

Abstract
Pancreatic beta-cell function and mass are markedly adaptive to compensate for the changes in insulin requirement observed during several situations such as pregnancy, obesity, glucocorticoids excess, or administration. This requires a beta-cell compensation which is achieved through a gain of beta-cell mass and function. Elucidating the physiological mechanisms that promote functional beta-cell mass expansion and that protect cells against death, is a key therapeutic target for diabetes. In this respect, several recent studies have emphasized the instrumental role of microRNAs in the control of beta-cell function. MicroRNAs are negative regulators of gene expression, and are pivotal for the control of beta-cell proliferation, function, and survival. On the one hand, changes in specific microRNA levels have been associated with beta-cell compensation and are triggered by hormones or bioactive peptides that promote beta-cell survival and function. Conversely, modifications in the expression of other specific microRNAs contribute to beta-cell dysfunction and death elicited by diabetogenic factors including, cytokines, chronic hyperlipidemia, hyperglycemia, and oxidized LDL. This review underlines the importance of targeting the microRNA network for future innovative therapies aiming at preventing the beta-cell decline in diabetes.
1. Introduction
The concentration of glucose in the blood is tightly monitored by the pancreatic islet beta-cell production of insulin. The main function of insulin is to reduce blood glucose levels by triggering the uptake and the storage of this carbohydrate by the cells of the body. The quantity of insulin released by beta-cells varies according not only to secretagogues such as glucose but also as a function of the insulin demand from target tissues. A feedback loop also exists between insulin sensitivity and insulin secretion, such that changes in sensitivity of peripheral tissues are balanced by corresponding increases in secretion, insuring preservation of euglycemia [1, 2]. A rise in the insulin demand occurs during normal body growth (from birth to early childhood periods), as a consequence of an increase in body weight and during pregnancy. To meet the requirement of insulin, beta-cells adapt both their mass and function to release sufficient insulin to maintain blood glucose homeostasis [1, 2]. Evidence for this compensatory process has been consistently provided by rodent models of obesity and diabetes and notably by the emerging availability of human pancreas necropsies [2, 3]. Indeed, beta-cell mass and function in pancreases of nondiabetic or prediabetic obese individuals is larger than in lean normoglycemic subjects [3–5]. In obesity beta-cell mass increases by 30–40% whereas insulin secretory output augments by 100% [6]. Conversely, diminished beta-cells mass and function contribute to the decrease in plasma insulin level in individuals with diabetes. Postmortem histology further a 20–65% decrement in beta-cell mass in islets from obese individuals with type 2 diabetes (T2D) when compared to BMI-matched nondiabetic subjects [3–5, 7–9]. This adaptive capacity of human islets to obesity has been confirmed in experimental murine models [10, 11]. In one study, human islets were grafted in an immunodeficient mouse strain sensitive to high fat-diet (HFD-)induced obesity [10]. This mice model is used for longitudinal studies of islets exposed to an obesogenic environment [10]. Enlarged volume of human beta-cells was observed in xenotransplanted mice fed with HFD for 12 weeks [10]. However, despite the gain of beta-cell mass and the increase in insulin expression, these mice displayed hyperglycemia. This study confirms the requirement for an appropriate number of functional beta-cells to circumvent insulin resistance [10]. Therefore, insulin deficiency in T2D may in part result from an insufficient number of functional beta-cells under conditions such as ageing, weight gain, or metabolic alterations [7, 12, 13].
Despite intensive research, current treatments of T2D do not prevent the appearance of long-term complications and, over time, can also become inefficient to insure appropriate glycemic control. This inefficacy may result from the fact that available strategies do not permit to protect beta-cells against their inescapable decline. The existing therapies with exogenous insulin or hypoglycemic agents for type 1 diabetes (T1D) are also unsatisfactory, since they do not offer a cure and are mostly insufficient for preventing the secondary complications associated with diabetes [14]. Transplantation of a sufficient number of pancreatic beta-cells can normalize blood glucose levels and may prevent the complications of diabetes [15]. However, immunosuppressive therapy is a current obstacle in transplantation and beta-cells from cadaveric donors are in such a short supply that transplants can be provided only to a limited number of patients. Regeneration of the functional beta-cell mass in patients could potentially represent an alternative to transplantation. In view of the inefficacy of the current treatments and the increasing global prevalence of diabetes [16], it is urgent to intensify efforts for developing new therapeutic strategies for both T1D and T2D. In this regard, it is tempting to postulate that strategies aiming at improving beta-cell function and mass plasticity as well as beta-cell survival under proapoptotic conditions could be of major interest for designing innovative therapeutics to prevent beta-cell decline and restore their functional adaptive ability in diabetes.
Adaptive capacity of beta-cell mass and function depends on the activity of transcriptional and translational regulators, which tightly and timely modulate the expression of genes in response to environmental cues. The noncoding microRNAs (miRNAs) are extremely important to accomplish this task [17]. MiRNAs act as translational repressors that bind to the 3′UTR of target mRNAs, leading to translational inhibition and/or messenger degradation [18, 19]. Each miRNA can have hundreds of targets, thereby triggering pleiotropic effects in beta-cells. This review provides insights into the pivotal role of miRNAs in beta-cell adaptation and failure during diabetes [20–26].
2. miRNAs Required for Beta-Cell Specification Fate
The regulation of the beta-cell mass in adult life results from the dynamic balance between proliferation, neogenesis, and apoptosis. The mechanisms underlying the control of these phenomena are participating also to normal pancreas development, and thereby can help in understanding the compensatory mechanisms elicited in response to environmental cues and metabolic changes [27, 28]. The pancreas derives from a pool of endodermal cells. At the initial stage, the proliferation of the progenitor cells is stimulated by growth factors and other signalling molecules produced by the surrounding mesenchyme. This process is governed by a sequential cascade including the appearance of neurogenin3 (Neurog3) [29]. The number of Neurog3 expressing cells increases and peaks at embryonic day e15.5, after which the level of this transcription factor gradually declines [29]. Neurog3 is undetectable in insulin- and glucagon-producing cells, suggesting that it is not necessary for postnatal islet function [29]. In fact, transient expression of Neurog3 is critical for temporarily allowing the lineage-committed transcription factors required for the differentiation of the endocrine progenitor cells, which will give rise to the endocrine cell subtypes within the islets [29–31]. Ablation of Neurog3 prevents the generation of all pancreatic endocrine cells in mice. Evidence for a role of miRNAs in the control of Neurog3 during pancreas development has been provided by a mice model in which the pancreatic expression of the large majority of the RNAs has been abolished. miRNAs are usually generated by RNA polymerase II. This enzyme initially yields pre-miRNA molecules containing a hairpin loop, which undergoes sequential processing including cytosolic excision of the hairpin by the ribonuclease (RNase) type III Dicer1 [32]. In mammals, the loss of the RNase III domain of Dicer1 blocks the formation of most miRNAs [32]. The islet-specific Dicer1 knockout mice generated using the Pdx1-Cre transgene survive until birth but fail to grow and die by P3 [20]. The pancreas of Dicer1-null mice displays an almost absolute loss of insulin-producing cells and there is a marked decrease in other cell types at e18.5 [20]. The defect of endocrine cells observed in the Dicer1 knockout mice is associated with an increase in Hes1 level and a reduction in the formation of endocrine progenitor cells expressing Neurog3 [20]. Besides the induction of Notch signaling by Hes1, a possible synergistic mechanism accounting for Neurog3 inhibition during pancreas development could be a direct control by miRNAs. Demonstration of this hypothesis has been attempted in a model for pancreatic regeneration [33]. Regeneration of beta-cells following a 50 or 70% pancreatectomy is not associated with induction of Neurog3 protein in progenitor cells despite the presence of the transcript. This result prompted the authors to propose a posttranslational control of Neurog3 expression mediated by miRNAs [33, 34]. Results from global miRNA profiling in regenerating pancreas after partial pancreatectomy have highlighted upregulation of 4 miRNAs including miR-15a, miR-15b, miR-16, and miR-195 (Table 1) [33]. All the four miRNAs are predicted to target the Neurog3 mRNA, suggesting that they could contribute to the posttranslational regulation of the transcription factor [33]. Whether these miRNAs individually contribute to pancreas development has not yet been investigated.
Table 1.
miRNAs required for beta cell specification fate and pancreas development.
| miRNAs | Known functional effect | Targets | References |
|---|---|---|---|
| mir-15a, miR-15b, miR-16, and miR-195 | Pancreas development, beta-cells fate and regeneration | Neurog3 | [33] |
| miR-375 | Beta- and alpha-cells expansion | [42] | |
| miR-7a | Beta-cell proliferation | mTOR pathway components | [44, 45, 48] |
| miR-124a | Pancreas development | Foxa2 | [33] |
3. miRNAs Are Required for Proliferation of Progenitor Cells and Mature Beta-Cells
When progenitor cells start expressing insulin they stop dividing. However, the beta-cell mass continues to expand during fetal and postnatal growth [28, 35–37]. A process that could account for beta-cell mass expansion in rodent is replication. In normal rats, the beta-cell population approximately doubles each day starting from the 16th day after conception [36]. After birth the beta-cell population still grows but during adult life at a much slower pace [37–39]. A role for miRNAs in the control of differentiated beta-cells has been highlighted by the generation of a mice model with beta-cell specific ablation of Dicer1. Disruption of the enzyme using the rat insulin promoter 2 (RIP-)Cre transgene leads to alteration in islet morphology, reduction in beta-cell number, and impairment in glucose-induced insulin secretion [40, 41]. Marked perturbations in beta-cell expansion and mass have been reported in knockout mice for individual miRNAs. The first one for which a major role in pancreatic development has been demonstrated is miR-375 (Table 1) [22, 42]. This miRNA is highly enriched in human and mice beta-cells [22]. The importance of miR-375 in pancreatic endocrine cell development has emerged from studies in zebrafish embryos [43]. Injection of anti-miR-375 morpholinos into one- or two-stage embryos resulted in disruption of the islet cell phenotype [43]. The miR-375 KO mice have been instrumental for unveiling a role for this miRNA in beta-cell expansion besides its involvement in the control of glucose-induced insulin secretion [42]. A 30–40% decrease in beta-cell mass has been measured within islets from these mice and, strikingly, a 1.7-fold increase in alpha-cells [42]. The combined hyperglucagonemia and hypoinsulinemia in miR-375 KO animals led them to develop hyperglycemia [42]. Other miRNAs such as miR-7a have been shown to potentially contribute to beta-cell expansion during pancreatic organogenesis. miR-7a belongs to the evolutionarily conserved miR-7a/b family and is abundant in beta-cells of rodent and human islets [44]. Inhibition of miR-7a activates the mammalian target of rapamycin (mTOR, a.k.a FRAP, RAFT, or RAPT) pathway in the mouse MIN6 insulin-producing cells and in primary mouse islets [45]. Activation of this evolutionarily conserved serine/threonine protein kinase promotes beta-cell replication and expansion of the beta-cell mass [46, 47]. The mTOR pathway can be divided into two biochemically and functionally distinct multicomponent complexes termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [48]. The two complexes are pivotal for the control of beta-cell mass although their downstream targets are distinct [48]. Disruption of miR-7a leads to upregulation of the downstream targets of mTORC1, p70S6 K, eukaryotic translation initiation factor 4E (eIF4E) and two MAPK-interacting kinases that phosphorylate eIF4E, as well as one of the essential TORC2 components, Mapkap1 [48]. Consequently, activation of the mTOR pathway caused by the suppression of miR-7a results in increased proliferation of beta-cells. So far, independent studies have shown that human beta-cells can proliferate within islets or under in vitro condition but the rate is extremely low [3, 49–53]. It is noteworthy that the authors have observed a nearly 30-fold increase in human beta-cell proliferation upon the reduction of miR-7a level [48].
4. miRNAs Regulating Nutrient-Induced Insulin Secretion and Insulin Production
The function of mature beta-cells is to release appropriate amounts of insulin in response to its main physiological stimulus, glucose, and to other secretagogues, including the incretin hormone glucagon like peptide-1 (GLP-1) and its mimetics [54]. In fact, GLP-1 potentiates glucose-induced insulin secretion by interacting with the GLP-1 receptor [54]. In human islet beta-cells, efficient incretin-stimulated insulin secretion relies on the recruitment of a highly coordinated subnetwork of beta-cells [55]. Activation of GLP-1R elevates cAMP levels, which in turn promote insulin secretion via both protein kinase A (PKA-)dependent and PKA-independent mechanisms [54]. A downstream long-term effect resulting from PKA activation consists of the modification of the expression of several genes [54, 56]. Such modulation is deemed to contribute to the plasticity of the secretory response to GLP-1 and glucose. The expression of miR-375 has been shown to be controlled by the activation of the cAMP/PKA pathway [57]. Indeed, incubation of rat insulin-producing cells with the GLP-1 mimetic exendin-4 leads to a decrease in miR-375 levels in a mechanism that involves PKA [57]. Reduction of miR-375 occurs also in response to glucose but in this case in a cAMP/PKA-independent manner [57, 58]. Decrease of miR-375 in the mouse insulin-producing MIN6 cells enhances insulin secretion, whereas overexpression of this miRNA hampers the ability of the cells to secrete in response to glucose [22]. One of the targets of miR-375 that accounts for glucose-induced insulin secretion is myotrophin (Table 2) [22]. Silencing of the latter mimics the effect of miR-375 on insulin secretion [22]. Thus, the drop of miR-375 caused by glucose and exendin-4 could be beneficial for insulin secretion by increasing the expression of myotrophin [22]. Another miRNA required for insulin secretion is miR-9 (Table 2). This miRNA is upregulated during differentiation of human embryonic stem cells and during the formation of cells of both neuronal and pancreatic lineages [21, 59]. Moreover, appropriate expression of this miRNA is required for mature beta-cell tasks and probably during development. Either overexpression or silencing of miR-9 is deleterious for the secretory capacity of beta-cells [26]. In fact, manipulation of miR-9 level impinges the expression of the Onecut2 (Oc2) transcription factor, which in turn hampers the content of the secretory machinery component Slp4 [26]. As consequence of the increased level of Slp4, beta-cells insulin secretion in response to secretagogues is impaired [26].
Table 2.
miRNAs regulating nutrient-induced insulin secretion and insulin gene expression.
| miRNAs | Known functional effect | Targets | References |
|---|---|---|---|
| miR-9 | Insulin secretion | Onecut-2, Sirt1 | [26] |
| miR-21 | Insulin secretion | VAMP2, Rab3a | [93] |
| miR-29a, b | Insulin secretion | Mctl1 | [71] |
| miR-30d | Insulin transcription | [63] | |
| miR-34a | Insulin secretion | VAMP2, Rab3a | [93] |
| miR-96 | Insulin secretion | Noc2 | [24] |
| miR-124a | Insulin secretion | Rab27a, Noc2, MCT1 | [24, 71] |
| miR-204 | Insulin transcription | MafA | [65] |
| miR-375 | Insulin transcription, insulin secretion | PDK1, myotrophin | [22, 58] |
Slp4 belongs to the Rab GTPase effector family that includes also RIM2, MyRIP/Slac2c, and Noc2 [60]. In beta-cells, these effectors are associated with Rab3a and/or Rab27a and regulate the assembly of the SNARE proteins SNAP25, Syntaxin-1, and VAMP-2, thereby controlling insulin exocytosis [60]. The expression of SNAP25, Rab3a, Rab27, and Noc2 are regulated by miR-124a in mouse insulin-producing cells [24]. Overexpression of miR-124 increases SNAP25 and Rab3a levels but reduces those of Rab27 and Noc2 [24]. miR-96 is also expressed in beta-cells and controls the expression of Slp4 and Noc2 [24]. The level of Slp4 increases and this of Noc2 decreases in cells that overexpress miR-96 [24]. The key role of miR-124a and miR-96 in the control of the level of several critical components of machinery governing insulin secretion suggests a potential participation of these miRNAs in the terminal differentiation of beta-cells.
Mature beta-cells have the exclusive task to produce insulin. At normal glucose concentrations, insulin represents approximately 1/3 of the synthesized proteins [61]. However, this ratio can reach almost 1/2 at 7 mmol/L glucose [61]. The increase of insulin production occurring within the first minutes to hours upon glucose exposure is mostly achieved through enhanced protein synthesis and mRNA stabilization. In contrast, at later time points it is mainly due to transcriptional and posttranscriptional mechanisms [62]. The long-term control of insulin mRNA levels triggered by glucose contributes to the replenishment of the hormone content and is achieved through the activation of transcriptional regulators and miRNAs. Culture of insulin-producing cells at high glucose concentration affects the expression of more than hundred miRNAs, including miR-30d which is able to increase insulin gene expression [63]. Key transcription factors involved in glucose-mediated control of insulin expression have been described in detail elsewhere [64]. These factors include v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), pancreatic and duodenal homeobox 1 (PDX1), and neurogenic differentiation 1 (NeuroD) [64]. MAFA abundance is under the control of miR-204. In diabetes, the beta-cell expression of this miRNA increases in response to the elevation of the cellular redox regulator thioredoxin-interacting protein. In turn miR-204 reduces the expression of insulin [65]. Regulation of insulin transcription during development involves additional transcription factors including the members of the Onecut (OC) family [66]. In view of these findings, we tested the role of Oc2 and indirectly miR-9 in the control of insulin production. We found that overexpression or silencing of miR-9 decreases insulin expression (Figure 1(a)), indicating that adequate levels of this miRNA are required for maintaining optimal insulin mRNA levels. In addition, elevated amounts of miR-9 reduce the activity of a luciferase reporter construct driven by the rat insulin promoter (Figure 1(b)), suggesting a role for miR-9 in the control of insulin gene expression. Inactivation of OC-2 using a dominant negative construct mimics the effect of miR-9 on insulin promoter (Figure 1(b)). Thus, besides regulating glucose-induced insulin secretion, miR-9 appears also to be crucial for maintaining insulin mRNA levels in a mechanism probably involving Oc2. Accumulation of insulin mRNA in response to glucose relies on nuclear translocation of PDX1. Glucose-induced nuclear import of the transcription factor is triggered by activation of the phosphatidylinositol 3-kinase (PI3K) pathway [67]. This signaling cascade results in the phosphorylation of protein kinase B by the 3-phosphoinositide-dependent kinase 1 (PDK1) [68]. Beta-cell specific knockout of PDK-1 leads to a reduction in islet cell mass and the development of overt diabetes [69]. PDK1 has been identified as a target of miR-375 [58]. Overexpression of miR-375 in rat insulin-producing INS-1E cells decreases the expression of PDK-1, leading to reduction of insulin mRNA level [58].
Figure 1.
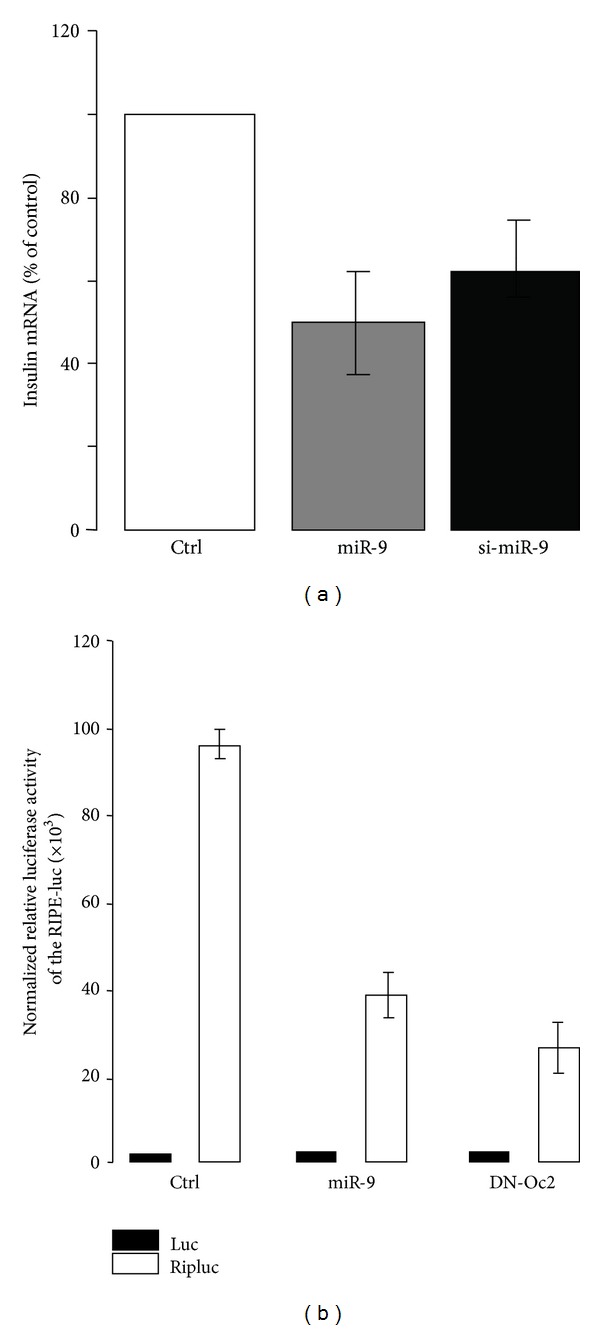
Role of miR-9 in insulin mRNA and promoter activity. (a) Effect of miR-9 on insulin mRNA. The RNA duplex containing the mature form of miR-9 [26] and a siRNA directed against miR-9 (si-miR-9) or a control oligonucleotide was transfected in MIN6 cells for 48 hrs. The expression of the preproinsulin mRNA was measured by quantitative PCR. The mRNA level was normalized against the housekeeping acidic ribosomal phosphoprotein P0 gene (Rplp0) and the expression level in cells transfected with the control siRNA was set to 100%. Data are the mean of ± SEM of 3 independent experiments. (b) Effects of miR-9 and dominant negative Oc2 mutant on the activity of an insulin reporter construct in MIN6 cells. MIN6 cells were transiently transfected with miR-9 RNA duplexes containing the mature form of miR-9 [26] or the dominant negative Oc2 mutant [26]. The cells were cotransfected with a luciferase reporter construct driven by a 600 bp fragment of the rat insulin promoter (Ripluc) and with pRLSV40, a construct producing a renilla luciferase activity under the control of the constitutive SV40 promoter. The firefly luciferase activity produced by Ripluc was normalized to the renilla luciferase activity to rule out differences in the transfection efficiency. The empty pGL3 basic (luc) was used as control. Each experiment was performed at least three times in triplicate.
Numerous genes that are required for glucose-induced insulin secretion and cells survival are highly or selectively expressed in beta-cells [60]. In addition, proper control of glucose-induced insulin secretion involves the absence or the low expression of “disallowed genes” including those coding for lactate dehydrogenase A (Ldha) and monocarboxylate transporter-1 (Mct1) [70]. Overexpression of ldha in insulin secreting cells affects glucose-induced insulin secretion. Islets of individuals with diabetes display an increase in the expression of LDHA when compared to controls [70]. Beta-cells elevation of MCT1 in mice fosters pyruvate-stimulated insulin secretion, thus leading to hyperinsulinism during exercise [71]. The absence of MCT1 in beta-cells could hence prevent inappropriate insulin secretion elicited by pyruvate. A mechanism that accounts for repression of the “disallowed” MCT1 in beta-cells could involve some miRNAs. The MCT1 mRNA is a direct target of miR-29a, miR-29b, and miR-124 [71]. From this example, it is possible that the miRNAs contribute to the silencing of “disallowed” genes in beta-cells.
5. miRNAs Associated with Compensatory Beta-Cell Mass Expansion in Pregnancy and Obesity
Pregnancy is the strongest physiological stimulus inducing beta-cell mass plasticity. The mass of beta-cells and their secretory activity returns to prepregnancy levels within the first 10 days following parturition in rodents [72, 73]. The levels of four miRNAs including miR-144, miR-218, miR-338-3p, and miR-451 are modified in islets from pregnant rats when compared to age-matched animals (Table 3) [74]. The expression of these miRNAs returned to resting levels after parturition [74]. In vitro experiments have confirmed a role for miR-338-3p and miR-451 in the control of beta-cell tasks [74]. While miR-338-3p levels are diminished, those of miR-451 are increased during pregnancy [74, 75]. Rodent and human beta-cell expansion during pregnancy is likely to occur thanks to enhanced proliferation combined with a minimal rate of apoptosis [72, 73, 76]. Overexpression of miR-451 does not increase the proliferation rate of insulin-producing cells but this miRNA protects the cells against apoptosis elicited by palmitate and cytokines [74]. Downregulation of miR-338-3p appears to be even more important in the adaptation of beta-cells during gestation. Indeed, reduction of miR-338-3p expression leads to a specific increase in proliferation of cultured insulin-producing islet cells and transplanted pseudoislets cells [74, 75]. Furthermore, as is the case for miR-451 overexpression, reduction of miR-338-3p protects the beta-cells against apoptosis evoked by diabetogenic conditions such as chronic exposure to elevated palmitate or cytokines, indicating that the decrease of miR-338-3p is pivotal for compensatory beta-cell mass expansion during pregnancy. Despite these proproliferative and antiapoptotic effects, miR-338-3p downregulation or miR-451 overexpression did not significantly impact insulin content and glucose-induced insulin secretion, indicating that upon changes in the level of these miRNAs the beta-cells retain a fully differentiated phenotype. The beta-cell mass not only increases during pregnancy but also during insulin-resistance and obesity [1]. The gain of beta-cell mass compensates for the increased insulin demand from peripheral tissues, thereby maintaining euglycemia [1]. An increase of miR-451 and a decrease of miR-338-3p analogous to those observed in islets of pregnant rats are also detected in islets of obese mice fed with high fat diet [74]. Moreover, diminution of miR-338-3p occurs in young still normoglycemic but already obese db/db mice, indicating a broader role for this miRNA in physiological islet adaptation [74]. The exact mechanisms governing the expression of miR-338-3p remain to be defined. During pregnancy, the level of estradiol and incretins such as GLP-1 is elevated [77, 78] and may be responsible for beta-cell proliferation [77, 79, 80]. GLP-1 is also increased in obese individuals possibly contributing to beta-cell mass expansion [77]. Interestingly, agonists of the GPR30 estradiol receptor and of the GLP-1 receptor are able to decrease miR-338-3p levels in beta-cells via a signalling cascade involving a rise in cAMP and the activation of PKA [74].
Table 3.
miRNAs associated with compensatory beta-cells.
| miRNAs | Cell types/models | Expression change | Known functional effect | References |
|---|---|---|---|---|
| miR-132 | Islets of prediabetic db/db mice | Up | Beta-cells proliferation | [84] |
| miR-184 | Down | |||
| miR-338-3p | Islets of pregnant rats and islets of prediabetic db/db mice and obese mice fed with a high fat diet | Down | Beta-cells proliferation/antiapoptotic | [74] |
| Cells cultured with estradiol or incretins | ||||
| miR-451 | Islets of pregnant rats, islets of prediabetic db/db mice and obese mice fed with a high fat diet | Up | Antiapoptotic | [74] |
6. miRNAs Associated with Beta-Cell Dysfunction under Diabetogenic Condition
The compensatory processes described above precede beta-cell decline during the development of diabetes [2, 81]. Failure in mechanisms that maintain the adaptive capacity of islet beta-cells may account for impaired beta-cell function and mass. This hypothesis has been tested by measuring the expression of miRNAs in islets of leptin receptor deficient db/db mice of different ages. The db/db mice at 6 weeks of age are obese and insulin resistant [82, 83]. However, normoglycemia is preserved and manifestation of diabetes is delayed because of increased functional beta-cell mass. Besides the decreased expression of miR-338-3p [74], the adaptive islets of these mice display variations in other miRNAs (Table 4) [84]. These include an increase in miR-132 and a decrease in miR-184, miR-203, and miR-210 [84]. Overexpression of miR-132 and inactivation of miR-184 trigger proliferation in dispersed beta-cells from rat islets [84]. In contrast, in vitro reduction of miR-203 and miR-210 increases rat beta-cell apoptosis [84]. The reduction of miR-210 and miR-184 is more pronounced in isolated islets from overtly diabetic db/db mice, suggesting that an unbalance in the level of these miRNAs can result in a switch from beta-cell adaptation to programmed cell death [84]. In addition, changes in the expression of miR-199a-3p and miR-383 appear to contribute to beta-cell failure in diabetic db/db mice. Indeed, upregulation of miR-199a-3p and diminution in miR-383 increase rat beta-cell apoptosis in vitro [84]. At the present time, the miRNAs that are associated with compensatory human islets remain to be identified. So far, one study has quantified the miRNAs level in a small group of islets of individuals with and without type 2 diabetes [85]. Only an increase in the miR-187 level is associated with beta-cell failure in diabetes [85]. This result suggests that different miRNAs account for adaptation and decline of beta-cells in human and rodents during diabetes.
Table 4.
miRNAs associated with beta-cell failure.
| miRNAs | Cells type/models | Expression change | Known functional effect | References |
|---|---|---|---|---|
| miR-21 | Cells cultured with cytokines | Up | Glucose-induced insulin secretion and proapoptotic | [93] |
| miR-34a and miR-146a, b | Islets of diabetic db/db mice, cells cultured with cytokines or palmitate | Up | Glucose-induced insulin secretion and proapoptotic | [91, 93] |
| miR-184 | Islets of diabetic db/db mice, cells cultured with glucolipotoxic condition | Down | Glucose-induced insulin secretion | [84, 90] |
| miR-187 | Islets of individuals with type 2 diabetes | Up | Glucose-induced insulin secretion | [85] |
| miR-199a-3p | Islets of diabetic db/db mice | Up | Proapoptotic | [74] |
| miR-203 and miR-383 | Islets of diabetic db/db mice, cells cultured with glucolipotoxic condition | Down | Proapoptotic | [90, 93] |
| miR-210 | Islets of diabetic db/db mice | Down | Proapoptotic | [84] |
Chronic elevation in circulating levels of nonesterified free fatty acids (NEFAs) is associated with obesity and is an independent predictor of T2D development [86, 87]. Numerous studies have highlighted palmitate, the most abundant NEFA in blood, as a detrimental factor promoting insulin resistance and beta-cell dysfunction. db/db mice display an abnormally increased blood NEFA concentration [88]. Beta-cell failure elicited by this lipid includes a decrease of insulin expression, impaired secretory capacity in response to nutrients and/or loss of beta-cell mass via apoptosis [3, 4, 89]. Elevated palmitate levels are thought to synergize with chronic hyperglycemia in promoting beta-cell failure in obesity-associated diabetes [90]. The decrease in miR-184, miR-203 and miR-383 is mimicked by chronic exposure of beta-cells to palmitate and/or glucose, suggesting a role for glucolipotoxicity in the variation of these miRNAs observed in islets of diabetic mice [90]. Additional miRNAs are changed in db/db mice, probably contributing to beta-cell dysfunction and death [91]. The expression of miR-34a and miR-146 is indeed augmented in islets of these mice [91]. Elevation in their levels causes dysfunction and apoptosis and mimics the harmful effects of palmitate in cultured islets [91]. Palmitate also triggers beta-cell dysfunction by an indirect mechanism that involves activation of the inflammatory process [92]. Continuous infusion of palmitate in mice evokes an increase in M1-type proinflammatory monocyte/macrophages infiltration within islets [92]. Several studies contend a role for low grade inflammation as a major issue, which links obesity to the development of diabetes [7, 89]. Interestingly, the levels of miR-34a and miR-146 are elevated by proinflammatory cytokines in isolated human islets and insulin-producing cells, indicating that the signaling cascades causing beta-cell failure elicited by palmitate and cytokines may converge and result in the activation of the same miRNAs [93]. Beside these two miRNAs, cytokines induce also the expression of miR-21 [93]. This miRNA plays a role in cell survival and can also affect glucose-induced insulin secretion by modulating the levels of components of the secretory machinery [93].
Besides chronic hyperlipidemia and hyperglycemia, patients with diabetes display an increased ratio of oxidized LDL over native LDL [94–96]. The concentration of oxidized LDL is already elevated in prediabetic individuals [97] and increases throughout the duration of the disease [98]. The rise of oxidized LDL is thought to result, in part, from the reduced antioxidant property of HDL [94, 98–100]. Elevation of oxidized LDL apparently correlates with reduction of plasma HDL concentration, a hallmark of metabolic syndrome [101]. Importantly, infusion of recombinant HDL in patients with T2D reduces glycemia [101]. The beneficial effect of HDL relies on both improved insulin secretion and glucose uptake in muscles [101]. Several independent groups, including ours, have confirmed the protective effect of HDL against the harmful effects evoked by oxidized LDL in beta-cells [94, 95, 101, 102]. Coincubation of islets and insulin-producing cells with HDL prevents the defective insulin production and glucose-induced insulin secretion observed in the presence of 2 mM oxidized human LDL cholesterol [94, 95, 101, 102]. Moreover, cell survival is strongly improved in the presence of HDL [94, 95, 101, 102]. Although native LDL above 3.1 mM cholesterol perturbs insulin secretion and cell survival [103, 104], at 2 mM cholesterol the lipoprotein does not affect the accomplishment of the tasks and the viability of beta-cells [94, 95, 101, 102]. A global microarray profiling was done to investigate the contribution of miRNAs in the adverse effects elicited by oxidized LDL. The modified lipoprotein modified the expression of a set of 10 miRNAs (Table 5). The expression changes were further prevented by coincubation with HDL (Table 5). However, quantitative PCR analysis confirmed the variation for only 5 of them (Figure 2). The expression of miR-9 was decreased, whereas that of miR-21 was increased in insulin-secreting cells cultured with oxidized LDL particles (Figure 2). As already mentioned, upregulation of miR-21 hampers glucose-induced insulin secretion by modifying the expression of components of the secretory machinery [26, 93]. Moreover, appropriate levels of miR-9 level are required to achieve optimal insulin expression (Figure 1(b)). Thus, the changes in these two miRNAs may contribute to beta-cell dysfunction provoked by the oxidized lipoprotein. Further studies will be needed to determine whether the decrease of miR-98, miR-325, and miR-374 (Figure 2) also contributes to the loss of specific beta-cell tasks and increased death caused by oxidized LDL.
Table 5.
Global miRNA profiling of MIN6 cells cultured with human native and oxidized LDL with or without HDL. We compared by microarray analysis the expression of 350 miRNAs in MIN6 cells that were incubated with 2 mmol/L of human native (Na LDL) or oxidized LDL (oxLDL) cholesterol plus or minus 1 mmol/L of HDL for 72 hrs.
| Microarray | Name | NaLDL | oxLDL | Change (log2) | Expression change | oxLDL | oxLDL + HDL | Change (log2) |
|---|---|---|---|---|---|---|---|---|
| NaLDL versus oxLDL | mmu-miR-9 | 1 622.36 | 739.3 | −1.16 | Down | 2 417.08 | 3 407.71 | 0.46 |
| mmu-miR-21 | 6 073.84 | 15 447.81 | 1.37 | Up | 23 042.48 | 11 193.24 | −1.01 | |
| mmu-miR-98 | 7 909.01 | 2 817.75 | −1.5 | Down | 12 432.59 | 16 691.57 | 0.42 | |
| mmu-miR-192 | 222.59 | 514.66 | 1.02 | Up | 1 076.26 | 732.38 | −0.51 | |
| mmu-miR-325 | 1 018.38 | 462.41 | −1.16 | Down | 1 757.68 | 2 642.59 | 0.65 | |
| mmu-miR-342-3p | 2 181.11 | 1 326.71 | −0.73 | Down | 2 483.59 | 4 337.97 | 0.79 | |
| mmu-miR-346 | 904.18 | 505.43 | −0.85 | Down | 485.7 | 770.91 | 0.64 | |
| mmu-miR-374 | 5 082.44 | 1 887.82 | −1.43 | Down | 5 022.55 | 7 143.28 | 0.54 | |
| Mmu-miR-708 | 366.85 | 712.44 | 0.94 | up | 825.11 | 402.52 | −1.06 | |
| mmu-miR-801 | 488.33 | 915.52 | 0.85 | up | 521.23 | 216.79 | −1.29 |
Figure 2.
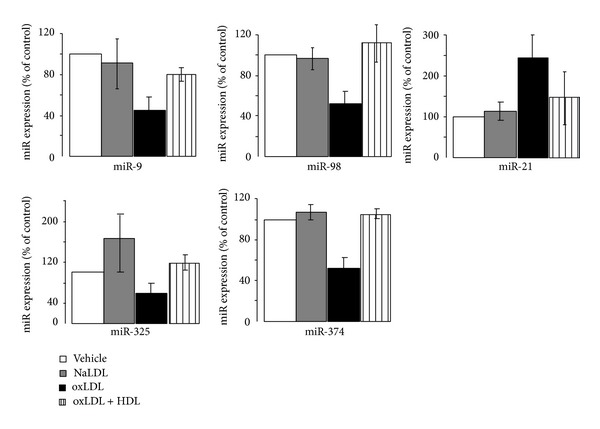
Identification of miRNAs differentially expressed in MIN6 cells cultured with human native and oxidized LDL. The expression of the indicated miRNAs was measured by quantitative RT-PCR in MIN6 cells that were cultured with vehicle, 2 mmol/L of oxidized LDL (oxLDL), and native LDL (NaLDL), plus or minus 1 mmol/L HDL-cholesterol for 72 hrs. Human plasma LDL and HDL fractions were isolated by sequential ultracentrifugation (LDL density, 1.063) as described [95]. Oxidation of LDL particles was done by incubation of 1 mg LDL protein/mL PBS with 5 μmol/L CuSO4 at 37°C for 6–8 h [95]. The oxidation reaction was verified as previously described by determining the lipid peroxide content [95]. The results are expressed as fold changes and correspond to the mean ± SD.
It is widely accepted that the beta-cell decline in diabetes relies on genetic factors [105]. The contribution of genetic factors varies according to the forms of diabetes. In monogenic and dominant forms of diabetes, mutations in a single gene can lead to beta-cell failure and thereby to the development of the disease [105, 106]. The maturity-onset diabetes of the young (MODY) is a familial monogenic form of early-onset type 2 diabetes, which usually develops in childhood, adolescence, or young adulthood [105]. MODY is now classified in the group of “genetic defect in beta-cell function” with a subclassification according to the gene involved [105]. The most common mutation in the gene encoding transcription factor 1 (TCF-1)/hepatocyte nuclear factor 1a (HNF1A) that causes MODY3 is a frame shift mutation in exon 4, Pro291fsinsC-HNF1A [105–107]. The mutation within the gene results in a truncated protein that plays as a dominant negative action. Overexpression of this mutant in insulin-producing cells hampers glucose-induced insulin secretion [108]. Impaired insulin secretion caused by the mutated protein is associated with an elevation in the levels of miR-103 and miR-224. Thus, genetic variation may impact the expression of miRNAs, potentially synergizing with environmental stressors in triggering islet beta-cell dysfunction in diabetes [107].
7. Conclusion and Perspectives
miRNAs are essential regulators of beta-cell function as evidenced by the growing number of these small RNA molecules, which play a central role in normal development, plasticity, and dysfunction of insulin-secreting cells. Besides their intracellular function, a large set of miRNAs are released in stable form in body fluids including blood and urine. Variations in the blood miRNA pool are emerging as promising biomarkers of several diseases including diabetes [109]. Indeed, circulating miRNAs including miR-103 and miR-224 have been found in the blood of patients with diabetes [108]. Transport of miRNAs within blood is achieved through different pathways involving the association with HDL particles, exosomes, and other proteins such as argonaute 2 or nucleophosmin 1 [109, 110]. Defective beta-cells can release miRNAs into bloodstream following pathophysiological conditions. Future investigations should puzzle out the physiological meaning of these circulating RNAs and determine whether the pool of miRNAs released in the blood differs according to the activation state of beta-cells. If so, monitoring these miRNAs would be insightful for monitoring whether beta-cells are in a compensatory or a failure condition. The extraordinary amount of new information provided by the discovery of the miRNAs has drawn researchers and clinical diabetologist to explore the potential involvement of another emerging class of noncoding RNAs, the long noncoding RNAs (lncRNAs) [111]. LncRNAs represent a heterogeneous population of RNA molecules longer than 200 nucleotides. The function of most of them remains unknown although several lncRNAs exert nonredundant roles in processes such as transcriptional regulation and survival [112–114]. A large number of lncRNAs is also present in human islets and some of them have their expression modified in diabetes [111]. There is no doubt that the coming years will witness the emergence of lncRNAs as additional players in the control of beta-cell function and/or in the regulation of lineage plasticity. The discovery of the regulatory potential of this emerging RNA world promises to unveil new opportunities for developing drugs capable of protecting beta-cells in the context of diabetes.
Acknowledgments
This work was supported by the Chair of Excellence from the French National Agency for Research N°ANR-10-CEXC-005-01, the Swiss National Science Foundation (Romano Regazzi, Gérard Waeber, Amar Abderrahmani), the Regional Council Nord Pas de Calais, and the European Regional Development Fund and an interdisciplinary grant from the Faculty of Biology and Medicine of the University of Lausanne (Romano Regazzi).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. The Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weir GC, Bonner-Weir S. Five of stages of evolving β-cell dysfunction during progression to diabetes. Diabetes. 2004;53(supplement 3):S16–S21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- 3.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 4.Meier JJ, Breuer TGK, Bonadonna RC, et al. Pancreatic diabetes manifests when beta cell area declines by approximately 65% in humans. Diabetologia. 2012;55(5):1346–1354. doi: 10.1007/s00125-012-2466-8. [DOI] [PubMed] [Google Scholar]
- 5.Meier JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51(5):703–713. doi: 10.1007/s00125-008-0936-9. [DOI] [PubMed] [Google Scholar]
- 6.Camastra S, Manco M, Mari A, et al. β-cell function in morbidly obese subjects during free living: long-term effects of weight loss. Diabetes. 2005;54(8):2382–2389. doi: 10.2337/diabetes.54.8.2382. [DOI] [PubMed] [Google Scholar]
- 7.Donath MY, Ehses JA, Maedler K, et al. Mechanisms of β-cell death in type 2 diabetes. Diabetes. 2005;54(supplement 2):S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 8.Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes, Obesity & Metabolism. 2008;10(supplement 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- 9.Marselli L, Suleiman M, Masini M, et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia. 2014;57(2):362–365. doi: 10.1007/s00125-013-3098-3. [DOI] [PubMed] [Google Scholar]
- 10.Gargani S, Thevenet J, Yuan JE, et al. Adaptive changes of human islets to an obesogenic environment in the mouse. Diabetologia. 2013;56(2):350–358. doi: 10.1007/s00125-012-2775-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyrberg B, Ustinov J, Otonkoski T, Andersson A. Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes. 2001;50(2):301–307. doi: 10.2337/diabetes.50.2.301. [DOI] [PubMed] [Google Scholar]
- 12.Maedler K, Donath MY. Beta-cells in type 2 diabetes: a loss of function and mass. Hormone Research. 2004;62(supplement 3):67–73. doi: 10.1159/000080503. [DOI] [PubMed] [Google Scholar]
- 13.Maedler K, Schumann DM, Schulthess F, et al. Aging correlates with decreased β-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for fas and pancreatic duodenal homeobox-1. Diabetes. 2006;55(9):2455–2462. doi: 10.2337/db05-1586. [DOI] [PubMed] [Google Scholar]
- 14.Nathan DM. Long-term complications of diabetes mellitus. The New England Journal of Medicine. 1993;328(23):1676–1685. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- 15.Street CN, Lakey JRT, Shapiro AMJ, et al. Islet graft assessment in the Edmonton Protocol: implications for predicting long-term clinical outcome. Diabetes. 2004;53(12):3107–3114. doi: 10.2337/diabetes.53.12.3107. [DOI] [PubMed] [Google Scholar]
- 16.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 17.Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57BI/6J male mice. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005610.e5610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455(7209):64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. 2007;56(12):2938–2945. doi: 10.2337/db07-0175. [DOI] [PubMed] [Google Scholar]
- 21.Joglekar MV, Joglekar VM, Hardikar AA. Expression of islet-specific microRNAs during human pancreatic development. Gene Expression Patterns. 2009;9(2):109–113. doi: 10.1016/j.gep.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 22.Poy MN, Eliasson L, Krutzfeldt J, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432(7014):226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 23.Guay C, Roggli E, Nesca V, Jacovetti C, Regazzi R. Diabetes mellitus, a microRNA-related disease? Translational Research. 2011;157(4):253–264. doi: 10.1016/j.trsl.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 24.Lovis P, Gattesco S, Regazzi R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biological Chemistry. 2008;389(3):305–312. doi: 10.1515/BC.2008.026. [DOI] [PubMed] [Google Scholar]
- 25.Baroukh N, Ravier MA, Loder MK, et al. MicroRNA-124a regulates foxa2 expression and intracellular signaling in pancreatic β-cell lines. The Journal of Biological Chemistry. 2007;282(27):19575–19588. doi: 10.1074/jbc.M611841200. [DOI] [PubMed] [Google Scholar]
- 26.Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. The Journal of Biological Chemistry. 2006;281(37):26932–26942. doi: 10.1074/jbc.M601225200. [DOI] [PubMed] [Google Scholar]
- 27.Pagliuca FW, Melton DA. How to make a functional beta-cell. Development. 2013;140:2472–2483. doi: 10.1242/dev.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bouwens L, Rooman I. Regulation of pancreatic beta-cell mass. Physiological Reviews. 2005;85(4):1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 29.Rukstalis JM, Habener JF. Neurogenin3: a master regulator of pancreatic islet differentiation and regeneration. Islets. 2009;1(3):177–184. doi: 10.4161/isl.1.3.9877. [DOI] [PubMed] [Google Scholar]
- 30.Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes & Development. 2002;16(12):1488–1497. doi: 10.1101/gad.985002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Apelqvist Å, Li H, Sommer L, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 32.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nature Cell Biology. 2009;11(3):228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 33.Joglekar MV, Parekh VS, Mehta S, Bhonde RR, Hardikar AA. MicroRNA profiling of developing and regenerating pancreas reveal post-transcriptional regulation of neurogenin3. Developmental Biology. 2007;311(2):603–612. doi: 10.1016/j.ydbio.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Lee CS, de León DD, Kaestner KH, Stoffers DA. Regeneration of pancreatic islets after partial pancreatectomy in mice does not involve the reactivation of neurogenin-3. Diabetes. 2006;55(2):269–272. [PubMed] [Google Scholar]
- 35.Kaung H-LC. Growth dynamics of pancreatic islet cell populations during fetal and neonatal development of the rat. Developmental Dynamics. 1994;200(2):163–175. doi: 10.1002/aja.1002000208. [DOI] [PubMed] [Google Scholar]
- 36.McEvoy RC, Madson KL. Pancreatic insulin-, glucagon-, and somatostatin-positive islet cell populations during the perinatal development of the rat. I. Morphometric quantitation. Biology of the Neonate. 1980;38(5-6):248–254. doi: 10.1159/000241372. [DOI] [PubMed] [Google Scholar]
- 37.McEvoy RC. Changes in the volumes of the A-, B-, and D-cell populations in the pancreatic islets during the postnatal development of the rat. Diabetes. 1981;30(10):813–817. doi: 10.2337/diab.30.10.813. [DOI] [PubMed] [Google Scholar]
- 38.Montanya E, Nacher V, Biarnes M, Soler J. Linear correlation between β-cell mass and body weight throughout the lifespan in Lewis rats: role of β-cell hyperplasia and hypertrophy. Diabetes. 2000;49(8):1341–1346. doi: 10.2337/diabetes.49.8.1341. [DOI] [PubMed] [Google Scholar]
- 39.Wang RN, Bouwens L, Klöppel G. Beta-cell growth in adolescent and adult rats treated with streptozotocin during the neonatal period. Diabetologia. 1996;39(5):548–557. doi: 10.1007/BF00403301. [DOI] [PubMed] [Google Scholar]
- 40.Kalis M, Bolmeson C, Esguerra JLS, et al. Beta-cell specific deletion of dicer1 leads to defective insulin secretion and diabetes mellitus. PLoS ONE. 2011;6(12) doi: 10.1371/journal.pone.0029166.e29166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mandelbaum AD, Melkman-Zehavi T, Oren R, et al. Dysregulation of Dicer1 in beta cells impairs islet architecture and glucose metabolism. Experimental Diabetes Research. 2012;2012:8 pages. doi: 10.1155/2012/470302.470302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poy MN, Hausser J, Trajkovski M, et al. miR-375 maintains normal pancreatic α- and β-cell mass. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(14):5813–5818. doi: 10.1073/pnas.0810550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kloosterman WP, Lagendijk AK, Ketting RF, Moulton JD, Plasterk RHA. Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biology. 2007;5(8) doi: 10.1371/journal.pbio.0050203.e203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Correa-Medina M, Bravo-Egana V, Rosero S, et al. MicroRNA miR-7 is preferentially expressed in endocrine cells of the developing and adult human pancreas. Gene Expression Patterns. 2009;9(4):193–199. doi: 10.1016/j.gep.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 45.Wang Y, Liu J, Liu C, Naji A, Stoffers DA. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes. 2013;62(3):887–895. doi: 10.2337/db12-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rachdi L, Balcazar N, Osorio-Duque F, et al. Disruption of Tsc2 in pancreatic β cells induces β cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(27):9250–9255. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hamada S, Hara K, Hamada T, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic β-cells leads to increased β-cell mass and prevention of hyperglycemia. Diabetes. 2009;58(6):1321–1332. doi: 10.2337/db08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie J, Herbert TP. The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic β-cell mass: implications in the development of type-2 diabetes. Cellular and Molecular Life Sciences. 2012;69(8):1289–1304. doi: 10.1007/s00018-011-0874-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parnaud G, Bosco D, Berney T, et al. Proliferation of sorted human and rat beta cells. Diabetologia. 2008;51(1):91–100. doi: 10.1007/s00125-007-0855-1. [DOI] [PubMed] [Google Scholar]
- 50.Meier JJ, Butler AE, Saisho Y, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57(6):1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veld PI, de Munck N, van Belle K, et al. β-cell replication is increased in donor organs from young patients after prolonged life support. Diabetes. 2010;59(7):1702–1708. doi: 10.2337/db09-1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bar Y, Russ HA, Knoller S, Ouziel-Yahalom L, Efrat S. HES-1 is involved in adaptation of adult human β-cells to proliferation in vitro. Diabetes. 2008;57(9):2413–2420. doi: 10.2337/db07-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0035801.e35801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Drucker DJ. The biology of incretin hormones. Cell Metabolism. 2006;3(3):153–165. doi: 10.1016/j.cmet.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 55.Hodson DJ, Mitchell RK, Bellomo EA, et al. Lipotoxicity disrupts incretin-regulated human beta cell connectivity. The Journal of Clinical Investigation. 2013;123(10):4182–4194. doi: 10.1172/JCI68459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferdaoussi M, Abdelli S, Yang J-Y, et al. Exendin-4 protects β-cells from interleukin-1β-induced apoptosis by interfering with the c-Jun NH2-terminal kinase pathway. Diabetes. 2008;57(5):1205–1215. doi: 10.2337/db07-1214. [DOI] [PubMed] [Google Scholar]
- 57.Keller DM, Clark EA, Goodman RH. Regulation of microRNA-375 by cAMP in pancreatic beta-cells. Molecular Endocrinology. 2012;26(6):989–999. doi: 10.1210/me.2011-1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, van Obberghen E. MiR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic β-Cells. Diabetes. 2008;57(10):2708–2717. doi: 10.2337/db07-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krichevsky AM, Sonntag K-C, Isacson O, Kosik KS. Specific MicroRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells. 2006;24(4):857–864. doi: 10.1634/stemcells.2005-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abderrahmani A, Plaisance V, Lovis P, Regazzi R. Mechanisms controlling the expression of the components of the exocytotic apparatus under physiological and pathological conditions. Biochemical Society Transactions. 2006;34(5):696–700. doi: 10.1042/BST0340696. [DOI] [PubMed] [Google Scholar]
- 61.Schuit FC, Veld PAI, Pipeleers DG. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(11):3865–3869. doi: 10.1073/pnas.85.11.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suckale J, Solimena M. Pancreas islets in metabolic signaling—focus on the beta-cell. Frontiers in Bioscience. 2008;13(18):7156–7171. doi: 10.2741/3218. [DOI] [PubMed] [Google Scholar]
- 63.Tang X, Muniappan L, Tang G, Özcan S. Identification of glucose-regulated miRNAs from pancreatic β cells reveals a role for miR-30d in insulin transcription. RNA. 2009;15(2):287–293. doi: 10.1261/rna.1211209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Andrali SS, Smapley ML, Vanderford NL, Özcan S. Glucose regulation of insulin gene expression in pancreatic β-cells. Biochemical Journal. 2008;415(1):1–10. doi: 10.1042/BJ20081029. [DOI] [PubMed] [Google Scholar]
- 65.Xu G, Chen J, Jing G, Shalev A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nature Medicine. 2013;19:1141–1146. doi: 10.1038/nm.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tweedie E, Artner I, Crawford L, et al. Maintenance of hepatic nuclear factor 6 in postnatal islets impairs terminal differentiation and function of β-cells. Diabetes. 2006;55(12):3264–3270. doi: 10.2337/db06-0090. [DOI] [PubMed] [Google Scholar]
- 67.Rafiq I, da Silva Xavier G, Hooper S, Rutter GA. Glucose-stimulated preproinsulin gene expression and nuclear trans-location of pancreatic duodenum homeobox-1 require activation of phosphatidylinositol 3-kinase but not p38 MAPK/SAPK2. The Journal of Biological Chemistry. 2000;275(21):15977–15984. doi: 10.1074/jbc.275.21.15977. [DOI] [PubMed] [Google Scholar]
- 68.Rhodes CJ, White MF, Leahy JL, Kahn SE. Direct autocrine action of insulin on beta-cells: does it make physiological sense? Diabetes. 2013;62(7):2157–2163. doi: 10.2337/db13-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hashimoto N, Kido Y, Uchida T, et al. Ablation of PDK1 in pancreatic β cells induces diabetes as a result of loss of β cell mass. Nature Genetics. 2006;38(5):589–593. doi: 10.1038/ng1774. [DOI] [PubMed] [Google Scholar]
- 70.Pullen TJ, Rutter GA. When less is more: the forbidden fruits of gene repression in the adult beta-cell. Diabetes, Obesity & Metabolism. 2013;15(6):503–512. doi: 10.1111/dom.12029. [DOI] [PubMed] [Google Scholar]
- 71.Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic beta-cells leads to relative hyperinsulinism during exercise. Diabetes. 2012;61(7):1719–1725. doi: 10.2337/db11-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology. 1992;130(3):1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 73.Rieck S, Kaestner KH. Expansion of β-cell mass in response to pregnancy. Trends in Endocrinology and Metabolism. 2010;21(3):151–158. doi: 10.1016/j.tem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacovetti C, Abderrahmani A, Parnaud G, et al. MicroRNAs contribute to compensatory beta cell expansion during pregnancy and obesity. The Journal of Clinical Investigation. 2012;122(10):3541–3551. doi: 10.1172/JCI64151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jacovetti C, Regazzi R. Compensatory beta-cell mass expansion: a big role for a tiny actor. Cell Cycle. 2013;12(2):197–198. doi: 10.4161/cc.23378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Butler AE, Cao-Minh L, Galasso R, et al. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010;53(10):2167–2176. doi: 10.1007/s00125-010-1809-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brubaker PL, Drucker DJ. Minireview: glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145(6):2653–2659. doi: 10.1210/en.2004-0015. [DOI] [PubMed] [Google Scholar]
- 78.Valsamakis G, Margeli A, Vitoratos N, et al. The role of maternal gut hormones in normal pregnancy: fasting plasma active glucagon-like peptide 1 level is a negative predictor of fetal abdomen circumference and maternal weight change. European Journal of Endocrinology. 2010;162(5):897–903. doi: 10.1530/EJE-10-0047. [DOI] [PubMed] [Google Scholar]
- 79.Nadal A, Alonso-Magdalena P, Soriano S, Ropero AB, Quesada I. The role of oestrogens in the adaptation of islets to insulin resistance. The Journal of Physiology. 2009;587(21):5031–5037. doi: 10.1113/jphysiol.2009.177188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vasavada RC, Garcia-Ocaña A, Zawalich WS, et al. Targeted expression of placental lactogen in the beta cells of transgenic mice results in beta cell proliferation, islet mass augmentation, and hypoglycemia. The Journal of Biological Chemistry. 2000;275(20):15399–15406. doi: 10.1074/jbc.275.20.15399. [DOI] [PubMed] [Google Scholar]
- 81.Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. β-cell adaptation and decompensation during the progression of diabetes. Diabetes. 2001;50(supplement 1):S154–S159. doi: 10.2337/diabetes.50.2007.s154. [DOI] [PubMed] [Google Scholar]
- 82.Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in beta-cell gene expression and progression to diabetes. Diabetes. 2013;62(5):1557–1568. doi: 10.2337/db12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, Chan L. The db/dbdb/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of Western diet feeding. Metabolism. 2000;49(1):22–31. doi: 10.1016/s0026-0495(00)90588-2. [DOI] [PubMed] [Google Scholar]
- 84.Nesca V, Guay C, Jacovetti C, et al. Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia. 2013;56(10):2203–2212. doi: 10.1007/s00125-013-2993-y. [DOI] [PubMed] [Google Scholar]
- 85.Locke JM, da Silva Xavier G, Dawe HR, Rutter GA, Harries LW. Increased expression of miR-187 in human islets from individuals with type 2 diabetes is associated with reduced glucose-stimulated insulin secretion. Diabetologia. 2014;57(1):122–128. doi: 10.1007/s00125-013-3089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boden G. Obesity and free fatty acids. Endocrinology and Metabolism Clinics of North America. 2008;37(3):635–646. doi: 10.1016/j.ecl.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Charles MA, Eschwège E, Thibult N, et al. The role of non-esterified fatty acids in the deterioration of glucose tolerance in Caucasian subjects: results of the Paris prospective study. Diabetologia. 1997;40(9):1101–1106. doi: 10.1007/s001250050793. [DOI] [PubMed] [Google Scholar]
- 88.Kjørholt C, Åkerfeldt MC, Biden TJ, Laybutt DR. Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of β-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes. 2005;54(9):2755–2763. doi: 10.2337/diabetes.54.9.2755. [DOI] [PubMed] [Google Scholar]
- 89.Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care. 2008;31(supplement 2):S161–S164. doi: 10.2337/dc08-s243. [DOI] [PubMed] [Google Scholar]
- 90.Poitout V. Glucolipotoxicity of the pancreatic β-cell: myth or reality? Biochemical Society Transactions. 2008;36(5):901–904. doi: 10.1042/BST0360901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lovis P, Roggli E, Laybutt DR, et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic β-Cell dysfunction. Diabetes. 2008;57(10):2728–2736. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eguchi K, Manabe I, Oishi-Tanaka Y, et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metabolism. 2012;15(4):518–533. doi: 10.1016/j.cmet.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 93.Roggli E, Britan A, Gattesco S, et al. Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic β-cells. Diabetes. 2010;59(4):978–986. doi: 10.2337/db09-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abderrahmani A, Niederhauser G, Favre D, et al. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia. 2007;50(6):1304–1314. doi: 10.1007/s00125-007-0642-z. [DOI] [PubMed] [Google Scholar]
- 95.Favre D, Niederhauser G, Fahmi D, et al. Role for inducible cAMP early repressor in promoting pancreatic beta cell dysfunction evoked by oxidative stress in human and rat islets. Diabetologia. 2011;54(9):2337–2346. doi: 10.1007/s00125-011-2165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haefliger JA, Martin D, Favre D, et al. Reduction of connexin36 content by ICER-1 contributes to insulin-secreting cells apoptosis induced by oxidized LDL particles. PloS ONE. 2013;8(1) doi: 10.1371/journal.pone.0055198.e55198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Holvoet P, Kritchevsky SB, Tracy RP, et al. The metabolic syndrome, circulating oxidized LDL, and risk of myocardial infarction in well-functioning elderly people in the health, aging, and body composition cohort. Diabetes. 2004;53(4):1068–1073. doi: 10.2337/diabetes.53.4.1068. [DOI] [PubMed] [Google Scholar]
- 98.Nakhjavani M, Khalilzadeh O, Khajeali L, et al. Serum oxidized-LDL is associated with diabetes duration independent of maintaining optimized levels of LDL-cholesterol. Lipids. 2010;45(4):321–327. doi: 10.1007/s11745-010-3401-8. [DOI] [PubMed] [Google Scholar]
- 99.Bellomo G, Maggi E, Poli M, Agosta FG, Bollati P, Finardi G. Antoantibodies against oxidatively modified low-density lipoproteins in NIDDM. Diabetes. 1995;44(1):60–66. doi: 10.2337/diab.44.1.60. [DOI] [PubMed] [Google Scholar]
- 100.Brunham LR, Kruit JK, Verchere CB, Hayden MR. Cholesterol in islet dysfunction and type 2 diabetes. The Journal of Clinical Investigation. 2008;118(2):403–408. doi: 10.1172/JCI33296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Drew BG, Duffy SJ, Formosa MF, et al. High-density lipoprotein modulates glucose metabolism in patients with type 2 diabetes mellitus. Circulation. 2009;119(15):2103–2111. doi: 10.1161/CIRCULATIONAHA.108.843219. [DOI] [PubMed] [Google Scholar]
- 102.Okajima F, Kurihara M, Ono C, et al. Oxidized but not acetylated low-density lipoprotein reduces preproinsulin mRNA expression and secretion of insulin from HIT-T15 cells. Biochimica et Biophysica Acta. 2005;1687(1–3):173–180. doi: 10.1016/j.bbalip.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 103.Rütti S, Ehses JA, Sibler RA, et al. Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic β-cells. Endocrinology. 2009;150(10):4521–4530. doi: 10.1210/en.2009-0252. [DOI] [PubMed] [Google Scholar]
- 104.Roehrich M-E, Mooser V, Lenain V, et al. Insulin-secreting β-cell dysfunction induced by human lipoproteins. The Journal of Biological Chemistry. 2003;278(20):18368–18375. doi: 10.1074/jbc.M300102200. [DOI] [PubMed] [Google Scholar]
- 105.Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends in Molecular Medicine. 2010;16(9):407–416. doi: 10.1016/j.molmed.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 106.Vaxillaire M, Abderrahmani A, Boutin P, et al. Anatomy of a homeoprotein revealed by the analysis of human MODY3 mutations. The Journal of Biological Chemistry. 1999;274(50):35639–35646. doi: 10.1074/jbc.274.50.35639. [DOI] [PubMed] [Google Scholar]
- 107.Bonner C, Nyhan KC, Bacon S, et al. Identification of circulating microRNAs in HNF1A-MODY carriers. Diabetologia. 2013;56(8):1743–1751. doi: 10.1007/s00125-013-2939-4. [DOI] [PubMed] [Google Scholar]
- 108.Wang H, Antinozzi PA, Hagenfeldt KA, Maechler P, Wollheim CB. Molecular targets of a human HNF1α mutation responsible for pancreatic β-cell dysfunction. The EMBO Journal. 2000;19(16):4257–4264. doi: 10.1093/emboj/19.16.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guay C, Jacovetti C, Nesca V, Motterle A, Tugay K, Regazzi R. Emerging roles of non-coding RNAs in pancreatic beta-cell function and dysfunction. Diabetes, Obesity & Metabolism. 2012;14(supplement 3):12–21. doi: 10.1111/j.1463-1326.2012.01654.x. [DOI] [PubMed] [Google Scholar]
- 110.Zampetaki A, Kiechl S, Drozdov I, et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circulation Research. 2010;107(6):810–817. doi: 10.1161/CIRCRESAHA.110.226357. [DOI] [PubMed] [Google Scholar]
- 111.Moran I, Akerman I, van de Bunt M, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metabolism. 2012;16(4):435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wahlestedt C. Targeting long non-coding RNA to therapeutically upregulate gene expression. Nature Reviews Drug Discovery. 2013;12:433–446. doi: 10.1038/nrd4018. [DOI] [PubMed] [Google Scholar]
- 113.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152(6):1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 114.Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]