative tumor resection at the initial operation is critical.
ACCs can invade through the tumor capsule and are frequently microscopically present at the surface of the gland. Application of laparoscopic instruments to the tumor can result in shedding of malignant cells that is undetectable to the operating surgeon. Laparoscopy does not allow for optimal exploration of the peritoneal cavity, and tactile sensation is limited compared with an open approach. It is nonexistent during robotically performed procedures (250). Minimizing direct contact with the tumor surface is important so as not to abrade cells from the tumor surface or enter the tumor capsule. Tumors should not be morcellated before removal for reasons related to later pathological review, and often a larger incision needs to be made, defeating the purpose of small laparoscopic incisions.
Some surgeons compromise by initiating adrenalectomies laparoscopically to assess for evidence of intraperitoneal metastasis or invasion of the adrenal gland into other organs (246, 251). However, this direct exploration of the tumor violates oncological principles of resection. Frank penetration of the tumor capsule may occur early on as may microscopic abrasion, leading to spread of tumor cells.
A recurring argument is that in expert hands, LA may be appropriate for certain malignant adrenal tumors. However, there is no consensus definition of what constitutes adequate expertise. Thirty to 40 LAs are generally sufficient to reach some level of proficiency with the technique (252). This does not translate to expertise for biologically aggressive, often invasive, larger adrenal cancers. Unfortunately, most ACCs are removed by low-volume and less experienced adrenal surgeons (253).
Published data comparing the efficacy of LA vs OA for ACC are limited. All large series are retrospective, include fewer than 200 patients (with most reports including fewer than 10 patients), provide limited or no follow-up, are hampered by referral bias, and include patients who did not undergo their initial surgical resection at the referral center. The initial recommendations published after the first International Adrenal Cancer Symposium held in Ann Arbor, Michigan, in 2003 stated there was no role for laparoscopic removal of a known or likely ACC, but controversy existed regarding the role of laparoscopic removal of indeterminate lesions (15). Recent recommendations by the American Association of Clinical Endocrinologists and the American Association of Endocrine Surgeons advocate OA by an experienced surgeon as the procedure of choice (254). Conversely, the European Society of Endocrine Surgeons and European Society for Medical Oncology suggest LA could be performed for stage 1 and 2 ACC tumors less than 8 or 10 cm if an R0 resection is performed and surrounding periadrenal tissue removed (255, 256). Neither guideline addresses differentiating stage 1 and 2 ACCs from microscopic or unappreciated stage 3 ACC preoperatively or how to ensure an R0 resection at the conclusion of an operation, because no surgeon begins an operation intending to perform an R1 or R2 resection.
At least 7 studies have been published since 2010 that specifically address the topic of LA vs OA for ACC. Two studies published by the M.D. Anderson Cancer Center reported a recurrence rate of 86% in the OA group (154 patients) and 100% in the LA group (6 patients) (251, 257). Local recurrence and peritoneal carcinomatosis was more common in the LA group. In a study by Leboulleux et al (258), peritoneal carcinomatosis occurred in only 25% of patients treated by OA, as opposed to 60% of patients who underwent LA.
In contrast, other studies reported evidence that LA may be comparable to OA in patients with stage 1 and 2 ACC based on no significant difference in recurrence-free survival (259, 260). However, patients who had macroscopically incomplete resection, tumor capsule violation, and conversion from laparoscopic to open surgery and those found to have stage 3 tumors on final pathology were excluded from the study. This limits analysis to patients with true stage 1 or stage 2 disease, which cannot be determined definitively before surgery.
A case-control study from the German ACC Registry Group reported no difference in overall or disease-free survival, tumor capsule violation, or peritoneal carcinomatosis among 117 patients undergoing OA and 35 patients undergoing LA for stage 1 to 3 ACCs less than 10 cm (261). However, 3 times as many patients in the OA group had stage 3 disease and only 4 patients (11%) undergoing LA were found to have stage 3 disease, potentially introducing a bias toward more advanced disease in the OA group, and 37% of all patients had no data regarding margin status.
Surgical studies should focus on local and peritoneal recurrence as indicators of quality of surgical resection, because type of operative approach likely has a much smaller role in the development of distant metastases. A retrospective study from the University of Michigan reviewed 88 ACC patients, 17 of whom underwent LA, and 79% of the operations were performed at outside facilities, and no laparoscopic operations for ACC were performed at the University of Michigan, potentially introducing a referral bias (262). Although overall recurrence rates were similar and despite on average smaller tumors in the LA group (7.0 cm) compared with the OA group (12.3 cm), the LA group had a significantly earlier recurrence (9.2 vs 19.2 months). Furthermore, there were more R1 or R2 resections or notation of intraoperative tumor spill (50% vs 18%). These data suggest that although LA may be technically feasible (even for large tumors) (263, 264), the use of LA in ACC leads to a shorter disease-free interval and a higher incidence of incomplete resections. These results were confirmed in an extended follow-up study of 110 patients undergoing OA and 46 undergoing LA. After LA, 30% had positive margins or intraoperative tumor spill compared with 16% of OA patients despite larger tumors and more stage 3 tumors. Overall survival for patients with stage 2 ACC was longer in those undergoing OA, and time to visible tumor bed recurrence or peritoneal recurrence in stage 2 patients was shorter in LA patients.
In summary, existing data are inconclusive and more studies are needed to better judge the equivalence of LA to OA. In accordance with the experience gained at the authors' institution, a conservative approach using an open approach is recommended for all adrenocortical lesions that cannot be classified as benign before surgery.
4. Surgery for recurrent disease
Extent of disease and tempo of disease progression guide the decision for reoperation in the setting of recurrence. The number of organs involved by tumor at the time of the first metastasis is a predictor of survival (243, 265). In addition, University of Michigan data show the site of first metastasis can also be used to predict survival, with those having tumor recurrence in the peritoneum outside the tumor bed having the worst survival. Surgery is indicated in those patients with disease confined to 1 site or organ. Beyond that, decisions regarding resection must be individualized. The type of initial operative resection is important to the decision-making process for reoperation. Patients with tumor bed recurrence who have undergone LA are much more likely to have disease too small to be detected by imaging elsewhere in the peritoneal cavity compared with those having undergone OA based on our experience.
A 1999 study reported median survival of 74 months (5-year survival, 57%) in those undergoing complete second resections vs a median survival of 16 months (5-year survival, 0%) in those undergoing incomplete second resection. Although neither tumor grade nor additional nonsurgical treatment received was discussed in this study (266), data from other studies note their influence on outcome, although subsequent recurrence is expected (267, 268).
Tumor grade influences the decision for reoperation because it correlates with survival (243, 245). In those with low-grade tumors, tempo of disease progression can be slower and lead to longer survival with resection of sites of recurrence or metastasis. In contrast, those patients with high-grade tumors benefit less from reresection, because other sites of disease often appear quickly. It is not uncommon for the authors to wait 3 months while treating with chemotherapy to assess for tumor responsiveness and/or tempo of progression. If progression is not rapid, surgery may proceed with greater benefit, whereas those with evidence of marked progression of disease do not undergo surgery.
B. Adjuvant therapy
The outcomes after surgical resection alone have remained suboptimal (11, 244, 267, 269–273). The evidence that patients with ACC remain at high risk for tumor recurrence despite complete surgical tumor excision has fueled the search for adjuvant therapies. Even with ostensibly complete resections, rates of local recurrence have typically ranged from at least 19% to 34% in those patients with no residual disease after surgery (244, 267, 270). High local recurrence rates after seemingly complete resections underscore the difficulty of achieving adequate margins during surgery. This fact is well illustrated by an analysis of almost 4000 patients from the National Cancer Data Base, which found that 9% of ACC patients treated with surgery had microscopically positive margins (R1), whereas a further 10% had macroscopically positive margins (R2) (11). Not surprisingly, patients with positive margins had a dismal prognosis compared with those with uninvolved margins; 5-year survival was 10% after R2 resection, 21% after R1, and 49% after R0 (11). These results closely correspond with the surgical outcomes from a variety of other solid malignancies, in which a survival benefit is often seen in patients after a complete resection with negative margins but not in those with positive margins (19, 91, 244, 267, 273–275).
For patients with an increased risk of local recurrence, eg, R1 resection with microscopic rests, radiation therapy to the tumor bed had long been employed. The adjuvant use of mitotane had also been practiced at several centers for decades. However, until recently, conclusive data regarding the efficacy of both of these adjuvant treatment modalities had been missing, and adjuvant therapy remains a controversy. Adjuvant treatment modalities are discussed in the respective treatment chapters below.
C. Medical therapy
1. Mitotane
The adrenolytic activity of derivates of the insecticide dichlorodiphenyltrichloroethane was first described in dogs in 1948 (276). Early trials in human patients with hypercortisolism failed because the original insecticide DDD (dichlorodiphenyldichloroethane) (Rothane) was a mixture of several isomers (277, 278). In 1960 Bergenstal et al reported responses to therapy with the isolated 1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane isomer (o,p′DDD, mitotane) that harbors the adrenolytic activity (278). Since then, further modifications and isolations of enantiomers have aimed to improve the adrenolytic activity, improve pharmacokinetics and reduce side effects, unfortunately with only marginal improvement (279, 280). However, some compounds, such as the dichlorodiphenyltrichloroethane-metabolite, 3-methylsulphonyl-DDE (dichlorodiphenyldichloroethylene) have extended the adrenolytic activity to other species, such as rodents, but been of limited value in human cell line xenografts (281, 282).
Mitotane remains the only drug approved by the U.S. Food and Drug Administration and European Medicine Executive Agency for treatment of ACC (15). The pharmacological mechanism by which mitotane exerts its adrenolytic effect is still not completely understood. Mitotane leads with relative specificity to a destruction of the inner zones of the adrenal cortex, the zona fasciculata, and zona reticularis. In dog adrenal glands, mitotane leads to cell death, most likely via necrosis, and is followed by the emergence of a dense inflammatory infiltrate (283). In ex vivo adrenal perfusion experiments, it was shown that mitotane can be extracted in the adrenal gland and further metabolized (284). Active metabolites produced by adrenal mitochondria, in turn, covalently bind to mitochondrial proteins hypothesized to inhibit mitochondrial respiration (285). Furthermore, mitotane metabolites inhibit several enzymes in the adrenocortical steroidogenesis pathway, mainly at the level of the cholesterol side-chain cleavage enzymes CYP11A1 (which appears to be one of the covalently bound mitotane targets) and CYP11B1 (286, 287).
Roughly 40% of mitotane is absorbed from the GI tract, and a significant amount is distributed to fatty tissues. After a usual daily dose of 5 to 15 g/d, plasma levels range between 0 and 90 mg/L. Doses greater than 20 g regularly result in neurological side effects, which are reversible with normalization of plasma levels (288).
Several studies have evaluated the efficacy of mitotane as an adjuvant therapy or for advanced ACC as a single treatment or in combination with chemotherapy (Tables 6 and 7). However, all studies are retrospective, and older studies lack the advantage of cross-sectional imaging. Therefore, all disadvantages of retrospective studies need to be taken into account when interpreting the large variation in responses. In addition, most studies were single-center studies, often including only a relatively small number of patients. These shortcomings have led to the currently only prospective randomized multicenter study for mitotane as an adjuvant therapy for low to moderate risk for recurrence ACC (ADIUVO, Efficacy of Adjuvant Mitotane Treatment) (289).
Table 6.
Studies Evaluating Mitotane as an Adjuvant Therapy (All Retrospective)
| Study | Year | Number With/Without Mitotane | DFS | OS |
|---|---|---|---|---|
| Bodie (340) | 1989 | 21/25 | Not significant | Not significant |
| Pommier (275) | 1992 | 10/43 | Significantly better | |
| Vassiloupolou-Sellin (369) | 1993 | 8/6 | Significantly worse | Not favorable |
| Haak (269) | 1994 | 11/36 | Not significant | Not significant |
| Barzon (370) | 1997 | 7/11 | Not significant | Not significant |
| Terzolo (290) | 2007 | 47/130 | Significantly better | Significantly better/favorable |
| Bertherat (291) | 2007 | 86/80 | Not significant | NA |
| Grubbs (257) | 2010 | 22/196 | Favorable | No difference |
Abbreviations: DFS, disease-free survival; OS, overall survival.
Table 7.
Studies Using Mitotane as a Therapeutic Agent (Nonadjuvant)
| Study | Year | Design | Number of Patients | Observed Responses | Number of Responders | Percent |
|---|---|---|---|---|---|---|
| Van Slooten (371) | 1984 | Retrospective | 34 | PR | 8 | 23.53 |
| Venkatesh (26) | 1989 | Retrospective | 64 | PR | 21 | 32.81 |
| Luton (10) | 1990 | Retrospective | 37 | SD (2), PR (8) | 10 | 27.03 |
| Decker (372) | 1991 | Prospective | 36 | CR (2), PR (6) | 8 | 22.22 |
| Pommier (275) | 1992 | Retrospective | 29 | PR (7) | 7 | 24.14 |
| Haak (269) | 1994 | Retrospective | 52 | PR (7), CR (8) | 15 | 28.85 |
| Barzon (370) | 1997 | Retrospective | 11 | PR (2) | 2 | 18.18 |
| Williamson (373) | 2000 | Prospective | 16 | SD (2), PR (2) | 4 | 25.00 |
| Baudin (374) | 2001 | Prospective | 13 | CR (1), PR (3) | 4 | 30.77 |
| Gonzalez (244) | 2007 | Retrospective | 67 | CR (4), SD (10), PR (9) | 23 | 34.33 |
| Total | 359 | 102 | 28.41 | |||
| Average % | 26.01 |
Abbreviations: PR, partial remission; SD, stable disease; CR, complete remission.
a. Mitotane for adjuvant therapy.
Adjuvant treatment is routinely started within 3 months after surgery. The advantage of starting mitotane as early as possible after surgery was recently confirmed in mouse experiments, in which mitotane was significantly more successful in preventing the growth of xenotransplants when given at the time of tumor cell inoculation rather than at the time of visible tumor growth (282). A recent large retrospective study suggests a benefit of adjuvant mitotane therapy. In this study, adjuvant mitotane therapy showed significant improvement in median tumor-free survival in patients with completely resected ACCs (42 vs 10 and 25 months in 2 control groups). Median overall survival was significant only in comparison with one of the control groups (110 vs 52 and 67 months) (290). It seems that only a subgroup of patients may benefit. Analysis from other centers suggest probable benefits only for cortisol-producing tumors (291). Although often usual practice, no study has formally evaluated the combination of mitotane and radiation therapy. This approach is supported by in vitro findings of mitotane acting as a radiation sensitizer (292, 293).
b. Mitotane for recurrent and advanced disease.
The efficacy of mitotane therapy in the setting of not completely resectable, metastasized, or recurrent ACC is well established. Overall, 30% of patients show stable disease or partial remission after treatment with mitotane (Table 7). Some trials report an occasional complete remission, but these are rare events (294). All studies have been retrospective and uncontrolled, and it is the general experience that a subgroup of patients shows a very slow disease progress, possibly confounding interpretation of these results. Despite the fact that at best only one-third of patients will have a response to mitotane, only very few studies have analyzed patient-, tumor-, or drug-related factors that may influence patient outcome and predict patients who may respond to mitotane therapy. The most important prognostic factor is the mitotane plasma level (295). Most studies, including a large retrospective analysis, have defined the therapeutic mitotane level to be 14 to 20 mg/L (296). Some case series have argued for an effectiveness of low-dose mitotane therapy (297). However, until further evidence for the effectiveness of low levels, mitotane treatment in the adjuvant or therapeutic setting should aim for the established therapeutic target range. On the molecular level, RRM1 expression has been found to be inversely correlated with mitotane response. Low RRM1 expression was a predictor of response to mitotane therapy with prolonged tumor-free survival (298).
c. Mitotane management.
Managing mitotane therapy is a fairly intensive process and requires experience. The authors prefer a slight modification of the protocol as outlined by Terzolo et al (241). The dose is initiated at 1 g twice daily and increased every 4 to 7 days by 0.5 to 1 g/d until a daily dose of 5 to 7 g is reached. A low-dose loading protocol has also been described, probably leading to fewer side effects, the same efficacy, and increased patient compliance. Regardless of the initial protocol, appropriate monitoring of blood levels is key and readily available in most countries. After the initial loading phase, the mitotane dose is titrated to a blood level of 14 to 20 mg/L. Side effects are mainly GI, neurological, and metabolic/endocrinological in nature and can usually be managed when mild (Table 8).
Table 8.
Mitotane Therapy Side Effects
| Organ System | Symptoms and Signs | Frequency | Action Required |
|---|---|---|---|
| GI tract | |||
| General | Nausea, vomiting, diarrhea | Very common | Supportive therapy |
| Increased ALT and AST | None | Common | Hold mitotane and evaluate for causes with fast increase or enzymes >3- to 4-fold |
| Increased ALP and GGT | None | Very common | None |
| Autoimmune or drug-induced hepatitis | Cholestasis, liver failure | Rare | Stop mitotane |
| CYP3A4 induction | Increased hepatic drug metabolism | Very common | Evaluate all drugs for CYP3A4 metabolism, consider measuring levels |
| Central nervous system | |||
| General | Fatigue, somnolence, stupor, ataxia, balance disorder, decreased memory, depression, dysphasia | Very common | Obtain mitotane level and hold mitotane until symptoms resolve |
| Endocrine system | |||
| Adrenal insufficiency | Fatigue, nausea, abdominal pain, increased ACTH | Very common | Always start hydrocortisone with mitotane (minimum 30 mg/d, but may need substantially more); consider fludrocortisone |
| Hypogonadism | Loss of libido, fatigue, muscle weakness, low bioavailable testosterone | Common | Initiate testosterone replacement |
| Hypothyroidism | Weight gain, fatigue, dry skin, depression | Common | Initiate thyroid hormone replacement |
| Gynecomastia | Painful breast growth | Common | Consider radiation therapy or pharmacotherapy with aromatase inhibitor or antiestrogen. Replace testosterone in case of hypogonadism |
| Lab abnormalities | Increased SHBG, CBG, low TSH, low free T4 | Very common | None |
| Hypercholesterolemia | High cholesterol | Very common | Treat with statin (choose statin not metabolized by CYP3A4, eg, pravastatin) |
| Skin | |||
| Rash | Rash | Common | Hold mitotane, may try restart depending on severity of reaction |
| Blood | |||
| Leukopenia, thromocytopenia | Rare | Depending on severity, hold mitotane |
The GI side effects (nausea and diarrhea) are most commonly dependent on the actual dose in contact with the lumen of the GI tract. These effects are rarely dose limiting and can be attenuated by distributing the mitotane amount into 3 or 4 daily doses. GI side effects are also often ameliorated by taking mitotane with food, specifically lipid-rich foods, such as dairy (eg, a milkshake) or peanut butter. Mild to moderate side effects can also be treated with antiemetic and antidiarrheal medications. Nausea can be treated with ondansetron, prochlorperazine, or metoclopramide. Because all of these drugs are metabolized by CYP3A4, increased doses may be necessary. Diarrhea can be treated with loperamide and in severe cases with opium tincture. Most importantly, patients should be carefully evaluated whether GI symptoms could be due to adrenal insufficiency, in which case a hydrocortisone increase may ameliorate symptoms.
Neurological side effects have a wide range from minor mental slowing, ataxia, and dysphasia to severe somnolence and lethargy. Some patients on mitotane with significant neurological side effects receive a work-up for a presumed stroke when treating physicians are not familiar with mitotane therapy or the information on mitotane therapy was not readily available. Neurological side effects are dependent on plasma mitotane levels and usually do not occur until blood levels rise higher than 20 mg/L. In the case of worsening or intolerable side effects, dose reductions or withholding medication for 1 to 4 months is sometimes necessary. Neurological side effects are the main limiting side effect.
Several biochemical abnormalities occur, but most of them can be tolerated and do not require dose adjustments. Mitotane therapy almost invariably leads to an increase in liver enzymes and hypercholesterolemia. Alkaline phosphatase and γ-glutamyl transferase (GGT) can increase significantly and the rise is usually of no clinical significance, but aspartate aminotransferase (AST) and alanine aminotransferase (ALT) show only mild elevation. Usually no adjustment of mitotane dose is necessary. However, with rapidly rising levels of AST and ALT or levels greater than 3-fold the normal range, mitotane therapy should be temporarily withheld and evaluation for mitotane-induced hepatotoxicity or other liver pathologies initiated. Hypercholesterolemia is best treated with a statin, preferably pravastatin or another compound that is not metabolized by CYP3A4.
Major endocrine abnormalities result from the effect of mitotane on steroid hormone biosynthesis. Three main mechanisms lead to adrenal insufficiency and decreased bioavailability of cortisol: 1) inhibition of steroid hormone biosynthesis at the level of CYP11B1 and CYP11A1, 2) induction of CYP3A4 and increased 6β-hydroxylation of cortisol, and 3) induction of cortisol binding globulin (CBG). Adrenal insufficiency occurs invariably and is treated preemptively. All patients are started on a minimum of 30- to 40-mg daily dose of hydrocortisone. Supraphysiological hydrocortisone doses up to 50 to 100 mg daily may be necessary because of the increased cortisol catabolism. Dose adjustments are mainly made based on clinical findings and evaluation of ACTH, morning serum cortisol, and 24-hour urine free cortisol levels. Due to CBG induction and increased cortisol metabolism, neither plasma level nor 24-hour urine excretion are entirely reliable for treatment monitoring. Hydrocortisone therapy needs to be continued after cessation of mitotane until the patient does not show any clinical or biochemical evidence of adrenal insufficiency. Even after discontinuation of mitotane therapy, CYP3A4 induction and mitotane levels persist up to several months. Occasionally, mitotane may affect mineralocorticoid synthesis and replacement therapy with fludrocortisone therapy may become necessary. Replacement with mineralocorticoids should be considered in cases with hypotension or symptomatic orthostatic hypotension and hyperkalemia. Doses of 0.05 to 0.2 mg fludrocortisone are usually sufficient, and monitoring can be done by following renin activity targeted to the normal range.
Hypogonadism in male patients often requires replacement therapy. Mitotane induces SHBG and increases total testosterone but decreases the bioavailable fraction. Furthermore, mitotane has recently been shown to inhibit 5α-reductase, reducing the generation of more potent androgens (299). Possibly due to the relative increase in SHBG as well as inhibition of 5α-reductase by mitotane, male patients might develop gynecomastia. Gonadotropins are unchanged. In the presence of hypogonadal symptoms, testosterone replacement therapy is recommended.
Although TSH and free T3 levels are often unchanged, free T4 levels decrease. It has been determined that this is not a laboratory artifact because mitotane does not interfere with thyroid hormone measurements (300). One interpretation is that the changes are due to partial central hypothyroidism. This is further backed up by experiments in cell lines, where mitotane directly induces apoptosis in thyrotrope cells (300). The decision for replacement therapy with levothyroxine should be made in the presence of clinical symptoms and signs of hypothyroidism.
Metabolism of the patient's nonendocrine medication should also be evaluated. It has recently become clear that mitotane is a very strong inducer of the drug-metabolizing microsomal liver and gut enzyme CYP3A4 (299, 301–303). Common drugs metabolized by this enzyme are statins, opiates, benzodiazepines, warfarin, and some antibiotics (for a complete list see Ref. 301). Other drugs regularly used in combination with mitotane, such as platinum-based cytotoxic drugs, doxorubicin, and etoposide are also metabolized by CYP3A4, potentially reducing their antineoplastic effect. This is especially important when evaluating new drugs and targeted agents. A study using sunitinib, which is metabolized by CYP3A4, raised concerns that several of the study subjects did not reach therapeutic levels of this drug (302, 304). This is of concern because many new drugs and experimental regimens are evaluated in patients that have failed other therapies, such as mitotane. As mentioned above, mitotane levels can persist for up to 1 year after therapy, and the effect on CYP3A4 can last at least as long. This observation gave rise to the discussion to evaluate new agents as a frontline therapy rather than after failing traditional schemes.
Greater than half of the women with ACC are diagnosed during childbearing age. There is a general concern that pregnancy may lead to increased likelihood of relapse and increased tumor growth. However, supporting evidence is minimal. During mitotane therapy, we generally recommend contraception, preferably barrier methods. With regard to fetal health, data are conflicting. Although there is a clear theoretical concern for disorders of sexual development in the developing fetus, there is little supporting evidence. At least 6 pregnancies with conception during or shortly after mitotane therapy have been reported. Four children were born evidently healthy, and no birth defects were encountered (305–307). In 1 case, an abortion was performed and minor abnormalities of the morphology of the adrenal gland were described (308). A recent case report analyzed blood levels in a 21-week gestation normally developed fetus from a pregnancy terminated for ACC recurrence (309). In this study, amnion fluid and cord blood levels of mitotane were undetectable, arguing that there may be minimal exposure to the fetus.
A summary of suggested laboratory surveillance with mitotane therapy is shown in Table 9.
Table 9.
Surveillance During Mitotane Therapy
| Regular Laboratory Surveillance for Mitotane |
| Mitotane level (monthly until therapeutic, then every 3 mo) |
| Every 3 mo |
| Complete blood count |
| Liver function tests (AST, ALT) |
| Cholestatic parameters (ALP, GGT, bilirubin) |
| Free T4, TSH |
| Cholesterol |
| Renin |
| ACTH, cortisol, 24-h urine free cortisola |
| Males: bioavailable testosterone, LH, FSH |
Use of these parameters is limited by the effects of mitotane on cortisol levels and cortisol metabolism and need to be interpreted with caution.
2. Cytotoxic chemotherapy
Cytotoxic chemotherapy is currently a mainstay of treatment for advanced and metastasized ACC. Initial studies in the 1970s and 1980s evaluated single-compound regimens with minor success and a response rate ranging from 10% to 20%. These early studies led to the currently used combination regimens. Of the single-compound studies, at least 2 are worthwhile mentioning because they are somewhat unique to ACC. Suramin, a compound used traditionally for the treatment of sleeping sickness, had been found to induce adrenal insufficiency in humans and to have adrenolytic activity in animal experiments, where it led to inflammatory changes of the adrenal cortex (310, 311). Subsequently, suramin has been tested in 2 studies where it led to an overall response rate of ∼20%. However, treatment had a narrow therapeutic range and serious side effects (312, 313). Another compound that had been evaluated as a single agent is gossypol, a natural phenol from the cotton plant. An initial study suggested a response rate of 14%, but the results of repeat studies are pending (314).
Several studies have evaluated chemotherapeutic regimens with or without concurrent mitotane (Table 10). The interpretation of early studies is difficult due to different study populations, ranging from microscopically remaining tumor to advanced pretreated metastasized disease. Responses are defined differently in these studies, and it is an ongoing discussion whether stable disease is a meaningful endpoint in otherwise noncurable cancers. The overall response to chemotherapeutic regimens is 30% and 50%, when counting stable disease as a response. However, the response is invariably transient and short-lived (6–18 months). To establish a gold standard of cytotoxic chemotherapy for ACC, a recent phase 3 trial (FIRM-ACT, First International Randomized Trial in Locally Advanced and Metastatic Adrenocortical Carcinoma Treatment) compared the most promising regimens (etoposide, doxorubicin, cislatin, mitotane [EDPM] vs streptozotocin, mitotane). This study confirmed the efficacy of chemotherapy and proved the superiority of EDPM. The response rate was 20% and 50%, when stable disease was included. However, the median progression-free survival, again, was short with a median of 5 months (and even 2 months in the streptozotocin, mitotane group). Although this study established EDPM as an efficacious therapy, it also underscored the limitations of chemotherapy for ACC. All study patients received concurrent mitotane therapy and with the recent finding of increased metabolism of other drugs due to CYP3A4 induction (eg, affecting cisplatin metabolism), there is criticism regarding whether chemotherapy without mitotane may be more successful. Subanalyses from the FIRM-ACT trial using mitotane levels and mitotane treatment duration may be helpful.
Table 10.
Multidrug Chemotherapy in ACC (All Prospective)
| Study | Year | Regimen | Disease | Total Number | CR | PR | SD | % CR, PR | % CR, PR, SD | Mitotane | Response Duration, mo |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Van Slooten (375) | 1983 | Cyclo, Cis, Dox | Advanced | 11 | 0 | 2 | 6 | 18 | 73 | No | 10–23 |
| Schlumberger (376) | 1991 | 5FU, Cis, Dox | Advanced | 13 | 1 | 3 | 3 | 31 | 54 | No | 6–42 |
| Burgess (377) | 1993 | Cis, Eto | Advanced | 11 | 0 | 6 | NA | 55 | No | Median 9 | |
| Bukowski (378) | 1993 | Cis | Advanced | 37 | 1 | 10 | NA | 30 | Yes | Median 7.9 | |
| Bonacci (379) | 1998 | Cis, Eto | Advanced | 18 | 3 | 3 | 2 | 33 | 44 | Some | 9–26 |
| Berruti (380) | 1998 | Cis, Eto, Dox | Advanced | 28 | 2 | 13 | 8 | 54 | 82 | Yes | Median 24.4 |
| Williamson (373) | 2000 | Cis, Eto | Advanced | 37 | 0 | 5 | NA | 14 | No | NA | |
| Khan (381) | 2000 | Sz | Advanced | 23 | 1 | 6 | 5 | 30 | 52 | Yes | Median 7 |
| Abraham (382) | 2002 | Dox, Eto, Vin | Advanced | 35 | 1 | 7 | 23 | Yes | Mean 12.4 | ||
| Baudin (383) | 2002 | Irinotecan | Advanced/failed | 12 | 0 | 0 | 3 | 0 | 25 | No | NA |
| Khan (384) | 2004 | Cyclo, Vin, Cis, Ten | Advanced/failed | 11 | 0 | 2 | 7 | 18 | 82 | No | Median 6.75 |
| Berruti (385) | 2005 | Cis, Eto, Dox | Advanced | 72 | 5 | 30 | NA | 49 | Yes | Median 18 | |
| Sperone (386) | 2010 | Gem, 5FU or Cap | Advanced/failed | 28 | 1 | 1 | 11 | 7 | 46 | Yes | Median 9.8 |
| Fassnacht (387) | 2012 | Cis, Eto, Dox | Advanced | 151 | 2 | 29 | 53 | 21 | 56 | Yes | Median 5.1 |
| Fassnacht (387) | 2012 | Sz | Advanced | 153 | 1 | 11 | 34 | 8 | 30 | Yes | Median 2.5 |
| Total | 640 | 18 | 128 | 132 | 23 | 43 | |||||
| Median | 30 | 54 |
Abbreviations: Cyclo, cyclophosphamide; Cis, cisplatin; 5FU, fluorouracil; Eto, etopside; Dox, doxorubicin; Sz, streptozotocin; Vin, vincristine; Ten, teniposide; Gem, gemcitabine; PR, partial remission; SD, stable disease; CR, complete remission; mo, months.
3. Targeted therapy
The poor prognosis of ACC despite traditional therapies has led to the exploration of new modes of treatment, using targeted agents. The term targeted therapy refers to pharmacological compounds with defined molecular targets, such as receptors or intracellular enzymes. Unfortunately, results of these studies have been disappointing so far (Table 11). They all share the criticism that they have been evaluated in heavily pretreated patients, who often have been or were on concurrent mitotane therapy, which may have blurred some of their potential effects. This is an argument to try some of these agents in treatment-naive patients. The most data for targeted therapy exist for the IGF-1R antagonists. These studies had been initiated with great hopes and were based on the knowledge that children with BWS have higher levels of IGF-2 and that IGF2 is the most highly expressed gene in sporadic ACCs when compared with ACAs or normal tissues. Drugs targeting the IGF-1R system are currently being tried in phase 1 and phase 2 trials in several tumor entities (315). Several studies investigated drugs targeting IGF-1R in patients with stage 4 disease. The first study investigated figtilimumab, a fully human monoclonal antibody directed toward IGF-1R (316). Treatment-related toxicities were generally mild and mainly included hyperglycemia, nausea, fatigue, and anorexia. Eight of 14 patients had stable disease as their best response. However, after 7 cycles (∼6 months), all patients showed disease progression. Another phase 2 study used IMCA12 (cixutumab), a fully humanized IGF-1R antibody, in patients with stage 4 ACC that were treatment-naive. Although the initial trial design was to compare mitotane plus IMCA12 vs mitotane alone, a first phase of this trial enrolled 19 patients to mitotane and IMCA12 to assess toxicity. However, due to the lack of overall response and recruitment difficulties, the trial was terminated early (G.D.H., unpublished results). In another study, cixutumab in combination with temsirolimus was evaluated in 10 patients with ACC. Stable disease in 4 patients, at a maximum lasting for greater than 8 months, was the best response (317). The phase 3 trial GALACCTIC compared OSI906, a small-molecule inhibitor of IGF-1R and insulin receptor, in patients with stage 4 disease in a placebo-controlled fashion, with two-thirds receiving study drug and one-third receiving placebo. The results of this study as well as potential subanalyses of patients benefiting from the trial drug are pending. The generally disappointing results of studies targeting the IGF-1R system together with a critical review of in vitro results and studies in transgenic mice have sparked a recent discussion on the contribution of IGF-2 to adrenal tumorigenesis (318).
Table 11.
Targeted Therapy in ACC (All Advanced Disease/Failed Therapy)
| Study | Year | Regimen | Disease | Total Number | CR | PR | SD | %CR, PR | %CR, PR, SD | Response Duration, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| Rustin (388) | 2003 | Combretastatin | Advanced/failed | 1 | 0 | 1 | 0 | 100 | 100 | Continued after initial PD |
| Shah (389) | 2005 | Irinotecan, flavopiridol, CDK inhibitor | Advanced/failed | 2 | 0 | 0 | 2 | 0 | 100 | 8.1–15.4 |
| Gross (390) | 2006 | Imatinib | Advanced/failed | 4 | 0 | 0 | 0 | 0 | 0 | NA |
| Quinkler (391) | 2008 | Erlotinib, gemcitabine | Advanced/failed | 10 | 0 | 0 | 1 | 0 | 10 | 8 |
| Hong (392) | 2009 | Sorafenib, tipifarnib | Advanced/failed | 2 | 0 | 0 | 2 | 0 | 100 | 4 and 7 |
| Wortmann (239) | 2010 | Bevacizumab, gemcitabine | Advanced/failed | 10 | 0 | 0 | 0 | 0 | 0 | |
| Haluska (316) | 2010 | Figitimumab | Advanced/failed | 14 | 0 | 0 | 8 | 0 | 57 | 3–5.5 |
| Naing (317) | 2011 | Cixutumab, temserolimus | Advanced/failed | 10 | 0 | 0 | 4 | 0 | 40 | 8+ |
| Berruti (393) | 2012 | paclitaxel, sorafenib | Advanced/failed | 25 | 0 | 0 | 0 | 0 | 0 | Progression in 9 patients, early termination |
| Kroiss (304) | 2012 | Sunitinib | Advanced/failed | 35 | 0 | 0 | 5 | 0 | 14 | 5.6–11.2 |
| Total | 113 | 0 | 1 | 22 | 1 | 20 | ||||
| Median | 0 | 27 |
Abbreviations: PR, partial remission; SD, stable disease; CR, complete remission; mo, months.
A study using the multikinase inhibitor sunitinib led to stable disease in 5 of 35 patients (304). Interestingly, concomitant mitotane treatment negatively affected patient response, and sunitinib and mitotane levels were anticorrelated. The reasons for these findings are induction of CYP3A4 by mitotane and metabolism of sunitinib by the same enzyme (301, 304). This observation has focused the discussion on drug-drug interactions when using mitotane and led to the conclusion that newer substances need to be evaluated either as a frontline therapy or after washout of mitotane.
Although molecular targeted therapy based on basic research findings is intriguing, unfortunately, none of the studies so far have shown definitive effectiveness that would make any of the substances a good candidate for further exploration or routine use in ACC therapy. However, trials with new targeted substances are under way, and altered regimens and combination therapies may hold some promise.
A radionucleotide-based approach to therapy of ACC is the use of [131I]IMTO. [123I]IMTO single-photon emission CT imaging showed high tracer uptake in tissue of adrenocortical origin (110), suggesting that [131I]IMTO represents a suitable compound for targeted radionuclide therapy. [131I]IMTO treatment in 11 patients with advanced ACC resulted in median progression-free survival for 14 months in 6 patients who responded to therapy (319). The clinical utility of this technology, however, needs further evaluation with prospective clinical trials
4. Therapy for hormone excess
Control of the deleterious effects of elevated hormone levels in ACC patients is important. In general, several inhibitors of steroidogenesis as well as direct hormone receptor antagonists can be used to achieve this goal. With the exception of mifepristone and mitotane, treatment of hormone excess with all substances mentioned below is regarded as off-label treatment in most countries. Adjunct therapy to control side effects with nonspecific pharmacotherapy, such as the use of drugs to prevent osteoporosis, antihyperglycemic drugs, or antihypertensive drugs, may be necessary as well.
Several inhibitors of steroidogenesis are in use and summarized in Table 12 and Figure 1. During treatment with any of the steroidogenesis inhibitors, patients need to be regularly evaluated for adrenal insufficiency and should be regarded as adrenal-insufficient in times of physical stress (febrile illness or significant injury/surgery). Mitotane inhibits CYP11A1 and CYP11B1 and together with its adrenolytic effects may lead to some control of hormone levels. Ketoconazole and metyrapone are commonly used to control glucocorticoid excess. Ketoconazole inhibits CYP17A1, CYP11A1, and to some extent CYP11B1 (320). The usual starting dose is 200 mg twice daily and can be increased to 1200 mg/d. During treatment with ketoconazole, liver enzymes need to be carefully watched. Because it is an inhibitor of several hepatic drug-metabolizing enzymes (eg, CYP3A4, CYP2C9, and CYP1A2), drug interactions need to be carefully reviewed. Another powerful inhibitor of steroidogenesis at the level of CYP11B1 is metyrapone (321), and 250 mg twice daily is the usual starting dose and can be increased to 2 to 3 g/d in 250-mg intervals. Due to the inhibition of CYP11B1, a relative increase in adrenal androgens may occur, possibly worsening symptoms related to hyperandrogenemia. Other steroidogenesis inhibitors such as aminoglutethimide or etomidate are not in widespread use. Aminoglutethimide is an inhibitor of CYP11A1 and CYP11B1 and was initially introduced as an antiepileptic medication (322, 323). However, aminoglutethimide is no longer available in most countries. Etomidate is an anesthetic compound often used for rapid induction for intubation or short-term procedures. Even at doses much lower than those used for anesthesia, etomidate is a powerful inhibitor of CYP11B1 and CYP11B2 (324, 325). For this effect, it can be used in the inpatient setting. Some centers have experience with a steady low-dose perfusor, which is a last-resort option. Steady infusion can be safe because doses used are only 1/10 of the anesthetic dose (2–3 vs 20–30 mg/h).
Table 12.
Therapeutic Agents for Hormonal Control
| Hormonal Derangement | Medication | Dose per Day | Side Effects |
|---|---|---|---|
| Hypercortisolism | Mitotane | 2–12 g, to target level 14–20 mg/L | See Table 7, use mainly for adrenolytic effects |
| Mifepristone | 300–1200 mg | Fatigue, nausea, headache, hypokalemia, arthralgia, vomiting, edema, and endometrial thickening | |
| Metyrapone | 250–3000 mg | Hyperandrogenemia | |
| Ketoconazole | 200–1200 mg | Nausea, vomiting, abdominal pain, hepatotoxicity | |
| Etomidate | 0.02–0.03 mg/kg/h | Sedation (usually occurs at significantly higher doses) | |
| Aminoglutethemidea | 250–1000 mg | Rash, nausea, vomiting, anorexia, sedation, lethargy | |
| Mineralocorticoid excess | Spironolactone | 25–200 mg | Hyperkalemia, gynecomastia |
| Eplerenone | 50–200 mg | Hyperkalemia | |
| Hyperandrogenemia | Spironolactone | Hyperkalemia, gynecomastia | |
| Estrogen excess | Aromatase inhibitors | Anastrazole 1 mg | Bone loss, nausea |
| Letrozole 2.5 mg | |||
| Exemestane 25 mg | |||
| SERMs (tamoxifen, raloxifene) | Tamoxifen 20–40 mg, raloxifen 60 mg | Deep venous thrombosis |
Abbreviation: SERM, selective estrogen receptor modulator.
Not commonly available anymore.
A direct antagonist used for glucocorticoid excess is mifepristone. Treatment can be initiated with 300 mg daily and titrated up to 1200 mg daily. Overt adrenal insufficiency is rare under treatment with mifepristone (326). However, neither ACTH nor glucocorticoid levels can be used to guide therapy. The most common side effects are hypokalemia and hypertension due to the direct effects of the very high cortisol levels on the renal mineralocorticoid receptors. This effect can be further controlled with the addition of spironolactone or eplerenone.
Spironolactone can also be used to control androgen effects in women with androgen-secreting tumors and mineralocorticoid effects in patients with mineralocorticoid-secreting tumors. Doses may need to be as high as 200 to 400 mg/d. For the rare cases of male patients with gynecomastia, aromatase inhibitors (eg, anastrozole and letrozole) as well as estrogen receptor antagonists (eg, tamoxifen and raloxifene) can be used.
Because cortisol production is regarded as an adverse prognostic factor, one can speculate that treatment to normalize hormone effects may not only improve QOL but also positively affect survival parameters.
D. Radiation therapy
As with other malignancies, local control of ACC is important both for effecting the possibility of a disease cure and for improving symptomatic outcomes. Although traditionally considered ineffective for ACC (327–331), radiotherapy has been shown in several recent series to offer a significant improvement in disease control in both the adjuvant and palliative settings (247, 332–337), although such an improvement has not been universally demonstrated (338).
1. Radiation therapy in nonmetastatic ACC
The high rates of local failure after complete resection, in addition to the resultant positive margins when such a resection is not possible, emphasize the potential role for radiotherapy in sterilizing the adrenal fossa after surgery. Despite this, there has been a long-held resistance to using radiotherapy mainly based on a small number of anecdotal experiences that concluded radiation to be ineffective. The shortcomings of such conclusions were accurately detailed by Percarpio and Knowlton (335), who described how these studies relied on poorly documented results in a low number of heterogeneous patients. For example, 2 early reports state that radiotherapy is ineffective, but they offer no details regarding patient or treatment characteristics (266, 339). Three other studies found radiotherapy to be ineffective on the basis of experiences in 2, 3, and 5 patients, respectively (275, 340, 341).
Since the publication of those aforementioned studies, the field of radiation oncology has witnessed tremendous advances in the ability to accurately deliver higher, more efficacious doses of radiation to the clinical target while sparing normal tissues. This has resulted in improved outcomes in a number of different cancers (2, 342–350). Nonetheless, in the setting of ACC, radiotherapy is infrequently employed. In the United States, it is used in approximately 10% of cases, according to analyses of both the National Cancer Data Base and the SEER program (4, 11, 90). Similarly, in Europe, a recent analysis of the Dutch Adrenal Network Registry found that radiotherapy is employed in only 8% of patients (296).
Despite this, there is emerging evidence that radiotherapy might be effective (Table 13). Fassnacht et al (332) retrospectively analyzed the use of radiotherapy after a complete resection in 14 patients with nonmetastatic disease. After surgery, patients were treated to a mean dose of 49 Gy. Their outcomes were then case-matched to controls for stage, tumor size, margin status, and adjuvant mitotane treatment. Although radiotherapy did not improve survival, it did significantly improve local control: at 5 years, local recurrence-free survival was 86% in the group that received radiation compared with 11% in those that did not (332).
Table 13.
Summary of Studies Evaluating the Role of Radiotherapy in ACC
| Study | Year | Adjuvant |
Palliative |
||||||
|---|---|---|---|---|---|---|---|---|---|
| No. of Cases | RT Dose (Gy) | Chemotherapy | Local Control | No. of Cases | RT Dose (Gy) | Chemotherapy | Response | ||
| Percarpio (335) | 1976 | 4 | 28–40 | NR | 1/4 (25%) | 12 | 15–51 | NR | 12/12 (100%) |
| King (351) | 1979 | 4 | 42–55 | 0/4 (0%) | NR | 12 | NR | NR | 6/12 (50%) |
| Didoklar (272) | 1981 | 10 | NR | NR | 4/10 (40%) | ||||
| Henley (274) | 1983 | 10 | NR | NR | 4/10 (40%) | ||||
| Venkatesh (26) | 1989 | 26 | NR | 10/26 (39%) | 6/26 (23%) | ||||
| Markoe (334) | 1991 | 5 | 42–60 | 2/5 (40%) | 3/5 (60%) | 5 | 30–50 | 1/5 (20%) | 5/5 (100%) |
| Pommier (275) | 1992 | 3 | 39–45 | NR | 0/3 (0%) | 5 | NR | NR | 5/5 (100%) |
| Fassnacht (332) | 2006 | 14 | 40–54 | 5/14 (36%) | 12/14 (86%) | ||||
| Polat (336) | 2009 | 22 | 10–60 | NR | 17/22 (77%) | ||||
| Hermsen (333) | 2010 | 3 | NR | NR | 3/3 (100%) | 8 | Up to 50 | NR | 8/8 (100%) |
| Sabolch (247) | 2011 | 10 | 45–57 | 8/10 (80%) | 8/10 (80%) | ||||
| Habra (338) | 2012 | 16 | 36–59.4 | 4/16 (25%) | 9/16 (56%) | ||||
Abbreviation: NR, not reported; RT, radiotherapy.
Other studies have demonstrated similar findings. Sabolch et al (247) compared 38 cases of surgery without radiotherapy with 10 cases of adjuvant radiotherapy and 16 cases of definitive radiotherapy for unresectable disease. Adjuvant cases used dosages between 45 and 57 Gy, and definitive cases used a median dose of 39.2 Gy (22.5–73.5 Gy). Results from this study showed that whereas radiotherapy was not associated with a survival benefit, it did significantly improve local control. Local failure occurred in 33% of those treated with surgery alone but only in 12% of those whose regimen involved radiation. Patients treated with adjuvant radiotherapy after surgery had a 20% local recurrence rate. Importantly, this study also demonstrated the feasibility of treating unresectable disease with definitive radiotherapy (247).
In contrast, Habra et al (338) present results from 16 patients treated in the community setting with primary resection followed by adjuvant radiotherapy. These patients were matched on the basis of margin status and disease stage to 32 patients who received surgery alone. The median dose delivered was 50.4 Gy. There were no significant differences found in local control or survival between these 2 groups (338). Interestingly, patients who received adjuvant radiotherapy after surgery seemed to do substantially worse in the series by Habra et al (338) when compared with results for similarly treated patients reported by the 2 other series (247, 332). More specifically, although Habra et al (338) found a local recurrence rate of 44% for such patients, Fassnacht et al (332) showed a 14% local failure rate and Sabolch et al (247) found a 20% rate. Although it is difficult to determine from retrospective series the reasons for such discordant outcomes, the patients reported on by Habra et al (338) were treated outside of an academic center and might not have benefited from the multidisciplinary approach that such centers offer, particularly for rare cancers. Regardless, it is clear that prospective studies are needed to assess the efficacy of radiotherapy in the adjuvant setting.
2. Radiation therapy for palliation
For palliative cases, there are numerous although primarily anecdotal reports that radiotherapy can adequately relieve patient symptoms. One of the earliest of such studies was published by Percarpio et al in 1976 (335). This detailed 12 instances of palliative radiotherapy, all of which resulted in relief of symptoms or reduction in palpable tumor size, regardless of the substantial variation in the location and characteristics of their metastatic disease (335). Other early reports have shown good palliative responses in approximately half of the patients: King and Lack (351) described a decrease in pain in half of the 12 patients treated, whereas Henley et al (274) found that palliative radiotherapy reduced symptoms or tumor burden in 4 of 10 patients. However, in neither of these studies are the details of radiotherapy provided (274, 351). A study that stands in contrast to these others is that of Venkatesh et al (26), who found that palliative radiotherapy was effective in only 6 of 26 patients. This was an update of an earlier publication (24), and the determination of treatment efficacy included criteria for patient survival but not for relief of pain, which is a dubious definition in the setting of palliation (26).
More modern studies have consistently shown adequate palliation in most patients treated. Magee et al (337) found that palliative radiotherapy was effective in 4 of 6 patients, either reducing the mass of their tumors or ameliorating symptoms of hormone excess associated with a functional tumor. Even more recently, good results were achieved by Markoe et al (334), using doses of 30 to 50 Gy to treat bone metastases and unresectable tumors. They reported that all 5 patients that they treated received adequate relief of their pain (334). Hermsen et al (333) found similar outcomes, as all 6 patients that they treated for bone metastases were successfully palliated. However, the largest series in the literature on palliative radiotherapy was recently published by Polat et al (336). In this series, 22 patients had painful osseous metastases, and 17 patients (77%) experienced adequate palliation of pain after radiotherapy with dosages totaling 10 to 60 Gy.
3. Radiation therapy techniques
With regard to the adjuvant radiotherapy technique at our institution, we use radiotherapy for patients who have undergone an R1 or R2 resection. Patients who have had a complete (R0) resection are considered for adjuvant radiotherapy, although this decision is personalized on a case-by-case basis, accounting for individual disease and patient characteristics. Typically, intensity-modulated radiotherapy is used to deliver 55 Gy in 2.2-Gy fractions to the tumor bed. Simultaneously, the bilateral para-aortic lymph nodes are covered with 45 Gy in 1.8-Gy fractions.
The lymphatic drainage of the adrenal gland is routinely covered in the target volumes (247, 334, 336). ACC has a well-documented propensity for lymphatic dissemination (11, 91, 351), with involved nodal disease in approximately 15% to 27% of patients at the time of surgery (11, 91). Furthermore, there is emerging evidence from the surgical literature that adequate lymph node dissection at the time of adrenalectomy likely improves both rates of disease recurrence and survival (249). Extrapolating both from these data (249) and the documented pattern of spread (11, 91, 351), we routinely include the lymphatic basin in radiotherapeutic treatment fields. However, whether there is a significant benefit to such an approach is a potential area of future study.
For cases of definitive radiotherapy, it is appropriate to target the gross disease and associated lymphatic basin. Using a 4-dimensional CT scan during radiation treatment planning, the motion of the tumor bed with breathing is routinely assessed. Every attempt should be made to escalate doses to the gross disease to levels of 60 Gy or greater, as tolerated by nearby organs at risk, which are typically the ipsilateral kidney, bowels, and liver. Such an approach usually requires intensity-modulated radiotherapy.
The use of palliative radiation is appropriately based on individual patients' palliative needs and disease burden. A single 8-Gy fraction is typically employed to palliate osseous metastases. In patients with a better prognosis or in those with metastatic disease involving the spinal cord or soft tissue, the use 3-dimensional conformal therapy is helpful to deliver a total 30 Gy in 3-Gy fractions.
Other future areas of study in the adjuvant setting include whether dose escalation might deliver better rates of local control while still sparing the associated organs at risk by using highly conformal techniques. In cases of small tumors, this might be accomplished with ablative dosages delivered via a stereotactic body radiotherapy technique. Another area of study is the proper sequencing and timing of radiotherapy with mitotane and other systemic therapies.
Finally, and most importantly, a prospective, multicenter trial is necessary to definitively establish whether adjuvant radiotherapy is effective in reducing local recurrences. Until such time as this is established, we agree with recent expert consensus opinions and practice guidelines in concluding that the current evidence warrants recommending that adjuvant radiotherapy be performed in the postsurgical setting, especially in the case of R1 and R2 resection (15, 255).
E. Other local therapies
In case of inoperable metastatic disease, palliation is possible with local treatment modalities, such as radiofrequency ablation (RFA) or transarterial chemoembolization (TACE). None of these methods have been explored in clinical trials. However, both methods are an alternative to surgery, when surgery is not desired or contraindicated. Both TACE and RFA have a tolerable side effect profile and often less negative impact on well-being than surgery. The use of these treatment modalities largely follows the general use of these procedures for the reduction of tumor load and treatment of metastasis in other tumor entities. RFA has been successfully employed in the palliative setting, rendering patients free of liver metastasis (352). RFA may also be used complementarily during surgical procedures. There are general limitations for RFA, such as in cases of highly vascularized metastasis or proximity to large vessels, because both function as a heat sink preventing the full destructive effect of RFA (353). TACE, localized chemoembolization, is another alternative. The advantage is that with selective embolization, high intratumor levels of cytotoxic substances can be achieved and at the same time systemic effects can be minimized. Although it is a standard alternative option for treatment of metastasis at many centers, published experiences are limited (354–356). In the largest series to date, 103 lesions in 29 patients were treated with response in 21% of patients and stabilization in 62% (357). Predictors of response were a size of <3 cm and high lipidol uptake. Although treatment of metastasis is fairly safe, caution is demanded when treating primary adrenal tumors. Adrenal tumors, including ACC, have a tendency to undergo hemorrhage and might lead to bleeding complications (356).
X. Surveillance
As a rule of thumb, ACC patients should be followed in 3-month intervals during and after initial treatment. Only after a recurrence-free time of 2 to 3 years may surveillance intervals be increased to 6 months until a completion of surveillance for a total of 5 years. Although there are relapses in patients after more than 5 years, this is rare and, in our experience, occurs in <3% of patients that were treated with an initial curative approach.
Patients should undergo a thorough physical examination and interview, specifically aiming to identify recurrence or new onset of hormone excess syndromes. Surveillance should also include a full restaging, including cross-sectional imaging of chest, abdomen, and pelvis. For lesions of an unclear nature, the use of [18F]FDG-PET may be considered. Laboratory evaluation should include steroid hormones that can serve as tumor markers and may therefore diagnose disease recurrence. A promising future surveillance tool is the method of steroid profiling, which may exhibit great sensitivity in detecting small changes in steroid metabolites and precursors. Particularly, the reoccurrence of presurgical steroid profiles may raise concern of relapse (95). Other imaging modalities should be used only in case of clinical suspicion for specific lesions that are best detected by this modality (eg, bone scan for osseous metastases). In case of adjuvant mitotane therapy, evaluation for side effects is also important.
XI. Future Perspectives
Over recent decades, basic science and clinical ACC research have significantly increased our understanding of the pathology of the disease and set standards in clinical care. However, ACC remains a disease with a dismal prognosis. Current large-scale high-resolution analysis will hopefully increase our understanding of the genetic and epigenetic changes underlying ACC pathogenesis. The Cancer Genome Atlas initiative recently decided to conduct a detailed analysis of rare cancers, and an international effort to combine tissue and data resources made it possible for ACC to be one of the tumor types to be analyzed. The expected results hopefully will also give new leads for possible targeted therapies. On the clinical side, the main international expertise centers have united in large-scale trials, such as the FIRM-ACT and GALACCTIC trial, providing a unique platform for future trials. The main future goals for trials are 2-fold: 1) prospectively evaluate traditional therapies, specifically adjuvant mitotane and radiation therapy, and of course 2) to investigate new treatment agents. The current situation is a fertile ground for international collaborative studies but also may be a time of rethinking of established therapies, such as mitotane therapy. Although it is true that it is the only approved drug to treat ACC, it is also of limited efficacy and may interfere with trials using new therapeutic agents. Therefore, it may be beneficial to start evaluating new drugs at the front end of therapy rather than after failure of several lines of established therapy. However, we will await future discussions on the scientific and ethical perspectives of this issue.
Acknowledgments
We thank our colleagues in the University of Michigan Comprehensive Cancer Center Endocrine Oncology Program who have been instrumental to the research and clinical care of ACC patients over the years. In particular, we thank David (Ed) Schteingart, whose longstanding interest in adrenal research and dedication to the care of patients with adrenal diseases were instrumental in bringing this clinic to life. His support of the program's growth over the last 3 decades has been invaluable. We thank Beth Hesseltine for her insight into the practical management of mitotane therapy and Joanne Heaton and Roy Lirov for the careful editorial review of this manuscript.
This work was supported by: NIH T32-DK007245 (to T.E.) and NIH-R01-CA134606 (to G.H.D.).
Disclosure Summary: G.D.H. consults for ISIS, Orphagen, Embara, and Atterocor; holds stocks in Orphagen, Embara, and Atterocor; and is a partial owner of Atterocor. All other authors do not have any disclosures relevant for this publication.
Footnotes
- ACA
- adrenocortical adenoma
- ACC
- adrenocortical carcinoma
- ALT
- alanine aminotransferase
- APC
- adenomatous polyposis coli
- AST
- aspartate aminotransferase
- BWS
- Beckwith-Wiedemann syndrome
- CAH
- congenital adrenal hyperplasia
- CBG
- cortisol binding globulin
- CGH
- comparative genomic hybridization
- CT
- computed tomography
- CYP11B1
- cytochrome P450 family 11 subfamily B1
- DHEAS
- dehydroepiandrosterone sulfate
- DST
- dexamethasone suppression test
- EDPM
- etoposide, doxorubicin, cislatin, mitotane
- ENSAT
- European Network for the Study of Adrenal Tumors
- FAP
- familial adenomatous polyposis
- FDG
- fluorodeoxyglucose
- GGT
- γ-glutamyl transferase
- GI
- gastrointestinal
- HPF
- high-power field
- HU
- Hounsfield units
- IGF-1R
- IGF-1 receptor
- IMTO
- iodometomidate
- LA
- laparoscopic adrenalectomy
- LFS
- Li Fraumeni syndrome
- LOH
- loss of heterozygosity
- MEN1
- multiple endocrine neoplasia type 1
- miRNA
- microRNA
- MRI
- magnetic resonance imaging
- MTO
- metomidate
- OA
- open adrenalectomy
- PET
- positron emission tomography
- QOL
- quality of life
- RFA
- radiofrequency ablation
- SEER
- Surveillance, Epidemiology, and End Results
- SUV
- standardized uptake value
- TACE
- transarterial chemoembolization
- TP53
- tumor protein 53
- VEGF
- vascular endothelial growth factor
- VEGFR
- VEGF receptor
- WNT
- wingless-type.
References
- 1. Mansmann G, Lau J, Balk E, Rothberg M, Miyachi Y, Bornstein SR. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004;25:309–340 [DOI] [PubMed] [Google Scholar]
- 2. Viani GA, Stefano EJ, Afonso SL. Higher-than-conventional radiation doses in localized prostate cancer treatment: a meta-analysis of randomized, controlled trials. Int J Radiat Oncol Biol Phys. 2009;74:1405–1418 [DOI] [PubMed] [Google Scholar]
- 3. Allolio B, Fassnacht M. Clinical review: Adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91:2027–2037 [DOI] [PubMed] [Google Scholar]
- 4. Kebebew E, Reiff E, Duh QY, Clark OH, McMillan A. Extent of disease at presentation and outcome for adrenocortical carcinoma: have we made progress? World J Surg. 2006;30:872–878 [DOI] [PubMed] [Google Scholar]
- 5. Pianovski MA, Maluf EM, de Carvalho DS, et al. Mortality rate of adrenocortical tumors in children under 15 years of age in Curitiba, Brazil. Pediatr Blood Cancer. 2006;47:56–60 [DOI] [PubMed] [Google Scholar]
- 6. Figueiredo BC, Sandrini R, Zambetti GP, et al. Penetrance of adrenocortical tumours associated with the germline TP53 R337H mutation. J Med Genet. 2006;43:91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garritano S, Gemignani F, Palmero EI, et al. Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum Mutat. 2010;31:143–150 [DOI] [PubMed] [Google Scholar]
- 8. Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonça BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol. 2004;48:647–650 [DOI] [PubMed] [Google Scholar]
- 9. Fassnacht M, Allolio B. Clinical management of adrenocortical carcinoma. Best Pract Res Clin Endocrinol Metab. 2009;23:273–289 [DOI] [PubMed] [Google Scholar]
- 10. Luton JP, Cerdas S, Billaud L, et al. Clinical features of adrenocortical carcinoma, prognostic factors, and the effect of mitotane therapy. N Engl J Med. 1990;322:1195–1201 [DOI] [PubMed] [Google Scholar]
- 11. Bilimoria KY, Shen WT, Elaraj D, et al. Adrenocortical carcinoma in the United States: treatment utilization and prognostic factors. Cancer. 2008;113:3130–3136 [DOI] [PubMed] [Google Scholar]
- 12. Wajchenberg BL, Albergaria Pereira MA, et al. Adrenocortical carcinoma: clinical and laboratory observations. Cancer. 2000;88:711–736 [PubMed] [Google Scholar]
- 13. Wooten MD, King DK. Adrenal cortical carcinoma. Epidemiology and treatment with mitotane and a review of the literature. Cancer. 1993;72:3145–3155 [DOI] [PubMed] [Google Scholar]
- 14. Wasserman JD, Zambetti GP, Malkin D. Towards an understanding of the role of p53 in adrenocortical carcinogenesis. Mol Cell Endocrinol. 2012;351:101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schteingart DE, Doherty GM, Gauger PG, et al. Management of patients with adrenal cancer: recommendations of an international consensus conference. Endocr Relat Cancer. 2005;12:667–680 [DOI] [PubMed] [Google Scholar]
- 16. Crucitti F, Bellantone R, Ferrante A, Boscherini M, Crucitti P. The Italian Registry for Adrenal Cortical Carcinoma: analysis of a multiinstitutional series of 129 patients. The ACC Italian Registry Study Group. Surgery. 1996;119:161–170 [DOI] [PubMed] [Google Scholar]
- 17. Michalkiewicz E, Sandrini R, Figueiredo B, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22:838–845 [DOI] [PubMed] [Google Scholar]
- 18. Hsing AW, Nam JM, Co Chien HT, McLaughlin JK, Fraumeni JF., Jr Risk factors for adrenal cancer: an exploratory study. Int J Cancer. 1996;65:432–436 [DOI] [PubMed] [Google Scholar]
- 19. Icard P, Goudet P, Charpenay C, et al. Adrenocortical carcinomas: surgical trends and results of a 253-patient series from the French Association of Endocrine Surgeons study group. World J Surg. 2001;25:891–897 [DOI] [PubMed] [Google Scholar]
- 20. Sirianni R, Zolea F, Chimento A, et al. Targeting estrogen receptor-α reduces adrenocortical cancer (ACC) cell growth in vitro and in vivo: potential therapeutic role of selective estrogen receptor modulators (SERMs) for ACC treatment. J Clin Endocrinol Metab. 2012;97:E2238–E2250 [DOI] [PubMed] [Google Scholar]
- 21. Kerber RA, O'Brien E. A cohort study of cancer risk in relation to family histories of cancer in the Utah population database. Cancer. 2005;103:1906–1915 [DOI] [PubMed] [Google Scholar]
- 22. Dong C, Hemminki K. Modification of cancer risks in offspring by sibling and parental cancers from 2,112,616 nuclear families. Int J Cancer. 2001;92:144–150 [PubMed] [Google Scholar]
- 23. Fraumeni JF, Jr, Miller RW. Adrenocortical neoplasms with hemihypertrophy, brain tumors, and other disorders. J Pediatr. 1967;70:129–138 [DOI] [PubMed] [Google Scholar]
- 24. Nader S, Hickey RC, Sellin RV, Samaan NA. Adrenal cortical carcinoma. A study of 77 cases. Cancer. 1983;52:707–711 [DOI] [PubMed] [Google Scholar]
- 25. Fassnacht M, Allolio B. Epidemiology of adrenocortical carcinoma. In: Hammer G, Else T, eds. Adrenocortical Carcinoma. 1st ed New York, NY: Springer; 2010:23–29 [Google Scholar]
- 26. Venkatesh S, Hickey RC, Sellin RV, Fernandez JF, Samaan NA. Adrenal cortical carcinoma. Cancer. 1989;64:765–769 [DOI] [PubMed] [Google Scholar]
- 27. Lima Lde O, Lerario AM, Alencar GA, et al. Clinical and molecular aspects of a pediatric metachronous adrenocortical tumor. Arq Bras Endocrinol Metabol. 2011;55:72–77 [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez-Galindo C, Figueiredo BC, Zambetti GP, Ribeiro RC. Biology, clinical characteristics, and management of adrenocortical tumors in children. Pediatr Blood Cancer. 2005;45:265–273 [DOI] [PubMed] [Google Scholar]
- 29. Varley JM, McGown G, Thorncroft M, et al. Are there low-penetrance TP53 alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65:995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wagner J, Portwine C, Rabin K, Leclerc JM, Narod SA, Malkin D. High frequency of germline p53 mutations in childhood adrenocortical cancer. J Natl Cancer Inst. 1994;86:1707–1710 [DOI] [PubMed] [Google Scholar]
- 31. Li FP, Fraumeni JF, Jr, Mulvihill JJ, et al. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362 [PubMed] [Google Scholar]
- 32. Bougeard G, Sesboüé R, Baert-Desurmont S, et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45:535–538 [DOI] [PubMed] [Google Scholar]
- 33. Chompret A, Abel A, Stoppa-Lyonnet D, et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet. 2001;38:43–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chompret A, Brugières L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herrmann LJ, Heinze B, Fassnacht M, et al. TP53 germline mutations in adult patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2012;97:E476–E485 [DOI] [PubMed] [Google Scholar]
- 36. Raymond VM, Else T, Everett JN, Long JM, Gruber SB, Hammer GD. Prevalence of germline TP53 mutations in a prospective series of unselected patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:E119–E125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Custódio G, Komechen H, Figueiredo FR, Fachin ND, Pianovski MA, Figueiredo BC. Molecular epidemiology of adrenocortical tumors in southern Brazil. Mol Cell Endocrinol. 2012;351:44–51 [DOI] [PubMed] [Google Scholar]
- 38. Latronico AC, Pinto EM, Domenice S, et al. An inherited mutation outside the highly conserved DNA-binding domain of the p53 tumor suppressor protein in children and adults with sporadic adrenocortical tumors. J Clin Endocrinol Metab. 2001;86:4970–4973 [DOI] [PubMed] [Google Scholar]
- 39. Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330–9335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Custódio G, Parise GA, Kiesel Filho N, et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J Clin Oncol. 2013;31:2619–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weksberg R, Shuman C, Smith AC. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:12–23 [DOI] [PubMed] [Google Scholar]
- 42. Beckwith J. Extreme cytomegaly of the adrenal fetal cortex, omphalocele, hyperplasia of kidneys and pancreas, and Leydig-cell hyperplasia: another syndrome? Paper presented at: 11th Annual Meeting of the Western Society for Pediatric Research; November 11–12, 1963, Los Angeles, CA [Google Scholar]
- 43. Lapunzina P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am J Med Genet C Semin Med Genet. 2005;137C:53–71 [DOI] [PubMed] [Google Scholar]
- 44. Wiedemann H. Tumours and hemihypertrophy associated with Wiedemann-Beckwith syndrome. Eur J Pediatr. 1983;141:129 [Google Scholar]
- 45. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome: a prospective study of 107 cases and comparison with 1009 cases from the literature. Medicine (Baltimore). 2004;83:43–83 [DOI] [PubMed] [Google Scholar]
- 46. Skogseid B, Rastad J, Gobl A, et al. Adrenal lesion in multiple endocrine neoplasia type 1. Surgery. 1995;118:1077–1082 [DOI] [PubMed] [Google Scholar]
- 47. Waldmann J, Bartsch DK, Kann PH, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg. 2007;392:437–443 [DOI] [PubMed] [Google Scholar]
- 48. Gatta-Cherifi B, Chabre O, Murat A, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol. 2012;166:269–279 [DOI] [PubMed] [Google Scholar]
- 49. Griniatsos JE, Dimitriou N, Zilos A, et al. Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol. 2011;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Langer P, Cupisti K, Bartsch DK, et al. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg. 2002;26:891–896 [DOI] [PubMed] [Google Scholar]
- 51. Skogseid B, Larsson C, Lindgren PG, et al. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1992;75:76–81 [DOI] [PubMed] [Google Scholar]
- 52. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97:2990–3011 [DOI] [PubMed] [Google Scholar]
- 53. Karamurzin Y, Zeng Z, Stadler ZK, et al. Unusual DNA mismatch repair-deficient tumors in Lynch syndrome: a report of new cases and review of the literature. Hum Pathol. 2012;43:1677–1687 [DOI] [PubMed] [Google Scholar]
- 54. Medina-Arana V, Delgado L, González L, et al. Adrenocortical carcinoma, an unusual extracolonic tumor associated with Lynch II syndrome. Fam Cancer. 2011;10:265–271 [DOI] [PubMed] [Google Scholar]
- 55. Berends MJ, Cats A, Hollema H, et al. Adrenocortical adenocarcinoma in an MSH2 carrier: coincidence or causal relation? Hum Pathol. 2000;31:1522–1527 [DOI] [PubMed] [Google Scholar]
- 56. Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ. Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Mod Pathol. 2004;17:981–989 [DOI] [PubMed] [Google Scholar]
- 57. Stoffel E, Mukherjee B, Raymond VM, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009;137:1621–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Weissman SM, Burt R, Church J, et al. Identification of individuals at risk for Lynch syndrome using targeted evaluations and genetic testing: National Society of Genetic Counselors and the Collaborative Group of the Americas on Inherited Colorectal Cancer joint practice guideline. J Genetic Couns. 2012;21:484–493 [DOI] [PubMed] [Google Scholar]
- 59. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–1860 [DOI] [PubMed] [Google Scholar]
- 60. Dinh TA, Rosner BI, Atwood JC, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res. 2011;4:9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834 [DOI] [PubMed] [Google Scholar]
- 62. Raymond VM, Everett JN, Furtado LV, et al. Adrenocortical carcinoma is a Lynch syndrome-associated cancer. J Clin Oncol. 2013;31:3012–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takazawa R, Ajima J, Yamauchi A, Goto M. Unusual double primary neoplasia: adrenocortical and ureteral carcinomas in werner syndrome. Urol Int. 2004;72:168–170 [DOI] [PubMed] [Google Scholar]
- 64. Seki M, Tanaka K, Kikuchi-Yanoshita R, et al. Loss of normal allele of the APC gene in an adrenocortical carcinoma from a patient with familial adenomatous polyposis. Hum Genet. 1992;89:298–300 [DOI] [PubMed] [Google Scholar]
- 65. Wakatsuki S, Sasano H, Matsui T, Nagashima K, Toyota T, Horii A. Adrenocortical tumor in a patient with familial adenomatous polyposis: a case associated with a complete inactivating mutation of the APC gene and unusual histological features. Hum Pathol. 1998;29:302–306 [DOI] [PubMed] [Google Scholar]
- 66. Painter TA, Jagelman DG. Adrenal adenomas and adrenal carcinomas in association with hereditary adenomatosis of the colon and rectum. Cancer. 1985;55:2001–2004 [DOI] [PubMed] [Google Scholar]
- 67. Fienman NL, Yakovac WC. Neurofibromatosis in childhood. J Pediatr. 1970;76:339–346 [DOI] [PubMed] [Google Scholar]
- 68. Sørensen SA, Mulvihill JJ, Nielsen A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N Engl J Med. 1986;314:1010–1015 [DOI] [PubMed] [Google Scholar]
- 69. Else T. Overview of genetic syndromes associated with adrenocortical cancer. In: Hammer GD, Else T, eds. Adrenocortical Carcinoma. 1st ed New York, NY: Springer; 2010:153–172 [Google Scholar]
- 70. Marshall WH, Martin FI, Mackay IR. Gardner's syndrome with adrenal carcinoma. Australas Ann Med. 1967;16:242–244 [DOI] [PubMed] [Google Scholar]
- 71. Traill Z, Tuson J, Woodham C. Adrenal carcinoma in a patient with Gardner's syndrome: imaging findings. AJR Am J Roentgenol. 1995;165:1460–1461 [DOI] [PubMed] [Google Scholar]
- 72. Wagner AS, Fleitz JM, Kleinschmidt-Demasters BK. Pediatric adrenal cortical carcinoma: brain metastases and relationship to NF-1, case reports and review of the literature. J Neurooncol. 2005;75:127–133 [DOI] [PubMed] [Google Scholar]
- 73. Anselmo J, Medeiros S, Carneiro V, et al. A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer. J Clin Endocrinol Metab. 2012;97:351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Morin E, Mete O, Wasserman JD, Joshua AM, Asa SL, Ezzat S. Carney complex with adrenal cortical carcinoma. J Clin Endocrinol Metab. 2012;97:E202–206 [DOI] [PubMed] [Google Scholar]
- 75. Bauman A, Bauman CG. Virilizing adrenocortical carcinoma. Development in a patient with salt-losing congenital adrenal hyperplasia. JAMA. 1982;248:3140–3141 [DOI] [PubMed] [Google Scholar]
- 76. Barzon L, Maffei P, Sonino N, Pilon C, Baldazzi L, Balsamo A, Del Maschio O, Masi G, Trevisan M, Pacenti M, Fallo F. The role of 21-hydroxylase in the pathogenesis of adrenal masses: review of the literature and focus on our own experience. J Endocrinol Invest. 2007;30:615–623 [DOI] [PubMed] [Google Scholar]
- 77. Nermoen I, Rorvik J, Holmedal SH, et al. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2011;75:753–759 [DOI] [PubMed] [Google Scholar]
- 78. NIH ClinicalTrials.gov. In:
- 79. Ishikura K, Takamura T, Takeshita Y, et al. Cushing's syndrome and big IGF-II associated hypoglycaemia in a patient with adrenocortical carcinoma. BMJ Case Rep. 2010;2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Anderson H. A tumor of the adrenal gland with fatal hypoglycemia. Am J Med Sci. 1930;180:71–79 [Google Scholar]
- 81. Rustin MH, Bowcock SJ, Coomes EN. Adrenocortical carcinoma with tumour-induced hypoglycaemia. J R Soc Med. 1983;76:74–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yamanaka K, Iitaka M, Inaba M, Morita T, Sasano H, Katayama S. A case of renin-producing adrenocortical cancer. Endocr J. 2000;47:119–125 [DOI] [PubMed] [Google Scholar]
- 83. Oka T, Onoe K, Nishimura K, et al. Erythropoietin-producing adrenocortical carcinoma. Urol Int. 1996;56:246–249 [DOI] [PubMed] [Google Scholar]
- 84. Schteingart DE, Giordano TJ, Benitez RS, et al. Overexpression of CXC chemokines by an adrenocortical carcinoma: a novel clinical syndrome. J Clin Endocrinol Metab. 2001;86:3968–3974 [DOI] [PubMed] [Google Scholar]
- 85. Abiven G, Coste J, Groussin L, et al. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91:2650–2655 [DOI] [PubMed] [Google Scholar]
- 86. Seccia TM, Fassina A, Nussdorfer GG, Pessina AC, Rossi GP. Aldosterone-producing adrenocortical carcinoma: an unusual cause of Conn's syndrome with an ominous clinical course. Endocr Relat Cancer. 2005;12:149–159 [DOI] [PubMed] [Google Scholar]
- 87. Egoshi K, Masai M, Nagao K, Ito H. 11-Deoxycorticosterone-producing adrenocortical carcinoma. Urol Int. 1998;61:251–253 [DOI] [PubMed] [Google Scholar]
- 88. Müssig K, Wehrmann M, Horger M, Maser-Gluth C, Häring HU, Overkamp D. Adrenocortical carcinoma producing 11-deoxycorticosterone: a rare cause of mineralocorticoid hypertension. J Endocrinol Invest. 2005;28:61–65 [DOI] [PubMed] [Google Scholar]
- 89. Sturgeon C, Shen WT, Clark OH, Duh QY, Kebebew E. Risk assessment in 457 adrenal cortical carcinomas: how much does tumor size predict the likelihood of malignancy? J Am Coll Surg. 2006;202:423–430 [DOI] [PubMed] [Google Scholar]
- 90. Paton BL, Novitsky YW, Zerey M, et al. Outcomes of adrenal cortical carcinoma in the United States. Surgery 2006;140:914–920; discussion 919–920 [DOI] [PubMed] [Google Scholar]
- 91. Fassnacht M, Johanssen S, Quinkler M, et al. Limited prognostic value of the 2004 International Union Against Cancer staging classification for adrenocortical carcinoma: proposal for a Revised TNM Classification. Cancer. 2009;115:243–250 [DOI] [PubMed] [Google Scholar]
- 92. Allolio B, Hahner S, Weismann D, Fassnacht M. Management of adrenocortical carcinoma. Clin Endocrinol (Oxf). 2004;60:273–287 [DOI] [PubMed] [Google Scholar]
- 93. Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93:1526–1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Grondal S, Eriksson B, Hagenas L, Werner S, Curstedt T. Steroid profile in urine: a useful tool in the diagnosis and follow up of adrenocortical carcinoma. Acta Endocrinol (Copenh). 1990;122:656–663 [DOI] [PubMed] [Google Scholar]
- 95. Arlt W, Biehl M, Taylor AE, et al. Urine steroid metabolomics as a biomarker tool for detecting malignancy in adrenal tumors. J Clin Endocrinol Metab. 2011;96:3775–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Song G, Joe BN, Yeh BM, Meng MV, Westphalen AC, Coakley FV. Risk of catecholamine crisis in patients undergoing resection of unsuspected pheochromocytoma. Int Braz J Urol. 2011;37:35–40; discussion 40–41 [DOI] [PubMed] [Google Scholar]
- 97. Johnson PT, Horton KM, Fishman EK. Adrenal mass imaging with multidetector CT: pathologic conditions, pearls, and pitfalls. Radiographics. 2009;29:1333–1351 [DOI] [PubMed] [Google Scholar]
- 98. Bharwani N, Rockall AG, Sahdev A, et al. Adrenocortical carcinoma: the range of appearances on CT and MRI. AJR Am J Roentgenol. 2011;196:W706–W714 [DOI] [PubMed] [Google Scholar]
- 99. Dunnick NR, Heaston D, Halvorsen R, Moore AV, Korobkin M. CT appearance of adrenal cortical carcinoma. J Comput Assist Tomogr. 1982;6:978–982 [DOI] [PubMed] [Google Scholar]
- 100. Berland LL, Silverman SG, Gore RM, et al. Managing incidental findings on abdominal CT: white paper of the ACR incidental findings committee. J Am Coll Radiol. 2010;7:754–773 [DOI] [PubMed] [Google Scholar]
- 101. Hussain S, Belldegrun A, Seltzer SE, Richie JP, Gittes RF, Abrams HL. Differentiation of malignant from benign adrenal masses: predictive indices on computed tomography. AJR Am J Roentgenol. 1985;144:61–65 [DOI] [PubMed] [Google Scholar]
- 102. Haider MA, Ghai S, Jhaveri K, Lockwood G. Chemical shift MR imaging of hyperattenuating (>10 HU) adrenal masses: does it still have a role? Radiology. 2004;231:711–716 [DOI] [PubMed] [Google Scholar]
- 103. Caoili EM, Korobkin M, Francis IR, Cohan RH, Dunnick NR. Delayed enhanced CT of lipid-poor adrenal adenomas. AJR Am J Roentgenol. 2000;175:1411–1415 [DOI] [PubMed] [Google Scholar]
- 104. Slattery JM, Blake MA, Kalra MK, et al. Adrenocortical carcinoma: contrast washout characteristics on CT. AJR Am J Roentgenol. 2006;187:W21–W24 [DOI] [PubMed] [Google Scholar]
- 105. Elsayes KM, Mukundan G, Narra VR, et al. Adrenal masses: MR imaging features with pathologic correlation. Radiographics. 2004;24(Suppl 1):S73–S86 [DOI] [PubMed] [Google Scholar]
- 106. Fishman EK, Deutch BM, Hartman DS, Goldman SM, Zerhouni EA, Siegelman SS. Primary adrenocortical carcinoma: CT evaluation with clinical correlation. AJR Am J Roentgenol. 1987;148:531–535 [DOI] [PubMed] [Google Scholar]
- 107. Egbert N, Elsayes KM, Azar S, Caoili EM. Computed tomography of adrenocortical carcinoma containing macroscopic fat. Cancer Imaging. 2010;10:198–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Groussin L, Bonardel G, Silvéra S, et al. 18F-Fluorodeoxyglucose positron emission tomography for the diagnosis of adrenocortical tumors: a prospective study in 77 operated patients. J Clin Endocrinol Metab. 2009;94:1713–1722 [DOI] [PubMed] [Google Scholar]
- 109. Sundin A. Imaging of adrenal masses with emphasis on adrenocortical tumors. Theranostics. 2012;2:516–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hahner S, Stuermer A, Kreissl M, et al. [123I]Iodometomidate for molecular imaging of adrenocortical cytochrome P450 family 11B enzymes. J Clin Endocrinol Metab. 2008;93:2358–2365 [DOI] [PubMed] [Google Scholar]
- 111. Mackie GC, Shulkin BL, Ribeiro RC, et al. Use of [18F]fluorodeoxyglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:2665–2671 [DOI] [PubMed] [Google Scholar]
- 112. Leboulleux S, Dromain C, Bonniaud G, et al. Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. J Clin Endocrinol Metab. 2006;91:920–925 [DOI] [PubMed] [Google Scholar]
- 113. Khan TS, Sundin A, Juhlin C, Långström B, Bergström M, Eriksson B. 11C-Metomidate PET imaging of adrenocortical cancer. Eur J Nucl Med Mol Imaging. 2003;30:403–410 [DOI] [PubMed] [Google Scholar]
- 114. Terzolo M, Stigliano A, Chiodini I, et al. ; Italian Association of Clinical Endocrinologists AME position statement on adrenal incidentaloma. Eur J Endocrinol. 2011;164:851–870 [DOI] [PubMed] [Google Scholar]
- 115. Funder JW, Carey RM, Fardella C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:3266–3281 [DOI] [PubMed] [Google Scholar]
- 116. Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85:637–644 [DOI] [PubMed] [Google Scholar]
- 117. Song JH, Chaudhry FS, Mayo-Smith WW. The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol. 2008;190:1163–1168 [DOI] [PubMed] [Google Scholar]
- 118. Zhang LJ, Yang GF, Shen W, Qi J. Imaging of primary adrenal lymphoma: case report and literature review. Acta Radiol. 2006;47:993–997 [DOI] [PubMed] [Google Scholar]
- 119. Hough AJ, Hollifield JW, Page DL, Hartmann WH. Prognostic factors in adrenal cortical tumors. A mathematical analysis of clinical and morphologic data. Am J Clin Pathol. 1979;72:390–399 [DOI] [PubMed] [Google Scholar]
- 120. van Slooten H, Schaberg A, Smeenk D, Moolenaar AJ. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985;55:766–773 [DOI] [PubMed] [Google Scholar]
- 121. Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. Am J Surg Pathol. 1984;8:163–169 [DOI] [PubMed] [Google Scholar]
- 122. Aubert S, Wacrenier A, Leroy X, et al. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002;26:1612–1619 [DOI] [PubMed] [Google Scholar]
- 123. Giordano TJ. Adrenocortical tumors: an integrated clinical, pathologic, and molecular approach at the University of Michigan. Arch Pathol Lab Med. 2010;134:1440–1443 [DOI] [PubMed] [Google Scholar]
- 124. Volante M, Bollito E, Sperone P, et al. Clinicopathological study of a series of 92 adrenocortical carcinomas: from a proposal of simplified diagnostic algorithm to prognostic stratification. Histopathology. 2009;55:535–543 [DOI] [PubMed] [Google Scholar]
- 125. Papotti M, Libè R, Duregon E, Volante M, Bertherat J, Tissier F. The Weiss score and beyond–histopathology for adrenocortical carcinoma. Horm Cancer. 2011;2:333–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cibas ES, Medeiros LJ, Weinberg DS, Gelb AB, Weiss LM. Cellular DNA profiles of benign and malignant adrenocortical tumors. Am J Surg Pathol. 1990;14:948–955 [DOI] [PubMed] [Google Scholar]
- 127. Arola J, Salmenkivi K, Liu J, Kahri AI, Heikkilä P. p53 and Ki67 in adrenocortical tumors. Endocr Res. 2000;26:861–865 [DOI] [PubMed] [Google Scholar]
- 128. Sasano H, Imatani A, Shizawa S, Suzuki T, Nagura H. Cell proliferation and apoptosis in normal and pathologic human adrenal. Mod Pathol. 1995;8:11–17 [PubMed] [Google Scholar]
- 129. Morimoto R, Satoh F, Murakami O, et al. Immunohistochemistry of a proliferation marker Ki67/MIB1 in adrenocortical carcinomas: Ki67/MIB1 labeling index is a predictor for recurrence of adrenocortical carcinomas. Endocr J. 2008;55:49–55 [DOI] [PubMed] [Google Scholar]
- 130. Giordano TJ. The argument for mitotic rate-based grading for the prognostication of adrenocortical carcinoma. Am J Surg Pathol. 2011;35:471–473 [DOI] [PubMed] [Google Scholar]
- 131. Zhang H, Bu H, Chen H, et al. Comparison of immunohistochemical markers in the differential diagnosis of adrenocortical tumors: immunohistochemical analysis of adrenocortical tumors. Appl Immunohistochem Mol Morphol. 2008;16:32–39 [DOI] [PubMed] [Google Scholar]
- 132. Arola J, Liu J, Heikkilä P, et al. Expression of inhibin alpha in adrenocortical tumours reflects the hormonal status of the neoplasm. J Endocrinol. 2000;165:223–229 [DOI] [PubMed] [Google Scholar]
- 133. Arola J, Liu J, Heikkilä P, Voutilainen R, Kahri A. Expression of inhibin alpha in the human adrenal gland and adrenocortical tumors. Endocr Res. 1998;24:865–867 [DOI] [PubMed] [Google Scholar]
- 134. Jorda M, De MB, Nadji M. Calretinin and inhibin are useful in separating adrenocortical neoplasms from pheochromocytomas. Appl Immunohistochem Mol Morphol. 2002;10:67–70 [DOI] [PubMed] [Google Scholar]
- 135. Schröder S, Padberg BC, Achilles E, Holl K, Dralle H, Kloppel G. Immunocytochemistry in adrenocortical tumours: a clinicomorphological study of 72 neoplasms. Virchows Arch A Pathol Anat Histopathol. 1992;420:65–70 [DOI] [PubMed] [Google Scholar]
- 136. Ghorab Z, Jorda M, Ganjei P, Nadji M. Melan A (A103) is expressed in adrenocortical neoplasms but not in renal cell and hepatocellular carcinomas. Appl Immunohistochem Mol Morphol. 2003;11:330–333 [DOI] [PubMed] [Google Scholar]
- 137. Sbiera S, Schmull S, Assie G, et al. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J Clin Endocrinol Metab. 2010;95:E161–E171 [DOI] [PubMed] [Google Scholar]
- 138. Duregon E, Volante M, Giorcelli J, Terzolo M, Lalli E, Papotti M. Diagnostic and prognostic role of steroidogenic factor 1 in adrenocortical carcinoma: a validation study focusing on clinical and pathologic correlates. Hum Pathol. 2013;44:822–828 [DOI] [PubMed] [Google Scholar]
- 139. Weiss LM, Medeiros LJ, Vickery AL., Jr Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol. 1989;13:202–206 [DOI] [PubMed] [Google Scholar]
- 140. Ohgaki H, Kleihues P, Heitz PU. p53 mutations in sporadic adrenocortical tumors. Int J Cancer. 1993;54:408–410 [DOI] [PubMed] [Google Scholar]
- 141. Lin SR, Lee YJ, Tsai JH. Mutations of the p53 gene in human functional adrenal neoplasms. J Clin Endocrinol Metab. 1994;78:483–491 [DOI] [PubMed] [Google Scholar]
- 142. Reincke M, Karl M, Travis WH, Mastorakos G, Allolio B, Linehan HM, Chrousos GP. p53 mutations in human adrenocortical neoplasms: immunohistochemical and molecular studies. J Clin Endocrinol Metab. 1994;78:790–794 [DOI] [PubMed] [Google Scholar]
- 143. Heaton JH, Wood MA, Kim AC, et al. Progression to adrenocortical tumorigenesis in mice and humans through insulin-like growth factor 2 and β-catenin. Am J Pathol. 2012;181:1017–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Berthon A, Martinez A, Bertherat J, Val P. Wnt/β-catenin signalling in adrenal physiology and tumour development. Mol Cell Endocrinol. 2012;351:87–95 [DOI] [PubMed] [Google Scholar]
- 145. Leal LF, Mermejo LM, Ramalho LZ, et al. Wnt/β-catenin pathway deregulation in childhood adrenocortical tumors. J Clin Endocrinol Metab. 2011;96:3106–3114 [DOI] [PubMed] [Google Scholar]
- 146. Gaujoux S, Grabar S, Fassnacht M, et al. β-Catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res. 2011;17:328–336 [DOI] [PubMed] [Google Scholar]
- 147. El Wakil A, Lalli E. The Wnt/β-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol. 2011;332:32–37 [DOI] [PubMed] [Google Scholar]
- 148. Masi G, Lavezzo E, Iacobone M, Favia G, Palù G, Barzon L. Investigation of BRAF and CTNNB1 activating mutations in adrenocortical tumors. J Endocrinol Invest. 2009;32:597–600 [DOI] [PubMed] [Google Scholar]
- 149. Tissier F, Cavard C, Groussin L, et al. Mutations of β-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7627 [DOI] [PubMed] [Google Scholar]
- 150. Assie G, Giordano TJ, Bertherat J. Gene expression profiling in adrenocortical neoplasia. Mol Cell Endocrinol. 2012;351:111–117 [DOI] [PubMed] [Google Scholar]
- 151. Ribeiro RC, Michalkiewicz EL, Figueiredo BC, et al. Adrenocortical tumors in children. Braz J Med Biol Res. 2000;33:1225–1234 [DOI] [PubMed] [Google Scholar]
- 152. Cagle PT, Hough AJ, Pysher TJ, et al. Comparison of adrenal cortical tumors in children and adults. Cancer. 1986;57:2235–2237 [DOI] [PubMed] [Google Scholar]
- 153. Dehner LP, Hill DA. Adrenal cortical neoplasms in children: why so many carcinomas and yet so many survivors? Pediatr Dev Pathol. 2009;12:284–291 [DOI] [PubMed] [Google Scholar]
- 154. Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27:867–881 [DOI] [PubMed] [Google Scholar]
- 155. Magro G, Esposito G, Cecchetto G, et al. Pediatric adrenocortical tumors: morphological diagnostic criteria and immunohistochemical expression of matrix metalloproteinase type 2 and human leucocyte-associated antigen (HLA) class II antigens. Results from the Italian Pediatric Rare Tumor (TREP) Study project. Hum Pathol. 2012;43:31–39 [DOI] [PubMed] [Google Scholar]
- 156. West AN, Neale GA, Pounds S, et al. Gene expression profiling of childhood adrenocortical tumors. Cancer Res. 2007;67:600–608 [DOI] [PubMed] [Google Scholar]
- 157. Faria AM, Almeida MQ. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol Cell Endocrinol. 2012;351:52–57 [DOI] [PubMed] [Google Scholar]
- 158. Erlandson RA, Reuter VE. Oncocytic adrenal cortical adenoma. Ultrastruct Pathol. 1991;15:539–547 [DOI] [PubMed] [Google Scholar]
- 159. Macchi C, Rebuffat P, Blandamura S, et al. Adrenocortical oncocytoma: case report and review of the literature. Tumori. 1998;84:403–407 [DOI] [PubMed] [Google Scholar]
- 160. Hoang MP, Ayala AG, Albores-Saavedra J. Oncocytic adrenocortical carcinoma: a morphologic, immunohistochemical and ultrastructural study of four cases. Mod Pathol. 2002;15:973–978 [DOI] [PubMed] [Google Scholar]
- 161. Song SY, Park S, Kim SR, Suh YL. Oncocytic adrenocortical carcinomas: a pathological and immunohistochemical study of four cases in comparison with conventional adrenocortical carcinomas. Pathol Int. 2004;54:603–610 [DOI] [PubMed] [Google Scholar]
- 162. Ohtake H, Kawamura H, Matsuzaki M, et al. Oncocytic adrenocortical carcinoma. Ann Diagn Pathol. 2010;14:204–208 [DOI] [PubMed] [Google Scholar]
- 163. Wong DD, Spagnolo DV, Bisceglia M, Havlat M, McCallum D, Platten MA. Oncocytic adrenocortical neoplasms–a clinicopathologic study of 13 new cases emphasizing the importance of their recognition. Hum Pathol. 2011;42:489–499 [DOI] [PubMed] [Google Scholar]
- 164. Lin BT, Bonsib SM, Mierau GW, Weiss LM, Medeiros LJ. Oncocytic adrenocortical neoplasms: a report of seven cases and review of the literature. Am J Surg Pathol. 1998;22:603–614 [DOI] [PubMed] [Google Scholar]
- 165. Bisceglia M, Ludovico O, Di Mattia A, et al. Adrenocortical oncocytic tumors: report of 10 cases and review of the literature. Int J Surg Pathol. 2004;12:231–243 [DOI] [PubMed] [Google Scholar]
- 166. Brown FM, Gaffey TA, Wold LE, Lloyd RV. Myxoid neoplasms of the adrenal cortex: a rare histologic variant. Am J Surg Pathol. 2000;24:396–401 [DOI] [PubMed] [Google Scholar]
- 167. Suresh B, Kishore TA, Albert AS, Joy A. Myxoid adrenal cortical carcinoma–a rare variant of adrenocortical carcinoma. Indian J Med Sci. 2005;59:505–507 [PubMed] [Google Scholar]
- 168. Karim RZ, Wills EJ, McCarthy SW, Scolyer RA. Myxoid variant of adrenocortical carcinoma: report of a unique case. Pathol Int. 2006;56:89–94 [DOI] [PubMed] [Google Scholar]
- 169. Raparia K, Ayala AG, Sienko A, Zhai QJ, Ro JY. Myxoid adrenal cortical neoplasms. Ann Diagn Pathol. 2008;12:344–348 [DOI] [PubMed] [Google Scholar]
- 170. Papotti M, Volante M, Duregon E, et al. Adrenocortical tumors with myxoid features: a distinct morphologic and phenotypical variant exhibiting malignant behavior. Am J Surg Pathol. 2010;34:973–983 [DOI] [PubMed] [Google Scholar]
- 171. Hsieh MS, Chen JH, Lin LW. Myxoid adrenal cortical carcinoma presenting as primary hyperaldosteronism: case report and review of the literature. Int J Surg Pathol. 2011;19:803–807 [DOI] [PubMed] [Google Scholar]
- 172. Zhang J, Sun J, Liang Z, Gao J, Zeng X, Liu T. Myxoid adrenocortical neoplasms: a study of the clinicopathologic features and EGFR gene status of ten Chinese cases. Am J Clin Pathol. 2011;136:783–792 [DOI] [PubMed] [Google Scholar]
- 173. Sheng JY, He HC, Zhu Y, et al. Myxoid adrenal cortical tumor: report of four cases. Chin Med J (Engl). 2012;125:1672–1674 [PubMed] [Google Scholar]
- 174. Giordano TJ, Thomas DG, Kuick R, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol. 2003;162:521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Coli A, Di Giorgio A, Castri F, Destito C, Marin AW, Bigotti G. Sarcomatoid carcinoma of the adrenal gland: a case report and review of literature. Pathol Res Pract. 2010;206:59–65 [DOI] [PubMed] [Google Scholar]
- 176. Ragazzon B, Assié G, Bertherat J. Transcriptome analysis of adrenocortical cancers: from molecular classification to the identification of new treatments. Endocrine-related cancer. 2011;18:R15–R27 [DOI] [PubMed] [Google Scholar]
- 177. Beuschlein F, Reincke M, Karl M, et al. Clonal composition of human adrenocortical neoplasms. Cancer Res. 1994;54:4927–4932 [PubMed] [Google Scholar]
- 178. Gicquel C, Leblond-Francillard M, Bertagna X, et al. Clonal analysis of human adrenocortical carcinomas and secreting adenomas. Clin Endocrinol (Oxf). 1994;40:465–477 [DOI] [PubMed] [Google Scholar]
- 179. Blanes A, Diaz-Cano SJ. DNA and kinetic heterogeneity during the clonal evolution of adrenocortical proliferative lesions. Hum Pathol. 2006;37:1295–1303 [DOI] [PubMed] [Google Scholar]
- 180. Klein FA, Kay S, Ratliff JE, White FK, Newsome HH. Flow cytometric determinations of ploidy and proliferation patterns of adrenal neoplasms: an adjunct to histological classification. J Urol. 1985;134:862–866 [DOI] [PubMed] [Google Scholar]
- 181. Bowlby LS, DeBault LE, Abraham SR. Flow cytometric analysis of adrenal cortical tumor DNA. Relationship between cellular DNA and histopathologic classification. Cancer. 1986;58:1499–1505 [DOI] [PubMed] [Google Scholar]
- 182. Amberson JB, Vaughan ED, Jr, Gray GF, Naus GJ. Flow cytometric determination of nuclear DNA content in benign adrenal pheochromocytomas. Urology. 1987;30:102–104 [DOI] [PubMed] [Google Scholar]
- 183. Kjellman M, Kallioniemi OP, Karhu R, et al. Genetic aberrations in adrenocortical tumors detected using comparative genomic hybridization correlate with tumor size and malignancy. Cancer Res. 1996;56:4219–4223 [PubMed] [Google Scholar]
- 184. Gicquel C, Bertagna X, Gaston V, et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res. 2001;61:6762–6767 [PubMed] [Google Scholar]
- 185. Kjellman M, Roshani L, Teh BT, et al. Genotyping of adrenocortical tumors: very frequent deletions of the MEN1 locus in 11q13 and of a 1-centimorgan region in 2p16. J Clin Endocrinol Metab. 1999;84:730–735 [DOI] [PubMed] [Google Scholar]
- 186. Zhao J, Speel EJ, Muletta-Feurer S, et al. Analysis of genomic alterations in sporadic adrenocortical lesions. Gain of chromosome 17 is an early event in adrenocortical tumorigenesis. Am J Pathol. 1999;155:1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Dohna M, Reincke M, Mincheva A, Allolio B, Solinas-Toldo S, Lichter P. Adrenocortical carcinoma is characterized by a high frequency of chromosomal gains and high-level amplifications. Genes Chromosomes Cancer. 2000;28:145–152 [PubMed] [Google Scholar]
- 188. Barreau O, Assié G, Wilmot-Roussel H, et al. Identification of a CpG Island Methylator Phenotype in Adrenocortical Carcinomas. J Clin Endocrinol Metab. 2013;98:E174–E184 [DOI] [PubMed] [Google Scholar]
- 189. Stephan EA, Chung TH, Grant CS, et al. Adrenocortical carcinoma survival rates correlated to genomic copy number variants. Mol Cancer Ther. 2008;7:425–431 [DOI] [PubMed] [Google Scholar]
- 190. de Fraipont F, El Atifi M, Cherradi N, et al. Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic acid microarrays identifies several candidate genes as markers of malignancy. J Clin Endocrinol Metab. 2005;90:1819–1829 [DOI] [PubMed] [Google Scholar]
- 191. de Reyniès A, Assié G, Rickman DS, et al. Gene expression profiling reveals a new classification of adrenocortical tumors and identifies molecular predictors of malignancy and survival. J Clin Oncol. 2009;27:1108–1115 [DOI] [PubMed] [Google Scholar]
- 192. Giordano TJ, Kuick R, Else T, et al. Molecular classification and prognostication of adrenocortical tumors by transcriptome profiling. Clin Cancer Res. 2009;15:668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Fragoso MC, Almeida MQ, Mazzuco TL, et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: validation in a Brazilian cohort of adult and pediatric patients. Eur J Endocrinol. 2012;166:61–67 [DOI] [PubMed] [Google Scholar]
- 194. Czech B, Hannon GJ. Small RNA sorting: matchmaking for Argonautes. Nat Rev Genet. 2011;12:19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Tömböl Z, Szabó PM, Molnár V, et al. Integrative molecular bioinformatics study of human adrenocortical tumors: microRNA, tissue-specific target prediction, and pathway analysis. Endocr Relat Cancer. 2009;16:895–906 [DOI] [PubMed] [Google Scholar]
- 198. Soon PS, Tacon LJ, Gill AJ, et al. miR-195 and miR-483-5p Identified as Predictors of Poor Prognosis in Adrenocortical Cancer. Clin Cancer Res. 2009;15:7684–7692 [DOI] [PubMed] [Google Scholar]
- 199. Doghman M, El Wakil A, Cardinaud B, et al. Regulation of insulin-like growth factor-mammalian target of rapamycin signaling by microRNA in childhood adrenocortical tumors. Cancer Res. 2010;70:4666–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Patterson EE, Holloway AK, Weng J, Fojo T, Kebebew E. MicroRNA profiling of adrenocortical tumors reveals miR-483 as a marker of malignancy. Cancer. 2011;117:1630–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Veronese A, Lupini L, Consiglio J, et al. Oncogenic role of miR-483-3p at the IGF2/483 locus. Cancer Res. 2010;70:3140–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Barzon L, Chilosi M, Fallo F, et al. Molecular analysis of CDKN1C and TP53 in sporadic adrenal tumors. Eur J Endocrinol. 2001;145:207–212 [DOI] [PubMed] [Google Scholar]
- 203. Gicquel C, Le Bouc Y. Molecular markers for malignancy in adrenocortical tumors. Horm Res. 1997;47:269–272 [DOI] [PubMed] [Google Scholar]
- 204. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756 [DOI] [PubMed] [Google Scholar]
- 205. Pilon C, Pistorello M, Moscon A, et al. Inactivation of the p16 tumor suppressor gene in adrenocortical tumors. J Clin Endocrinol Metab. 1999;84:2776–2779 [DOI] [PubMed] [Google Scholar]
- 206. Gaujoux S, Tissier F, Groussin L, et al. Wnt/β-catenin and 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase A signaling pathways alterations and somatic β-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab. 2008;93:4135–4140 [DOI] [PubMed] [Google Scholar]
- 207. Tadjine M, Lampron A, Ouadi L, Horvath A, Stratakis CA, Bourdeau I. Detection of somatic β-catenin mutations in primary pigmented nodular adrenocortical disease (PPNAD). Clin Endocrinol (Oxf). 2008;69:367–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Tadjine M, Lampron A, Ouadi L, Bourdeau I. Frequent mutations of β-catenin gene in sporadic secreting adrenocortical adenomas. Clin Endocrinol (Oxf). 2008;68:264–270 [DOI] [PubMed] [Google Scholar]
- 209. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47 [DOI] [PubMed] [Google Scholar]
- 210. Voutilainen R, Miller WL. Coordinate tropic hormone regulation of mRNAs for insulin-like growth factor II and the cholesterol side-chain-cleavage enzyme, P450scc [corrected], in human steroidogenic tissues. Proc Natl Acad Sci U S A. 1987;84:1590–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. l'Allemand D, Penhoat A, Lebrethon MC, et al. Insulin-like growth factors enhance steroidogenic enzyme and corticotropin receptor messenger ribonucleic acid levels and corticotropin steroidogenic responsiveness in cultured human adrenocortical cells. J Clin Endocrinol Metab. 1996;81:3892–3897 [DOI] [PubMed] [Google Scholar]
- 212. Pham-Huu-Trung MT, Villette JM, Bogyo A, Duclos JM, Fiet J, Binoux M. Effects of insulin-like growth factor I (IGF-I) on enzymatic activity in human adrenocortical cells. Interactions with ACTH. J Steroid Biochem Mol Biol. 1991;39:903–909 [DOI] [PubMed] [Google Scholar]
- 213. Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev. 1997;18:378–403 [DOI] [PubMed] [Google Scholar]
- 214. Han VK, Lu F, Bassett N, Yang KP, Delhanty PJ, Challis JR. Insulin-like growth factor-II (IGF-II) messenger ribonucleic acid is expressed in steroidogenic cells of the developing ovine adrenal gland: evidence of an autocrine/paracrine role for IGF-II. Endocrinology. 1992;131:3100–3109 [DOI] [PubMed] [Google Scholar]
- 215. Gaston V, Le Bouc Y, Soupre V, et al. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2001;9:409–418 [DOI] [PubMed] [Google Scholar]
- 216. DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859 [DOI] [PubMed] [Google Scholar]
- 217. Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Tumour-suppressor activity of H19 RNA. Nature. 1993;365:764–767 [DOI] [PubMed] [Google Scholar]
- 218. Ilvesmäki V, Kahri AI, Miettinen PJ, Voutilainen R. Insulin-like growth factors (IGFs) and their receptors in adrenal tumors: high IGF-II expression in functional adrenocortical carcinomas. J Clin Endocrinol Metab. 1993;77:852–858 [DOI] [PubMed] [Google Scholar]
- 219. De Fraipont F, Le Moigne G, Defaye G, et al. Transcription profiling of benign and malignant adrenal tumors by cDNA macro-array analysis. Endocr Res. 2002;28:785–786 [DOI] [PubMed] [Google Scholar]
- 220. Erickson LA, Jin L, Sebo TJ, et al. Pathologic features and expression of insulin-like growth factor-2 in adrenocortical neoplasms. Endocr Pathol. 2001;12:429–435 [DOI] [PubMed] [Google Scholar]
- 221. Logié A, Boulle N, Gaston V, et al. Autocrine role of IGF-II in proliferation of human adrenocortical carcinoma NCI H295R cell line. 1999;23:23–32 [DOI] [PubMed] [Google Scholar]
- 222. Weber MM, Fottner C, Wolf E. The role of the insulin-like growth factor system in adrenocortical tumourigenesis. Eur J Clin Invest. 2000;30(Suppl 3):69–75 [DOI] [PubMed] [Google Scholar]
- 223. Almeida MQ, Fragoso MC, Lotfi CF, et al. Expression of insulin-like growth factor-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab. 2008;93:3524–3531 [DOI] [PubMed] [Google Scholar]
- 224. Wolf E, Kramer R, Blum WF, Föll J, Brem G. Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: endocrine changes and effects on body and organ growth. Endocrinology. 1994;135:1877–1886 [DOI] [PubMed] [Google Scholar]
- 225. Cecim M, Ghosh PK, Esquifino AI, et al. Elevated corticosterone levels in transgenic mice expressing human or bovine growth hormone genes. Neuroendocrinology. 1991;53:313–316 [DOI] [PubMed] [Google Scholar]
- 226. Libé R, Bertherat J. Molecular genetics of adrenocortical tumours, from familial to sporadic diseases. Eur J Endocrinol. 2005;153:477–487 [DOI] [PubMed] [Google Scholar]
- 227. Barlaskar FM, Spalding AC, Heaton JH, et al. Preclinical targeting of the type I insulin-like growth factor receptor in adrenocortical carcinoma. J Clin Endocrinol Metab. 2009;94:204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Kim AC, Reuter AL, Zubair M, et al. Targeted disruption of β-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development. 2008;135:2593–2602 [DOI] [PubMed] [Google Scholar]
- 229. Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665 [DOI] [PubMed] [Google Scholar]
- 230. Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600 [DOI] [PubMed] [Google Scholar]
- 231. Chapman A, Durand J, Ouadi L, Bourdeau I. Identification of genetic alterations of AXIN2 gene in adrenocortical tumors. J Clin Endocrinol Metab. 2011;96:E1477–E1481 [DOI] [PubMed] [Google Scholar]
- 232. Ragazzon B, Libé R, Gaujoux S, et al. Transcriptome analysis reveals that p53 and β-catenin alterations occur in a group of aggressive adrenocortical cancers. Cancer Res. 2010;70:8276–8281 [DOI] [PubMed] [Google Scholar]
- 233. Drelon C, Berthon A, Ragazzon B, et al. Analysis of the role of Igf2 in adrenal tumour development in transgenic mouse models. PLoS One. 2012;7:e44171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70 [DOI] [PubMed] [Google Scholar]
- 235. Affara NI, Robertson FM. Vascular endothelial growth factor as a survival factor in tumor-associated angiogenesis. In Vivo. 2004;18:525–542 [PubMed] [Google Scholar]
- 236. Bagri A, Kouros-Mehr H, Leong KG, Plowman GD. Use of anti-VEGF adjuvant therapy in cancer: challenges and rationale. Trends Mol Med. 2010;16:122–132 [DOI] [PubMed] [Google Scholar]
- 237. de Fraipont F, El Atifi M, Gicquel C, Bertagna X, Chambaz EM, Feige JJ. Expression of the angiogenesis markers vascular endothelial growth factor-A, thrombospondin-1, and platelet-derived endothelial cell growth factor in human sporadic adrenocortical tumors: correlation with genotypic alterations. J Clin Endocrinol Metab. 2000;85:4734–4741 [DOI] [PubMed] [Google Scholar]
- 238. Kolomecki K, Stepien H, Bartos M, Kuzdak K. Usefulness of VEGF, MMP-2, MMP-3 and TIMP-2 serum level evaluation in patients with adrenal tumours. Endocr Regul. 2001;35:9–16 [PubMed] [Google Scholar]
- 239. Wortmann S, Quinkler M, Ritter C, et al. Bevacizumab plus capecitabine as a salvage therapy in advanced adrenocortical carcinoma. Eur J Endocrinol. 2010;162:349–356 [DOI] [PubMed] [Google Scholar]
- 240. Mariniello B, Rosato A, Zuccolotto G, et al. Combination of sorafenib and everolimus impacts therapeutically on adrenocortical tumor models. Endocr Relat Cancer. 2012;19:527–539 [DOI] [PubMed] [Google Scholar]
- 241. Terzolo M, Ardito A, Zaggia B, et al. Management of adjuvant mitotane therapy following resection of adrenal cancer. Endocrine. 2012;42:521–525 [DOI] [PubMed] [Google Scholar]
- 242. Hermsen IG, Gelderblom H, Kievit J, Romijn JA, Haak HR. Extremely long survival in six patients despite recurrent and metastatic adrenal carcinoma. Eur J Endocrinol. 2008;158:911–919 [DOI] [PubMed] [Google Scholar]
- 243. Assié G, Antoni G, Tissier F, et al. Prognostic parameters of metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2007;92:148–154 [DOI] [PubMed] [Google Scholar]
- 244. Gonzalez RJ, Tamm EP, Ng C, et al. Response to mitotane predicts outcome in patients with recurrent adrenal cortical carcinoma. Surgery. 2007;142:867–875; discussion 867–875 [DOI] [PubMed] [Google Scholar]
- 245. Miller BS, Gauger PG, Hammer GD, Giordano TJ, Doherty GM. Proposal for modification of the ENSAT staging system for adrenocortical carcinoma using tumor grade. Langenbecks Arch Surg. 2010;395:955–961 [DOI] [PubMed] [Google Scholar]
- 246. Miller BS, Gauger PG, Hammer GD, Doherty GM. Resection of adrenocortical carcinoma is less complete and local recurrence occurs sooner and more often after laparoscopic adrenalectomy than after open adrenalectomy. Surgery. 2012;152:1150–1157 [DOI] [PubMed] [Google Scholar]
- 247. Sabolch A, Feng M, Griffith K, Hammer G, Doherty G, Ben-Josef E. Adjuvant and definitive radiotherapy for adrenocortical carcinoma. Int J Radiat Oncol Biol Phys. 2011;80:1477–1484 [DOI] [PubMed] [Google Scholar]
- 248. Gaujoux S, Brennan MF. Recommendation for standardized surgical management of primary adrenocortical carcinoma. Surgery. 2012;152:123–132 [DOI] [PubMed] [Google Scholar]
- 249. Reibetanz J, Jurowich C, Erdogan I, et al. Impact of lymphadenectomy on the oncologic outcome of patients with adrenocortical carcinoma. Ann Surg. 2012;255:363–369 [DOI] [PubMed] [Google Scholar]
- 250. Zafar SS, Abaza R. Robot-assisted laparoscopic adrenalectomy for adrenocortical carcinoma: initial report and review of the literature. J Endourol. 2008;22:985–989 [DOI] [PubMed] [Google Scholar]
- 251. Gonzalez RJ, Shapiro S, Sarlis N, et al. Laparoscopic resection of adrenal cortical carcinoma: a cautionary note. Surgery 2005;138:1078–1085; discussion 1085–1076 [DOI] [PubMed] [Google Scholar]
- 252. Guerrieri M, Campagnacci R, De Sanctis A, Baldarelli M, Coletta M, Perretta S. The learning curve in laparoscopic adrenalectomy. J Endocrinol Invest. 2008;31:531–536 [DOI] [PubMed] [Google Scholar]
- 253. Park HS, Roman SA, Sosa JA. Outcomes from 3144 adrenalectomies in the United States: which matters more, surgeon volume or specialty? Arch Surg. 2009;144:1060–1067 [DOI] [PubMed] [Google Scholar]
- 254. Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinologists and American Association of Endocrine Surgeons Medical Guidelines for the Management of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15:450–453 [DOI] [PubMed] [Google Scholar]
- 255. Berruti A, Baudin E, Gelderblom H, et al. Adrenal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii131–138 [DOI] [PubMed] [Google Scholar]
- 256. Henry JF, Peix JL, Kraimps JL. Positional statement of the European Society of Endocrine Surgeons (ESES) on malignant adrenal tumors. Langenbecks Arch Surg. 2012;397:145–146 [DOI] [PubMed] [Google Scholar]
- 257. Grubbs EG, Callender GG, Xing Y, et al. Recurrence of adrenal cortical carcinoma following resection: surgery alone can achieve results equal to surgery plus mitotane. Ann Surg Oncol. 2010;17:263–270 [DOI] [PubMed] [Google Scholar]
- 258. Leboulleux S, Deandreis D, Al Ghuzlan A, et al. Adrenocortical carcinoma: is the surgical approach a risk factor of peritoneal carcinomatosis? Eur J Endocrinol. 2010;162:1147–1153 [DOI] [PubMed] [Google Scholar]
- 259. Lombardi CP, Raffaelli M, De Crea C, et al. Open versus endoscopic adrenalectomy in the treatment of localized (stage I/II) adrenocortical carcinoma: results of a multiinstitutional Italian survey. Surgery. 2012;152:1158–1164 [DOI] [PubMed] [Google Scholar]
- 260. Porpiglia F, Garrone C, Giraudo G, Destefanis P, Fontana D, Morino M. Transperitoneal laparoscopic adrenalectomy: experience in 72 procedures. J Endourol. 2001;15:275–279 [DOI] [PubMed] [Google Scholar]
- 261. Brix D, Allolio B, Fenske W, et al. Laparoscopic versus open adrenalectomy for adrenocortical carcinoma: surgical and oncologic outcome in 152 patients. Eur Urol. 2010;58:609–615 [DOI] [PubMed] [Google Scholar]
- 262. Miller BS, Ammori JB, Gauger PG, Broome JT, Hammer GD, Doherty GM. Laparoscopic resection is inappropriate in patients with known or suspected adrenocortical carcinoma. World J Surg. 2010;34:1380–1385 [DOI] [PubMed] [Google Scholar]
- 263. Castillo OA, Vitagliano G, Secin FP, Kerkebe M, Arellano L. Laparoscopic adrenalectomy for adrenal masses: does size matter? Urology. 2008;71:1138–1141 [DOI] [PubMed] [Google Scholar]
- 264. Palazzo FF, Sebag F, Sierra M, Ippolito G, Souteyrand P, Henry JF. Long-term outcome following laparoscopic adrenalectomy for large solid adrenal cortex tumors. World J Surg. 2006;30:893–898 [DOI] [PubMed] [Google Scholar]
- 265. Erdogan I, Deutschbein T, Jurowich C, et al. The role of surgery in the management of recurrent adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:181–191 [DOI] [PubMed] [Google Scholar]
- 266. Schulick RD, Brennan MF. Long-term survival after complete resection and repeat resection in patients with adrenocortical carcinoma. Ann Surg Oncol. 1999;6:719–726 [DOI] [PubMed] [Google Scholar]
- 267. Bellantone R, Ferrante A, Boscherini M, et al. Role of reoperation in recurrence of adrenal cortical carcinoma: results from 188 cases collected in the Italian National Registry for Adrenal Cortical Carcinoma. Surgery. 1997;122:1212–1218 [DOI] [PubMed] [Google Scholar]
- 268. Jensen JC, Pass HI, Sindelar WF, Norton JA. Recurrent or metastatic disease in select patients with adrenocortical carcinoma. Aggressive resection vs chemotherapy. Arch Surg. 1991;126:457–461 [DOI] [PubMed] [Google Scholar]
- 269. Haak HR, Hermans J, van de Velde CJ, et al. Optimal treatment of adrenocortical carcinoma with mitotane: results in a consecutive series of 96 patients. Br J Cancer. 1994;69:947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Icard P, Chapuis Y, Andreassian B, Bernard A, Proye C. Adrenocortical carcinoma in surgically treated patients: a retrospective study on 156 cases by the French Association of Endocrine Surgery. Surgery 1992;112:972–979; discussion 979–980 [PubMed] [Google Scholar]
- 271. Kendrick ML, Lloyd R, Erickson L, Farley DR, Grant CS, Thompson GB, Rowland C, Young WF, Jr., van Heerden JA. Adrenocortical carcinoma: surgical progress or status quo? Arch Surg. 2001;136:543–549 [DOI] [PubMed] [Google Scholar]
- 272. Didolkar MS, Bescher RA, Elias EG, Moore RH. Natural history of adrenal cortical carcinoma: a clinicopathologic study of 42 patients. Cancer. 1981;47:2153–2161 [DOI] [PubMed] [Google Scholar]
- 273. Zografos GC, Driscoll DL, Karakousis CP, Huben RP. Adrenal adenocarcinoma: a review of 53 cases. J Surg Oncol. 1994;55:160–164 [DOI] [PubMed] [Google Scholar]
- 274. Henley DJ, van Heerden JA, Grant CS, Carney JA, Carpenter PC. Adrenal cortical carcinoma–a continuing challenge. Surgery. 1983;94:926–931 [PubMed] [Google Scholar]
- 275. Pommier RF, Brennan MF. An eleven-year experience with adrenocortical carcinoma. Surgery. 1992;112:963–970; discussion 970–971 [PubMed] [Google Scholar]
- 276. Nelson AA, Woodard G. Adrenal cortical atrophy and liver damage produced in dogs by feeding 2,2-bis-(parachloro-phenyl)-1,1-dichloroethane. Fed Proc. 1948;7:277. [PubMed] [Google Scholar]
- 277. Sheehan HL, Summers VK, Nichols J. D.D.D. therapy in Cushing's syndrome. Lancet. 1953;1:312–314 [DOI] [PubMed] [Google Scholar]
- 278. Bergenstal DM, Hertz R, Lipsett MB, Moy RH. Chemotherapy of adrenocortical cancer with o,p'DDD. Ann Intern Med. 1960;53:672–682 [Google Scholar]
- 279. Asp V, Cantillana T, Bergman A, Brandt I. Chiral effects in adrenocorticolytic action of o,p'-DDD (mitotane) in human adrenal cells. Xenobiotica. 2010;40:177–183 [DOI] [PubMed] [Google Scholar]
- 280. Schteingart DE, Sinsheimer JE, Benitez RS, et al. Structural requirements for mitotane activity: development of analogs for treatment of adrenal cancer. Anticancer Res. 2012;32:2711–2720 [PubMed] [Google Scholar]
- 281. Lund BO, Bergman A, Brandt I. Metabolic activation and toxicity of a DDT-metabolite, 3-methylsulphonyl-DDE, in the adrenal zona fasciculata in mice. Chem Biol Interact. 1988;65:25–40 [DOI] [PubMed] [Google Scholar]
- 282. Lindhe O, Skogseid B. Mitotane effects in a H295R xenograft model of adjuvant treatment of adrenocortical cancer. Horm Metab Res. 2010;42:725–730 [DOI] [PubMed] [Google Scholar]
- 283. Schteingart DE, Sinsheimer JE, Counsell RE, et al. Comparison of the adrenalytic activity of mitotane and a methylated homolog on normal adrenal cortex and adrenal cortical carcinoma. Cancer Chemother Pharmacol. 1993;31:459–466 [DOI] [PubMed] [Google Scholar]
- 284. Martz F, Straw JA. Metabolism and covalent binding of 1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane (o,p,'-DDD). Correlation between adrenocorticolytic activity and metabolic activation by adrenocortical mitochondria. Drug Metab Dispos. 1980;8:127–130 [PubMed] [Google Scholar]
- 285. Sinsheimer JE, Freeman CJ. Mitotane (1-(o-chlorophenyl)-1-(p-chlorophenyl)-2,2-dichloroethane) metabolism in perfusion studies with dog adrenal glands. Drug Metab Dispos. 1987;15:267–269 [PubMed] [Google Scholar]
- 286. Cai W, Counsell RE, Schteingart DE, Sinsheimer JE, Vaz AD, Wotring LL. Adrenal proteins bound by a reactive intermediate of mitotane. Cancer Chemother Pharmacol. 1997;39:537–540 [DOI] [PubMed] [Google Scholar]
- 287. Lindhe O, Skogseid B, Brandt I. Cytochrome P450-catalyzed binding of 3-methylsulfonyl-DDE and o,p'-DDD in human adrenal zona fasciculata/reticularis. J Clin Endocrinol Metab. 2002;87:1319–1326 [DOI] [PubMed] [Google Scholar]
- 288. Terzolo M. Mitotane. In: Hammer G, Else T, eds. Adrenocortical Carcinoma. 1st ed New York, NY: Springer; 2010:369–381 [Google Scholar]
- 289. Efficacy of adjuvant mitotane treatment in prolonging recurrence-free survival in patients with adrenocortical carcinoma at low-intermediate risk of recurrence submitted to radical resection. Adiuvo website. http://www.adiuvo-trial.org
- 290. Terzolo M, Angeli A, Fassnacht M, et al. Adjuvant mitotane treatment for adrenocortical carcinoma. N Engl J Med. 2007;356:2372–2380 [DOI] [PubMed] [Google Scholar]
- 291. Bertherat J, Coste J, Bertagna X. Adjuvant mitotane in adrenocortical carcinoma. N Engl J Med. 2007;357:1256–1257; author reply 1259 [DOI] [PubMed] [Google Scholar]
- 292. Cerquetti L, Bucci B, Marchese R, et al. Mitotane increases the radiotherapy inhibitory effect and induces G2-arrest in combined treatment on both H295R and SW13 adrenocortical cell lines. Endocr Relat Cancer. 2008;15:623–634 [DOI] [PubMed] [Google Scholar]
- 293. Cerquetti L, Sampaoli C, Amendola D, et al. Mitotane sensitizes adrenocortical cancer cells to ionizing radiations by involvement of the cyclin B1/CDK complex in G2 arrest and mismatch repair enzymes modulation. Int J Oncol. 2010;37:493–501 [DOI] [PubMed] [Google Scholar]
- 294. Boven E, Vermorken JB, van Slooten H, Pinedo HM. Complete response of metastasized adrenal cortical carcinoma with o,p'-DDD. Case report and literature review. Cancer. 1984;53:26–29 [DOI] [PubMed] [Google Scholar]
- 295. Terzolo M, Baudin AE, Ardito A, et al. Mitotane levels predict the outcome of patients with adrenocortical carcinoma treated adjuvantly following radical resection. Eur J Endocrinol. 2013;169:263–270 [DOI] [PubMed] [Google Scholar]
- 296. Hermsen IG, Fassnacht M, Terzolo M, et al. Plasma concentrations of o,p'DDD, o,p'DDA, and o,p'DDE as predictors of tumor response to mitotane in adrenocortical carcinoma: results of a retrospective ENS@T multicenter study. J Clin Endocrinol Metab. 2011;96:1844–1851 [DOI] [PubMed] [Google Scholar]
- 297. Dickstein G, Shechner C, Arad E, Best LA, Nativ O. Is there a role for low doses of mitotane (o,p'-DDD) as adjuvant therapy in adrenocortical carcinoma? J Clin Endocrinol Metab. 1998;83:3100–3103 [DOI] [PubMed] [Google Scholar]
- 298. Volante M, Terzolo M, Fassnacht M, et al. Ribonucleotide reductase large subunit (RRM1) gene expression may predict efficacy of adjuvant mitotane in adrenocortical cancer. Clin Cancer Res. 2012;18:3452–3461 [DOI] [PubMed] [Google Scholar]
- 299. Chortis V, Taylor AE, Schneider P, et al. Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5α-reductase, explaining the need for personalized glucocorticoid and androgen replacement. J Clin Endocrinol Metab. 2013;98:161–171 [DOI] [PubMed] [Google Scholar]
- 300. Zatelli MC, Gentilin E, Daffara F, et al. Therapeutic concentrations of mitotane (o,p'-DDD) inhibit thyrotroph cell viability and TSH expression and secretion in a mouse cell line model. Endocrinology. 2010;151:2453–2461 [DOI] [PubMed] [Google Scholar]
- 301. Kroiss M, Quinkler M, Lutz WK, Allolio B, Fassnacht M. Drug interactions with mitotane by induction of CYP3A4 metabolism in the clinical management of adrenocortical carcinoma. Clin Endocrinol (Oxf). 2011;75:585–591 [DOI] [PubMed] [Google Scholar]
- 302. van Erp NP, Guchelaar HJ, Ploeger BA, Romijn JA, Hartigh Jd, Gelderblom H. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur J Endocrinol. 2011;164:621–626 [DOI] [PubMed] [Google Scholar]
- 303. van Herwaarden AE, Wagenaar E, van der Kruijssen CM, et al. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Invest. 2007;117:3583–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Kroiss M, Quinkler M, Johanssen S, et al. Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. J Clin Endocrinol Metab. 2012;97:3495–3503 [DOI] [PubMed] [Google Scholar]
- 305. Baszko-Błaszyk D, Błaszyk K, Wasko R, Sowinski J. Pregnancy in a patient with adrenocortical carcinoma during treatment with Mitotane - a case report. Endokrynologia Polska. 2011;62:186–188 [PubMed] [Google Scholar]
- 306. Coonrod DV, Rizkallah TH. Virilizing adrenal carcinoma in a woman of reproductive age: a case presentation and literature review. Am J Obstet Gynecol. 1995;172:1912–1914; discussion 1914–1915 [DOI] [PubMed] [Google Scholar]
- 307. Kojori F, Cronin CM, Salamon E, Burym C, Sellers EA. Normal adrenal function in an infant following a pregnancy complicated by maternal adrenal cortical carcinoma and mitotane exposure. J Pediatr Endocrinol Metab. 2011;24:203–204 [DOI] [PubMed] [Google Scholar]
- 308. Leiba S, Weinstein R, Shindel B, et al. The protracted effect of o,p'-DDD in Cushing's disease and its impact on adrenal morphogenesis of young human embryo. Ann Endocrinol. 1989;50:49–53 [PubMed] [Google Scholar]
- 309. Tripto-Shkolnik L, Blumenfeld Z, Bronshtein M, Salmon A, Jaffe A. Pregnancy in a patient with adrenal carcinoma treated with mitotane: a case report and review of literature. J Clin Endocrinol Metab. 2013;98:443–447 [DOI] [PubMed] [Google Scholar]
- 310. Feuillan P, Raffeld M, Stein CA, et al. Effects of suramin on the function and structure of the adrenal cortex in the cynomolgus monkey. J Clin Endocrinol Metab. 1987;65:153–158 [DOI] [PubMed] [Google Scholar]
- 311. Kaplan LD, Wolfe PR, Volberding PA, et al. Lack of response to suramin in patients with AIDS and AIDS-related complex. Am J Med. 1987;82:615–620 [DOI] [PubMed] [Google Scholar]
- 312. Arlt W, Reincke M, Siekmann L, Winkelmann W, Allolio B. Suramin in adrenocortical cancer: limited efficacy and serious toxicity. Clin Endocrinol (Oxf). 1994;41:299–307 [DOI] [PubMed] [Google Scholar]
- 313. LaRocca RV, Cooper MR, Uhrich M, et al. Use of suramin in treatment of prostatic carcinoma refractory to conventional hormonal manipulation. Urol Clin North Am. 1991;18:123–129 [PubMed] [Google Scholar]
- 314. Flack MR, Pyle RG, Mullen NM, et al. Oral gossypol in the treatment of metastatic adrenal cancer. J Clin Endocrinol Metab. 1993;76:1019–1024 [DOI] [PubMed] [Google Scholar]
- 315. Arnaldez FI, Helman LJ. Targeting the insulin growth factor receptor 1. Hematol Oncol Clin North Am. 2012;26:527–542, vii–viii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Haluska P, Worden F, Olmos D, et al. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother Pharmacol. 2010;65:765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Naing A, LoRusso P, Fu S, et al. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res. 2012;18:2625–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Drelon C, Berthon A, Val P. Adrenocortical cancer and IGF2: is the game over or our experimental models limited? J Clin Endocrinol Metab. 2013;98:505–507 [DOI] [PubMed] [Google Scholar]
- 319. Hahner S, Kreissl MC, Fassnacht M, et al. [131I]Iodometomidate for targeted radionuclide therapy of advanced adrenocortical carcinoma. J Clin Endocrinol Metab. 2012;97:914–922 [DOI] [PubMed] [Google Scholar]
- 320. Auchus R. Pharmacotherapy for hormone excess in adrenocortical carcinoma. In: Hammer G, Else T, eds. Adrenocortical Carcinoma. 1st ed New York, NY: Springer; 2010:383–402 [Google Scholar]
- 321. Hartzband PI, Van Herle AJ, Sorger L, Cope D. Assessment of hypothalamic-pituitary-adrenal (HPA) axis dysfunction: comparison of ACTH stimulation, insulin-hypoglycemia and metyrapone. J Endocrinol Invest. 1988;11:769–776 [DOI] [PubMed] [Google Scholar]
- 322. Santen RJ, Wells SA, Runic S, et al. Adrenal suppression with aminoglutethimide. I. Differential effects of aminoglutethimide on glucocorticoid metabolism as a rationale for use of hydrocortisone. J Clin Endocrinol Metab. 1977;45:469–479 [DOI] [PubMed] [Google Scholar]
- 323. Schteingart DE, Conn JW. Effects of aminoglutethimide upon adrenal function and cortisol metabolism in Cushing's syndrome. J Clin Endocrinol Metab. 1967;27:1657–1666 [DOI] [PubMed] [Google Scholar]
- 324. Drake WM, Perry LA, Hinds CJ, Lowe DG, Reznek RH, Besser GM. Emergency and prolonged use of intravenous etomidate to control hypercortisolemia in a patient with Cushing's syndrome and peritonitis. J Clin Endocrinol Metab. 1998;83:3542–3544 [DOI] [PubMed] [Google Scholar]
- 325. Schulte HM, Benker G, Reinwein D, Sippell WG, Allolio B. Infusion of low dose etomidate: correction of hypercortisolemia in patients with Cushing's syndrome and dose-response relationship in normal subjects. J Clin Endocrinol Metab. 1990;70:1426–1430 [DOI] [PubMed] [Google Scholar]
- 326. Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2012;97:2039–2049 [DOI] [PubMed] [Google Scholar]
- 327. Hajjar RA, Hickey RC, Samaan NA. Adrenal cortical carcinoma. A study of 32 patients. Cancer. 1975;35:549–554 [DOI] [PubMed] [Google Scholar]
- 328. Hutter AM, Jr, Kayhoe DE. Adrenal cortical carcinoma. Clinical features of 138 patients. Am J Med. 1966;41:572–580 [DOI] [PubMed] [Google Scholar]
- 329. Huvos AG, Hajdu SI, Brasfield RD, Foote FW., Jr Adrenal cortical carcinoma. Clinicopathologic study of 34 cases. Cancer 1970;25:354–361 [DOI] [PubMed] [Google Scholar]
- 330. Macfarlane DA. Cancer of the adrenal cortex; the natural hisotry, prognosis, and treatment in a study of 55 cases. Ann R Coll Surg Engl. 1958;23:155–186 [PMC free article] [PubMed] [Google Scholar]
- 331. Murphy WT. Radiation Therapy. 2nd ed Philadelphia, PA: WB Saunders; 1967 [Google Scholar]
- 332. Fassnacht M, Hahner S, Polat B, et al. Efficacy of adjuvant radiotherapy of the tumor bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91:4501–4504 [DOI] [PubMed] [Google Scholar]
- 333. Hermsen IG, Groenen YE, Dercksen MW, Theuws J, Haak HR. Response to radiation therapy in adrenocortical carcinoma. J Endocrinol Invest. 2010;33:712–714 [DOI] [PubMed] [Google Scholar]
- 334. Markoe AM, Serber W, Micaily B, Brady LW. Radiation therapy for adjunctive treatment of adrenal cortical carcinoma. Am J Clin Oncol. 1991;14:170–174 [DOI] [PubMed] [Google Scholar]
- 335. Percarpio B, Knowlton AH. Radiation therapy of adrenal cortical carcinoma. Acta Radiol Ther Phys Biol. 1976;15:288–292 [DOI] [PubMed] [Google Scholar]
- 336. Polat B, Fassnacht M, Pfreundner L, et al. Radiotherapy in adrenocortical carcinoma. Cancer. 2009;115:2816–2823 [DOI] [PubMed] [Google Scholar]
- 337. Magee BJ, Gattamaneni HR, Pearson D. Adrenal cortical carcinoma: survival after radiotherapy. Clin Radiol. 1987;38:587–588 [DOI] [PubMed] [Google Scholar]
- 338. Habra MA, Ejaz S, Feng L, et al. A retrospective cohort analysis of the efficacy of adjuvant radiotherapy after primary surgical resection in patients with adrenocortical carcinoma. J Clin Endocrinol Metab. 2013;98:192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Cohn K, Gottesman L, Brennan M. Adrenocortical carcinoma. Surgery. 1986;100:1170–1177 [PubMed] [Google Scholar]
- 340. Bodie B, Novick AC, Pontes JE, et al. The Cleveland Clinic experience with adrenal cortical carcinoma. J Urol. 1989;141:257–260 [DOI] [PubMed] [Google Scholar]
- 341. Kasperlik-Załuska AA, Migdalska BM, Zgliczynski S, Makowska AM. Adrenocortical carcinoma. A clinical study and treatment results of 52 patients. Cancer. 1995;75:2587–2591 [DOI] [PubMed] [Google Scholar]
- 342. Arriagada R, Lê M, Rochard F, Contesso G. Conservative treatment versus mastectomy in early breast cancer: patterns of failure with 15 years of follow-up data. Institut Gustave-Roussy Breast Cancer Group. J Clin Oncol. 1996;14:1558–1564 [DOI] [PubMed] [Google Scholar]
- 343. Blichert-Toft M, Rose C, Andersen J, et al. Danish randomized trial comparing breast conservation therapy with mastectomy: six years of life-table analysis. Danish Breast Cancer Cooperative Group. J Natl Cancer Inst. 1992;19–25 [PubMed] [Google Scholar]
- 344. Fisher B, Anderson S, Bryant J, et al. Twenty-year follow-up of a randomized trial comparing total mastectomy, lumpectomy, and lumpectomy plus irradiation for the treatment of invasive breast cancer. N Engl J Med. 2002;347:1233–1241 [DOI] [PubMed] [Google Scholar]
- 345. Johansen H, Kaae S, Jensen M. Extended radical mastectomy versus simple mastectomy followed by radiotherapy in primary breast cancer. A fifty-year follow-up to the Copenhagen Breast Cancer randomised study. Acta Oncol. 2008;47:633–638 [DOI] [PubMed] [Google Scholar]
- 346. Kuban DA, Tucker SL, Dong L, et al. Long-term results of the M.D. Anderson randomized dose-escalation trial for prostate cancer. Int J Radiat Oncol Biol Phys. 2008;70:67–74 [DOI] [PubMed] [Google Scholar]
- 347. Poggi MM, Danforth DN, Sciuto LC, et al. Eighteen-year results in the treatment of early breast carcinoma with mastectomy versus breast conservation therapy: the National Cancer Institute Randomized Trial. Cancer. 2003;98:697–702 [DOI] [PubMed] [Google Scholar]
- 348. van Dongen JA, Voogd AC, Fentiman IS, et al. Long-term results of a randomized trial comparing breast-conserving therapy with mastectomy: European Organization for Research and Treatment of Cancer 10801 trial. J Natl Cancer Inst. 2000;92:1143–1150 [DOI] [PubMed] [Google Scholar]
- 349. Veronesi U, Cascinelli N, Mariani L, et al. Twenty-year follow-up of a randomized study comparing breast-conserving surgery with radical mastectomy for early breast cancer. N Engl J Med. 2002;347:1227–1232 [DOI] [PubMed] [Google Scholar]
- 350. Zietman AL, DeSilvio ML, Slater JD, et al. Comparison of Conventional-dose vs high-dose conformal radiation therapy in clinically localized adenocarcinoma of the prostate: a randomized controlled trial. JAMA. 2005;294:1233–1239 [DOI] [PubMed] [Google Scholar]
- 351. King DR, Lack EE. Adrenal cortical carcinoma: a clinical and pathologic study of 49 cases. Cancer. 1979;44:239–244 [DOI] [PubMed] [Google Scholar]
- 352. Ripley RT, Kemp CD, Davis JL, et al. Liver resection and ablation for metastatic adrenocortical carcinoma. Ann Surg Oncol. 2011;18:1972–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Bauditz J, Quinkler M, Wermke W. Radiofrequency thermal ablation of hepatic metastases of adrenocortical cancer–a case report and review of the literature. Exp Clin Endocrinol Diabetes. 2009;117:316–319 [DOI] [PubMed] [Google Scholar]
- 354. Li SH, Huang CH, Ko SF, Chou FF, Huang SC. Extended survival in a patient with recurrent and metastatic adrenal cortical carcinoma by aggressive transarterial embolization–a case report. J Surg Oncol. 2005;90:101–105 [DOI] [PubMed] [Google Scholar]
- 355. Soga H, Takenaka A, Ooba T, et al. A twelve-year experience with adrenal cortical carcinoma in a single institution: long-term survival after surgical treatment and transcatheter arterial embolization. Urol Int. 2009;82:222–226 [DOI] [PubMed] [Google Scholar]
- 356. Thacker PG, Friese JL, Loe M, Biegler P, Larson M, Andrews J. Embolization of nonliver visceral tumors. Semin Intervent Radiol. 2009;26:262–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. de Baere T, Teriitehau C, Deschamps F, et al. Predictive factors for hypertrophy of the future remnant liver after selective portal vein embolization. Ann Surg Oncol. 2010;17:2081–2089 [DOI] [PubMed] [Google Scholar]
- 358. Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27:1250–1256 [DOI] [PubMed] [Google Scholar]
- 359. Schussheim DH, Skarulis MC, Agarwal SK, et al. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178 [DOI] [PubMed] [Google Scholar]
- 360. Higurashi M, Iijima K, Sugimoto Y, et al. The birth prevalence of malformation syndromes in Tokyo infants: a survey of 14,430 newborn infants. Am J Med Genet. 1980;6:189–194 [DOI] [PubMed] [Google Scholar]
- 361. Thorburn MJ, Wright ES, Miller CG, Smith-Read EH. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child. 1970;119:316–321 [DOI] [PubMed] [Google Scholar]
- 362. Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010;18:8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Bülow S, Faurschou Nielsen T, et al. The incidence rate of familial adenomatous polyposis. Results from the Danish Polyposis Register. Int J Colorectal Dis. 1996;11:88–91 [DOI] [PubMed] [Google Scholar]
- 364. Burn J, Chapman P, Delhanty J, et al. The UK Northern region genetic register for familial adenomatous polyposis coli: use of age of onset, congenital hypertrophy of the retinal pigment epithelium, and DNA markers in risk calculations. J Med Genet. 1991;28:289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Järvinen HJ. Epidemiology of familial adenomatous polyposis in Finland: impact of family screening on the colorectal cancer rate and survival. Gut. 1992;33:357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Lammert M, Friedman JM, Kluwe L, Mautner VF. Prevalence of neurofibromatosis 1 in German children at elementary school enrollment. Arch Dermatol. 2005;141:71–74 [DOI] [PubMed] [Google Scholar]
- 367. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40 [DOI] [PubMed] [Google Scholar]
- 368. Stratakis CA, Horvath A. Carney complex. In: Pagon RA, Adam MP, Bird TD, Dolan CR, et al. eds. GeneReviews. Seattle, WA: University of Washington; 2003. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1286/ [PubMed] [Google Scholar]
- 369. Vassilopoulou-Sellin R, Guinee VF, Klein MJ, et al. Impact of adjuvant mitotane on the clinical course of patients with adrenocortical cancer. Cancer. 1993;71:3119–3123 [DOI] [PubMed] [Google Scholar]
- 370. Barzon L, Fallo F, Sonino N, Daniele O, Boscaro M. Adrenocortical carcinoma: experience in 45 patients. Oncology. 1997;54:490–496 [DOI] [PubMed] [Google Scholar]
- 371. van Slooten H, Moolenaar AJ, van Seters AP, Smeenk D. The treatment of adrenocortical carcinoma with o,p'-DDD: prognostic implications of serum level monitoring. Eur J Cancer Clin Oncol. 1984;20:47–53 [DOI] [PubMed] [Google Scholar]
- 372. Decker RA, Elson P, Hogan TF, et al. Eastern Cooperative Oncology Group study 1879: mitotane and adriamycin in patients with advanced adrenocortical carcinoma. Surgery. 1991;110:1006–1013 [PubMed] [Google Scholar]
- 373. Williamson SK, Lew D, Miller GJ, Balcerzak SP, Baker LH, Crawford ED. Phase II evaluation of cisplatin and etoposide followed by mitotane at disease progression in patients with locally advanced or metastatic adrenocortical carcinoma: a Southwest Oncology Group Study. Cancer. 2000;88:1159–1165 [PubMed] [Google Scholar]
- 374. Baudin E, Pellegriti G, Bonnay M, et al. Impact of monitoring plasma 1,1-dichlorodiphenildichloroethane (o,p'DDD) levels on the treatment of patients with adrenocortical carcinoma. Cancer. 2001;92:1385–1392 [DOI] [PubMed] [Google Scholar]
- 375. van Slooten H, van Oosterom AT. CAP (cyclophosphamide, doxorubicin, and cisplatin) regimen in adrenal cortical carcinoma. Cancer Treat Rep. 1983;67:377–379 [PubMed] [Google Scholar]
- 376. Schlumberger M, Brugieres L, Gicquel C, Travagli JP, Droz JP, Parmentier C. 5-Fluorouracil, doxorubicin, and cisplatin as treatment for adrenal cortical carcinoma. Cancer. 1991;67:2997–3000 [DOI] [PubMed] [Google Scholar]
- 377. Burgess MA, Legha SS, Sellin RV. Chemotherapy with cis-platinum and etoposide (VP-16) for patients with advanced adrenal cortical carcinoma. Proc Am Soc Clin Oncol. 1993;12:188 [Google Scholar]
- 378. Bukowski RM, Wolfe M, Levine HS, et al. Phase II trial of mitotane and cisplatin in patients with adrenal carcinoma: a Southwest Oncology Group study. J Clin Oncol. 1993;11:161–165 [DOI] [PubMed] [Google Scholar]
- 379. Bonacci R, Gigliotti A, Baudin E, et al. Cytotoxic therapy with etoposide and cisplatin in advanced adrenocortical carcinoma. Br J Cancer. 1998;78:546–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Berruti A, Terzolo M, Pia A, Angeli A, Dogliotti L. Mitotane associated with etoposide, doxorubicin, and cisplatin in the treatment of advanced adrenocortical carcinoma. Italian Group for the Study of Adrenal Cancer. Cancer. 1998;83:2194–2200 [PubMed] [Google Scholar]
- 381. Khan TS, Imam H, Juhlin C, et al. Streptozocin and o,p'DDD in the treatment of adrenocortical cancer patients: long-term survival in its adjuvant use. Ann Oncol. 2000;11:1281–1287 [DOI] [PubMed] [Google Scholar]
- 382. Abraham J, Bakke S, Rutt A, et al. A phase II trial of combination chemotherapy and surgical resection for the treatment of metastatic adrenocortical carcinoma: continuous infusion doxorubicin, vincristine, and etoposide with daily mitotane as a P-glycoprotein antagonist. Cancer. 2002;94:2333–2343 [DOI] [PubMed] [Google Scholar]
- 383. Baudin E, Docao C, Gicquel C, et al. Use of a topoisomerase I inhibitor (irinotecan, CPT-11) in metastatic adrenocortical carcinoma. Ann Oncol. 2002;13:1806–1809 [DOI] [PubMed] [Google Scholar]
- 384. Khan TS, Sundin A, Juhlin C, Wilander E, Oberg K, Eriksson B. Vincristine, cisplatin, teniposide, and cyclophosphamide combination in the treatment of recurrent or metastatic adrenocortical cancer. Med Oncol. 2004;21:167–177 [DOI] [PubMed] [Google Scholar]
- 385. Berruti A, Terzolo M, Sperone P, et al. Etoposide, doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr Relat Cancer. 2005;12:657–666 [DOI] [PubMed] [Google Scholar]
- 386. Sperone P, Ferrero A, Daffara F, et al. Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr Relat Cancer. 2010;17:445–453 [DOI] [PubMed] [Google Scholar]
- 387. Fassnacht M, Terzolo M, Allolio B, et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med. 2012;366:2189–2197 [DOI] [PubMed] [Google Scholar]
- 388. Rustin GJ, Galbraith SM, Anderson H, et al. Phase I clinical trial of weekly combretastatin A4 phosphate: clinical and pharmacokinetic results. J Clin Oncol. 2003;21:2815–2822 [DOI] [PubMed] [Google Scholar]
- 389. Shah MA, Kortmansky J, Motwani M, et al. A phase I clinical trial of the sequential combination of irinotecan followed by flavopiridol. Clin Cancer Res. 2005;11:3836–3845 [DOI] [PubMed] [Google Scholar]
- 390. Gross DJ, Munter G, Bitan M, et al. The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer. 2006;13:535–540 [DOI] [PubMed] [Google Scholar]
- 391. Quinkler M, Hahner S, Wortmann S, et al. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008;93:2057–2062 [DOI] [PubMed] [Google Scholar]
- 392. Hong DS, Sebti SM, Newman RA, et al. Phase I trial of a combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in advanced malignancies. Clin Cancer Res. 2009;15:7061–7068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Berruti A, Sperone P, Ferrero A, et al. Phase II study of weekly paclitaxel and sorafenib as second/third-line therapy in patients with adrenocortical carcinoma. Eur J Endocrinol. 2012;166:451–458 [DOI] [PubMed] [Google Scholar]