

Abstract
Cells apoptosis induced by intense heat stress is the prominent feature of heat-related illness. However, little is known about the biological effects of heat stress on cells apoptosis. Herein, we presented evidence that intense heat stress could induce early apoptosis of HUVEC cells through activating mitochondrial pathway with changes in mitochondrial membrane potential(ΔΨm), release of cytochrome c, and activation of caspase-9 and -3. We further revealed that p53 played a crucial role in heat stress-induced early apoptosis, with p53 protein rapidly translocated into mitochondria. Using pifithrin-α(PFT), a p53's mitochondrial translocation inhibitor, we found that pretreated with PFT, heat stress induced mitochondrial p53 translocation was significantly suppressed, accompanied by a significant alleviation in the loss of ΔΨm, cytochrome c release and caspase-9 activation. Furthermore, we also found that generation of reactive oxygen species (ROS) was a critical mediator in heat stress-induced apoptosis. In addition, the antioxidant MnTMPyP significantly decreased the heat stress-induced p53's mitochondrial translocation, followed by the loss of ΔΨm, cytochrome c release, caspase-9 activation and heat stress-mediated apoptosis. Conclusively, these findings indicate the contribution of the transcription-independent mitochondrial p53 pathway to early apoptosis in HUVEC cells induced by oxidative stress in response to intense heat stress.
Heat is likely to be gained by way of infrared radiation from the sun, or direct contact with hot objects, and absorbed when the ambient temperature rises above body temperature, as well as generated as a byproduct of cellular metabolism and the mechanical work of skeletal muscle1. Thus, heat stress is widely considered as a major extracellular stimulus2. Moreover, an increase in cellular temperature incurs protein denaturation and interrupts critical cellular processes, thus resulting in apoptosis and cell death3. It is well established that intense heat stress wrecks damages to the organism mainly by direct toxicity to cell4 and radiation5.Temperaturesgreater than 41.6°C to 42°C,the critical human body temperature point, can lead to cell death from apoptosis even in a few hours6,whereas extreme high temperatures of 49°C to 50°C can give rise to cellular death from necrosis in less than 5 minutes7. Therefore, excessive gain of heat from the elevated ambient temperature has been proposed to be a risk factor for the severely life-threatening disorder known as heat stroke characterized by an increased core body temperature that rapidly rises to above 40°C and engenders central nervous system dysfunction8. Besides, the mortality thereof is found very high even after adequate hypothermia or other intensive care therapy9. For the heat wave that affected Europe in the summer of 2003, 45 000 deaths have been reported of which one-third was attributed to heatstroke10,11. With the prospect of increased global temperatures in near future, the mortality and morbidity may continue to rise11. However, the pathogenesis of tissue injury and cell death in heat-stroke attributed to heat stress is not well understood. Studies in cell lines and animal models have suggested that endothelial-cell is an early target of heat stress injury and also is a prominent feature of severe heat-stroke12,13. Recent studies suggest that endothelial-cell can induce significant apoptosis during acute-phase response to heat stress14.Thus,endothelial-cell apoptosis appears to be a mechanism of heat-stroke,but little is known about the biological effects of heat stress on endothelial-cell apoptosis.
The tumor suppressor gene p53 is a key regulator of apoptosis, which has proapoptotic activity through transcription-dependent or independent pathway15. Under stressed conditions, p53 protein works as a transcription factor, activating a series of genes such as Puma, Noxa, Bax, and Bid16. On the other hand, cytoplasmic p53 directly activates mitochondrial death pathway through a transcription-independent manner17. It recently becomes clear that early in the course of p53-dependent apoptosis, caused by death signal stimuli, a fraction of p53 proteins rapidly undergo translocation to mitochondria(detectable at 30 min-1 h), and trigger an early first wave of marked caspase 3 activation, followed by an early wave of apoptosis18. This rapid first wave of apoptosis is transcription-independent, and precedes a second slower wave that is transcription-dependent18. Besides, such death signal inducers, which induce mitochondrial p53 accumulation and rapid apoptosis, have been confirmed including exposure to oxidative stress, DNA-damaging drugs, γ-irradiation or hypoxia19. Oxidative stress have also been indentified in numerous studies correlated with p53-directed cell apoptosis20,21. In addition, preliminary data have indicated that ROS plays a signaling role in mitochondrial migration of p5322 and oxidative stress is thought to play a pivotal role in heat stress induced apoptosis23. However, the field about how ROS regulates p53-mediated cell apoptosis and heat stress induced apoptosis remains rarely trodden.
Therefore, in this study, we investigated the molecular mechanism of apoptosis induced by intense heat stress. We reported for the first time that intense heat stress can induce early wave apoptosis in HUVEC cells which occurs through the mechanism that intense heat stress induces p53 translocation to mitochondria, and triggers mitochondrial apoptotic pathways. We also found that ROS produced by heat stress might be involved in transcription-independent p53-mediated mitochondrial pathways.
Results
Heat shock inhibits cell proliferation in HUVEC cells
To investigate the effect of heat stress on inhibition of HUVEC cells proliferation, we initially examined the effect of heat stress on the proliferation of HUVEC cells. HUVEC cells were maintained in culture media for 48 h and the culture dishes were sealed with parafilm and immersed for 2 h in a circulating water bath thermo-regulated at 37°C, 39°C, 41°C, 43°C, or 45°C for the heat stress treatments2. Culture media were replaced with fresh media and the cells were further incubated at 37°C for 6 h. As shown in (Figure 1), exposure of HUVEC cells to heat stress for 2 h and further incubation for 6 h registered an inhibition of the growth of HUVEC cells in a temperature-dependent manner.
Figure 1. The effects of heat stress on cell viability in HUVECs cells.
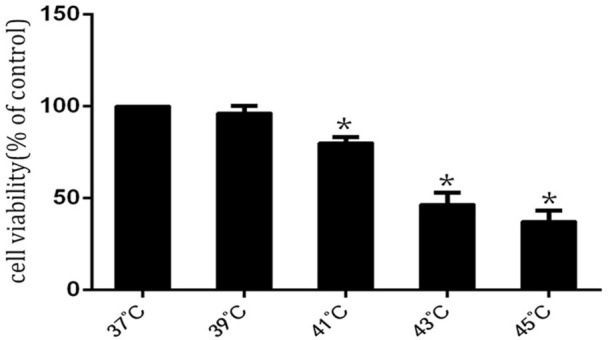
HUVEC cells viability after heat stress was tested by WST-1. Results are expressed as the percentage of cell proliferation relative to the proliferation of control treatment. The data shown are the mean from three independent experiments. Each value is the mean ± SD of three determinations. The asterisk indicates a significant difference between control and test groups, *p < 0.05.
Intense heat stress induces early wave of apoptosis by triggering the mitochondrial pathway in HUVEC cells
A quantitative evaluation was made, with flow cytometry adopted to assess the apoptotic cells. When cells were exposed to intense heat stress, the number of early apoptotic cells increased markedly under the treatment of HUVEC cells at 43°C in 2 h and apoptosis induced amounted to 27.7% by 6 h (Figure 2). In order to determine the apoptotic pathways involved in intense heat stress-induced early wave of apoptosis,we examined the changes in the activities of initiator caspase (caspase-8,-9, and-4) and effector caspase (caspase-3). As shown in Figure 3a,d caspase-9 and -3 activation were detected at 3 h after heat stress treatment. The levels of caspase-9 and -3 activities were increased by more than 2 fold and 3 fold, respectively. Caspase-8 and -4 were not activated. We also analyzed the changes in mitochondrial membrane potential(ΔΨm) and release of cytochrome c from mitochondria in heat stress-treated HUVEC cells. As shown in Figure 3e the exposure of HUVEC cells to intense heat stress resulted in the reduction of mitochondrial membrane potential in a time-dependent manner followed by significant increases in the release of cytochrome c from mitochondria(Figure 3f). These results indicated that intense heat stress might induce the mitochondrial pathway leading to caspase-9/caspase-3-mediated early apoptosis in HUVEC cells. To further explore whether heat stress induces apoptosis through the mitochondrial pathway, we infected HUVEC cells stably over-expressBcl-2 or the vector alone (control cells) with several strains. As shown in Figure 4a,b and c, the overexpressing Bcl-2 of cells significantly decreased the heat stress-induced apoptosis, the loss of ΔΨm and cytochrome c release. These data further supports the hypothesis that the mitochondrial pathway may mediate the intense heat stress-induced early wave of apoptosis of HUVEC ells.
Figure 2. Intense heat stress-induced apoptosis.
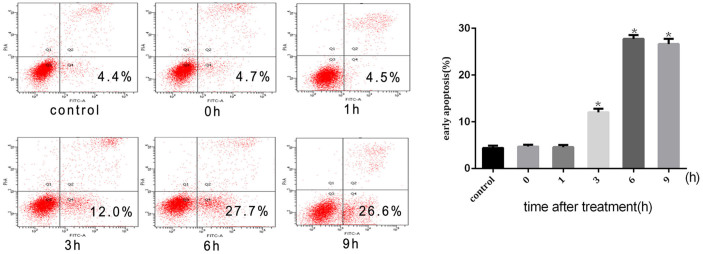
HUVEC cells were treated with 43°C for 2 h, culture media were replaced with fresh media and the cells were further incubated for different times as indicated, and apoptosis was analyzed by flow cytometry using Annexin V-FITC/PI staining. Data are presented as means ± SD of three separate experiments. The asterisk indicates a significant difference between control(37°C) and test groups(43°C), *p < 0.05.
Figure 3. Heat stress-induced apoptosis through the initiation of the mitochondrial pathway.

HUVEC cells were treated with 43°C for 2 h, culture media were replaced with fresh media and the cells were further incubated for different times as indicated. (a–d) Enzymatic activity of caspase-9,-3,-8 and -4 was measured in cell lysates using fluorogenic substrate Ac-LEHD-AFC,Ac-DEVD-AMC, Ac-ATAD-AFC and Ac-IETD-pNA, respectively, and was expressed relative to the control at 37°C (100%). (e) The loss of ΔΨm was measured by JC-1 and flow cytometry. (f) The expression levels of cytochrome C were determined by Western Blots(cropped).COX IV, mitochondrial loading control. Each value is the mean ± SD of three separate determinations, *P < 0.05 compared with control group(37°C).
Figure 4. Heat stress-induced apoptosis in HUVEC cells over-expressing protein Bcl-2.

HUVEC cells stably over-expressing the anti-apoptotic protein Bcl-2 or vector alone were treated with 43°C for 2 h, the cells further incubated for 6 h, and(a) Western Blots of bcl-2 protein expressed(cropped) in HUVEC transfectant cells. β-actin was run as an internal control.(b) apoptosis was analyzed by flow cytometry using Annexin V-FITC/PI staining. (c) The loss of ΔΨm was measured by JC-1 and flow cytometry. (d)Intracellular location of cytochrome C was determined by Western Blots(cropped). COX IV, mitochondrial loading control. Each value is the mean ± SD of three separate determinations. *P < 0.05 versus vector alone cells exposed to heat stress.
P53 activation of the mitochondrial apoptosis pathway
Previous study has reported heat stress induces apoptotic death in cells through mitochondrial pathway24. Our study has also found that intense heat stress results in caspase-9 activation. However, the upstream of mitochondrial apoptotic pathway induced by intense heat stress has not been fully illustrated. Nevertheless, It is well known that p53 protein plays a central role in the regulation of apoptosis. To investigate the involvement of p53 in heat stress-induced apoptosis, the effects of the transfection of the P53 siRNA on the cellular features of apoptosis were studied. As shown in Figure 5a, the transfected cells were assayed for P53 protein expression by western blot assay. Of the several tested siRNA target sequences, one siRNA was effective in decreasing protein levels. P53 mRNA level measured by RT- PCR was significantly decreased in P53 siRNA- transfected cells compared to control cells (scrambled siRNA) (Figure 5b). The P53 siRNA-transfected cells expressed reduced level of p53 protein, and were less susceptible to intense heat stress-induced early apoptosis compared with control cells (Figure 5c). The caspase-9 activation, release of cytochrome c and mitochondrial membrane potential(ΔΨm) in response to intense heat stress were also studied in P53 siRNA transfected HUVEC cells. When compared with control cells, P53 siRNA transfected HUVEC cells showed a drastic reduction in caspase-9 activity (Figure 5d) and the release of cytochrome c (Figure 5f). The loss of ΔΨm was alleviated in P53 siRNA transfected HUVEC cells compared with control cells (Figure 5e). These results indicate that signaling of heat stress-induced apoptosis is in part dependent on the expression of p53.
Figure 5. Heat stress-induced apoptosis in P53 siRNA transfectant HUVEC cells.

(a) HUVEC cells were transfected with scrambled siRNA (Scr) or P53 siRNA (P53). Western Blots of P53 protein expressed(cropped) in HUVEC transfectant cells.β-actin was run as an internal control. (b) RT-PCR analysis of P53 in HUVEC transfectant cells. Total RNA was isolated from HUVEC transfectant cells and P53 mRNA levels were determined by RT-PCR and analyzed on agarose gel electrophoresis. β-actin from the same samples was amplified as control. (c–f) HUVEC transfectant cells were treated with 43°C for 2 h with the cells further incubated for 6 h, and (c) apoptosis was analyzed by flow cytometry using Annexin V-FITC/PI staining. (d) Enzymatic activity of caspase-9 was measured in cell lysates using fluorogenic substrate Ac-LEHD-AFC and was expressed relative to the control at 37°C (100%).(e) The loss of ΔΨm was measured by JC-1 and flow cytometry. (f) Intracellular location of cytochrome C was determined by Western Blots(cropped). COX IV, mitochondrial loading control. Each value is the mean ± SD of three separate determinations. *P < 0.05 versus scrambled siRNA-transfected cells exposed to heat stress.
Translocation of p53 to Mitochondria in heatstress-treated HUVEC cells
Based on evidence for the crucial role of transcription-independent p53-mediated cells apoptosis in this process of early wave apoptosis25, we analyzed further the subcellular distribution of p53 in HUVEC cells during early apoptosis induced by intense heat stress. Highly enriched mitochondria fractions e purified from untreated or heat-treated HUVEC cells were prepared by sucrose gradient centrifugation26 and analyzed by Western blot. Exposure of HUVEC cells to heat stress for 2 h, and further incubation for different times exhibited a significant amount of p53 accumulation at the mitochondria in a time -dependent manner, which occurs at 0 h after cells were treated with 43°C and peaked at 6 h (Figure 6a). To directly localize p53 in mitochondria of early apoptosing cells, we performed indirect immunofluorescence. P53 mitochondrial translocation was detected by immunofluorescence staining (Figure 6b). These findings thus suggest that intense heat stress induces rapid mitochondrial translocation of p53, which may activate the apoptotic program.
Figure 6. Intense heat stress induces translocation of p53 from nucleus to mitochondria.
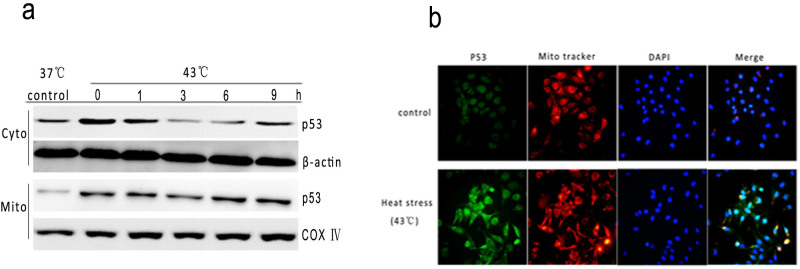
(a) To determine localization of p53, HUVEC cells were treated with 43°C for 2 h, further incubated for different time as indicated, and the levels of p53 (mitochondrial and cytosolic fraction) proteins were assessed by Western blot using corresponding antibodies(cropped). COX IV, mitochondrial loading control. (b) immunofluorescence localization of p53 to mitochondria in heat-treated cells. HUVEC cells were treated with 43°C for 2 h, and stained with anti-p53 antibody (green), MitoTracker (red) and nuclear(blue). Merged images are shown in the right panels.
Mitochondrial localization of p53 promotes mitochondrial apoptosis pathways
To verify further that mitochondrial p53 accumulation is associated with heat stress induced cells apoptotic death, we administered pifithrin-α(PFT), a highly selective and potent synthetic p53 inhibitor to show that PFT can block p53's mitochondrial translocation27. HUVEC cells were pretreated with PFT for 1.5 h, then treated with intense heat stress (43°C) for 2 h. As shown in Figure 7a, after pretreatment with PFT the heat stress induced mitochondrial p53 translocation displayed a significant reduction. To investigate the downstream of the mitochondrial p53 pathway,we examined cytochrome c release to the cytosol, caspase-9 activation and mitochondrial membrane potential(ΔΨm)in PFT treated HUVEC cells. A significant decrease in cytosolic cytochrome c (Figure 7b), and caspase-9 activation (Figure 7c) were observed. The loss of ΔΨm was also alleviated in pretreated with PFT HUVEC cells compared with control cells (figure 7d). These results indicate that p53 molecules accumulated in response to heat stress translocate into the mitochondria and activate mitochondrial apoptosis pathway.
Figure 7. Mitochondrial p53 activates mitochondrial apoptosis pathway in PFT treated HUVEC cells.

HUVEC cells were pretreated with or without pifithrin-α for 1.5 h followed by exposure to 43°C for 2 h, and the cells further incubated for 6 h. (a–b) Western blot(cropped) analysis of the effect of PFT on heat stress-induced P53 mitochondrial translocation and release of cytochrome c in HUVEC cells. COX IV, mitochondrial loading control. (c) The effect of PFT on enzymatic activity of caspase-9 was measured in cell lysate using fluorogenic substrate Ac-LEHD-AFC and was expressed relative to the control at 37°C (100%). (d) Flow cytometry and JC-1 measure the effet of PFT on ΔΨm. Data are presented as mean ± SD for three independent experiments, *P < 0.05 compared with HS group.
Intense heat stress increases the generation of ROS in HUVEC cells
Based on the evidence that ROS generation plays an important role in heat stress28, we further validated the possibility that heat stress-induced apoptosis prompts ROS accumulation. We utilized the fluorescent dye DCFH-DA, which produces enhanced fluorescence when cells generate ROS. H2O2-treated HUVEC cells were used as a positive control. DCFH-DA–based detection revealed that intracellular ROS production in HUVEC cells increased due to the treatment with heat stress. As shown in Figure 8, we observed an increase in a time-dependent manner in intracellular ROS levels in HUVEC cells after the treatment with heat stress which peaked at 2 h.
Figure 8. The effect of heat stress on the production of ROS in HUVEC cells.
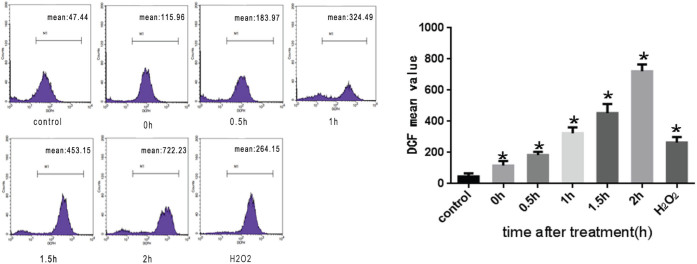
HUVEC cells were treated for 2 h in a water bath at 37°C or 43°C. Culture media were replaced with fresh media and the cells were further incubated for the indicated time. The amounts of ROS were assayed by DCFH staining. H2O2 was used as a positive control. Data are presented as mean ± SD for three independent experiments, *P < 0.05 compared with control group(37°C).
The role of ROS on heat stress-induced apoptosis
To investigate whether ROS generation via heat stress also involves p53-mediated mitochondrial pathways, we assessed mitochondrial apoptosis related proteins in HUVEC cells which had been pretreated with antioxidant MnTMPyP (10 umol/l) for 1 h, then treated with intense heat stress (43°C). The depletion of ROS by the cell-permeable ROS scavenger MnTMPyP significantly decreased the heatstress-mediated translocation of p53 proteins to mitochondria, cytochrome c release from mitochondria, caspase-9 activity, and apoptosis (Figure 9). These data confirm that heat stress treatment of HUVEC cells activates the mitochondria via release of ROS.
Figure 9. ROS involved in P53-mediated mitochondrial apoptotic pathway.

HUVEC cells were pretreated with or without MnTMPyP (10 umol/l) for 1 h before exposure to heat stress. (a) the effect of MnTMPyP on the production of ROS in HUVEC cells were assayed by DCFH staining.(b)Flow cytometry analysis of the effect of MnTMPyP on heat stress-induced cells apoptosis. H2O2 was used as a positive control. (c)The effect of MnTMPyP on enzymatic activity of caspase-9 was measured in cell lysates using fluorogenic substrate Ac-LEHD-AFC and was expressed relative to the control at 37°C (100%). (d) Flow cytometry and JC-1 to measure the effect of MnTMPyP on ΔΨm. (e–f): Westen blots analysis(cropped) on the effects of MnTMPyP on P53 mitochondrial translocation, and cytochrome c release from mitochondria induced by intense heat stress. COX IV, mitochondrial loading control. Data are presented as mean ± SD for three independent experiments, *P < 0.05 compared with HS group.
Discussion
Exposure to high ambient temperature and an elevation of body temperature has been proposed to be a risk factor for heat-related illness representing a continuum of disorders from minor syndromes such as heat cramps, heat syncope, and heat exhaustion to the severely life-threatening disorder known as heat stroke29. Although lots of researches were performed on this during the past decade, the mechanism of heat-related illnesses is not clear30. Studies in animal models found that there was widespread apoptosis in spleen, lymph node, thymus, small bowel, and cells of lymphoid origin in the lamina propria12. Using in vitro approach, numerous studies have established that apoptosis can be promoted by application of heat stress31,32,33. These data imply that apoptosis may play an important role in the physiology/pathophysiology of heat-related illness attributed to heat stress. In our study, we focused on endothelial cells because (1) endothelial cells are exposed to diverse stressors,which serve not only as a barrier between the bloodstream, its elements and toxins, and tissue parenchyma but also as generalized transducing cells that modulate the parenchymal responses to those stimuli34; (2) Endothelial-cell injury in acute-phase response to heat stress are the prominent features of heat stroke8; and (3) Endothelial-cell apoptosis has been shown to increase the extent of injury in cell lines and animal models attributable to heat stress12,35. In this work, we investigated the biological effects of intense heat stress on cells apoptosis, and showed that intense heat stress could induce early apoptosis through the transcription-independent mitochondrial p53 pathway, which might involve reactive oxygen species(ROS) attributed to intense heat stress.
Mitochondria are central integrators and transducers for pro-apoptotic signals, forming the nexus between the non-specific inducer phase and the final execution phase of apoptosis36. Among studies about cell lines, Hsu YL et al found that heat stress triggered the mitochondrial apoptotic pathway resulting in caspase-9 activity24. A previous study by Milleron RS et al. also found that under intense heat stress, cells underwent early phase of apoptosis (within 4 h of heat stress),which triggered the mitochondrial apoptotic pathway by activating the apical protease that induced mitochondrial outer membrane permeabilization(MOMP), a loss in ΔΨm and caspase-3 activation37. Consistently, our results showed that intense heat stress results in a significant increase of release of cytochrome c from mitochondria and decrease of ΔΨm. In addition, our results also revealed the degree of caspase-9 and caspase-3 activity after HUVEC cells were treated with intense heat stress. Caspase-8 and -4 was not activated in this study. Therefore, our results suggested that intense heat stress might initiate mitochondrial other than death receptors or the endoplasmic reticulum (ER) to promote the early wave of apoptosis of HUVEC cells.
P53 is a transcription factor which modulates expression of numerous target genes that control apoptosis,cell cycle arrest, senescence, DNA repair and genetic stability in response to death signal stressor38. In addition to its transcriptional route, P53 also promotes apoptosis through transcription-independent mechanisms, primarily signals through the mitochondrial pathway39. Mitochondrial p53 targeting occurs in a wide spectrum of cell types and after a variety of stress signals including oxidative stress, DNA damage, irradiation and hypoxic stress40. Previous studies demonstrated that early in the course of p53-dependent apoptosis, caused by such stressor, a fraction of wild-type p53 protein rapidly migrates to the mitochondria41. Once at the mitochondrion, p53 leads to mitochondrial dysfunction that can directly trigger the release of pro-apoptotic factors from the mitochondrial intermembrane space42. Tian XJ et al.43 recently demonstrated that at the population level, apoptosis appears in two waves, i.e the fast wave mediated by the mitochondrial p53 pathway and the slow wave by the nuclear p53 pathway. We have shown in this study that mitochondrial p53 translocation started within 1 h and was significant at 3 h after intense heat-treatment, followed by early cells apoptosis (peak at27.7% at 6 h after43°C for 2 h).To identify whether mitochondrial p53 accumulation is associated with heat stress-induced early cells apoptosis, we found that knockdown of p53 by P53 siRNA, reduced ΔΨm loss, release of cytochrome c and caspase-9 and -3 activation, as well as early wave of apoptosis (Figure 5).This shows that P53 plays an important role in intense heat stress-mediated HUVEC cellular early apoptosis. We also further evaluated the effect of Pifithrin-α(PFT) on apoptosis cells induced by intense heat stress. We found that PFT can significantly block p53's mitochondrial translocation, followed by decreased cytochrome c release (Figure 7), caspase-9 activation, as well as alleviated ΔΨm decrease in heat-treated cells. Therefore, it is plausible that the p53 in heat-treated HUVEC cells translocates to mitochondria where it triggers mitochondrial apoptotic pathways. These results are consistent with the notion that mitochondrial p53 can trigger fast apoptosis at higher damage levels43. Our data also explains the intriguing observation by Milleron RS et al37.who showed that intense heat stress-induced cells an early apoptosis through an unknown upstream of mitochondria.
Oxidative damage to cellular compartments has great implication for animal production as well as human health44. Under physiological conditions, the redox status of all aerobic cells is balanced by enzyme and non-enzyme systems. However, under certain stressful conditions, increases in generation of ROS and/or decreases in anti-oxidant capacity enhanced oxidative stress45. Pucciariello C et al.46 discovered that acute heat stress could disturb the balance between the ROS production and the antioxidant systems. Several reports link oxidative stress with heat stress and suggest synergistic augmentation of cell death, and report increased ROS generation in heat exposed cells47,48,49. What's more, recent studies have revealed that ROS acts as both an up-stream signal that triggers p53 activation and as a downstream factor that mediates apoptosis21. In other previous studies, ROS induced p53 (~2%) moves into mitochondria and initiates apoptosis50. Our study has shown that intense heat stress-mediated oxidative stress works primarily by increasing the production of ROS. We further observed that heat stress generates ROS, which activates caspase-9 rather than caspase-8 and -4. Furthermore, using MnTMPyP, an agent which is a cell-permeable ROS scavenger51, able to block caspase- 9 activity, P53 moves to mitochondria and engenders apoptosis. All in all, ROS, as the production of acute heat stress, might act as an upstream signal that triggers transcription-independent p53-mediated mitochondrial pathways to promote the early wave of apoptosis of intense heat stress on HUVEC cells.
In conclusion, our data indicate that HUVEC cells undergo apoptosis soon after intense heat stress. Intense heat stress-induced early wave of apoptosis is associated with the mitochondrial apoptosis pathway involving translocation of p53 to mitochondria, which are in turn mediated by ROS generation. Our study provides for the first time an evidence of ROS involved transcriptional-independent p53-mediated mitochondrial pathways in heat stress-mediated apoptosis of HUVEC cells.
Methods
Cell culture, treatments and cell proliferation assay
Human umbilical vein endothelial cells (HUVECs) were purchased from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. Cells passage 3, were grown in media recommended by the manufacturer. HUVEC cells maintained in culture media Culture dishes were sealed with parafilm and immersed for 2 h in a circulating water bath thermo-regulated at 37°C ± 0.5°C for the control treatment or at 39°C,41°C, 43°C,or45°C ± 0.5°C for the heat stress treatments. Culture media were replaced with fresh media and the cells further incubated for the indicated times. Cell proliferation was assessed by Premixed WST-1 Cell Proliferation Reagent (Clontech Laboratories Inc., Mountain View, CA, USA) according to the manufacturer's instructions.
Flow cytometric analysis of cell apoptosis using Annexin V-FITC/PI staining
Cell cycle phase were either kept untreated or exposed to 2 h 43°C before being analyzed by flow cytometry. The detection was performed according to the manual of AnnexinV-FITC apoptosis detection kit (invitrogen). About 1 × 106 cells were collected, washed with ice-cold PBS, and resuspended in the binding buffer containing suitable amount of Annexin V-FITC. After 10 min incubation in the dark at room temperature, the buffer was removed by centrifugation. The cells were resuspended in reaction buffer containing propidium iodide (PI). Then flow cytometric analysis was immediately performed to detect apoptosis.
Measurements of ROS
Levels of intracellular ROS were assessed by use of a reactive oxygen species assay kit (Beyotime, Nanjing, China). dichlorofluoresceindiacetate(DCFH-DA, Molecular Probes,Beyotime) was used. This reagent enters the cells and reacts with ROS, producing the fluorophoredichlorofluorescein (DCF). Briefly, cell cycle phase was either kept untreated or exposed to 2 h 43°C, and further incubated for different times, then about 3 × 105 were harvested, washed with serum free DMEM culture medium and the cells were stained with10 μM dichlorofluoresceindiacetate (DCFH-DA, Molecular Probes,Beyotime) for 30 min at 37°C in the darkness. Then the cells were harvested, washed and resuspended in serum-free DMEM culture medium for three times. The cells' fluorescence intensity was determined using flow cytometer52. 200 μM H2O2 –treated HUVEC cells were as a positive control.
Assay for caspase activity
After treatment with 43°C for 2 h, cells were harvested, and cell lysates were prepared at −80°C for 30 min and then incubated with the following appropriate caspase substrates at 37°C using a Quadruple MonochromatorMicroplate Reader (Infinite M1000, Tecan US, NC, USA). Caspase activities were measured by cleavage of the following fluorogenic peptide substrates53,54: Ac-LEHD-AFC for caspase-9, Ac-DEVD-AMC for caspase-3, Ac-ATAD-AFC for caspase-8 and Ac-LEVD-AFC for caspase-4. Activities of caspases are represented as relative cumulative fluorescence of the kinetic reaction and compared to untreated controls.
Immunofluorescence analysis
HUVEC cells were either left without or with heat stress for 2 h and further incubated for 3 h, then removed of the culture medium. All cells were labeled by adding pre-warmed (37°C) staining solution containing 200 nM MitoTracker® probe and incubated for 20 minutes under 37°C. The cells were washed with PBS twice, subsequently fixed in 3.5% paraformaldehyde, permeabilized in KB solution and 0.2% Triton X-100 at room temperature. After the blocking solution was washed out, cells were incubated with a primary antibody (rabbit anti-human p53, 1:500) (diluted in KB) for 30–45 min at 37°C and subsequently f washed with KB twice. After incubated for 30–45 min at 37°C with secondary antibody (diluted in KB), cells were washed with KB again before mounted onto slide with mounting solution containing 0.2 μg/ml DAPI and sealed with nail polisher. The slide were stored at 4°C in a dark box and observed under a fluorescent microscope.
Mitochondrial membrane potential assay
The mitochondrial membrane potential was detected using5,5′,6,6′-tetrachloro-1, 1′,3,3′ tetraethylbenzimidazolcarbocyanine iodide (JC-1; Molecular Probes, Eugene, OR, USA). Following treatment with heat stress, cells were rinsed once with complete culture medium and incubated with JC-1 for 30 min at 37°C, centrifuged, washed twice with cold PBS, transferred to a 96-well plate (105 cells/well), and assayed in a fluorescence plate reader with the following settings:excitation at 490 nm, emission at 540 nm and 590 nm, Changes in the ratio between the measurement at test wave lengths of 590 nm (red) and 540 nm (green). Fluorescence intensities are indicative of changes in the mitochondria membrane potential55.
Plasmid construction and stable transfection
Eukaryotic expression vectors of pcDNA3.1-Bcl-2 were constructed by Shanghai Genechem (Genechem Incorporation, Shanghai, China). HUVEC cells were transfected with empty vector(pcDNA3.1), Bcl-2 expression vector (pcDNA3.1-Bcl-2)by Lipofectamine 2000. Culture medium containing G418 was used to select stable transfectants.
siRNA transfection
Small-interfering RNA (siRNA) for P53 was designed and synthesized by Guangzhou RiboBio (RiboBioInc, China). The sequence of each gene and their scrambled are shown in follows. For p53,sense and antisense siRNAs are 5′-GAAAUUUGCGUGUGGAGUA (dTdT)-3′ and 5′ -UACUCCACACGCAAAUUUC(dTdT)-3′, respectively. For scrambled control, sense and antisense siRNAs are 5′-UGGUUUACAUGUCGACUAA(dTdT)-3′ and 5′-UUAGUCGACAUGUAAACCA(dTdT)-3′, respectively. Twenty-four hours prior to transfection, HUVEC cells were plated onto a 6-well plate (Nest, Biotech,China) at 30–50% confluence, which were then transfected into cells using TurboFect™ siRNA Transfection Reagent (Fermentas, Vilnius, Lithuania) according to the manufacturer's protocol. Cells were collected after 48–72 h for the further experiments. The mRNA and protein levels of P53 were estimated by RT-PCR and Western blotting. After 2 days of incubation at 37°C in a humidified atmosphere of 5% CO2, cells were exposed to heat stress.
Western blot analysis
HUVEC cells were pretreated with or without 43°C for 2 h, and further incubated for different times as indicated. Western blot was carried out as described56,57 with monoclonal Anti-p53 antibodies(1:1000; Cell signaling technology,Danvers, USA) and Polyclonal Anti-Cytochrome C antibodies(1:1000; Cell signaling technology,Danvers, USA). An HRP-conjugated anti-rabbit IgG antibody was used as the secondary antibody (ZhongshanInc, China). Signals were detected using enhanced chemiluminescence reagents (Pierce, Rockford, IL, USA).
RNA isolation, reverse transcription, and qRT-PCR
Total RNA was extracted from the p53siRNA and ScrsiRNA HUVEC cells using Trizol(Takara, Shiga, Japan). For P53, RNA was transcribed into cDNA and amplified with Specific sense: 5- ACACGCTTCCCTGGATTG-3; antisense primer: 5-CAAGAAGCCCAGACGGAAAC-3. Reverse transcription and QPCR were performed in accordance with manufacturer's instructions (Takara, Shiga, Japan). The PCR reaction for each gene was repeated three times. GAPDH was used as inner control for normalizing p53 expression. Differential expression of p53 was calculated using the 2-ΔΔCt method.
Statistical analysis
All data were analyzed for statistical significance using SPSS 13.0 software (SPSS, Chicago, IL, USA). Data were expressed as means ± SD from at least 3 independent experiments performed in duplicate. Statistical comparisons of the results were made using analysis of One-way ANOVA. p < 0.05 was considered statistically significant.
Author Contributions
Z.T.G. performed the study and composed this manuscript. H.W. and L.L. were responsible for primary data generation and analysis. Y.S.L., Z.F.L. and F.F.Y. participated in measurements of ROS and caspase activity. X.B.D. and S.F.H. performed apoptosis test. H.S.T. performed over-expressing protein Bcl-2. L.S. is the principal investigator and corresponding author for these studies.
Acknowledgments
This study was supported by National Natural Science Foundation of China (NO.81101467, 81101406), the project team of Natural Science Foundation of Guangdong Province(s2013030013217), the Project of Medical Research of PLABWS12J108 and Natural Science Foundation of Guangdong Province(NO.10151001002000001).
References
- Lugo-Amador N. M., Rothenhaus T. & Moyer P. Heat-related illness. Emerg Med Clin N Am. 22, 315–327 (2004). [DOI] [PubMed] [Google Scholar]
- Park C. H. et al. Heat shock-induced matrix metalloproteinase (MMP)-1 and MMP-3 are mediated through ERK and JNK activation and via an autocrine interleukin-6 loop. J Invest Dermatol. 123, 1012–1019 (2004). [DOI] [PubMed] [Google Scholar]
- Matsuki S. et al. Suppression of cytochrome c release and apoptosis in testeswith heat stress by minocycline. BBRC. 312, 843–849 (2003). [DOI] [PubMed] [Google Scholar]
- Johnson S. I., McMichael M. & White G. Heat stroke in small animal medicine: a clinical practice review. J Vet Emerg Crit Care. 16, 112–119 (2006). [Google Scholar]
- Ohtsubo T. et al. Acidic environment modifies heat or radiation-induced apoptosis in human maxillary cancer cells. Int J Radiation Oncology BiolPhys. 49, 1391–1399 (2001). [DOI] [PubMed] [Google Scholar]
- Sakaguchi Y. et al. Apoptosis in tumors andnormal tissues induced by whole body hyperthermia in rats. Cancer Res. 55, 5459–5464 (1995). [PubMed] [Google Scholar]
- Buckley I. K. A light and electron microscopic study of thermally in-jured cultured cells. Lab Invest. 26, 201–209 (1972). [PubMed] [Google Scholar]
- Bouchama A. & Knochel J. P. Heat stroke. N Engl J Med. 346, 1978–1988 (2002). [DOI] [PubMed] [Google Scholar]
- Bouchama A., Dehbi M. & Chaves-Carballo E. Cooling and hemodynamic management in heatstroke: practical recommendations. Crit Care. 11, R54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemon D. & Jougla E. The heat wave in France in August 2003. Rev Epidemiol Sante. 52, 3–5 (2004). [DOI] [PubMed] [Google Scholar]
- Patz J. A. et al. Impact of regional climate change on human health. Nature. 438, 310–317 (2005). [DOI] [PubMed] [Google Scholar]
- Roberts G. T. et al. Microvascular Injury, Thrombosis, Inflammation, andApoptosis in the Pathogenesis of Heatstroke A Study in Baboon Model. Arterioscl Throm Vas. 28, 1130–1136 (2008). [DOI] [PubMed] [Google Scholar]
- Bouchama A. et al. Evidence for endothelial cell activation/injury in heatstroke. Crit Care Med. 24, 1173–1178 (1996). [DOI] [PubMed] [Google Scholar]
- Brinton M. R. Thermal sensitivity of endothelial cells on synthetic vascular graft material. Int J Hyperther. 28, 163–174 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff S. et al. p53's mitochondrial translocation and MOMP action isindependent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 18, 733–744 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. K. et al. Stabilization and translocation of p53 to mitochondria is linked to Bax translocation to mitochondria in simvastatin-induced apoptosis. Biochem Bioph Res Co. 391, 1592–1597 (2010). [DOI] [PubMed] [Google Scholar]
- Chipuk J. E. & Green D. R. Dissecting p53-dependent apoptosis. Cell Death Differ. 13, 994–1002 (2006). [DOI] [PubMed] [Google Scholar]
- Moll U. M. et al. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol. 17, 631–636 (2005). [DOI] [PubMed] [Google Scholar]
- Vaseva A. V. & Moll U. M. The mitochondrial p53 pathway. BBA-Biomembranes. 1787, 414–420 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biroccio A. et al. C-myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in m14 human melanoma cells. Mol Pharmacol. 60, 174–182 (2001). [DOI] [PubMed] [Google Scholar]
- Liu B., Chen Y. & Clair D. K. ROS and p53: versatile partnership. Free Radic Biol Med. 44, 1529–1535 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithipongvanitch R. et al. Mitochondrial and nuclear p53 localization in cardiomyocytes: Redox modulation by doxorubicin (adriamycin)? Antioxid Redox Sign. 9, 1001–1008 (2007). [DOI] [PubMed] [Google Scholar]
- Lee S. J. et al. Regulation of heat shock-induced apoptosis by sensitive to apoptosis gene protein. Free Radic Biol Med. 45, 167–176 (2008). [DOI] [PubMed] [Google Scholar]
- Hsu Y. L. et al. Heat shock induces apoptosis through reactive oxygen species involving mitochondrial and death receptor pathways in corneal cells. Exp Eye Res. 93, 405–421 (2011). [DOI] [PubMed] [Google Scholar]
- Sorrentino G. et al. The prolyl-isomerase Pin1 activates the mitochondrial death program of p53. Cell Death Differ. 20, 198–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenhagen D. & Clayton D. A. The Number of Mitochondrial Deoxyribonucleic Acid Genomes in Mouse L and Human HeLa Cells. J Biol Chem. 249, 7991–7995 (1974). [PubMed] [Google Scholar]
- Kelly K. J., Plotkin Z., Vulgamott S. L. & Dagher P. C. P53 Mediates the Apoptotic Response to GTP Depletion after Renal Ischemia-Reperfusion: Protective Role of a p53 Inhibitor. J Am Soc Nephrol. 14, 128–138 (2003). [DOI] [PubMed] [Google Scholar]
- Pallepati P. & Averill-Bates D. A. Mild thermotolerance induced at 40°C protects HeLa cells against activation of death receptor-mediated apoptosis by hydrogen peroxide. Free Radical Bio Med. 50, 667–679 (2011). [DOI] [PubMed] [Google Scholar]
- Hall D. M. et al. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am J Physiol-Heart C. 280, H509–H521 (2001). [DOI] [PubMed] [Google Scholar]
- Zeller L. et al. Exertional heatstroke: clinical characteristics, diagnostic and therapeutic considerations. Eur J Intern Med. 22, 296–299 (2011). [DOI] [PubMed] [Google Scholar]
- Robertson J. D., Datta K. & Kehrer J. P. Bcl-Xl Overexpression Restricts Heat-Induced Apoptosis and Influences hsp70, bcl-2, and Bax Protein Levels in FL5.12 Cells. Biochem Bioph Res Co. 241, 164–168 (1997). [DOI] [PubMed] [Google Scholar]
- Moulin M. & Arrigo A. P. Long lasting heat shock stimulation of TRAIL-induced apoptosis in transformed T lymphocytes. Exp Cell Res. 312, 1765–1784 (2006). [DOI] [PubMed] [Google Scholar]
- Vogel P., Dux E. & Wiessner C. Evidence of apoptosis in primary neuronal cultures after heat shock. Brain Res. 764, 205–213 (1997). [DOI] [PubMed] [Google Scholar]
- Michiels C. Endothelial Cell Functions. J Cell Physiol. 196, 430–443 (2003). [DOI] [PubMed] [Google Scholar]
- Jacob A. K. et al. Endothelial cell apoptosis is accelerated by inorganicironandheatvia an oxygen radical dependent mechanism. Sugery. 122, 243–254 (1997). [DOI] [PubMed] [Google Scholar]
- Moll U. M. & Zaika A. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 493, 65–69 (2001). [DOI] [PubMed] [Google Scholar]
- Milleron R. S. & Bratton S. B. Heat Shock Induces Apoptosis Independently of Any Known Initiator Caspase-activating Complex. JBC. 281, 16991–17000 (2006). [DOI] [PubMed] [Google Scholar]
- Morselli E. et al. Mechanisms of p53-mediated mitochondrial membrane permeabilization. Cell Res. 18, 708–710 (2008). [DOI] [PubMed] [Google Scholar]
- Green D. R. & Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 458, 1127–1130 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko N. D., Zaika A. & Moll U. M. Death Signal-induced Localization of p53 Protein to Mitochondria. JBC. 275, 16202–16212 (2000). [DOI] [PubMed] [Google Scholar]
- Zhao Y. F. et al. p53 Translocation to Mitochondria Precedes Its Nuclear Translocation and Targets Mitochondrial Oxidative Defense Protein-Manganese Superoxide Dismutase. Cancer Res. 65, 3745–3750 (2005). [DOI] [PubMed] [Google Scholar]
- Erster S. et al. In Vivo Mitochondrial p53 Translocation Triggers a Rapid First Wave of Cell Death in Response to DNA Damage That Can Precede p53 Target Gene Activation. Mol Cell Biol. 24, p6728–6741 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X. J. et al. A Two-Step Mechanism for Cell Fate Decision byCoordination of Nuclear and Mitochondrial p53 Activities. PLoS ONE. 7, e38164 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P. et al. Dequalinium induces cell death in human leukemia cells by early mitochondrial alterations which enhance ROS production. Leukemia Res. 31, 969–978 (2007). [DOI] [PubMed] [Google Scholar]
- Circu M. L. & Aw T. Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Bio Med. 48, 749–762 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucciariello C., Banti V. & Perata P. ROS signaling as common element in low oxygen and heat stresses. Plant Physiol Bioch. 59, 3–10 (2012). [DOI] [PubMed] [Google Scholar]
- Burdon R. H., Gill V. M. & Rice-Evans C. Oxidative stress and heat shockprotein induction in human cells. Free Radic Res Commun. 3, 129–139 (1987). [DOI] [PubMed] [Google Scholar]
- Skibba J. L. et al. Oxidative stress as a precursor to the irreversible hepatocellular injury caused byhyperthermia. Int J yperther. 7, 749–761 (1991). [DOI] [PubMed] [Google Scholar]
- McAnulty S. R. et al. Hyperthermia increases exercise-induced oxidative stress. Int J Sports Med. 26, 188–192 (2005). [DOI] [PubMed] [Google Scholar]
- Marchenko N. D., Zaika A. & Moll U. M. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 275, 16202–16212 (2000). [DOI] [PubMed] [Google Scholar]
- Li Z. et al. ROS leads to MnSODupregulation through ERK2 translocation and p53 activation in selenite-induced apoptosis of NB4 cells. FEBS. 584, 2291–2297 (2010). [DOI] [PubMed] [Google Scholar]
- Liu L. et al. NG, a novel PABA/NO-based oleanolic acid derivative, induces Human hepatoma cell apoptosis via a ROS/MAPK-dependent mitochondrial pathway. Eur J Pharmacol. 691, 61–68 (2012). [DOI] [PubMed] [Google Scholar]
- Xu Y. F. et al. Corosolic acid induces apoptosis through mitochondrial pathway and caspases activation in human cervix adenocarcinoma HeLa cells. Cancer Lett. 284, 229–237 (2009). [DOI] [PubMed] [Google Scholar]
- Pallepati P. & Averill-Bates D. A. Mild thermotolerance induced at 40°C increases antioxidants and protectsHeLa cells against mitochondrial apoptosis induced by hydrogen peroxide:Role of p53. Arch Biochem Biophys. 495, 97–111 (2010). [DOI] [PubMed] [Google Scholar]
- Bettaieb A. & Averill-Bates D. A. Thermotolerance induced at a fever temperature of 40 C protects cells against hyperthermia-induced apoptosis mediated by death receptor signalling. Cell Biol. 86, 521–538 (2008). [DOI] [PubMed] [Google Scholar]
- Yamaguchi H. et al. p53 Acetylation Is Crucial for Its Transcription-independent ProapoptoticFunctions. JBC. 284, 11171–11183 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Camarillo C. et al. Hyperglycemia induces apoptosis and p53 mobilization to mitochondria in RINm5F cells. Mol Cell Biochem. 281, 163–171 (2006). [DOI] [PubMed] [Google Scholar]