

Abstract
A major focus of systems biology is to characterize interactions between cellular components, in order to develop an accurate picture of the intricate networks within biological systems. Over the past decade, protein microarrays have greatly contributed to advances in proteomics and are becoming an important platform for systems biology. Protein microarrays are highly flexible, ranging from large-scale proteome microarrays to smaller customizable microarrays, making the technology amenable for detection of a broad spectrum of biochemical properties of proteins. In this article, we will focus on the numerous studies that have utilized protein microarrays to reconstruct biological networks including protein–DNA interactions, posttranslational protein modifications (PTMs), lectin–glycan recognition, pathogen–host interactions and hierarchical signaling cascades. The diversity in applications allows for integration of interaction data from numerous molecular classes and cellular states, providing insight into the structure of complex biological systems. We will also discuss emerging applications and future directions of protein microarray technology in the global frontier.
Keywords: Protein microarray, Protein network, Biomarker, Interactome, Serum profiling, Systems biology
Introduction
Since the completion of major whole genome sequencing efforts, the scientific community has been faced with the challenge of identifying and characterizing the expressed gene products of given organisms [1]. The post-genomics era gave birth to the field of proteomics that aimed to systematically chart the biochemical properties and functions of all expressed proteins [2]. With a global view in mind, we now strive to integrate complex “omics”-data from all molecular ranks. The scope of proteomics is not limited to identifying protein–protein interactions, but also includes identification of protein posttranslational modifications (PTMs) and of interactions with DNA and RNA sequences, lipids and glycans. Weaving these layers together will allow us to construct the carefully tuned network that exists within live cells. Improvements in high throughput proteomic technologies coupled with advances in genomics and bioinformatics have laid a framework to enable this level of research.
Two of the most powerful platforms for proteomic studies are mass spectrometry and protein microarray technologies. Although mass spectrometry is well suited for high throughput protein identification, quantification and PTM site mapping [3], it still has its disadvantages such as bias against low abundance proteins and modifications, as well as undersampling of complex proteomes [4]. On the contrary, the protein microarray platform avoids these limitations and is particularly suited for unbiased global profiling [5].
A protein microarray, also termed a protein chip, is created by immobilization of thousands of different proteins (e.g., antigens, antibodies, enzymes and substrates, etc.) in discrete spatial locations at high density on a solid surface [6]. Depending on their applications, protein microarrays can be categorized into two varieties: analytical and functional protein microarrays. Analytical protein microarrays are usually composed of well-characterized biomolecules with specific binding activities, such as antibodies, to analyze the components of complex biological samples (e.g., serum and cell lysates) or to determine whether a sample contains a specific protein of interest [7]. They have been used for protein activity profiling, biomarker identification, cell surface marker/glycosylation profiling, clinical diagnosis and environmental/food safety analysis [8–10]. Alternatively, functional protein microarrays are constructed by printing a large number of individually purified proteins and are mainly used to comprehensively query biochemical properties and activities of those immobilized proteins. In principle, it is feasible to print arrays composed of virtually all annotated proteins of a given organism, effectively comprising a whole-proteome microarray [11].
In 2001 the Snyder group reported the fabrication of the first proteome microarray in the budding yeast, representing a major advance for the field [12]. In order to construct this array, approximately 5800 full-length yeast ORFs were individually expressed in yeast and their protein products purified as N-terminal GST-fusion proteins. Each purified protein was then robotically spotted on a single glass slide in duplicate at high density to form the first “proteome” microarray, covering more than 75% of the yeast proteome. More recently, proteome microarrays have been fabricated from the proteomes of viruses, bacteria, plants and humans [8,13–16]. Functional protein microarrays have been successfully applied to identify protein–protein, protein–lipid, protein–antibody, protein–small molecules, protein–DNA, protein–RNA, protein–lectin and lectin–cell interactions [8,9,12,14,16–19], to identify substrates or enzymes for phosphorylation, ubiquitylation, acetylation and nitrosylation [11,20–24], as well as to profile immune response [25]. In this review, we will focus on inventive applications for protein microarrays and the significant findings that contribute to understanding the complex interactomes within cells (Table 1).
Table 1.
Applications of protein microarrays in diverse biological network construction
| Assay type | Array content | Type of probe | Application | Ref |
|---|---|---|---|---|
| Network construction | ||||
| Protein–DNA interaction | 4191 Human proteins | DNA motif | Protein–DNA interaction network | [16] |
| Kinase assay | 2158 Arabidopsis proteins | Protein kinase | Signaling network | [23] |
| Ubiquitylation assay | Yeast proteome | Ubiquitylation enzymes | PTM network | [20] |
| ∼9000 Human proteins Human protoarray, invitrogen |
Concentrated cell extract | PTM network | [19] | |
| Acetylation assay | Yeast proteome | Acetyltransferase | PTM network | [21] |
| Pathogen–host interactions | ||||
| Viral kinase assay | 4191 Human proteins | Conserved viral kinases | Viral PTM target network | [31] |
| Protein–protein interaction | 60 EBV viral proteins | Human protein | Protein–protein interaction network | [13] |
| 4191 Human proteins | Viral protein | Protein–protein interaction network | [44] | |
| Protein–RNA interaction | Yeast proteome | BMV SLD RNA loop | Protein–RNA interaction network | [17] |
| Biomarker identification | ||||
| Antigen–antibody interaction | 5011 Human proteins | AIH patient sera | Biomarker identification | [47] |
| 82 Corona virus proteins | SARS patient sera | Antibody profiling | [48] | |
| E. coli K12 proteome | IBD patient sera | Biomarker identification | [8] | |
| Lectin–glycan interaction | Yeast proteome | Lectins | Protein glycosylation profiling | [53] |
| 94 Lectins | Live mammalian cells | Cell surface biomarker identification | [9] | |
Network construction
A solid understanding of the molecular mechanisms of biological functions requires systematic profiling of dynamic interactions between biomolecules. Processes such as transcriptional regulation, viral infection, numerous PTMs and protein–protein interactions account for a small fraction of the potential molecular interactions within a cell but highlight how fundamental these networks are for essential functions. High throughput technologies strive to provide an unbiased platform for charting these relationships at the proteome and genome scale. In this section we will review several studies that demonstrate the utility of protein microarrays in reconstructing interaction networks.
Protein–DNA interactions
With the completion of the human genome sequencing, decoding the functional elements is a major challenge. Computational approaches have the power to identify conserved DNA regulatory elements; however, computational strategies cannot confidently predict the proteins that bind to these elements. Identification of the interaction networks between the DNA functional elements and the human proteome requires extensive predictions and powerful high throughput techniques. Hu and colleagues undertook a large-scale analysis of protein–DNA interactions (PDIs) using a protein microarray composed of 4191 unique full length human proteins, encompassing ∼90% of the annotated transcriptions factors (TFs) and members of many other protein categories, such as RNA-binding proteins, chromatin-associated proteins, nucleotide-binding proteins, transcription co-regulators, mitochondrial proteins and protein kinases [18]. The protein microarrays were probed with 400 predicted and 60 known DNA motifs. As a result, a total of 17,718 PDIs were identified. Many known PDIs and a large number of new PDIs for both well characterized and predicted TFs were recovered, as well as new consensus sites for human TFs. Surprisingly, over 300 proteins that do not encode any known DNA-binding domains showed sequence-specific PDIs, suggesting that many human proteins may bind specific DNA sequences as a secondary function. To further investigate whether the DNA-binding activities of these unconventional DNA binding proteins (uDBPs) were physiologically relevant, Hu et al. carried out in-depth analysis on a well-studied protein kinase, Erk2, to determine the potential mechanism behind its DNA-binding activity [18]. Using a combination of in vitro and in vivo approaches, such as electrophoretic mobility shift assays (EMSA), luciferase assays, mutagenesis, and chromatin immunoprecipitation (chIP), they demonstrated that the DNA-binding activity of Erk2 is independent of its protein kinase activity and it acts as a transcription repressor of transcripts induced by interferon gamma signaling [18]. This approach allows for sophisticated network mapping of protein–DNA interactions and enables the discovery of the uncharacterized DNA-binding proteins. The emergence of uDBPs strengthens the ability to piece together the machinery involved in transcriptional regulation.
MAP kinase substrate phosphorylation network
The mitogen-activated protein kinase (MAPK) signaling cascade involves a hierarchy of kinases that activate one another through consecutive phosphorylation events in response to extracellular or intracellular signals [15]. Standard methods have only been able to establish a few combinatorial connections from upstream MKK-activating kinases (MKKKs) to downstream MPK-activating kinases (MKKs), MAPKs and their cytoplasmic and nuclear substrates [26,27]. Constructing this complicated interconnected network necessitates a systematic unbiased high-throughput approach to avoid confounding issues of redundancy and functional pleiotropy [15]. Akin to the protein microarray based kinase assays developed by Ptacek et al. [20], Popescu et al. employed high-density protein microarrays to identify novel MPK substrates. The authors first determined which Arabidopsis thaliana MKKs preferentially activate 10 different MPKs in vivo and used the activated MPKs to probe Arabidopsis protein microarrays containing 2158 unique proteins to reveal their phosphorylation substrates [15]. The initial screen identified 570 nonredundant MPK phosphorylation substrates with an average of 128 targets per activated MPK. With this data the authors were able to reconstruct a complex signaling cascade involving nine MKKs, 10 MPKs and 570 substrates [15]. Moreover, the resulting nodes and edges highlighted the specificity conserved within these interactions: 290 (51%) of MPK phosphorylation targets were hit by only one MPK and only 94 (16%) were phosphorylated by two or more MPKs [15]. Gene ontology (GO) analysis of effector substrates showed enrichment in TFs involved in the regulation of development, defense and stress responses [15]. The network that emerged from this study suggests the MAPK signaling cascade regulates transcription through combinatorial enzyme specificity and discrete phosphorylation events.
Ubiquitin E3 ligase substrate discovery
Ubiquitylation is one of the most widespread PTMs and mediates a huge range of cellular events and processes in eukaryotes [28]. Understanding ubiquitin substrate specificity is a complex combinatorial question, as it is conferred by unique permutations of E1, E2 and E3 enzymes. Lu et al. developed an assay to determine substrates of a HECT domain E3 ligase, Rsp5, using yeast proteome microarrays [22]. Over 90 novel proteins were found to be readily ubiquitylated by Rsp5, eight of which were validated as in vivo targets. Deeper in vivo characterization of two substrates, Sla1 and Rnr2, revealed that Rsp5-dependent ubiquitylation affects either the posttranslational process of the substrate or subcellular localization [22]. This design offers the ability to dissect the molecular mechanisms of a complex enzymatic cascade and gives the field a tool to understand how the system is organized globally.
Identification of non-histone substrates of protein acetyltransferases in yeast
Acetylation is a major epigenetic PTM widely known for its role in regulating chromatin state. However, it is suspected to regulate nonnuclear functions as well [29]. In yeast, no non-histone proteins were reported as substrates of histone acetyltransferases (HATs) and histone deacetylases (HDACs). The catalytic enzyme, Esa1, of the essential nucleosome acetyltransferase of the complex, NuA4, is the only essential HAT in yeast [30], strongly suggesting that it may mediate acetylation of non-histone proteins critical for cell survival. Another intriguing question was whether HATs could regulate activity of cytosolic proteins or even enzymes like protein kinases. To comprehensively discover the non-chromatin substrates of the NuA4 HAT complex in the yeast proteome, Lin et al. developed in vitro acetylation reactions on the yeast proteome microarrays, containing 5800 yeast proteins, using NuA4 and [14C]-acetyl-CoA [23]. Over 90 non-histone proteins were readily acetylated by the NuA4 complex. Although it was expected that the majority of the substrates would be involved in nucleosome assembly and histone binding categories, a significant number of the identified substrates were cytoplasmic proteins and metabolic enzymes [23]. Twenty proteins involved in a variety of cellular functions such as metabolism, transcription, cell cycle progression, RNA processing and stress response were selected for further validation. Standard double-immunoprecipitation techniques were used to validate 13 of the 20 substrates, including phosphoenolpyruvate carboxykinase (Pck1p). To understand the physiological relevance of non-chromatin acetylation, the authors focused on the cytosolic enzyme Pck1p to explore a connection between acetylation and metabolism. Tandem mass spectrometry (MS/MS) identified lysine 19 (K19) and K514 as the acetylation sites of Pck1p and site-directed mutagenesis revealed that acetylation of K514 is critical for its enzymatic activity and promotes extension of life span in yeast growing under starvation conditions. These findings demonstrate a functional role for non-chromatin acetylation in yeast metabolism and longevity.
Based on GO analysis, acetylation may regulate several other cellular processes as well. In a follow up study, Lu et al. investigated the impact of acetylation on another NuA4 substrate, Sip2, a regulatory subunit of the SNF1 kinase complex (yeast AMPK). Based on the MS/MS analysis and site-directed mutagenesis studies, the authors found that Sip2 acetylation enhances its interaction with the catalytic subunit Snf1 and inhibits Snf1’s kinase activity [31]. As a result, phosphorylation of one of Snf1’s downstream targets, Sch9 (homolog of Akt/S6K), is decreased, ultimately leading to slower growth but extended replicative life span. Finally, the authors demonstrated that the anti-aging effect of Sip2 acetylation is independent of extrinsic nutrient availability and TORC1 activity. These studies are now echoed by recent discoveries of many mitochondrial and cytosolic enzymes as substrates of acetyltransferases in higher eukaryotes via MS-based PTM profiling [32–34].
Global ubiquitylation substrate discovery from cell extracts
Readily generating a snapshot of global protein PTM profiles under various cellular conditions could be considered the Holy Grail for those researching PTMs. General PTM substrate identification strategies require enrichment from a cell extract sample followed by MS or in vitro assays using purified components. While both approaches have their strengths and weaknesses, a hybrid of the two is possible. The use of concentrated mammalian cell extracts in combination with protein microarrays can serve to identify PTM targets in a semi-in vivo setting while alleviating the challenge of analyzing a complex mixture. Merbl and Kirschner generated cell extracts that replicate the mitotic checkpoint and anaphase release to identify differentially regulated polyubiquitylation substrates [21]. The synchronized cell extracts were incubated with Invitrogen’s Human ProtoArray composed of 8000 proteins and the resulting polyubiquitylated proteins were detected with antibodies directed to ubiquitin chains [21]. The authors expected to recover substrates of the anaphase promoting complex (APC), the major ubiquitin ligase in mitosis and G1. To differentiate polyubiquitylation substrates of the APC from other ligases, Merbl and Kirschner designed three experimental set ups. All cell extracts were arrested with nocodozole as the control which inhibits the APC, in the second condition the sample was released from checkpoint arrest with the addition of UbcH10, an E2 ligase, and the final condition was supplemented with both UbcH10 and a specific inhibitor of APC. Approximately 132 proteins were differentially polyubiquitylated, 11 of which were known APC substrates, confirming the validity on the experimental design. Validation studies performed in rabbit reticulocyte lysate confirmed the degradation/ubiquitylation of 7 novel APC substrates [21]. This study demonstrates the efficacy of using protein microarrays in combination with cell extracts to recapitulate the global PTM signature in a specific cellular state.
Pathogen–host interactions
Protein microarrays allow for exploration of hypotheses that cannot be addressed by standard methods. Investigating the interactions between viral encoded proteins and the proteins within the infected host has been an important yet cumbersome task. Protein microarrays composed of either the host or the viral proteome can be fabricated and subsequently used to examine the relationships between the viral machinery and the host. This in vitro approach recapitulates viral infection in that the viral genome/proteome are allowed to physically interact with the host. The Hayward and Zhu groups have recently developed this new paradigm to examine direct interactions between viral and host proteins [14,35,36], leading to a deeper understanding of the mechanisms by which the viral proteins hijack the host as well as uncovering the direct targets of major viral enzymes.
Herpesvirus kinase-phosphorylome
The human α, β, and γ herpesviruses cause diseases distinct from one another, ranging from mild cold sores to pneumonitis, birth defects and cancers [35]. Although the viruses are different, once they enter the host cells they all must reprogram cellular gene expression, sense cell-cycle phase, modify cell-cycle progression and reactivate the lytic life cycle to produce new virions to spread infection [37]. Many lytic cycle genes involved in replication of the viral genomes are highly conserved across the herpesvirus family. For example, each herpesvirus encodes for an orthologous serine/threonine kinase [38] that shares structural similarity with human cyclin-dependent kinases (CDKs) [39] and phosphorylates the substrates of CDKs [38]. The ability of viral kinase to mimic host CDKs results in hijacking of key pathways to potentiate their own replication. Particular cellular phosphorylation events are observed during herpes infection and specific phosphorylation of antiviral drugs in infected cells are mediated by the conserved viral kinases [40]. Identifying the collective host targets of the viral kinases would reveal the commonly shared mechanisms and signaling pathways among different herpesviruses to promote their lytic replication. This knowledge will increase the therapeutic target options necessary for developing pan-antivirals.
To test this idea, Li et al. utilized the human transcription factor (TF) proteome array containing 4191 human proteins to identify commonly shared substrates of herpesvirus-encoded kinases [35]. Parallel kinases assays were performed using the four viral kinases, UL31, UL97, BGLF4 and ORF36, which is encoded by herpes simplex type 1 (HSV1), human cytomegalovirus (HCMV), Epstein-Barr virus (EBV) and Kaposi Sarcoma associated-virus (KSHV), respectively [38]. In total, 643 nonredundant substrates were identified across the four kinases and 110 substrates were targets of at least three kinases. GO analysis of the 110 shared substrates indicates that DNA damage functional class was significantly enriched. Among the DNA damage proteins, TIP60 was selected as a lead candidate for regulation of viral replication, due to its roles in DNA damage as well as transcriptional regulation through its HAT activity. Phosphorylation of TIP60 by BGLF4 in EBV-infected B cells was validated during further analysis. BGLF4 is known to phosphorylate multiple EBV proteins and only a small number of host proteins [38,41]. The functions of its previously-characterized targets are varied, implying that the kinase plays multiple roles to promote viral replication [41]. It is expressed in the early phase of the lytic infection cycle and is localized mainly in the nuclei of EBV-infected cells [42]. BGLF4 knockdown revealed that it is critical for release of infectious virus during viral lytic reactivation [41]. Subsequent experiments demonstrated that BGLF4-mediated phosphorylation enhanced TIP60 HAT activity by 10-fold, linking the phosphorylation event to viral replication. They also demonstrated the importance of phosphorylation of host DNA damage proteins for viral replication. More specifically, phosphorylation and activation of TIP60 by BGLF4 triggers EBV-induced DNA damage response (DDR) and promotes positive transcriptional regulation of critical lytic genes involved in viral replication. Lastly, the study confirmed that TIP60 was also required for efficient lytic replication in HCMV, KSHV and HSV-1. Taken together, this unbiased approach provides a novel paradigm for discovery of conserved targets of viral enzymes. While herpes kinases have been credible therapeutic candidates, knowing their targets and the signaling pathways they exploit will better enable the development of widely effective antiviral drugs.
BGLF4–SUMO2
In a follow up study, Li et al. took the inverse approach that employed a herpesvirus EBV protein microarray to assess human-host protein binding events [14]. Small ubiquitin-related modifier (SUMO) is covalently attached to proteins via an enzymatic cascade analogous to the ubiquitin pathway. SUMO is involved in a broad range of cellular processes including signal transduction, regulation of transcription, DNA damage response and mediation of protein–protein interactions [43,44]. Both latent and lytic EBV proteins interact with components of the SUMO machinery [14,44]. While covalent modification by SUMO is more commonly understood, noncovalent interactions with SUMO also contribute to SUMO effector signaling [43,44]. Noncovalent binding to SUMO is often mediated through SUMO-interaction motif (SIM) domains on target proteins [43,44]. To comprehensively identify the EBV proteins that bind to the SUMO moiety, the authors fabricated a protein microarray of full length proteins from EBV and KSHV individually purified from yeast. The array was used to perform a protein–protein binding assay using the SUMO2 paralog. They identified 11 EBV proteins as potential SUMO partners, including BGLF4, a conserved kinase [14]. As BGLF4 is known to play a multitude of roles in EBV, the authors pursued the importance of the cellular PTM in BGLF4 function. The BGLF4 SIM domains were mapped and when mutated at both the N- and C-terminal SIMs, the intracellular localization of the kinase shifted from nuclear to cytoplasmic. A mutation in the N-terminal SIM showed largely nuclear localization, whereas the C-terminal SIM mutation generated an intermediate phenotype with nuclear and cytoplasmic expression. The authors found that BGLF4 inhibits SUMOylation of lytic cycle transactivator ZTA and demonstrated that the SIM domains as well as kinase activity are required for inhibition [14]. SIM domains of BLGF4 were also shown to be necessary for suppressing global SUMOylation, inducing cellular DDR and promoting EBV lytic replication.
The virus takes advantage of the SUMOylation system by encoding proteins that are SUMO modified and those that bind to SUMO [14]. As previously mentioned SUMO is involved in DDR, which is further supported by the finding that BGLF4 appears to interact with sites of DNA damage via SUMO binding, revealing an additional mechanism promoting EBV-mediated DDR and lytic replication. SUMO interaction is as important as the kinase activity for the function of BGLF4.
LANA-interacting cellular protein
Another variation of protein microarray used for investigating pathogen–host interaction involves the human TF array to profile the interactions between KSHV latency proteins and host proteins. In KSHV-associated malignancies, majority of the tumor cells are latently-infected and express viral latency proteins including LANA [45]. LANA functions to maintain KSHV latency by driving viral replication [46,47], promoting dysregulated cell growth [48] and dynamically regulating both viral and cell gene transcription [49–51]. Identification of LANA’s interacting partners would provide new insights into the mechanisms LANA uses to maintain latent infection. LANA has been an attractive target and previous efforts to identify LANA binding proteins have attempted yeast two-hybrid screens [52], glutathione S-transferase (GST) affinity immunoprecipitation [53] and MS, resulting in apparent approach-dependent binding partners [54]. In a recent study, Shamay et al. purified FLAG-tagged LANA and probed it against the human TF array, which recovered 61 candidate binding partners [36]. Eight candidates validated by co-immunoprecipitation assays included TIP60, protein phosphatase 2A (PP2A), replication protein A (RPA) and XPA. LANA-associated TIP60 retained its acetyltransferase activity and showed enhanced stability, which is consistent with Li et al.’s finding that TIP60 in critical for KSHV lytic replication (see above). The binding interactions between LANA, RPA and XPA seem to echo LANA’s role in DNA damage, but further characterization of the LANA’s ability to bind to additional RPA complex members, RPA1 and RPA2, spawned a new hypothesis that LANA may also regulate host telomere length. To test this hypothesis, the authors performed ChIP assays with anti-RPA1 and -RPA2 antibodies using primers specific to the telomere regions and found that the presence of LANA drastically reduced the recruitment of both RPA1 and RPA2 to the host telomeres, while it had no impact on the protein level of the RPA complex. This observation raised the possibility that LANA might affect telomere length. Using Southern blot analysis of terminal restriction fragments, the standard method for quantifying telomere length, the authors demonstrated that the average length of telomeres was shortened by at least 50% in both LANA-expressing endothelial cells and KSHV-infected primary effusion lymphoma cells [55].
Biomarker identification
Biomarker identification represents a major effort in modern biomedical and clinical research, as it allows for better screening methods, diagnosis criteria, prognosis predictions and ultimately superior treatment for a broad range of diseases. Traditionally, biomarker discovery has utilized popular methods such as MS, ELISA, gene expression and antibody arrays to profile serum samples [56]. In recent years, protein microarray technology has extended into clinical proteomics and is becoming a powerful tool for biomarker discovery. Proteins on functional protein microarrays were originally viewed as substrates and binding partners, but when applied to immunology, the proteins on the array could be potential antigens associated with certain diseases. By comparison, protein microarray based-serum profiling is much more sensitive and can be performed at higher throughput while requiring less amount of sample. Here we will review a variety of clinically-relevant applications for protein microarrays in biomarker identification.
Autoantigen discovery for autoimmune hepatitis
In many autoimmune diseases, there is an unmet clinical need for cost-effective and accurate diagnostic methods. Improving upon the current standard requires discovery and characterization of reliable autoantigens coupled with sensitive and reproducible assays. Take autoimmune hepatitis (AIH) as an example: AIH is a chronic necroinflammatory disease of human liver with little known etiology. Detection of non-organ-specific and liver-related autoantibodies using immunoserological approaches has been widely used for diagnosis and prognosis [57]. However, these traditional autoantigens, such as anti-smooth muscle autoantibodies (SMA) and anti-antinuclear autoantibodies (ANA) are often mixtures of complex biological materials. Unambiguous and accurate detection of the disease demands identification and characterization of these autoantigens. Therefore, Song et al. fabricated a human protein microarray of 5011 non-redundant proteins that were expressed and purified as GST fusions in yeast [25]. There are several advantages associated with producing human proteins in yeast rather than bacteria: (1) higher solubility, (2) higher yields of large proteins (e.g., >50 kD), (3) better preserved conformation of proteins and (4) less immunogenicity of proteins when produced in yeast than in Escherichia coli [7,12,17]. However, unlike a viral or bacterial protein microarray, a significant obstacle to the use of a human protein microarray of high content is the high cost. For example, cost for a human protein array of 9000 proteins can exceed $1000 per array. In order to reduce the cost, Song et al. developed a two-phase strategy to identify new biomarkers in AIH. Phase I is designed for rapid selection of candidate biomarkers, which are then validated in Phase II (Figure 1). In Phase I, serum samples from 22 AIH patients and 30 healthy controls were selected and individually used to probe the human protein microarrays at a 1000-fold dilution, followed by detection of bound human autoantibodies using a Cy-5-conjugated anti-human IgG antibody. Statistical analysis revealed 11 candidate autoantigens. To validate these candidates and to avoid a potential overfitting problem (see below), which is especially likely when dealing with a small sample size, the 11 proteins and 3 positive controls were re-purified to build a large number of low-cost small arrays for Phase II validation. These arrays were then sequentially probed with serum samples used in Phase I and serum samples obtained from an additional 52 AIH, 50 primary biliary cirrhosis (PBC), 43 hepatitis B virus (HBV), 41 hepatitis C virus (HCV), 11 system lupus erythematosus (SLE) and 11 primary Sjögren’s syndrome (pSS) patients. As negative controls, they also included 26 serum samples from patients suffering from other types of severe diseases and 50 samples from healthy subjects. Three new antigens, RPS20, Alb2-like and dUTPase, were identified as highly AIH-specific biomarkers with sensitivity of 47.5%, 45.5% and 22.7%, respectively, which were further validated with additional AIH samples in a double-blind design. Finally, they demonstrated that these new biomarkers could be readily applied to ELISA-based assays for clinical diagnosis and prognosis [25].
Figure 1.
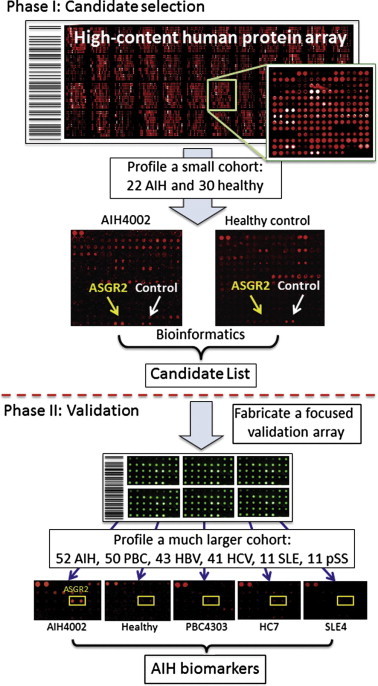
Scheme of the two-phase strategy for biomarker identification in human autoimmune diseases taking AIH as example In Phase I, a small cohort is used to rapidly identify a group of candidate biomarkers via serum profiling assays on a human protein microarray of high cost. Because a small number of microarrays are needed, cost of the experiments is relatively low. In Phase II, a focused protein microarray of low cost is fabricated by spotting down purified candidate proteins. A much larger cohort is then assayed on these arrays in a double blind fashion to validate the candidates identified in Phase I. AIH, autoimmune hepatitis; ASGR2, asialoglycoprotein receptor 2; PBC, primary biliary cirrhosis; HBV, hepatitis virus B; HCV, hepatitis C virus; SLE, system lupus erythematosus; pSS, primary Sjögren’s syndrome.
This study represents a new paradigm in biomarker identification using protein microarrays for three reasons. First, a manageable number of candidate biomarkers can be rapidly identified at low cost because fewer expensive protein microarrays of high-content are needed in the first phase of this two-phase strategy. Second, by using small arrays comprised of selected candidate proteins, the validation step can be rapidly carried out with a much larger cohort at low cost. This validation step is extremely important for avoiding the overfitting problem associated with statistical analysis in biomarker or classifier identification, especially when dealing with a small cohort (e.g., <40). Overfitting is a problem in which a statistical model describes random error or noise instead of the underlying relationship. It generally occurs in biomarker identification when the system is excessively complex, such as having too many individual-to-individual variations relative to the number of samples used. As a result, biomarkers that have been overfit generally have poor predictive performance. Therefore, testing an additional, larger cohort in a double-blind design is an effective way to rule out overfit biomarkers. Third, the authors developed ELISA-based assays to examine the performance of the validated biomarkers with additional samples. These newly identified biomarkers could serve as a translational step toward clinical practice.
SARS-CoV diagnosis
Protein microarrays can also be used as a diagnostic tool for infectious diseases. Severe acute respiratory syndrome (SARS) is an infectious disease, caused by a novel coronavirus (CoV), which appeared in Guangdong, China in November 2002. As of March 2003, the virus had spread globally and by July over 8000 SARS cases and approximately 800 deaths were reported worldwide [58]. At the time of the outbreak, no effective treatment of SARS was available, thus isolation and infection control were the best way to limit the spread of the virus. Therefore, rapid and reliable, early diagnosis is critical to control such an epidemic. Zhu et al. developed the first virus protein microarray, which included all the SARS-CoV proteins as well as proteins from five additional coronaviruses that can infect human (HCoV-299E and HCoV-OC43), cow (BCV), cat (FIPV) and mouse (MHVA59) [13]. The SARS microarray was used to screen sera from infected and noninfected individuals in a double-blind format. The samples were quickly distinguished as SARS positive or SARS negative based on the presence of human IgG and IgM antibodies against SARS-CoV proteins, with a 94% accuracy rate compared to a standard ELISA diagnostic test. The SARS microarray improved the sensitivity of the assay 50-fold over the ELISA and dramatically reduced the amount of sample required. This method may be suitable for diagnosis for many viral infections.
Novel serological biomarkers for inflammatory bowel disease
The two most common subtypes of inflammatory bowel disease (IBD) are Crohn’s disease (CD) and ulcerative colitis (UC). They are idiopathic in nature and are both characterized by an abnormal immunological response in the gut [59]. IBD is clinically thought to have autoimmune etiology, although, anti-microbial antibodies to normal bacteria are present in the sera of patients, leading to the pathogenesis of the disease [8]. The known serological antibodies are currently used as partial diagnostic criteria as they are not robust enough to stand alone [60]. Chen et al. elected to use an E. coli proteome microarray to characterize the differential immune response (serum anti-E. coli antibodies) in patients with CD and UC compared to healthy controls (HC). The microarray included 4256 E. coli proteins, encompassing the vast majority of the proteome of E. coli K12 strain. The sera from HC (n = 29), CD (n = 66) and UC (n = 39) were profiled using this array and the reactive anti-E. coli antibodies were detected with anti-human IgG antibodies. Data analysis revealed differential immunogenic response to 417 proteins between these three groups: 169, 186 and 19 were highly immunogenic in HC, CD and UC, respectively. Two robust sets of novel serological biomarkers were identified that can discriminate CD from HC or UC with >80% overall accuracy and sensitivity [8]. This is the first study to identify serological biomarkers in human immunological diseases with respect to the entire proteome of a microbial species. The underlying molecular pathology of other immune system related diseases can also be examined with this proteome microarray approach.
Lectin study: protein–glycan interaction
Cell surface glycosylation is a complex and highly-varied PTM that in turn is not amenable to standard high-throughput techniques. Glycosylation is present on the surface of all vertebrate cells, and it serves to distinguish cell types through very delicate differences [9]. It is also shown to be associated with cell differentiation, malignant transformation and subcellular localization [61–65]. Glycan binding proteins, known as lectins, are used to characterize glycosylation marks due to their ability to discriminate sugar isoforms [66]. Lectin microarrays have already been employed to characterize glycoproteins and lysates [67,68], however, they have not been used to systematically profile cell surface glycosylation signatures of mammalian cell types. Such studies have the potential to provide a tool for distinguishing normal versus abnormal cell surface profiles based on glycan–lectin interactions. Tao et al. fabricated a lectin microarray composed of 94 non-redundant lectins selected for defining cell surface glycan signatures [5]. Using 23 well-studied mammalian cell lines, the authors developed a systematic binary analysis of binding interactions of the selected lectins and cell types. They observed a broad range of binding potential and specificity across cell types, implying a high level of variation in cell surface glycans within mammalian cell types. For example, less than 20 lectins could capture the hESC, Caco-2, D407 and U937 cells, while more than 50 lectins captured the HEK293, K1106 and MCF7 cells [9]. Interestingly, similar cell types such as various breast cancer cell lines did not reveal overlapping lectin binding profiles, indicating lectins can discern subtle differences between physiologically-related cells.
To further test the utility of the lectin microarray for biomarker discovery, Tao et al. analyzed lectin binding in a model cancer stem-like system by comparing cell surface glycan signatures of all 24 cell types [9]. Focusing on MCF7, a breast cancer cell line that adopts cancer stem-like phenotypes when grown under specific conditions, the authors demonstrated that different growth conditions give rise to distinct lectin binding profiles that can distinguish these cancer cell subpopulations [9]. The lectin LEL was identified as a biomarker that can discriminate between MCF7 subpopulations. The authors propose that combined with other stem cell enrichment methods, lectin microarray technology is a potential tool for identifying cell surface markers in tumors, enabling the discovery of cancer stem cell-like targeted therapies.
Perspectives
Over the past decade, protein microarrays have evolved into a powerful and versatile tool for systems biology. They capitalize on femtomolar sensitivity, profiling full proteomes and high-throughput yet straightforward assays. We have described their utility for a myriad of applications that have resulted in impactful scientific findings including pathogen–host interactions, biomarker identification, unconventional transcription factors and PTM substrates (Figure 2).
Figure 2.
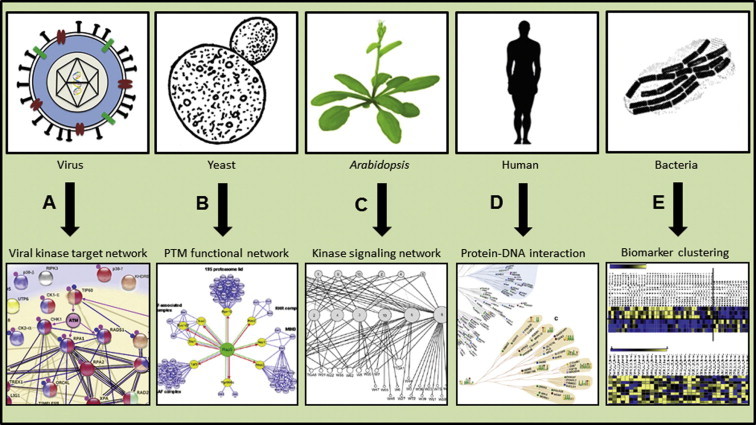
Reconstituted interaction networks in cellular systems generated through protein microarray studies Interaction mapping with protein microarrays has been applied to numerous organisms to achieve diverse representations of molecular networks. A. Li et al. probed a human transcription factor (TF) microarray with four conserved kinases encoded by herpesviruses to reveal the host targets of the viral kinases [35]. Verified interactions between the viral target host proteins are shown. B. Using a yeast proteome microarray, Lu et al. identified the substrates of the HECT E3 ligase Rsp5 [22]. Through gene ontology analysis Rsp5 was linked to subgroups of substrates based on function. C. The A. thaliana MAP kinase signaling network was reconstructed using an Arabidopsis protein microarray [15] (adapted with permission from Dr. Savithramma P. Dinesh-Kumar). The hierarchical phosphorylation network depicts the MKKs (upper nodes), MPKs (middle nodes) and substrates (bottom nodes). D. DNA binding specificity of unconventional DNA-binding proteins (uDBPs) was characterized using the TF microarray [18]. The uDBPs are clustered based on target sequence similarity and proteins of different functional classes are color-coded. “C” denotes consensus sequences for each sub-branch are shown. E. The E. coli proteome microarray was used to identify differentially immunogenic proteins between HC and CD patient samples depicted in the heat map [8]. The yellow and blue colors indicate high and low immunogenic responses, respectively. HC, healthy controls; CD, Crohn’s disease.
While protein microarrays leverage the advantage of uniform protein expression, for proteomics, their impact is limited by the extent of coverage. A remarkable advance was put forth by the Zhu laboratory with the construction of the first human proteome microarray containing over 17,000 full length proteins [16], the largest available to date (Figure 3). The discovery potential for this technology is dramatically increased by expanded proteome coverage. Multiple large-scale studies intended to link PTM substrates with their upstream enzymes, such as kinases, SUMO E3 ligases and ubiquitin ligases, are ongoing with the human proteome microarrays. As the number of bona fide PTMs increase and more substrates are found to acquire numerous modifications, we cannot ignore coregulation of PTMs. Directed studies to recapitulate crosstalk between enzymes, PTMs and their common substrates are possible with protein microarrays and may uncover key nodes of regulation and critical points where pathways converge. While MS is an ideal technology for the discovery of novel PTMs, such as the crotonylation PTM [69], it is not well suited to identify the enzymes responsible for novel modification. The richness of 17,000 natively-purified proteins on a single surface provides an ideal platform for discovery of novel enzyme function. The human proteome array can also be harnessed as a tool for high-throughput characterization of monoclonal antibody (mAb) specificity from hybridomas [16].
Figure 3.
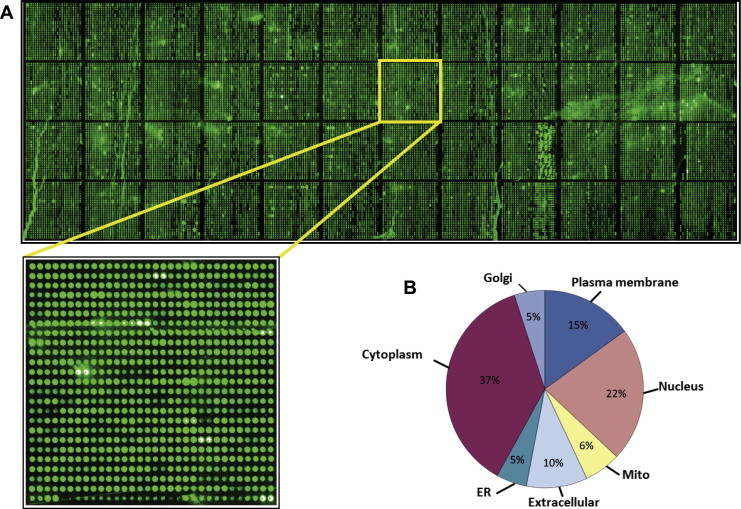
The human proteome microarray A. The human proteome microarray composed of 16,368 unique full-length recombinant proteins printed in duplicate on Full Moon glass slides. To monitor the quality, the microarray was probed with anti-GST monoclonal antibody, followed by Alexa-555 secondary antibody to visualize the signals. The proteins positively detected by the anti-GST antibody are represented in green. B. Cellular distribution of the proteins included in the human proteome microarray. ER, endoplasmic reticulum; Mito, mitochondria.
The capabilities of microarray technology are further expanding with the development of label-free optical techniques that monitor the real-time dynamics of biomolecular interactions. Oblique-incidence reflectivity difference (OIRD) is an emerging technique that measures the changes in reflectivity of polarized light [70,71]. OIRD has recently been applied to DNA and protein microarrays and has successfully determine association and dissociation rates of biomolecular interactions in a high-throughput format [72,73].
Constructing complex interaction networks involving the full range of cellular components is critical for deciphering how organisms are organized and is essential for understanding the aberrant changes that result in diseases. We have discussed the vast applications of protein microarrays for global characterization of interactomes and the significance of their findings for creating a comprehensive view of biological systems. In conclusion, protein microarray technology is no longer in its infancy and will undoubtedly serve as an invaluable tool for proteomics and systems biology.
Competing interests
The authors have declared no conflicts of interest.
Acknowledgements
We thank the NIH for supporting this work through the Grants awarded to HZ (Grant No. RR020839, DK082840, RO1GM076102, CA125807, CA160036 and HG006434) and an F31 NRSA Predoctoral Fellowship to IU (Grant No. 5F31GM096716).
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.

References
- 1.Kenyon G.L., DeMarini D.M., Fuchs E., Galas D.J., Kirsch J.F., Leyh T.S. Defining the mandate of proteomics in the post-genomics era: workshop report. Mol Cell Proteomics. 2002;1:763–780. [PubMed] [Google Scholar]
- 2.Neet K.E., Lee J.C. Biophysical characterization of proteins in the post-genomic era of proteomics. Mol Cell Proteomics. 2002;1:415–420. doi: 10.1074/mcp.r200003-mcp200. [DOI] [PubMed] [Google Scholar]
- 3.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 4.Desiere F., Deutsch E.W., Nesvizhskii A.I., Mallick P., King N.L., Eng J.K. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biol. 2005;6:R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang L., Guo S., Li Y., Zhou S., Tao S. Protein microarrays for systems biology. Acta Biochim Biophys Sin (Shanghai) 2011;43:161–171. doi: 10.1093/abbs/gmq127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith M.G., Jona G., Ptacek J., Devgan G., Zhu H., Zhu X. Global analysis of protein function using protein microarrays. Mech Ageing Dev. 2005;126:171–175. doi: 10.1016/j.mad.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 7.Zhu H., Qian J. Applications of functional protein microarrays in basic and clinical research. Adv Genet. 2012;79:123–155. doi: 10.1016/B978-0-12-394395-8.00004-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C.S., Sullivan S., Anderson T., Tan A.C., Alex P.J., Brant S.R. Identification of novel serological biomarkers for inflammatory bowel disease using Escherichia coli proteome chip. Mol Cell Proteomics. 2009;8:1765–1776. doi: 10.1074/mcp.M800593-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tao S.C., Li Y., Zhou J., Qian J., Schnaar R.L., Zhang Y. Lectin microarrays identify cell-specific and functionally significant cell surface glycan markers. Glycobiology. 2008;18:761–769. doi: 10.1093/glycob/cwn063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumble K.D. Protein microarrays: new tools for pharmaceutical development. Anal Bioanal Chem. 2003;377:812–819. doi: 10.1007/s00216-003-2088-6. [DOI] [PubMed] [Google Scholar]
- 11.Hu S., Xie Z., Qian J., Blackshaw S., Zhu H. Functional protein microarray technology. Wiley Interdiscip Rev Syst Biol Med. 2011;3:255–268. doi: 10.1002/wsbm.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 13.Zhu H., Hu S., Jona G., Zhu X., Kreiswirth N., Willey B.M. Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc Natl Acad Sci U S A. 2006;103:4011–4016. doi: 10.1073/pnas.0510921103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li R., Wang L., Liao G., Guzzo C.M., Matunis M.J., Zhu H. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J Virol. 2012;86:5412–5421. doi: 10.1128/JVI.00314-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Popescu S.C., Popescu G.V., Bachan S., Zhang Z., Gerstein M., Snyder M. MAPK target networks in Arabidopsis thaliana revealed using functional protein microarrays. Genes Dev. 2009;23:80–92. doi: 10.1101/gad.1740009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeong J.S., Jiang L., Albino E., Marrero J., Rho H.S., Hu J. Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol Cell Proteomics. 2012;11:1–10. doi: 10.1074/mcp.O111.016253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J., Zhu H., Haggarty S.J., Spring D.R., Hwang H., Jin F. Finding new components of the target of rapamycin (TOR) signaling network through chemical genetics and proteome chips. Proc Natl Acad Sci U S A. 2004;101:16594–16599. doi: 10.1073/pnas.0407117101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J. Profiling the human protein–DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139:610–622. doi: 10.1016/j.cell.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu J., Gopinath K., Murali A., Yi G., Hayward S.D., Zhu H. RNA-binding proteins that inhibit RNA virus infection. Proc Natl Acad Sci U S A. 2007;104:3129–3134. doi: 10.1073/pnas.0611617104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ptacek J., Devgan G., Michaud G., Zhu H., Zhu X., Fasolo J. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- 21.Merbl Y., Kirschner M.W. Large-scale detection of ubiquitination substrates using cell extracts and protein microarrays. Proc Natl Acad Sci U S A. 2009;106:2543–2548. doi: 10.1073/pnas.0812892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu J.Y., Lin Y.Y., Qian J., Tao S.C., Zhu J., Pickart C. Functional dissection of a HECT ubiquitin E3 ligase. Mol Cell Proteomics. 2008;7:35–45. doi: 10.1074/mcp.M700353-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin Y.Y., Lu J.Y., Zhang J., Walter W., Dang W., Wan J. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foster M.W., Forrester M.T., Stamler J.S. A protein microarray-based analysis of S-nitrosylation. Proc Natl Acad Sci U S A. 2009;106:18948–18953. doi: 10.1073/pnas.0900729106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song Q., Liu G., Hu S., Zhang Y., Tao Y., Han Y. Novel autoimmune hepatitis-specific autoantigens identified using protein microarray technology. J Proteome Res. 2010;9:30–39. doi: 10.1021/pr900131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asai T., Tena G., Plotnikova J., Willmann M.R., Chiu W.L., Gomez-Gomez L. MAP kinase signalling cascade in Arabidopsis innate immunity. Nature. 2002;415:977–983. doi: 10.1038/415977a. [DOI] [PubMed] [Google Scholar]
- 27.Ichimura K., Mizoguchi T., Yoshida R., Yuasa T., Shinozaki K. Various abiotic stresses rapidly activate Arabidopsis MAP kinases ATMPK4 and ATMPK6. Plant J. 2000;24:655–665. doi: 10.1046/j.1365-313x.2000.00913.x. [DOI] [PubMed] [Google Scholar]
- 28.VanDemark A.P., Hill C.P. Structural basis of ubiquitylation. Curr Opin Struct Biol. 2002;12:822–830. doi: 10.1016/s0959-440x(02)00389-5. [DOI] [PubMed] [Google Scholar]
- 29.Close P., Creppe C., Gillard M., Ladang A., Chapelle J.P., Nguyen L. The emerging role of lysine acetylation of non-nuclear proteins. Cell Mol Life Sci. 2010;67:1255–1264. doi: 10.1007/s00018-009-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allard S., Utley R.T., Savard J., Clarke A., Grant P., Brandl C.J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999;18:5108–5119. doi: 10.1093/emboj/18.18.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu J.Y., Lin Y.Y., Sheu J.C., Wu J.T., Lee F.J., Chen Y. Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell. 2011;146:969–979. doi: 10.1016/j.cell.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y., Zhao W., Yang J.S., Cheng Z., Luo H., Lu Z. Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol Cell Proteomics. 2012;11:1048–1062. doi: 10.1074/mcp.M112.019547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T., Wang S., Lin Y., Xu W., Ye D., Xiong Y. Acetylation negatively regulates glycogen phosphorylase by recruiting protein phosphatase 1. Cell Metab. 2012;15:75–87. doi: 10.1016/j.cmet.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li R., Zhu J., Xie Z., Liao G., Liu J., Chen M.R. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011;10:390–400. doi: 10.1016/j.chom.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shamay M., Liu J., Li R., Liao G., Shen L., Greenway M. A protein array screen for Kaposi’s sarcoma-associated herpesvirus LANA interactors links LANA to TIP60, PP2A activity and telomere shortening. J Virol. 2012;86:5179–5191. doi: 10.1128/JVI.00169-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arvin A., Campadelli-Fiume G., Mocarski E., Moore P.S., Roizman B., Whitley R., editors. Human herpesviruses: biology, therapy and immunoprophylaxis. Cambridge University Press; Cambridge: 2007. [PubMed] [Google Scholar]
- 38.Gershburg E., Pagano J.S. Conserved herpesvirus protein kinases. Biochim Biophys Acta. 2008;1784:203–212. doi: 10.1016/j.bbapap.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romaker D., Schregel V., Maurer K., Auerochs S., Marzi A., Sticht H. Analysis of the structure–activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J Med Chem. 2006;49:7044–7053. doi: 10.1021/jm060696s. [DOI] [PubMed] [Google Scholar]
- 40.Meng Q., Hagemeier S.R., Fingeroth J.D., Gershburg E., Pagano J.S., Kenney S.C. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J Virol. 2010;84:4534–4542. doi: 10.1128/JVI.02487-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gershburg E., Raffa S., Torrisi M.R., Pagano J.S. Epstein–Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J Virol. 2007;81:5407–5412. doi: 10.1128/JVI.02398-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gershburg E., Pagano J.S. Phosphorylation of the Epstein–Barr virus (EBV) DNA polymerase processivity factor EA-D by the EBV-encoded protein kinase and effects of the l-riboside benzimidazole 1263W94. J Virol. 2002;76:998–1003. doi: 10.1128/JVI.76.3.998-1003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gareau J.R., Lima C.D. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson E.S. Protein modification by SUMO. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 45.Ye F., Lei X., Gao S.J. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv Virol. 2011;2011:193860. doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grundhoff A., Ganem D. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J Virol. 2003;77:2779–2783. doi: 10.1128/JVI.77.4.2779-2783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu J., Garber A.C., Renne R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J Virol. 2002;76:11677–11687. doi: 10.1128/JVI.76.22.11677-11687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fakhari F.D., Jeong J.H., Kanan Y., Dittmer D.P. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest. 2006;116:735–742. doi: 10.1172/JCI26190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.An J., Sun Y., Rettig M.B. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood. 2004;103:222–228. doi: 10.1182/blood-2003-05-1538. [DOI] [PubMed] [Google Scholar]
- 50.Cai Q., Lan K., Verma S.C., Si H., Lin D., Robertson E.S. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: latency control under low oxygen conditions. J Virol. 2006;80:7965–7975. doi: 10.1128/JVI.00689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kusano S., Eizuru Y. Human I-mfa domain proteins specifically interact with KSHV LANA and affect its regulation of Wnt signaling-dependent transcription. Biochem Biophys Res Commun. 2010;396:608–613. doi: 10.1016/j.bbrc.2010.04.111. [DOI] [PubMed] [Google Scholar]
- 52.Krithivas A., Fujimuro M., Weidner M., Young D.B., Hayward S.D. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J Virol. 2002;76:11596–11604. doi: 10.1128/JVI.76.22.11596-11604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barbera A.J., Chodaparambil J.V., Kelley-Clarke B., Joukov V., Walter J.C., Luger K. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 54.Chen W., Dittmer D.P. Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2011;85:9495–9505. doi: 10.1128/JVI.02620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu H., Cox E., Qian J. Functional protein microarray as molecular decathlete: a versatile player in clinical proteomics. Proteomics Clin Appl. 2012;6:548–562. doi: 10.1002/prca.201200041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mattoon D., Michaud G., Merkel J., Schweitzer B. Biomarker discovery using protein microarray technology platforms: antibody–antigen complex profiling. Expert Rev Proteomics. 2005;2:879–889. doi: 10.1586/14789450.2.6.879. [DOI] [PubMed] [Google Scholar]
- 57.Manns M.P., Vogel A. Autoimmune hepatitis, from mechanisms to therapy. Hepatology. 2006;43:S132–S144. doi: 10.1002/hep.21059. [DOI] [PubMed] [Google Scholar]
- 58.Satija N., Lal S.K. The molecular biology of SARS coronavirus. Ann N Y Acad Sci. 2007;1102:26–38. doi: 10.1196/annals.1408.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thoreson R., Cullen J.J. Pathophysiology of inflammatory bowel disease: an overview. Surg Clin North Am. 2007;87:575–585. doi: 10.1016/j.suc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 60.Bossuyt X. Serologic markers in inflammatory bowel disease. Clin Chem. 2006;52:171–181. doi: 10.1373/clinchem.2005.058560. [DOI] [PubMed] [Google Scholar]
- 61.Hakomori S. Aberrant glycosylation in cancer cell membranes as focused on glycolipids: overview and perspectives. Cancer Res. 1985;45:2405–2414. [PubMed] [Google Scholar]
- 62.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dennis J.W., Granovsky M., Warren C.E. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta. 1999;1473:21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 64.Dwek M.V., Ross H.A., Leathem A.J. Proteome and glycosylation mapping identifies post-translational modifications associated with aggressive breast cancer. Proteomics. 2001;1:756–762. doi: 10.1002/1615-9861(200106)1:6<756::AID-PROT756>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 65.Kung L.A., Tao S.C., Qian J., Smith M.G., Snyder M., Zhu H. Global analysis of the glycoproteome in Saccharomyces cerevisiae reveals new roles for protein glycosylation in eukaryotes. Mol Syst Biol. 2009;5:308. doi: 10.1038/msb.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirabayashi J. Lectin-based structural glycomics: glycoproteomics and glycan profiling. Glycoconj J. 2004;21:35–40. doi: 10.1023/B:GLYC.0000043745.18988.a1. [DOI] [PubMed] [Google Scholar]
- 67.Ebe Y., Kuno A., Uchiyama N., Koseki-Kuno S., Yamada M., Sato T. Application of lectin microarray to crude samples: differential glycan profiling of LEC mutants. J Biochem. 2006;139:323–327. doi: 10.1093/jb/mvj070. [DOI] [PubMed] [Google Scholar]
- 68.Kuno A., Uchiyama N., Koseki-Kuno S., Ebe Y., Takashima S., Yamada M. Evanescent-field fluorescence-assisted lectin microarray: a new strategy for glycan profiling. Nat Methods. 2005;2:851–856. doi: 10.1038/nmeth803. [DOI] [PubMed] [Google Scholar]
- 69.Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen F., Lv H., Chen Z., Zhao T., Yang G. Optical real-time monitoring of the laser molecular-beam epitaxial growth of perovskite oxide thin films by an oblique-incidence reflectance-difference technique. J Opt Soc Am B. 2000;18:1031–1035. [Google Scholar]
- 71.Landry J.P., Zhu X.D., Gregg J.P. Label-free detection of microarrays of biomolecules by oblique-incidence reflectivity difference microscopy. Opt Lett. 2004;29:581–583. doi: 10.1364/ol.29.000581. [DOI] [PubMed] [Google Scholar]
- 72.Lu H., Wen J., Wang X., Yuan K., Li W., Lv H. Detection of the specific binding on protein microarrays by oblique-incidence reflectivity difference method. J Opt. 2010;12:1–5. [Google Scholar]
- 73.Wang X., Lu H., Wen J., Yuan K., Lv H., Jin K. Label-free and high-throughput detection of protein microarrays by oblique-incidence reflectivity difference method. Chinese Phys Lett. 2010;27:1–4. [Google Scholar]