

Abstract
Aims
Toll-interacting protein (Tollip) is a critical regulator of the Toll-like receptor-mediated signalling pathway. However, the role of Tollip in chronic pressure overload-induced cardiac hypertrophy remains unclear. This study aimed to determine the functional significance of Tollip in the regulation of aortic banding-induced cardiac remodelling and its underlying mechanisms.
Methods and results
First, we observed that Tollip was down-regulated in human failing hearts and murine hypertrophic hearts, as determined by western blotting and RT–PCR. Using cultured neonatal rat cardiomyocytes, we found that adenovirus vector-mediated overexpression of Tollip limited angiotensin II-induced cell hypertrophy; whereas knockdown of Tollip by shRNA exhibited the opposite effects. We then generated a transgenic (TG) mouse model with cardiac specific-overexpression of Tollip and subjected them to aortic banding (AB) for 8 weeks. When compared with AB-treated wild-type mouse hearts, Tollip-TGs showed a significant attenuation of cardiac hypertrophy, fibrosis, and dysfunction, as measured by echocardiography, immune-staining, and molecular/biochemical analysis. Conversely, a global Tollip-knockout mouse model revealed an aggravated cardiac hypertrophy and accelerated maladaptation to chronic pressure overloading. Mechanistically, we discovered that Tollip interacted with AKT and suppressed its downstream signalling pathway. Pre-activation of AKT in cardiomyocytes largely offset the Tollip-elicited anti-hypertrophic effects.
Conclusion
Our results provide the first evidence that Tollip serves as a negative regulator of pathological cardiac hypertrophy by blocking the AKT signalling pathway.
Keywords: Tollip, Cardiac remodelling, Pressure overload, AKT, Cardiomyocyte hypertrophy
1. Introduction
Cardiac hypertrophy is an adaptive response to a variety of mechanical and hormonal stimuli, such as hypertension, valvular heart disease, and ischaemic events.1,2 However, sustained hypertrophy of the myocardium can progress to heart failure. Despite great advances in pharmacological treatment and device therapy over the past decades, heart failure continues to be a major public health problem with high morbidity and mortality.3,4 Therefore, a better understanding of the factors that regulate cardiac hypertrophy is important for the development of new strategies to treat heart failure.
Toll-like receptor (TLR)-mediated signalling has been shown to play an essential role in the induction of innate immune responses.5,6 Upon activation, TLRs recruit the cytoplasmic protein MyD88 via its toll-interleukin-1 receptor (IL-1R) domain. MyD88, in turn, interacts with IL-1R-associated kinase (IRAK), which leads to the activation of various downstream signalling pathways.7,8 Recent evidence has suggested that TLR-mediated signalling is involved in several cardiovascular diseases, including atherosclerosis, ischaemia/reperfusion injury, and cardiac remodelling.9–11 For example, TLR4-mutant mice are resistant to ischaemia/reperfusion-triggered cardiac injury.12 Knockdown of TLR2 protects against ventricular remodelling after myocardial infarction.13 These observations prompted us to investigate whether specific blockade of the TLR signalling by a naturally existed molecule can inhibit cardiac hypertrophy and heart failure. To this end, Toll-interacting protein (Tollip), an endogenous negative modulator of TLR signalling,14,15 would be a good candidate. Tollip is expressed ubiquitously in various tissues including the heart.16 A previous study showed that Tollip interacts directly with TLR2/TLR4 and inhibits TLR-mediated cellular responses by suppressing the IRAK activity.14 Recently, Hu et al.17 utilized an IL-1β-induced myocyte hypertrophy model and observed that overexpression of Tollip attenuated the hypertrophic response of neonatal cardiomyocytes through down-regulation of the MyD88-dependent NF-κB pathway. However, whether Tollip plays a negative role in chronic pressure overload-induced cardiac hypertrophy and heart failure in vivo has not yet been investigated. Therefore, in the present study, we employed both Tollip-overexpressing and -absent mice to determine the role of Tollip in murine hearts in response to chronic aortic banding (AB).
Our results demonstrate that pressure overload-induced cardiac remodelling was limited in Tollip-overexpressing mice, whereas being exaggerated in Tollip-deficient mice. We further discovered that Tollip-mediated effects were dependent, at least partly, on the negative regulation of the Akt signalling pathway. Based on these data, we believe that up-regulation of Tollip in the heart would be a good therapeutic strategy for the treatment of pressure overload-induced cardiac hypertrophy and heart failure.
2. Methods
2.1. Animals and AB surgery
All protocols in this study were approved by the Animal Care and Use Committee of the Renmin Hospital of Wuhan University, China. Mice were anaesthetized intraperitoneally using sodium pentobarbital (50 mg/kg), and the adequacy of anaesthesia was confirmed by the absence of reflex response to foot squeeze. All surgeries and subsequent analyses were performed in a blinded fashion. Full-length human Tollip cDNA was cloned downstream of the cardiac myosin heavy chain (MHC) promoter. The α-MHC-Tollip construct was microinjected into fertilized mouse embryos (C57BL/6J background) to produce transgenic mice. Four independent transgenic lines were established and studied. Tail genomic DNA was used to identify transgenic mice by PCR analysis. Primers are as follows: 5′-ATCTCCCCCATAAGAGTTTGAGTC-3′ and 5′-CACAGTTGGCATCAGGACCAC AGGC-3′. Expected band size of PCR product will be 610 bp. We analysed α-MHC-Tollip (TG) and non-transgenic (NTG) male littermates ranging in age from 7 to 8 weeks to obtain functional data and gene expression levels. Male Tollip-knockout mice [C57BL/6 background, purchased from the European Mouse Mutant Archive (EMMA) (B6.Cg-Tolliptm1Kbns/Cnrm)] and their wild-type littermates (aged from 7 to 8 weeks) were used in this study.
AB surgery was performed as described previously.2 In addition, Doppler analysis was performed to ensure that adequate constriction of the aorta had been induced. The wall thickness and internal diameter of the left ventricle (LV) were assessed by echocardiography at the indicated time after surgery. At the end of these procedures, mice were sacrificed with an overdose of sodium pentobarbital (200 mg/kg) intraperitoneally injection. The heart weight (HW)/body weight (BW) (mg/g), HW/tibiae length (TL) (mg/mm), and lung weight (LW)/BW (mg/g) ratios of the sacrificed mice were measured in different groups.
2.2. Blood pressure and echocardiography
The blood pressure was assessed by inserting a microtip catheter transducer (SPR-839, Millar Instruments) into the right carotid artery and advanced into the left ventricle. After stabilization for 15 min, an ARIA pressure–volume conductance system coupled to a Powerlab/4SP A/D converter, stored, and displayed on a personal computer was used to record pressure signals and heart rate continuously as described previously.2 Echocardiography was performed via a MyLab 30CV ultrasound (BiosoundEsaote Inc.) with a 10 MHz linear array ultrasound transducer. The LV was assessed in both parasternal long-axis and short-axis views at a frame rate of 120 Hz. End-systole or end-diastole was defined as the phase in which the smallest or largest area of LV, respectively, was obtained. LV end-systolic diameter (LVESD), LV end-diastolic diameter (LVEDD), and LV wall thickness were measured from the LV M-mode tracing with a sweep speed of 50 mm/s at the mid-papillary muscle level. Per cent fractional shortening (%LVFS) and ejection fraction (%LVEF) were calculated as described previously.2
2.3. Histological analysis
Hearts were excised and placed immediately in 10% potassium chloride solution to ensure that they were stopped in diastole, washed with saline solution, and fixed with 10% neutral buffered formalin. Hearts were sectioned transversely close to the apex to visualize the left and right ventricles. Several sections (4–5 µm thick) were prepared. Haematoxylin–eosin (HE)-stained sections were used to determine the cross-sectional area of myocytes. A single myocyte was measured using an image quantitative digital analysis system (Image-Pro Plus 6.0). Between 100 and 200 myocytes in the left ventricles were outlined in each group. Evidence of interstitial and perivascular collagen deposition was visualized using Picrosirius red (PSR), and then high magnification light micrographs were taken by light microscopy.
2.4. Adenoviral vectors and cultured neonatal rat cardiomyocytes
To overexpress Tollip, we used replication-defective adenoviral vectors encompassing the entire coding region of rat Tollip gene under the control of the cytomegalovirus promoter. A similar adenoviral vector encoding the GFP gene was used as a control. To knock-down Tollip expression, three rat shTollip constructs were obtained from SABiosciences (KR51237G). Next, we generated three Ad-shTollip adenoviruses and selected the one that produced a significant decrease in Tollip levels for further experiments. Ad-shRNA was the non-targeting control.
Primary cultures of neonatal rat cardiomyocytes (NRCMs) were prepared as described previously.2,18 Briefly, cells from the hearts of 1- to 2-day-old Sprague-Dawley rats which were sacrificed by swift decapitation according to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health were seeded at a density of 1 × 106 cells/well in six-well culture plates coated with fibronectin in plating medium, which consisted of F10 medium supplemented with 10% FCS and penicillin/streptomycin. After 48 h, the culture medium was replaced with F10 medium containing 0.1% FCS and BrdU (0.1 mM). Cell viability was determined by measuring the cell number, frequency of contractions, cellular morphology, and trypan blue exclusion. We infected NRCMs with AdTollip, AdGFP, AdshTollip, or AdshRNA at a multiplicity of infection (MOI) of 10, which yielded transgene expression without toxicity in 95–100% of the cells. Additional treatments are described in the figure legends.
2.5. Immunofluorescent staining
Immunofluorescent staining for Tollip and α-actinin was performed in tissue sections or NRCMs for measuring the surface area of cardiomycytes. Briefly, NRCMs were infected with different adenoviruses for 24 h and subsequently stimulated with Ang II (1 μM) for another 48 h, the cells were then fixed with 3.7% formaldehyde in PBS, permeabilized in 0.1% Triton X-100 in PBS, and stained with α-actinin at a dilution of 1:100 using standard immunofluorescence staining techniques. For tissue sections, all the steps are same after de-parafinization steps.
2.6. Real-time quantitative RT–PCR
Total RNA was extracted from the frozen human tissues using TRIzol (Invitrogen, 15596–026). cDNA was synthesized from 2 mg RNA of each sample using the Transcriptor First Strand cDNA Synthesis Kit (Roche, 04896866001). To examine the relative mRNA expression of Tollip, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), β-MHC, CTGF, Collagen Iα, Collagen III, quantitative RT–PCR analysis was performed using the LightCycler 480 SYBR Green 1 Master Mix (Roche, 04707516001) and the LightCycler 480 QPCR System (Roche).
2.7. Western blotting and immunoprecipitation
Western blotting was conducted to determine: (i) protein levels of Tollip; (ii) the activation state of MAPK signalling (p-MEK/MEK, p-ERK/ERK, and p-P38/P38); and (iii) the activation state of AKT signalling (p-AKT/AKT and p-GSK3β/GSK3β). Quantification of western blots was performed by Odyssey infrared imaging system (Li-Cor Biosciences), which is detailed below. First, we opened the image of western blots within the Li-Cor system using the Odyssey V3.0 software. We then selected the ‘Add a rectangle to the image’. For each band, we drew a box compatible with the corresponding band, and then clicked the ‘export a report -based on the current report template’ to export the quantification data. The brightness values of all bands of interest were normalized to the GAPDH values of the corresponding samples (these raw values are included in the Supplementary material online, Excel file). Finally, the normalized values were shown as bar graphs.
For transient transfection and co-immunoprecipitation experiments, cultured HEK293 T cells were cotransfected with psicoR-flag-Akt and psicoR-EGFP-myc-Tollip for 48 h and lysed in immunoprecipitation buffer (20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40 supplemented with protease inhibitor cocktail) for 20 min and then centrifuged to remove cell debris. For each immunoprecipitation, 500 μL of the sample was incubated with 10 μL Protein A/G-agarose beads (11719394001, 11719386001,Roche) and 1 μg antibody on a rocking platform (overnight at 4°C), per the manufacturer's recommendations. Finally, immunoprecipitates were washed 5–6 times with cold immunoprecipitation buffer before adding 1× loading buffer. Cell lysates and immunoprecipitates were immunoblotted using the indicated primary antibodies, the corresponding secondary antibodies, and the Super Signal chemiluminescence kit (Millipore).
2.8. Human heart samples
Samples of human failing hearts were collected from the left ventricles of dilated cardiomyopathy (DCM) patients undergoing heart transplants. Control samples were obtained from the left ventricles of normal heart donors. Written informed consent was obtained from the family of prospective heart donors. All procedures involving human samples were approved by the Renming Hospital of Wuhan University Review Board. The study conforms to the principles outlined in the Declaration of Helsinki.
2.9. Statistical analysis
Data are presented as means ± SEM. Differences among groups were determined by two-way ANOVA followed by a post-hoc Tukey test. Comparisons between two groups were performed using an unpaired Student's t-test. A value of P < 0.05 was considered significant.
3. Results
3.1. Tollip expression is down-regulated in human failing hearts and murine hypertrophic hearts
To investigate the potential role of Tollip in cardiac hypertrophy, we first examined Tollip expression in the left ventricles of DCM patients undergoing heart transplantation because of end-stage heart failure. Real-time RT–PCR analysis showed that Tollip mRNA levels were reduced by 31 ± 2%, accompanied with increased mRNA levels of foetal genes including ANP, BNP, and β-MHC in failing hearts (n = 7), compared with donor hearts(n = 6) (seeSupplementary material online, Figure S1). Accordingly, results of western blotting revealed that protein levels of Tollip were decreased by 43 ± 4% in human failing samples accompanied by decreased ejection fraction (EF) values and cardiac function relative to normal donor hearts (Figure 1A and B). Similarly, levels of the Tollip protein and EF and FS values were progressively reduced in murine hearts over 4–8 weeks of AB (Figure 1C and D). These data implicate the possible involvement of Tollip in cardiac hypertrophy and cardiomyopathy.
Figure 1.

Tollip expression in human failing hearts and experimental hypertrophic models. (A) Representative western blots of Tollip in hearts of normal donors (n = 6) and patients with heart failure (n = 7). (B) The EF values of human donor and DCM hearts (n = 6). (C) Representative western blots of Tollip. (D) EF/FS values in WT mouse hearts after AB at the indicated timepoints (n = 6). *P < 0.05 vs. normal donors or shams.
3.2. Tollip negatively regulates Ang II-induced myocyte hypertrophy
Next, we determined the effect of Tollip on cardiomyocyte hypertrophy in vitro, using cultured NRCMs. The levels of Tollip was either reduced by infection of cardiomyocytes with AdshTollip or elevated by infection with AdTollip, which was determined by western blotting (Figure 2A). Notably, under basal conditions (PBS), either reduction or elevation of Tollip did not alter the size of cardiomyocytes. However, upon exposure to Ang II (1 μM) for 48 h, we observed that Ang II-caused hypertrophy was aggravated in Tollip-knockdown myocytes (Figure 2B and C), whereas it was alleviated in Tollip-overexpressing cells (Figure 2B and D), as measured by the cell surface area (CSA, Figure 2B–D) and mRNA levels of hypertrophy markers (ANP and β-MHC) (seeSupplementary material online, Figure S2A and B). These results indicate that Tollip negatively regulates Ang II-induced myocyte hypertrophy.
Figure 2.

Effects of Tollip on myocyte hypertrophy in vitro. (A) The protein expression level of Tollip after infection with AdshTollip or AdTollip (n = 4). (B) Representative images of NRCMs infected with AdshTollip or AdTollip in response to Ang II. (C and D) Quantitative results of the cell surface area in the indicated groups. n = 4, *P < 0.05 vs. Ad-shRNA or Ad-GFP. More than 100 cells for each group were analysed.
3.3. Loss of Tollip exaggerates pressure overload-induced cardiac hypertrophy in vivo
To define the outcome of reduced Tollip expression, observed in human DCM patients (Figure 1), in the regulation of cardiac hypertrophy in vivo, we utilized a knockout mouse model with a global deletion of Tollip. The absence of Tollip in murine hearts was confirmed by immune-blotting (Figure 3A). It is important to note here that, under basal conditions, Tollip-KO mice did not show any pathological/physiological alterations in their heart morphology and contractile function (data not shown). Four weeks after AB, wild-type (WT) mouse hearts exhibited a significant enlargement, wherein the size of cardiomyocytes was increased (Figure 3B and C), as well as increased ratios of heart weight/body weight (HW/BW), lung weight/body weight (LW/BW), and heart weight/tibia length (HW/TL), compared with those of sham-operated mice (Figure 3D–F). Remarkably, however, these parameters were more pronounced in Tollip-KO hearts upon 4-week AB, relative to AB-treated WT samples (Figure 3B–F). Consistently, the mRNA levels of several hypertrophy markers including ANP, BNP, and β-MHC, were significantly increased in KO hearts, compared with WTs after 4-week AB (Figure 3G). Moreover, Tollip-KO mice showed an aggravated cardiac dilation and dysfunction compared with WT mice in response to chronic pressure overloading, as evidenced by echocardiographic parameters (i.e. LVEDD, LVESD, and LVEF) (Figure 3H and seeSupplementary material online, Table S1). To further determine the effect of Tollip deletion on maladaptive cardiac remodelling, we examined cardiac fibrosis, a classical feature of developing pathological cardiac hypertrophy, in these pressure-overloading hearts. Fibrosis was quantified by visualizing the total amount of collagen present in the interstitial and perivascular spaces of the heart and by measuring the collagen volume. Our results showed that 4-week AB dramatically produced perivascular and interstitial fibrosis in WT hearts, which was further aggravated in KO hearts (Figure 3I). Accordingly, KO hearts exhibited significant increases in total collagen volume (Figure 3J), and mRNA levels of CTGF, collagen Iα, and collagen III, known mediators of fibrosis (Figure 3K), compared with WTs in response to 4-week AB. Collectively, these results provide first in vivo evidence that loss of Tollip in the heart would contribute to maladaptation to chronic pressure overloading.
Figure 3.

Effects of Tollip disruption on cardiac hypertrophy. (A) Representative western blots of Tollip expression in heart tissues from WT and Tollip knockout mice (n = 6). (B) Gross hearts, HE, and WGA-FITC staining are performed at 4 weeks after Sham or AB surgery (n = 5–7). (C) Statistical results of the myocyte cross-sectional areas (n = 100+ cells). (D–F) Ratios of HW/BW, LW/BW, and HW/TL of the indicated groups (n = 13–14). (G) mRNA levels of hypertrophy markers in AB-treated hearts were determined by real-time RT–PCR in WT and KO mice (n = 4). (H) Echocardiographic results of WT and KO mice (n = 13–14). (I) PSR staining on histological sections of the LV was performed on the indicated groups 4 weeks after AB (n = 5–7). (J) The fibrosis areas of histological sections were quantified using an image analysis system (n = 25–27 fields). (K) Real-time RT–PCR analysis for CTGF, collagen Iα, and collagen III to determine their mRNA expression levels in the indicated mice (n = 4). *P < 0.05 vs. WT/sham; #P < 0.05 vs. WT/AB.
3.4. Overexpression of Tollip attenuates AB-induced cardiac remodelling
To further determine whether elevation of Tollip could mitigate pressure overload-induced cardiac remodelling, we generated a transgenic mouse model with cardiac-specific overexpression of human Tollip (TG mice) using the α-myosin heavy chain promoter (Figure 4A). Western blotting results showed that Tollip was successfully overexpressed in the TG hearts of four lines (Figure 4B).The mouse line that had the highest level of Tollip expression in the heart (TG7) was selected for the following experiments. The human Tollip protein was robustly expressed in the heart but not in other organs, indicating cardiac specificity of the transgene expression (Figure 4C). Notably, Tollip-TG mice exhibited normal cardiac morphology and contractile function under basal conditions. However, TG mice demonstrated a significant attenuation of cardiac hypertrophy at 8 weeks after AB compared with NTG mice, as evidenced by a reduction of cardiomyocyte size (HE and WGA-FITC staining, Figure 4D and E) and lower ratios of HW/BW, HW/TL, and LW/BW(Figure 4F–H). In addition, AB-induced activation of hypertrophy markers (ANP, BNP, and β-MHC) was also markedly blunted in TG hearts, compared with NTGs (Figure 4I). Furthermore, AB-caused myocardial dysfunction (measured by echocardiography) was greatly improved in Tollip-TG mice, compared with NTGs (seeSupplementary material online, Figure S3A–D and Table S2). Accordingly, the extent of cardiac fibrosis triggered by chronic pressure overloading was remarkably reduced in TG mice (Figure 4J and K). Subsequent analysis of the expression of fibrosis markers including CTGF, collagen Iα, and collagen III, also demonstrated a blunted response in TG mice, compared with control NTGs (Figure 4L). Together, these gain-of-function data indicate that up-regulation of Tollip protects hearts against pressure overload-induced remodelling.
Figure 4.

Effects of Tollip overexpression on cardiac hypertrophy. (A) Schematic diagram of the αMHC-Tollip transgene construct. (B) Representative western blots of the human Tollip protein in heart tissues from four TG lines and WT mice (n = 6). (C) Representative western blots of the human Tollip protein in various tissues from the TG mice (n = 4), as indicated. (D) Gross hearts and HE and WGA-FITC staining were performed at 8 weeks after Sham or AB surgery (n = 6). Statistical results for (E) the myocyte cross-sectional areas (n = 100+ cells), (F) HW/BW, (G) LW/BW, and (H) HW/TL of the indicated groups (n = 11–13). (I) Expression of hypertrophic markers was determined by real-time RT–PCR in NTG and TG mice (n = 4). (J) PSR staining on histological sections of the LV was performed on the indicated groups 8 weeks after AB (n = 6). (K) Fibrosis areas from the histological sections were quantified using an image analysis system (n = 26–28 fields). (L) Real-time RT–PCR was performed to determine mRNA levels of CTGF, collagen Iα, and collagen III in the hearts of the indicated mice (n = 4). *P < 0.05 vs. NTG /sham; #P < 0.05 vs. NTG/AB.
3.5. Tollip negatively regulates Akt signalling pathway in hypertrophic hearts and cardiomyocytes
To dissect the possible mechanisms by which Tollip attenuates cardiac remodelling after AB, we next investigated the mitogen-activated protein kinases (MAPK) signalling and Akt signalling, two pathways known to be involved in pathological cardiac hypertrophy. Our western blotting results showed that AB caused a similar degree of activation of the MAPK signalling molecules (i.e. MEK1/2, ERK1/2, and p38) in both TG and NTG hearts, as evidenced by similar increases in the phosphorylation levels of MEK1/2, ERK1/2, and p38 (seeSupplementary material online, Figure S4A). In addition, elevation of the phosphorylated MEK1/2, ERK1/2, and p38 was also similar between Tollip-KOs and WTs (seeSupplementary material online, Figure S4B). These data indicate that anti-hypertrophic effects of Tollip may not be associated with the MAPK pathway. Therefore, we further examined the Akt-GSK3β axis and excitingly observed that AB-induced activation of the Akt pathway was more pronounced in KO mice than in WT mice, as evidenced by a significant increase of Akt, GSK3β, mTOR, 4E-BP1, P70S6 K, and S6 phosphorylation levels in KO hearts, compared with WTs (Figure 5A and C). Conversely, overexpression of Tollip dramatically reduced the levels of Akt, GSK3β, mTOR, 4E-BP1, P70S6K, and S6 phosphorylation, compared with those of NTGs after AB (Figure 5B and D). These results suggest that Tollip may exert its anti-hypertrophic effects by blocking Akt signalling activation. Indeed, our in vitro experiments confirmed such inhibitory effects of Tollip on the Akt signalling. As shown in Figure 5E and F, knockdown of Tollip in NRCMs by AdshTollip enhanced Akt and GSK3β phosphorylation in response to Ang II, whereas the overexpression of Tollip in NRCMs by AdTollip markedly suppressed the phosphorylation of AKT and GSK3β, compared with control cells, respectively. Taken together, both in vivo and in vitro data consistently indicate that Tollip-mediated inhibition of pathological cardiac hypertrophy is associated with in-activation of the Akt signalling pathway.
Figure 5.
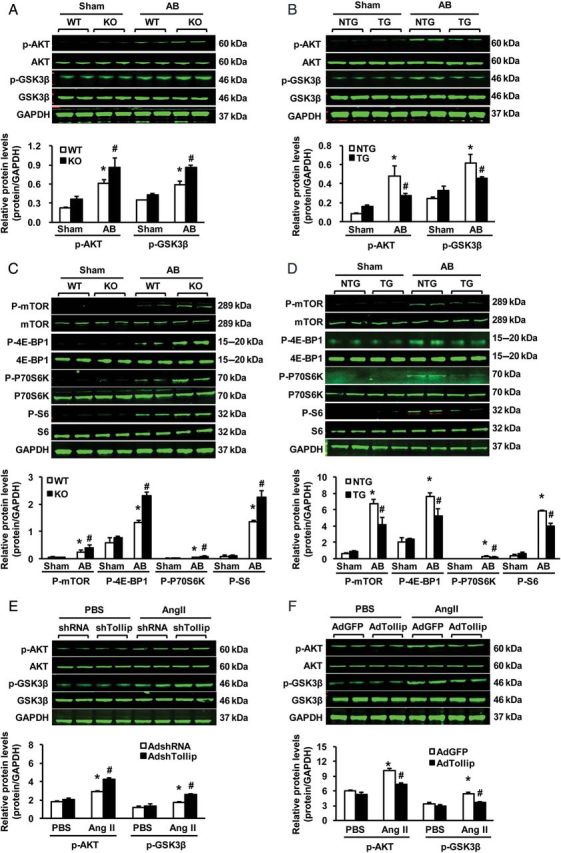
Effects of Tollip on the AKT signalling pathway. (A and C) Representative western blots and quantitative results showing the phosphorylation and total protein levels of AKT, GSK3β, mTOR, 4E-BP1, S6K, and S6 at 4 weeks after Sham or AB surgery in KO and WT mice (n = 4). *P < 0.05 vs. WT/sham; #P < 0.05 vs. WT/AB. (B and D) Representative western blots and quantitative results showing the phosphorylation and total protein levels of AKT, GSK3β, mTOR, 4E-BP1, S6K, and S6 at 8 weeks after Sham or AB surgery in TG and NTG mice (n = 4). * P < 0.05 vs. NTG/sham; #P < 0.05 vs. NTG/AB. (E) Representative blots and quantitative results showing the phosphorylation and total protein levels of AKT and GSK3β after infection with Ad-shTollip in response to PBS or Ang II (n = 4). *P < 0.05 vs. Ad-shRNA/PBS; #P < 0.05 vs. Ad-shRNA/Ang II. (F) Representative blots and quantitative results showing the phosphorylation and total protein levels of AKT and GSK3β after infection with Ad-Tollip in response to PBS or Ang II (n = 4). *P < 0.05 vs. Ad-GFP/PBS; #P < 0.05 vs. Ad-GFP/Ang II. GAPDH was used as a loading control.
3.6. Tollip-mediated anti-hypertrophic effects are largely dependent on the activation of Akt signalling
To determine whether Tollip-elicited negative effects on cardiac hypertrophy is dependent on the Akt activation, we co-infected NRMCs with AdshTollip plus AddnAkt (dominant negative mutation to inhibit Akt activation), or co-infected with AdTollip plus Adca-Akt (constitutively activation of Akt), respectively, followed by the addition of Ang II for 48 h. Our results of cell size analysis revealed that, Ang II-induced cell hypertrophy was significantly aggravated in Tollip-knockdown myocytes, as related to control AdshRNA-cells. However, this accelerative effect on cell hypertrophy was completely blocked by in-activation of Akt, as evidenced by no differences in the cell cross-section area (CSA) between AdshTollip- and AdshRNA-myocytes (Figure 6A). In contrast, we observed that the myocyte size was greatly increased in (Adca-Akt + AdTollip)-myocytes, compared with AdTollip-cells (Figure 6B).This suggests that Tollip-mediated suppression of myocyte hypertrophy is released by constitutively activation of Akt. Collectively, these data indicate that regulatory role of Tollip in pathological cardiac hypertrophy may be dependent, at least partly, on the activation of Akt.
Figure 6.
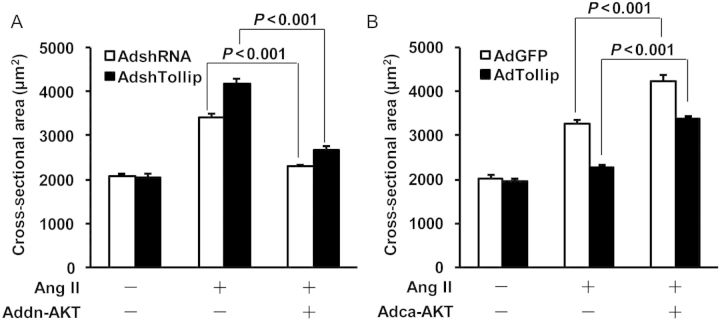
Regulation of Tollip-mediated anti-hypertrophy is largely dependent on the activation of Akt. (A) NRCMs were infected with AdshTollip and/or Addn-Akt, followed by the treatment with 1 μM of Ang II for 48 h. Surface areas of cardiomyocytes were then measured (see Methods) and revealed that knockdown of Tollip promoted Ang II-induced myocyte hypertrophy, which was blocked by the overexpression of dominant-negative Akt (Addn-Akt). n = 100+ cells, *P ≤ 0.001vs. controls, respectively. Similar results were observed in three independent experiments. (B) Tollip-mediated inhibition of Ang II-induced hypertrophy was released by overexpression of constitutively active Akt (Adca-Akt). n = 100+ cells, *P ≤ 0.001 vs. controls, respectively. Similar results were observed in three independent experiments.
4. Discussion
In this study, we demonstrate that cardiac-specific overexpression of Tollip protects hearts against maladaptive hypertrophy, dilatation, and fibrosis in response to chronic pressure overloading. Conversely, loss of Tollip results in an exaggerated response of pathological cardiac remodelling and fibrosis. These novel findings suggest that the cardiac-specific expression of Tollip is critically important in protecting the heart from hypertrophic stimuli. To our knowledge, these data provide the first direct evidence that Tollip negatively regulates the development of cardiac hypertrophy and failure.
Accumulating experimental literature suggests that sustained activation of TLR signalling following cardiac injury and stress is maladaptive and can lead to a heart failure phenotype.11 Once activated, TLRs serve as a docking site for downstream molecules and signalling cascades, leading to inflammation, cellular growth, hypertrophy, and survival.19,20 For example, Ha et al.21 demonstrated that the disruption of TLR4 attenuates cardiac hypertrophy following pressure overload in mice. These studies suggest that the molecular targets that block TLR signalling inhibits cardiac hypertrophy and improve cardiac function. Tollip is an ubiquitin-binding protein that interacts with several components of the TLR signalling cascade.22 Recent studies have shown that Tollip has a negative effect on TLR signalling, suggesting that Tollip may attenuate cardiac hypertrophy. Consistent with this notion, we found that transgenic mice with cardiac-specific overexpression of human Tollip were resistant to cardiac hypertrophy and dysfunction, whereas Tollip-knockout mice displayed the opposite phenotype in response to pressure overload. It is important to mention here that AB was performed on all four Tollip-TG lines. Our results indicated that ratios of HW/BW, LW/BW, HW/TL, and CSA of Tollip-TGs were decreased significantly compared with those of NTG mice 8 weeks after AB, but there was no significant difference among four Tollip-TG lines (seeSupplementary material online, Figure S5A–E).
The mechanism by which Tollip mediates its anti-hypertrophic effects remains unclear. Previous studies have implicated PI3K-Akt axis in the TLR signalling.23,24 Furthermore, we and others have demonstrated that the PI3K-Akt signalling pathway plays a key role in the progress of cardiac hypertrophy.25,26 Notably, DeBosch et al.27 have shown that Akt is a necessary factor for cardiac growth in response to IGF-1 stimulation or to exercise training, whereas Akt negatively modulates ET1- and pressure overload-induced cardiac hypertrophy. This study suggests that Akt promotes physiological hypertrophy but suppresses pathological hypertrophy. To investigate the molecular mechanism by which Tollip negatively regulates pathological cardiac hypertrophy, we therefore examined the status of the PI3K-Akt signalling pathway. An important finding of this study is that Akt activation was almost completely blocked by the cardiac expression of human Tollip, whereas the phosphorylation of Akt was enhanced by the loss of Tollip expression in response to chronic pressure overload. Using other pathological stress stimuli (i.e. isoproterenol and PE), we also observed that Akt activation was significantly attenuated in Tollip-overexpressing cardiomyocytes upon exposure to either isoproterenol or PE, compared with controls (data not shown). Together, these results indicate that in-activation of Akt is a major mechanism underlying the Tollip-elicited protective effects against pathological cardiac hypertrophy, consistent with previous reports.27
Given that the MAPK signalling cascade including p38, JNKs, and ERKs also play a key role in the development of cardiac hypertrophy,2,28 we therefore examined the status of MAPK signalling in our hypertrophic models. However, Tollip did not affect the increased phosphorylation levels of MEK1/2, ERK1/2, p38, and JNK1/2 induced by AB. Hence, Akt signalling should be a critical pathway by which Tollip exerts its anti-hypertrophic effects. Nevertheless, it remains unclear how Tollip negatively regulates the Akt activation. Our in vitro co-immunoprecipitation results showed that Flag-Akt co-precipitated with myc-Tollip in HEK293T cells (seeSupplementary material online, Figure S6A and B), demonstrating that Tollip may exert negative regulation of Akt by a direct interaction. Further experiments are needed to precisely determine this interaction in cardiomyocytes.
Cardiac fibrosis is a classic feature of pathological cardiac hypertrophy.2 Pandya et al.29 found that cardiac fibrosis following pressure overload is associated with increased collagen accumulation within the adventitia of the coronary arteries (perivascular fibrosis), which progressively extends into the neighbouring interstitial spaces (interstitial fibrosis). In the present study, we demonstrate that Tollip inhibits cardiac fibrosis and attenuates the expression of several fibrotic mediators induced by chronic pressure overload. The mechanism by which Tollip inhibits cardiac fibrosis remains elusive. Previous studies indicate that PI3K-AKT signalling is a key pathway involved in fibrosis.30,31 For example, Kang et al.30 found that TGF-β1 activates PI3K-AKT signalling and that AKT inhibition diminishes TGF-β1-induced fibrosis. Our previous study showed that the pharmacological inhibition of AKT/GSK3β signalling eliminates Smad2 phosphorylation and Smad2/3 translocation and reduces cardiac fibrosis in hypertrophied hearts.25 Thus, in the present study, the blockade of AKT signalling as a consequence of Tollip overexpression probably contributes, at least in part, to the lesser degree of cardiac fibrosis that was observed in response to pressure overload. In addition, recent studies have revealed that Tollip is a negative regulator of the IL-1 and TLR signallings.17 Zhu et al.32 also reported that Tollip plays a negative role in modulating TGF-beta signal pathway. Given that both TGF-beta and IRAK signalling pathways have been well recognized to be involved in myocardial inflammation and cardiomyocyte survival/growth, elevation of Tollip levels therefore may be implicated as a potential therapeutic approach to improving pathological cardiac remodelling.
In conclusion, our data demonstrate that Tollip attenuates pressure overload-induced cardiac hypertrophy, fibrosis, and cardiac dysfunction by blocking the AKT signalling pathway. Our study on Tollip may advance our understanding of the molecular mechanisms underlying chronic pressure-triggered cardiac remodelling and thereby, provide new strategy to prevent/treat pathological cardiac hypertrophy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by grants from the National Science and Technology Support Project (no. 2011BAI15B02 and no. 2012BAI39B05), the National Natural Science Foundation of China (nos 81070089, 81100230, and 81200071), the Key Project of the National Natural Science Foundation (no. 81330005), and the National Basic Research Program of China (no. 2011CB503902).
Conflict of interest: none declared.
Supplementary Material
References
- 1.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 2.Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z, et al. Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci USA. 2010;107:13818–13823. doi: 10.1073/pnas.1008397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickhout JG, Carlisle RE, Austin RC. Interrelationship between cardiac hypertrophy, heart failure, and chronic kidney disease: endoplasmic reticulum stress as a mediator of pathogenesis. Circ Res. 2011;108:629–642. doi: 10.1161/CIRCRESAHA.110.226803. [DOI] [PubMed] [Google Scholar]
- 4.Koitabashi N, Kass DA. Reverse remodeling in heart failure—mechanisms and therapeutic opportunities. Nat Rev Cardiol. 2012;9:147–157. doi: 10.1038/nrcardio.2011.172. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, et al. Mal (myd88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi K, Hernandez LD, Galan JE, Charles A, Janeway J, Medzhitov R, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 9.Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized ldl. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- 10.Arslan F, Smeets MB, O'Neill LA, Keogh B, McGuirk P, Timmers L, et al. Myocardial ischemia/reperfusion injury is mediated by leukocytic Toll-like receptor-2 and reduced by systemic administration of a novel anti-Toll-like receptor-2 antibody. Circulation. 2010;121:80–90. doi: 10.1161/CIRCULATIONAHA.109.880187. [DOI] [PubMed] [Google Scholar]
- 11.Topkara VK, Evans S, Zhang W, Epelman S, Staloch L, Barger PM, et al. Therapeutic targeting of innate immunity in the failing heart. J Mol Cell Cardiol. 2011;51:594–599. doi: 10.1016/j.yjmcc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, et al. Reduced myocardial ischemia-reperfusion injury in Toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 13.Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 14.Burns K, Clatworthy J, Martin L, Martinon F, Plumpton C, Maschera B, et al. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nat Cell Biol. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 15.Zhang G, Ghosh S. Negative regulation of Toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura M, Naito S. Tissue-specific mRNA expression profiles of human Toll-like receptors and related genes. Biol Pharm Bull. 2005;28:886–892. doi: 10.1248/bpb.28.886. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Li T, Wang Y, Li J, Guo L, Wu M, et al. Tollip attenuated the hypertrophic response of cardiomyocytes induced by IL-1beta. Front Biosci. 2009;14:2747–2756. doi: 10.2741/3411. [DOI] [PubMed] [Google Scholar]
- 18.Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C, et al. Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-beta-activated kinase 1-dependent signaling. Hypertension. 2010;56:232–239. doi: 10.1161/HYPERTENSIONAHA.110.149963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine. 2010;49:1–9. doi: 10.1016/j.cyto.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh MV, Swaminathan PD, Luczak ED, Kutschke W, Weiss RM, Anderson ME. Myd88 mediated inflammatory signaling leads to CaMKII oxidation, cardiac hypertrophy and death after myocardial infarction. J Mol Cell Cardiol. 2012;52:1135–1144. doi: 10.1016/j.yjmcc.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha T, Li Y, Hua F, Ma J, Gao X, Kelley J, et al. Reduced cardiac hypertrophy in Toll-like receptor 4-deficient mice following pressure overload. Cardiovasc Res. 2005;68:224–234. doi: 10.1016/j.cardiores.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 22.Lo YL, Beckhouse AG, Boulus SL, Wells CA. Diversification of Tollip isoforms in mouse and man. Mamm Genome. 2009;20:305–314. doi: 10.1007/s00335-009-9188-3. [DOI] [PubMed] [Google Scholar]
- 23.Troutman TD, Hu W, Fulenchek S, Yamazaki T, Kurosaki T, Bazan JF, et al. Role for B-cell adapter for PI3 K (BCAP) as a signaling adapter linking Toll-like receptors (TLRs) to serine/threonine kinases PI3 K/AKT. Proc Natl Acad Sci USA. 2012;109:273–278. doi: 10.1073/pnas.1118579109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ni M, MacFarlane AWt, Toft M, Lowell CA, Campbell KS, Hamerman JA. B-cell adaptor for PI3 K (BCAP) negatively regulates Toll-like receptor signaling through activation of PI3 K. Proc Natl Acad Sci USA. 2012;109:267–272. doi: 10.1073/pnas.1111957108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bian ZY, Wei X, Deng S, Tang QZ, Feng J, Zhang Y, et al. Disruption of mindin exacerbates cardiac hypertrophy and fibrosis. J Mol Med (Berl) 2012;90:895–910. doi: 10.1007/s00109-012-0883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, et al. Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radical Bio Med. 2005;39:1570–1580. doi: 10.1016/j.freeradbiomed.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 27.DeBosch B, Treskov I, Lupu TS, Weinheimer C, Kovacs A, Courtois M, et al. Akt1 is required for physiological cardiac growth. Circulation. 2006;113:2097–2104. doi: 10.1161/CIRCULATIONAHA.105.595231. [DOI] [PubMed] [Google Scholar]
- 28.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Bio. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 29.Pandya K, Kim HS, Smithies O. Fibrosis, not cell size, delineates beta-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc Natl Acad Sci USA. 2006;103:16864–16869. doi: 10.1073/pnas.0607700103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7a plays a critical role in TGF-beta1-induced pulmonary fibrosis. J Exp Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conte E, Fruciano M, Fagone E, Gili E, Caraci F, Iemmolo M, et al. Inhibition of PI3Kprevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: The role of class IP110 isoforms. PloS one. 2011;6:e24663. doi: 10.1371/journal.pone.0024663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu L, Wang L, Luo X, Zhang Y, Ding Q, Jiang X, et al. Tollip, an intracellular trafficking protein, is a novel modulator of the transforming growth factor-beta signaling pathway. J Biol Chem. 2012;287:39653–39663. doi: 10.1074/jbc.M112.388009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.