

Abstract
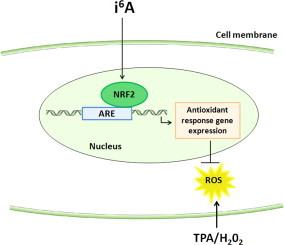
N6-isopentenyladenosine (i6A), a naturally occurring modified nucleoside, inhibits the proliferation of human tumor cell lines in vitro, but its mechanism of action remains unclear. Treatment of MCF7 human breast adenocarcinoma cells with i6A or with three synthetic analogs (allyl6A, benzyl6A, and butyl6A) inhibited growth and altered gene expression. About 60% of the genes that were differentially expressed in response to i6A treatment were also modulated by the analogs, and pathway enrichment analysis identified the NRF2-mediated oxidative stress response as being significantly modulated by all four compounds. Luciferase reporter gene assays in transfected MCF7 cells confirmed that i6A activates the transcription factor NRF2. Assays for cellular production of reactive oxygen species indicated that i6A and analogs had antioxidant effects, reducing basal levels and inhibiting the H2O2- or 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced production in MCF7 or dHL-60 (HL-60 cells induced to differentiate along the neutrophilic lineage) cell lines, respectively. In vivo, topical application of i6A or benzyl6A to mouse ears prior to TPA stimulation lessened the inflammatory response and significantly reduced the number of infiltrating neutrophils. These results suggest that i6A and analogs trigger a cellular response against oxidative stress and open the possibility of i6A and benzyl6A being used as topical anti-inflammatory drugs.
Keywords: Modified nucleosides, Gene expression, Reactive oxygen species, Anti-inflammatory drug, Pathway analysis
Abbreviations: allyl6A, N6-allyladenosine; benzyl6A, N6-benzyladenosine; butyl6A, N6-butyladenosine; i6A, N6-isopentenyladenosine
Highlights
-
•
i6A and analogs (allyl6A, benzyl6A and butyl6A) inhibit growth of MCF7 cells.
-
•
They activate NRF2-mediated oxidative stress response.
-
•
They inhibit ROS production in MCF7 or dHL-60 cells treated with H2O2 or TPA.
-
•
In vivo topical application of i6A or benzyl6A reduces TPA-induced inflammation.
-
•
i6A and benzyl6A have potential as topical antioxidant and anti-inflammatory drugs.
Graphical abstract
Introduction
The enzyme tRNA-isopentenyltransferase-1 (E.C. 2.5.1.75), encoded by the putative tumor suppressor gene TRIT1 [1], catalyzes the transfer of an isopentenyl group from isopentenyl diphosphate to the adenosine in position 37 of selenocysteine-specific transfer RNA (tRNA) [2,3]. The resulting isopentenyladenosine-tRNA (i6A-tRNA) improves the reading frame maintenance during the synthesis of selenoproteins [4]. N6-isopentenyladenosine (i6A), which is a breakdown product of i6A-tRNA turnover, is found in mammalian cells and is excreted in the urine [5,6].
As a modified nucleoside, i6A has been investigated from late sixties of last century for its effects on the inhibition of cell replication in different cancer cell lines [7]. These early works led to the study of its potential use as a natural antitumor drug in humans [8] and animal models [9]. Despite initial enthusiasm, the pilot clinical trial did not lead to convincing conclusions about therapeutic applications of i6A as an antitumor agent [10] and the molecule had only slight effects, if any, on tumor growth in rodents [9]. More recently, we did not observe any antitumor activity of i6A when injected intraperitoneally into Swiss nude mice bearing ascites of human ovarian cancer IGROV1 cells [11]. Only Laezza et al. reported that growth of a xenograft rat tumor, in nude mice, was inhibited after the subcutaneous injection of i6A directly at the tumor site [12]. These results, together with a growing body of evidence that i6A had antiproliferative effects in cell culture [[13], [14], [15]], suggested that i6A was ineffective in vivo as an anticancer agent because it was rapidly metabolized, as suggested in [16], becoming ineffective.
Based on biochemical research using cultured cells, different mechanisms of action for i6A have been suggested, including induction of apoptosis [14,17], inhibition of cell proliferation [15,18] or protein prenylation [12], and activation of the A3 adenosine receptor [19]. Using a different approach, gene expression analysis of i6A-treated MCF7 and A549 cells (from human breast adenocarcinoma and lung carcinoma, respectively), we found that the inhibitory effects of this compound were associated with the induction of several genes known to be activated during cell cycle arrest in response to stress [11]. Nonetheless, the precise mechanism by which i6A inhibits cancer cell growth remains unclear.
To help elucidate the mechanism of action of i6A, we previously synthesized and tested in vitro a large group of i6A analogs with N6-modifications [20]. Three compounds, namely allyl6A, benzyl6A, and butyl6A (with an allyl, benzyl, or butyl group, respectively, replacing the N6-isopentenyl group), strongly inhibited the proliferation of human T24 bladder cancer cells. The present study was conducted to determine if i6A and these three analogs exerted their cellular effects by acting on a common molecular pathway, in order to better understand their mechanism of action and to shed light on i6A involvement in the stress response, as suggested by our earlier study [11]. To this aim, we analyzed the transcriptomes of MCF7 cells treated (or not) with i6A or its analogs and used Ingenuity Pathways Analysis to identify the molecular pathways in which gene expression levels were most significantly altered.
Herein we show that i6A and its analogs altered the expression levels of genes involved in the NRF2-mediated oxidative stress response. The compounds directly activated the NRF2 transcription factor, leading to a cellular response against external oxidative stress stimuli, specifically H2O2 and 12-O-tetradecanoyl phorbol-13-acetate (TPA), in MCF7 cells and in the promyelocytic leukemia HL-60 cell line (differentiated to neutrophil lineage), respectively. Additionally, two of the studied compounds—i6A and benzyl6A—exhibited in vivo anti-inflammatory activity, when topically applied to the skin in a Car-S mouse model of TPA-induced oxidative stress leading to inflammation.
Material and methods
Cell lines, nucleosides and animals
Human breast adenocarcinoma MCF7 and promyelocytic leukemia HL-60 cell lines were purchased from the American Type Culture Collection (ATCC-LGC Standards, Teddington, UK) and propagated in the recommended culture media. HL-60 cells were induced to differentiate (dHL-60) along the neutrophilic lineage by culturing with 1.25% DMSO (Sigma-Aldrich, St. Louis, USA) for one week [21].
i6A and N6-benzyladenosine (benzyl6A) were purchased from OlChemIm (Olomouc, Czech Republic). We synthesized N6-allyladenosine (allyl6A) and N6-butyladenosine (butyl6A) as described [20]. Purified compounds were analyzed by 500-MHz 1H NMR to confirm the assigned structures and purity (≥ 99%).
Female Car-S mice [22] aged 8–12 weeks were used in ear inflammation assays. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” [23]. The study protocol was approved by the Ethics Committee for Animal Experimentation of Casaccia Research Center, ENEA, where the in vivo study was conducted.
Cell viability assay
MCF7 cells were plated at 700 cells per well in 96-well plates in culture medium containing resazurin (AlamarBlue®; Invitrogen; Life Technologies Italia, Monza, Italy; diluted according to the manufacturer’s instructions) and left to attach for 6 h. Then, to some wells, i6A or an analog was added to a final concentration of 10 µM (4 replicates per condition) and the cells were cultured for 4 days. Cell growth was determined from the metabolic conversion of resazurin into the fluorescent resorufin (excitation 535 nm, emission 590 nm) measured using an Ultra multiplate reader (Tecan Group, Mannedorf/Zurich, Switzerland).
For the dose–response experiment, cells were cultured for 4 days in the presence of the compounds at 0, 1, 3, 9, 27 or 81 μM. Each compound was tested at each concentration in six replicates. The concentrations at which the i6A analogs inhibited growth to the same extent as 10 μM i6A, i.e. equi-effective doses, were determined from the dose–response curves.
RNA extraction and microarray gene expression analysis
MCF7 cells were treated for 6 h with equi-effective doses of the nucleosides or left untreated (4 replicates per condition). Total RNA was extracted using Trizol Plus RNA Purification Kit (Invitrogen, Carlsbad, CA, USA), treated with deoxyribonuclease I (amplification grade, Invitrogen) and purified with RNeasy MinElute Cleanup kit (Qiagen, Hilden, Germany). The integrity of the RNA was verified using the RNA Nano Assay Kit on the 2100 Bioanalyzer microfluidics-based platform (Agilent Technologies, Santa Clara, CA, USA); all samples were determined to be of good quality, having an RNA integrity number >9.
Total RNA (500 ng) was used to synthesize biotinylated cRNA using the RNA Amplification Kit (Ambion, Austin, TX, USA). cRNA (1500 ng) was hybridized for 18 h to HumanHT-12 Expression BeadChips (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Fluorescence intensities were acquired with a BeadArray Reader (Illumina). Using BeadStudio v. 3 software, the dataset was normalized using a cubic spline algorithm, and a detection P-value <0.05 was set as a cut-off to select reliable expression data. After this quality control filtering, we used probes with a coefficient of variation >0.15 (n = 3286) for the subsequent analyses, in order to consider a smaller but more informative number of probes.
Unsupervised clustering and class comparison analyses of samples according to their gene expression profiles were done using BRB ArrayTools developed by Richard Simon and Amy Peng Lam1. Gene lists were generated by pair-wise comparison of untreated cells with cells treated with each of the four compounds. Genes that were differentially expressed between untreated and treated cells were identified using random variance t statistics [24]. Venn diagrams were drawn using Venny [25].
Pathway enrichment analysis of gene lists (containing genes differentially expressed at P< 1.0 × 10−4 and with fold-change ≥1.5) was performed using Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA, USA)2 to identify the canonical pathways modulated by treatment with i6A and its analogs. The four gene lists, containing gene symbols, fold-change, and P-values, were uploaded and the IPA Core Analysis was carried out using default settings. The resulting four datasets were then subject to Compare Analysis, carried out using default settings. Our attention was focused on the results obtained in the Canonical Pathways section, which showed the pathways that were most significantly altered across the four datasets. Pathway enrichment (i.e. statistically significant association between genes in our lists and pathways of the IPA knowledge base) was assessed using a right-tailed Fisher’s exact test and Benjamini–Hochberg multiple testing correction [26] 3. In particular, the number of genes in our dataset and the total number of the genes annotated in each pathway of the IPA knowledge base were taken into account to calculate a P-value that referred to the over-representation of genes in a given pathway. Therefore, enriched pathways are those having more genes belonging to our datasets than expected by chance.
Quantitative real-time PCR
Microarray results were validated by quantitative real-time PCR on 6 genes that were differentially expressed (P < 0.0001) in i6A-, allyl6A-, benzyl6A-, and butyl6A- treated cells (ATF3, CXCR7, HMOX1, IGDCC3, OSGIN1 and PPP1R3C) and on another 3 genes (DNAJB9, HBP1 and PPP1R15A) that were previously shown to be modulated by i6A treatment [11] and that here too were differentially expressed. Transcript levels of NFE2L2 gene were measured because of its involvement in the main i6A-affected pathway. Total RNA (1 μg) from untreated and nucleoside-treated cells, prepared as above, was reverse-transcribed using a 1:1 mixture of oligo(dT)18 and random hexamer primers, according to the protocol given in the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Science, Mannheim, Germany). cDNA (˜25 ng) was amplified in a 20 µl reaction volume containing 10 µl 2× Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) and 0.3 µM exon-spanning PCR primers (Supplementary Table 1). Reactions were run in duplicate on the 7900HT real-time PCR system (Applied Biosystems). Expression levels were normalized to those of human hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene. Relative quantities (RQ) in mRNA levels, with respect to a pool of RNA from untreated cells (used as calibrator), were assessed using the comparative cycle threshold (Ct) method.
Analysis of NRF2 signaling activity
The transcriptional activity of NRF2 at antioxidant response elements was measured using the Cignal Antioxidant Response Reporter (luc) kit (SABiosciences, Qiagen). Briefly, MCF7 cells were transiently transfected in 48-well plates with an NRF2-responsive firefly luciferase reporter plasmid and a control plasmid constitutively expressing Renilla luciferase (SABiosciences, Qiagen) in the presence of 1 µl FuGene HD transfection reagent (Promega), according to the manufacturer’s instructions. After 24 h, cells were treated with 10 µM i6A for 6 h or left untreated. Luciferase was assayed using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase signals were normalized to those of Renilla luciferase to control for transfection efficiency. Luminescence was measured on a Glomax® 20/20 luminometer (Promega). Six independent transfections were carried out in the luciferase assay.
ROS and superoxide anion assays
To measure the effects of i6A on basal ROS production, MCF7 cells were first treated with 1, 10 or 100 µM i6A for 6 h in complete medium and then loaded with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Life Technologies Italia, Monza, Italy) by incubating them with the indicator at 10 µM in PBS for 30 min. Instead, to measure the ability of i6A to inhibit ROS production induced by H2O2 treatment, MCF7 cells were pretreated with i6A (dose–response experiment: 1, 10, 100 μM i6A for 6 h; time-course experiment: 10 μM i6A for 1, 2, 6, 24 or 30 h), then loaded with H2DCFDA as above and finally stimulated to produce ROS with 1 mM H2O2 in PBS for 15 min. To test the effects of i6A analogs, cells were pretreated for 6 h with equi-effective concentrations of allyl6A, benzyl6A, and butyl6A prior to H2DCFDA labeling and H2O2 treatment. After the treatments, cells were washed with PBS and the fluorescence produced by the oxidized derivative of H2DCFDA, proportional to the quantity of ROS in the cells, was measured using an Ultra multiplate reader (Tecan Group; excitation, 485 nm; emission, 535 nm). At least 8 replicas were carried out for each condition.
Superoxide anion production by dHL-60 cells was measured in a luminol oxidation assay using the Superoxide Anion Detection kit (Calbiochem-Merck KGaA, Darmstadt, Germany). To measure the effects of i6A on basal production, cells were pretreated with 10 or 100 μM i6A in complete medium or left untreated for 6 h, then centrifuged and resuspended in the superoxide anion assay medium, containing luminol and enhancer solutions. Chemiluminescence from luminol oxidation was kinetically measured on a Tecan Ultra multiplate reader over 40 min. The same kit was used to measure the ability of i6A pretreatment to inhibit TPA-induced superoxide anion production. Briefly, cells were pretreated with i6A (dose–response experiment: 0, 1, 10, or 100 μM i6A for 6 h; time-course experiment: 10 μM i6A for 1, 2, 6, or 24 h), resuspended in assay medium, immediately stimulated with 8 μM TPA and assessed for chemiluminescence. To test the effects of i6A analogs, cells were pretreated with 10 μM of each compound for 6 h before TPA addition, and chemiluminescence was measured at 50–55 min. Each sample was read at least in triplicate and each experiment was carried out twice. Data were expressed as the mean and standard error of the relative luminescence units (RLU) normalized to the mean value of each experiment.
Mouse ear inflammatory model
We developed a mouse model of TPA-induced inflammation using Car-S mice, genetically susceptible to inflammation and skin tumorigenesis [27]. In this model, mice ears are treated once with 1 µg TPA in 20 µl acetone to induce skin inflammatory response (i.e. tissue edema and inflammatory cells infiltration) and then evaluated 24 h later macro- and microscopically. We used this model to test the in vivo anti-inflammatory effects of i6A and benzyl6A. In detail, the outer surface of the right ear of Car-S mice (n = 8 for each chemical) was pretreated with i6A or benzyl6A (10 mg/kg in 20 µl 95% ethanol), 24 and 1 h before a single treatment with TPA. The left ear was pretreated at the same time points with only 95% ethanol before TPA treatment, as a positive control for the induction of the inflammatory response. For negative controls, 4 additional mice received 95% ethanol (left ear) or i6A (right ear) according to the experimental schedule, followed by a single treatment with acetone. Then, 24 h after TPA (or acetone) treatment, ears were macroscopically examined before the mice were sacrificed. Ears were removed and cut in two parts for histological and immunohistochemical analyses.
For histological analysis, ears were fixed in 10% buffered formalin, paraffin-embedded, sectioned and stained with hematoxylin-eosin. For immunohistochemical analysis, 3-μm thick sections were de-waxed, rehydrated, then incubated in 0.3% H2O2 in methanol for 30 min to inhibit endogenous peroxidase. After antigen unmasking, sections were incubated with rat anti-mouse lymphocyte antigen 6 complex, locus G (Ly-6G) IgG (clone 1A8; 1:100; BD Biosciences, San Diego, USA) overnight at 4 °C. After incubation with biotinylated rabbit anti-rat IgG (1:500; Abcam, Cambridge, UK) for 1 h at room temperature, avidin DH and biotinylated horseradish peroxidase H were added (the Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, USA), and staining was carried out using 3-amino-9-ethylcarbazole (AEC) substrate kit for peroxidase (Vector Laboratories). Stained inflammatory cells in the dermis were quantified by collecting three digital images per tissue slice (NIS-Elements F 3.2 software, Nikon Instruments) and counting using the imaging software NIS-Elements BR 4.00.05 (Nikon).
Statistical analysis
Differences in quantitative measures were assessed using analysis of variance followed by Tukey’s test for multiple comparison, when appropriate. Correlation between microarray gene expression data and real-time PCR results was calculated using Pearson’s test. Statistical tests were done using the Rcmdr package in R [28]. All P-values were two-sided.
Results
i6A and its analogs inhibit MCF7 cell growth and alter the expression levels of genes involved in the NRF2-mediated antioxidant response
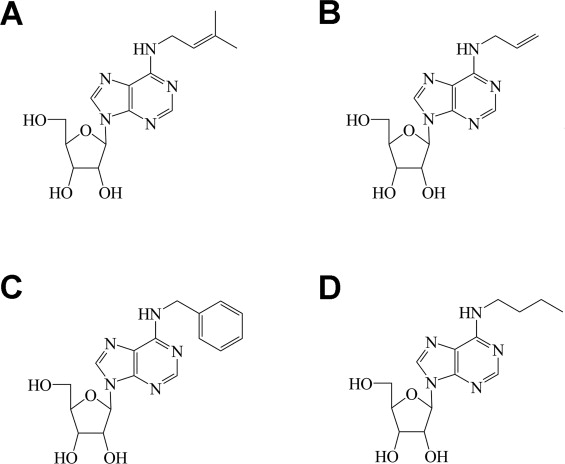
The nucleoside i6A and its analogs allyl6A, benzyl6A and butyl6A (whose chemical structures are shown in Supplementary Fig. 1) significantly inhibited the growth of MCF7 human breast adenocarcinoma cells when present in the culture medium at 10 µM for 4 days (P < 0.0001, Fig. 1). Dose–response experiments indicated that the concentrations of allyl6A, benzyl6A and butyl6A needed to inhibit growth to approximately the same extent as 10 µM i6A were 67, 11 and 44 µM, respectively (Supplementary Fig. 2).
Fig. 1.
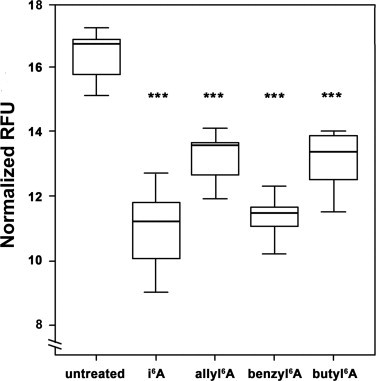
i6A and its analogs inhibited growth of MCF7 human breast adenocarcinoma cells. MCF7 cells were treated with a single dose of 10 μM i6A, allyl6A, benzyl6A or butyl6A in culture medium or left untreated for 4 days. Cell growth was measured with the AlamarBlue® assay and expressed as relative fluorescence units (RFU) at day 4 normalized to that at day 0. The line within each box represents the median fluorescence value of 8 replicates; upper and lower edges of each box represent the 75th and 25th percentile, respectively; upper and lower bars indicate the highest and lowest values less than one interquartile range from the extremes of the box. *** P < 0.0001 vs. untreated cells.
To investigate if i6A and its analogs inhibit cell growth by acting on the same cellular pathway, we treated MCF7 cells for 6 h with equi-effective concentrations of the nucleosides prior to extracting RNA for microarray analysis. Gene expression profiles were obtained using HumanHT-12 Expression BeadChips. Unsupervised clustering analysis of the samples according to their gene expression profiles revealed tight clustering of the untreated samples distinct from the treated samples, indicating that treatment with i6A or any of its analogs altered gene expression in MCF7 cells (Fig. 2A). Moreover, i6A-treated samples clustered together, showing that they differ somewhat from those treated with one of the other three compounds. Class comparison analysis of untreated vs. nucleoside-treated samples identified 232 differentially expressed genes (255 probes whose fluorescence intensities differed among the two classes) at a nominal P-value < 1.0 × 10−7 level, including 49 that were significant at nominal P < 1.0 × 10−10 (Fig. 2B); interestingly, the large majority (79%) of the 232 differentially expressed genes were up-regulated. Class comparison analyses were also done individually between untreated samples and those treated with i6A, allyl6A, benzyl6A and butyl6A (Fig. 2C). This analysis identified 451, 629, 508 and 527 differentially expressed genes, respectively, at nominal P < 1.0 × 10−4 and showing ≥1.5-fold change. Overall, 182 genes were modulated by all four compounds, suggesting that the nucleosides affect common molecular pathways. About 60% of the genes modulated by i6A were also modulated by the other compounds. These data are consistent with the unsupervised clustering of samples based on gene expression profiles where all treated samples were grouped together.
Fig. 2.
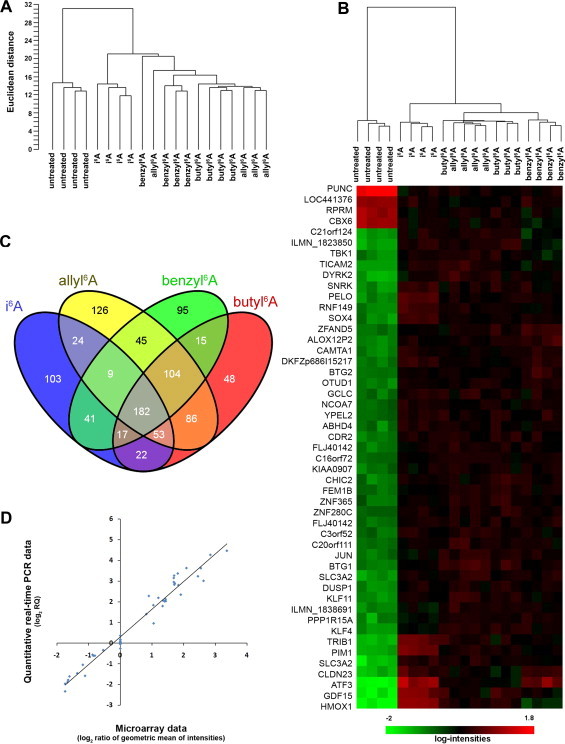
Gene expression profiles of untreated MCF7 cells and of cells treated for 6 h with 10 μM i6A or with equi-effective concentrations of allyl6A, benzyl6A or butyl6A. (A) Unsupervised clustering of samples (four replicates each) based on the expression levels of 3286 genes (detection P-value < 0.05 and coefficient of variation >0.15) revealed two main branches separating untreated from treated samples. Among treated cells, those treated with i6A clustered in a single branch distinct from those treated with the other three compounds. (B) Heatmap, resulted from the class comparison analysis, showing the first 49 most significantly (P < 1.0 × 10−10) differentially expressed genes in treated versus untreated cells and the clustering of samples (on top of the heatmap) based on the expression of these 49 genes only. Gene expression levels are indicated by the color bar: green, low; red, high. (C) Venn diagram of the numbers of differentially expressed genes (P < 1.0 × 10−4 and ≥1.5-fold) in MCF7 cells treated with i6A or one of its analogs, each compared to untreated cells. Overall, 182 genes were modified by all four nucleosides. (D) Correlation between microarray and quantitative PCR data for 9 genes measured under all five treatment conditions. Pearson’s r = 0.98, P < 0.0001.
To validate the microarray results, we selected nine differentially expressed genes (ATF3, CXCR7, DNAJB9, HBP1, HMOX1, IGDCC3, OSGIN1, PPP1R15A and PPP1R3C) and used quantitative real-time PCR (qPCR) to measure expression levels in untreated MCF7 cells and in cells treated with one of the four nucleosides. According to the microarray results, many of the genes (with the exception of CXCR7, IGDCC3 and PPP1R3C) were up-regulated by the nucleoside treatments, and these results were confirmed by qPCR. Correlation analysis comparing qPCR and microarray expression levels for these genes in the different samples gave Pearson’s r = 0.98 (P < 0.0001; Fig. 2D), indicating that the gene expression data were highly reliable.
The lists of 451, 629, 508 and 527 genes that were differentially expressed at the P < 1.0 × 10−4 level and with a fold-change ≥1.5 were then analyzed for pathway enrichment using Ingenuity Pathways Analysis (IPA) software. This analysis indicated that, among all the pathways defined in the IPA knowledge base, the “NRF2-mediated oxidative stress response” pathway was the most significantly associated with all four gene datasets, with a Fisher’s exact P < 0.001 for all four compounds (Table 1). After correction for multiple testing by the Benjamini–Hochberg method, significance was maintained at P < 0.01 for three of four datasets. Individually, the compounds altered the expression of 14–18 genes out of a total of 187 genes involved in this pathway; 11 genes were modulated by all four compounds. However, among these genes, NFE2L2, the gene coding for NRF2 protein, was not present although in allyl6A-treated cells it showed a statistically significant differential expression (P < 1.0 × 10−4). Nevertheless, by qPCR we measured the mRNA levels of NFE2L2 after treatment of MCF7 cells with all the four compounds and found that NFE2L2 expression was indeed significantly induced by all of them as compared to untreated MCF7 cells (Table 2).
Table 1.
Top enriched pathways, and genes belonging to them, modulated by i6A and its three analogs in MCF7 human breast adenocarcinoma cells.
| Ingenuity canonical pathway | Nucleoside | P-valuea | B-H P-valueb | Ratioc | Genes |
|---|---|---|---|---|---|
| NRF2-mediated oxidative stress response (187 genes) | i6A | 2.63E−06 | 8.51E−04 | 0.086 | ATF4, DNAJA4, DNAJB1 (alias Hsp40), DNAJB6, DNAJB9, GCLC, GCLM, GPX2, HERPUD1, HMOX1 (alias HO-1), JUN, JUND, KEAP1, MAFG, PIK3R1, TXNRD1 (alias TRXR1) |
| allyl6A | 1.32E−05 | 4.57E−03 | 0.096 | ATF4, CREBBP, DNAJB6, DNAJB9, EIF2AK3, ENC1, GCLC, GCLM, GPX2, HERPUD1, HMOX1, JUN, JUND, MAFG, NFE2L2, PIK3CA, PIK3R1, TXNRD1 | |
| benzyl6A | 3.80E−05 | 1.32E−02 | 0.080 | ACTG2, ATF4, DNAJB1, DNAJB6, DNAJB9, GCLC, GCLM, HMOX1, JUN, JUNB, JUND, MAFG, PIK3CA, PIK3R1, TXNRD1 | |
| butyl6A | 2.75E−04 | 0.0832 | 0.075 | ATF4, CREBBP, DNAJB6, DNAJB9, ENC1, GCLC, GCLM, HERPUD1, HMOX1, JUN, JUND, MAFG, PIK3R1, TXNRD1 | |
| p53 signaling (95 genes) | i6A | 2.51E−04 | 0.0282 | 0.095 | GADD45A, GNL3, JUN, PIK3R1, PMAIP1, RPRM, SERPINB5, TNFRSF10B, TP53BP2 |
| allyl6A | 9.12E−03 | 0.223 | 0.084 | ADCK3, HIPK2, JUN, PIK3CA, PIK3R1, PMAIP1, RPRM, SIRT1 | |
| benzyl6A | 2.24E−03 | 0.130 | 0.084 | ADCK3, APAF1, GADD45A, JUN, PIK3CA, PIK3R1, PMAIP1, RPRM | |
| butyl6A | 3.72E−02 | 0.402 | 0.063 | GADD45A, JUN, PIK3R1, PMAIP1, RPRM, SIRT1 | |
| Glucocorticoid receptor signaling (277 genes) | i6A | 2.63E−04 | 0.0282 | 0.058 | ANXA1, CDK7, CEBPB, CREB1, CREBZF, DUSP1, GTF2B, HSPA1A/HSPA1B, HSPA1L, HSPA4, HSPA6, JUN, NFKBIE, PIK3R1, PLAU, SOS1 |
| allyl6A | 2.14E−04 | 0.0372 | 0.072 | ANXA1, CEBPB, CREB1, CREBBP, DUSP1, FOXO3, GTF2B, HSPA1A/HSPA1B, HSPA4, JUN, NFAT5, NRIP1, PIK3CA, PIK3R1, PLAU, SMAD4, TAF4, TAF5, TAF6L, TRAF6 | |
| benzyl6A | 7.94E−04 | 0.0741 | 0.058 | CREB1, CREBZF, DUSP1, FOXO3, GTF2B, GTF2H1, HSPA1A/HSPA1B, HSPA4, HSPA6, JUN, NFAT5, NRIP1, PIK3CA, PIK3R1, TAF10, TAF5 | |
| butyl6A | 5.50E−04 | 0.0832 | 0.061 | ANXA1, CDK7, CEBPB, CREB1, CREBBP, CREBZF, DUSP1, GTF2B, HSPA1A/HSPA1B, HSPA4, JUN, NRIP1, PIK3R1, PLAU, TAF4B, TAF5, TRAF6 | |
Right-tailed Fisher’s exact test run in Ingenuity Pathway Analysis software.
Multiple testing correction with the Benjamini–Hochberg method run in Ingenuity Pathway Analysis software.
Ratio between the number of genes in the dataset (i.e. genes whose expression level changed by ≥1.5 fold and at P < 1.0 × 10−4) that map to the pathway and the total number of genes in the pathway.
In bold are the down-regulated genes.
Table 2.
NFE2L2 mRNA levels in MCF7 cells treated with the four compounds for 6 h, at equi-effective doses.
| Treatment | NFE2L2 RQ (SE)a | P-valueb |
|---|---|---|
| No | 1.08 (0.060) | |
| Allyl6A | 2.34 (0.063) | < 0.001 |
| Benzyl6A | 1.88 (0.083) | < 0.001 |
| Butyl6A | 1.83 (0.13) | < 0.001 |
| i6A | 2.12 (0.14) | < 0.001 |
Relative quantity (RQ) mean value of four replicas.
Analysis of variance, followed by Tukey’s test for multiple comparisons, versus untreated cells.
For two additional pathways (“p53 signaling” and “glucocorticoid receptor signaling”), gene expression was significantly modulated by all four compounds at the P < 0.05 and P < 0.001 levels, respectively, but, after correction for multiple testing, significance was lost for three or two datasets, respectively; therefore, these pathways were not further studied.
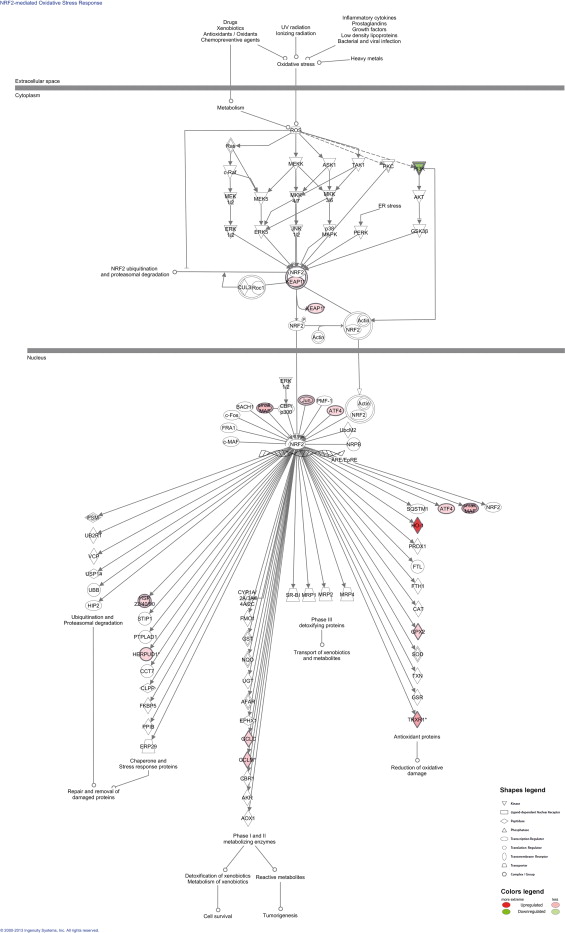
Focusing on the NRF2-mediated oxidative stress response, pathway analysis showed that i6A and its analogs had a prevalently up-regulatory effect, on genes both upstream and downstream of the transcription factor NRF2, suggesting that these modified nucleosides trigger a cellular response to oxidative stress. As illustrated in Supplementary Fig. 3, this response begins with an external stimulus that induces the intracellular production of reactive oxygen species (ROS), which activate cytoplasmic kinases, causing the transcription factor NRF2 to migrate to the nucleus and activate genes necessary for cellular protection. The figure also shows the position of genes modulated by i6A. Among the genes downstream of NRF2 and whose transcription depends on its activation by i6A treatment, some (e.g. HMOX1, alias HO-1) encode antioxidant proteins that reduce oxidative damage. Additionally, i6A treatment up-regulated some chaper (e.g. HERPUD1) involved in the repair and removal of damaged proteins; this observation is in agreement with our previous findings suggesting a role of i6A in the unfolded protein response [11]. Based on these results, we investigated the role of i6A and its analogs in the oxidative stress response mediated by the NRF2 pathway.
i6A activates the NRF2 transcription factor and reduces cellular ROS levels
NRF2 is a transcription factor that, in the presence of a cellular oxidative stress, binds to antioxidant response elements (AREs) in the promoters of genes involved in the cellular defense against this stress [29]. To functionally validate the expression data, we used a reporter gene assay to determine if i6A treatment activates NRF2, making it bind to promoters containing AREs. MCF7 cells were transiently transfected with a plasmid in which the luciferase reporter gene was controlled by a minimal CMV promoter containing multiple AREs and then cells were treated with i6A. Luciferase activity in i6A-treated cells was about 4-fold higher than that in untreated cells (P < 0.0001; Fig. 3). This result provided functional confirmation of the findings obtained from microarray analysis, supporting evidence that i6A activates the NRF2 pathway.
Fig. 3.
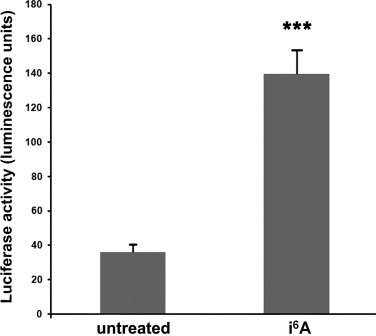
i6A treatment of MCF7 cells induced the NRF2 pathway. MCF7 cells were transiently transfected with a reporter gene plasmid in which the firefly luciferase gene was under the control of a minimal CMV promoter containing multiple antioxidant response elements (AREs). After 24 h, cells were treated with 10 µM i6A for 6 h or left untreated. Firefly luciferase activity was normalized to that of Renilla luciferase expressed constitutively from a control plasmid, to control for transfection efficiency. Values are mean and SE of six independent transfections. ***P < 0.0001 versus untreated cells.
To further investigate the role of i6A in the oxidative stress response, we examined its effects on cellular ROS production. First, we considered the possibility that i6A induces oxidative stress, by stimulating ROS production. MCF7 cells were therefore treated for 6 h with 1, 10, or 100 µM i6A or left untreated, and then labeled with 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA), a precursor molecule that in cells is cleaved by intracellular esterases and oxidized by ROS to form a fluorescent indicator. Surprisingly, we observed that i6A did not cause oxidative stress, but, on the contrary, reduced the basal amount of cellular ROS in a dose-dependent manner (Fig. 4A).
Fig. 4.
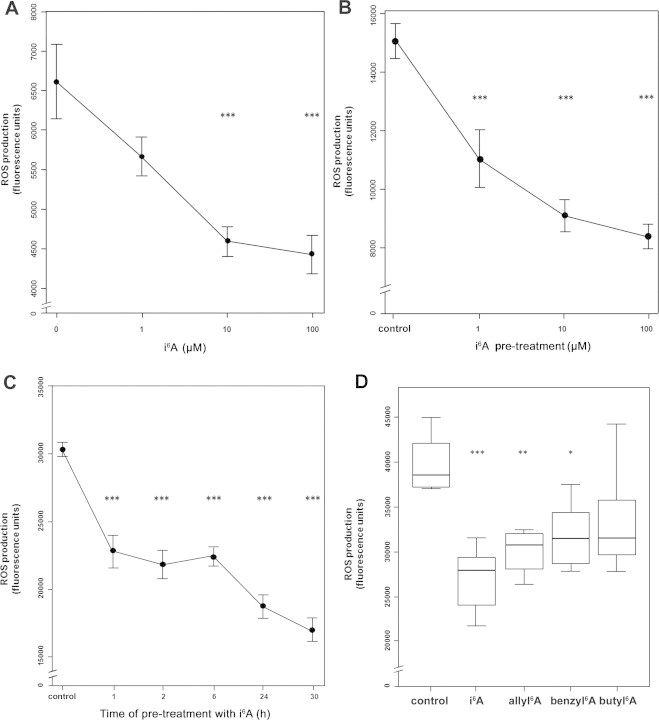
i6A inhibited ROS production in MCF7cells. (A) i6A reduced the basal production of reactive oxygen species (ROS) in a dose-dependent manner. Cells were treated with i6A for 6 h and then assayed for ROS production after labeling with H2DCFDA for 30 min. Data are shown as mean fluorescence units ± SE. (B) i6A inhibited H2O2-induced production of ROS in a dose-dependent manner. Cells were treated with i6A for 6 h before being loaded with H2DCFDA (30 min), stimulated with 1 mM H2O2 (15 min), and assayed for ROS production. Data are mean fluorescence units ± SE. (C) i6A inhibited H2O2-induced ROS production in a time-dependent manner. Cells were treated with 10 µM i6A for 1, 2, 6, 24 or 30 h before induction of ROS production with 1 mM H2O2 as above. Data are mean fluorescence units ± SE. (D) Equi-effective concentrations of i6A analogs also inhibit H2O2-induced ROS production. The line within each box represents the median fluorescence value; upper and lower edges of each box represent the 75th and 25th percentile, respectively; upper and lower bars indicate the highest and lowest values less than one interquartile range from the extremes of the box. Control: H2O2-only-treated cells. * P < 0.05; ** P < 0.01; *** P < 0.001. At least 8 replicas were carried out for each condition.
To further investigate the role of i6A in ROS production, we tested its effects in a cellular model of H2O2-induced oxidative stress. MCF7 cells were pretreated for 6 h with 1, 10, or 100 μM i6A, or left untreated, then labeled with H2DFCDA, stimulated with 1 mM H2O2, and assayed for ROS production. In agreement with our results on basal ROS production (Fig. 4A), i6A treatment also reduced the amount of ROS produced in response to H2O2 stimulation; this effect was both dose dependent (P < 0.001, Fig. 4B) and time dependent (P< 0.001, Fig. 4C). Finally, H2O2-induced ROS production was also significantly reduced in cells pretreated with equi-effective doses of allyl6A (P < 0.01) and benzyl6A (P < 0.05), but not with butyl6A (P > 0.05, Fig. 4D). These results suggest that i6A and its analogs activate NRF2 signaling and, therefore, trigger a cellular response against oxidative stress, induced by H2O2.
Then, we validated these results in another cellular model of oxidative stress, namely the production of superoxide anion by human promyelocytic leukemia HL-60 cells in response to stimulation by TPA. For these experiments, we used HL-60 cells differentiated along the neutrophil lineage (dHL-60) because of their high capacity to produce ROS [21]. First, to understand the extent to which the biological response to i6A was similar between the dHL-60 and MCF7 cell lines, we cultured dHL-60 cells in the presence or absence of 10 µM i6A for 6 h and then used qPCR to measure the expression levels of the same 10 genes tested in MCF7 cells. All 10 genes were upregulated by i6A treatment, with mean relative quantities ranging from 1.51–386-fold higher than untreated cells (Supplementary Table 2). This result indicates that there are some differences between this cell line and the MCF7 line (where three of these genes were down-regulated by i6A and its analogs); these differences may influence the effects of treatment with these compounds in these two cell lines. In any case, the NFE2L2 gene, the central gene of the NRF2 pathway, showed over-expression after treatment of both cell lines with i6A (Supplementary Table 2).
Next, we measured the effects of i6A on superoxide anion production by dHL-60 cells using a kinetic chemiluminescent assay (Fig. 5). In untreated cells the basal level of luminol oxidation was minimal, but it was even lower in cells pretreated with i6A (P < 0.0001, Fig. 5A). In cells stimulated with 8 μM TPA, the chemiluminescent signal increased rapidly over time, while in cells that had been pretreated with i6A the oxidation of luminol was reduced in both a dose-dependent (Fig. 5B) and time-dependent (Fig. 5C) manner. Finally, in cells pretreated with i6A analogs (6 h at 10 μM), the TPA-induced superoxide anion production at 50–55 min was significantly (P < 0.0001) reduced by benzyl6A and butyl6A, but not allyl6A (Fig. 5D). Therefore, in dHL-60 cells, like in MCF7 cells, i6A and its analogs were able to contrast oxidative stress induced, in this case, by a different chemical agent, i.e. TPA.
Fig. 5.
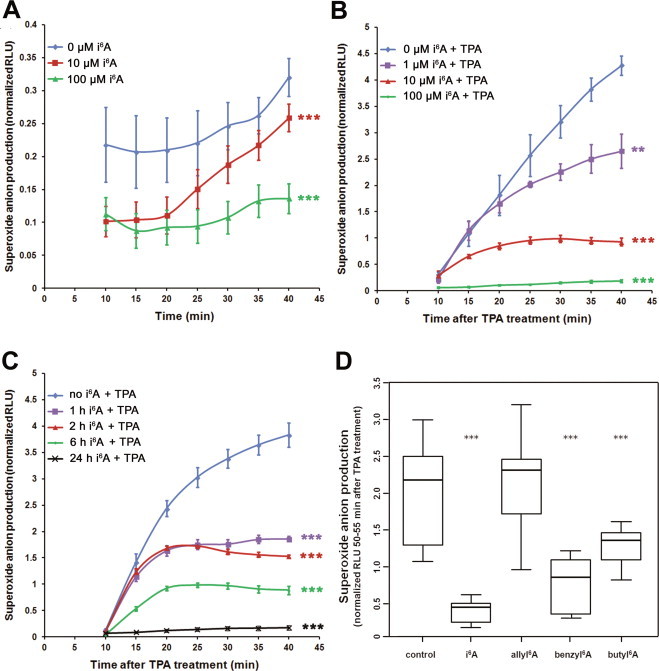
i6A inhibits superoxide anion production in dHL-60 cells. (A) Basal levels of superoxide anion production in dHL-60 cells are low and further reduced by treatment with 10 or 100 µM i6A for 6 h. Data are mean and SE. (B) i6A pretreatment inhibited TPA-induced superoxide anion production in a dose-dependent manner (1, 10, or 100 µM for 6 h before 8 μM TPA treatment). Data are mean and SE. (C) TPA-induced superoxide anion production was inhibited by pretreatment with 10 μM i6A in a time-dependent manner (1, 2, 6, or 24 h before TPA treatment). dHL-60 cells treated only with TPA were used as control. (D) Butyl6A and benzyl6A significantly inhibit TPA-induced superoxide oxidative stress. Control: dHL-60 cells treated only with TPA. The line within each box represents the median luminescence value; upper and lower edges of each box represent the 75th and 25th percentile, respectively; upper and lower bars indicate the highest and lowest values less than one interquartile range from the extremes of the box. Normalized RLU: relative luminescence units normalized to the mean value of each experiment. Data are from at least 5 replicates, from two independent experiments. ** P < 0.01, *** P < 0.001.
i6A and benzyl6A inhibit TPA-induced inflammation in vivo
To determine if the observed inhibitory effects of i6A on ROS production in cellular models could be reproduced in vivo, we used a mouse model of TPA-induced oxidative stress leading to inflammation. In particular, we measured the effects of i6A and benzyl6A pretreatment on the inflammatory response to TPA in Car-S mice, a strain that is genetically susceptible to inflammation and skin tumorigenesis [27]. In a split-body design, left ears served as the positive control group (pretreated only with vehicle before TPA) while right ears were the experimental group (pretreated with i6A or benzyl6A before TPA). In the macroscopic examination 24 h after TPA treatment (Fig. 6A), the left ears of 16 Car-S mice showed a typical inflammatory status, characterized by evident redness and tissue thickening. In contrast, the right ears of Car-S mice, pretreated with two doses of either i6A (n = 8; one animal is shown in Fig. 6A) or benzyl6A (n = 8) before TPA application, appeared macroscopically normal 24 h later.
Fig. 6.
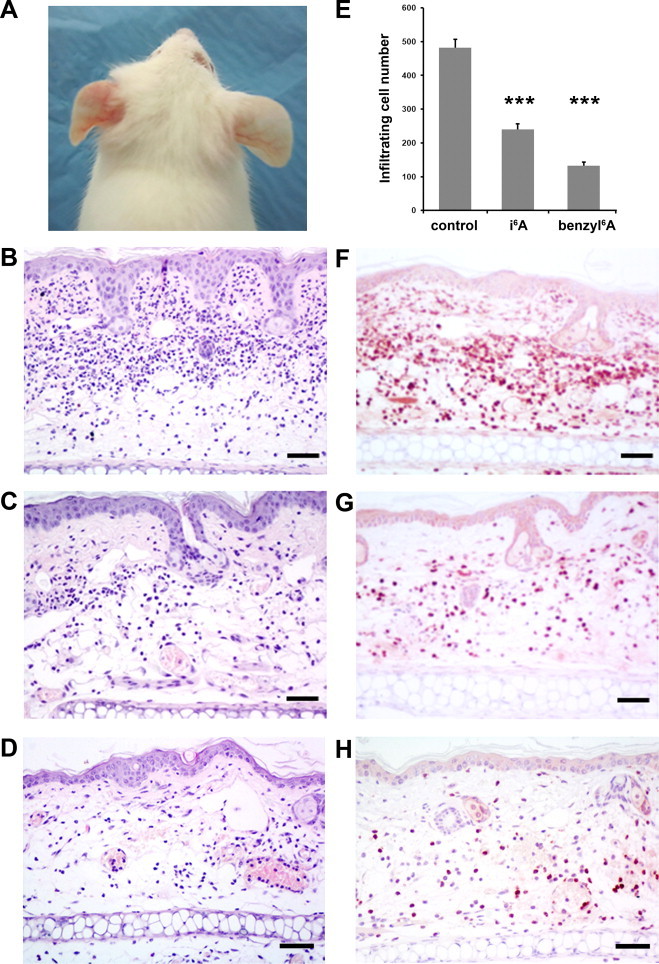
Topical application of i6A and benzyl6A reduced the inflammatory response to TPA treatment, on Car-S mice ears. (A) Photograph of one of the 8 Car-S mice pretreated twice on the right ear with i6A (in 95% ethanol) before a single treatment with TPA. The right ear appeared normal 24 h after TPA application, whereas the left ear, pretreated only with vehicle (ethanol) before TPA, showed a typical inflammatory status, characterized by evident redness and tissue thickening. (B) Hematoxylin-eosin staining of tissue slices from left ears (pretreated with ethanol alone before TPA) reveals massive infiltration of inflammatory cells in the dermal layer and tissue edema. (C, D) Stained tissue slices from right ears of mice pretreated with i6A (C) or benzyl6A (D) show a less severe inflammatory status caused by TPA application. (E) The number of infiltrating inflammatory cells, after TPA treatment, in i6A- and benzyl6A-pretreated ears was significantly lower than in vehicle-pretreated ears. *** P < 0.0001. (F, G and H) Immunohistochemical staining with anti-Ly-6G IgG showed that the infiltrating cells, in vehicle-, i6A- and benzyl6A-pretreated ears, respectively, were neutrophils. Scale bar: 50 µm.
At the microscopic examination, the positive control group showed a massive presence of inflammatory cells infiltrating the dermis together with increased vascular permeability and tissue edema (Fig. 6B). The altered histology is clearly seen when compared to tissue slices from negative control animals pretreated with vehicle or with i6A without a successive TPA treatment (Supplementary Fig. 4A and B). In the i6A- and benzyl6A-pretreated groups (Fig. 6C and D, respectively), we observed an evident reduced presence of inflammatory cells infiltrating the dermis, although the effects of TPA on vascular permeability and tissue edema were still evident. In the positive control and experimental groups, the infiltrating cells were classified as neutrophils, according to morphological criteria and immunostaining with an antibody against Ly-6G, an antigen expressed predominantly by peripheral neutrophils (Fig. 6F–H). Cell counting on three digital images per tissue slice revealed significantly fewer neutrophils in the ear dermis of i6A- (˜50%) and benzyl6A-pretreated mice (˜28%) than in samples that were not pretreated (Fig. 6E, P < 0.0001).
Discussion
This study showed that i6A and its synthetic analogs allyl6A, benzyl6A and butyl6A all inhibit the growth of MCF7 human breast adenocarcinoma cells and modulate their transcription profiles, in particular, by altering the expression of genes involved in the response to oxidative stress. Additionally, we found that these chemicals significantly reduced ROS production induced by two oxidants (H2O2 and TPA) in two cancer cell lines (MCF7 and dHL-60). Finally, we observed that i6A or benzyl6A pretreatment had an anti-inflammatory effect in vivo in a Car-S mouse model of TPA-induced inflammation.
In MCF7 cells treated or not with i6A or its analogs, microarray gene expression profiling revealed an overlapping pattern of differentially expressed genes, with 182 genes modified by all four compounds. Statistical analysis of the differentially expressed genes using IPA software resulted in the identification of the “NRF2-mediated oxidative stress response” pathway as most significantly associated to the lists of genes whose expression levels were significantly modulated by all four compounds. In the “NRF2-mediated oxidative stress response” pathway, under oxidative stress conditions, the transcription factor NRF2 (nuclear factor-erythroid 2-related factor 2) binds to the antioxidant response elements (ARE) within the promoters of genes that code for antioxidant proteins, activating their transcription and thus initiating a cellular defense response to oxidative stress (reviewed in [29]). This pathway is considered an important target for cancer chemoprevention, since many natural antioxidant and potential chemopreventive agents (e.g. isothiocyanates, indoles, terpenes and phenolic compounds) reportedly induce NRF2/ARE-dependent gene expression (reviewed in [30]). Moreover, the NRF2 pathway plays a relevant defensive role in pathologies such as chronic obstructive pulmonary disease, where its activation has been shown to inhibit oxidative stress, endoplasmic reticulum stress, and inflammation [31].
Although i6A has been studied for many years for its possible antineoplastic activity, we found here that it does not behave like standard chemotherapeutic agents, whose inhibition of cancer cell proliferation is associated with the generation of ROS and the consequent induction of oxidative stress [[32], [33], [34]]. In this study, i6A did not stimulate basal ROS production and instead had inhibitory effects. Moreover, i6A and one of its synthetic analogs, benzyl6A, had similar inhibitory effects on the production of ROS induced by both H2O2 and TPA treatment in MCF7 and dHL-60 cells, respectively. Thus these two molecules have particular interest as potential antioxidant agents. On the other hand, allyl6A and butyl6A behaved differently in the two cellular models: ally6A only reduced ROS production induced by H2O2 in MCF7 cells, whereas butyl6A only inhibited TPA-induced oxidative stress in dHL-60 cells. Additional studies are required to clarify these differences and to understand whether there is any molecular link between the anti-oxidant and antiproliferative effects of these molecules.
Considering the stimulating effects of i6A and its analogs on the NRF2-mediated oxidative stress response, as shown here, we hypothesize that these compounds act like dietary phytochemicals, non-nutritive compounds of edible plants some of which are classified as chemopreventive agents. Some phytochemicals block the initiation of carcinogenesis by activating the NRF2-mediated antioxidant response, and inhibit tumor progression via cell cycle arrest and induction of apoptosis after the activation of different cellular responses (reviewed in [35]). These compounds, like i6A and its analogs, induce detoxifying and antioxidant enzymes—such as heme oxygenase-1 (encoded by HMOX1) and glutamate-cysteine ligase (GCLC)—that protect cells against ROS and reactive metabolites of carcinogens, thus preventing tumorigenesis. Additionally, chemopreventive phytochemicals trigger several antineoplastic signaling pathways (e.g. induction of apoptosis via c-JUN NH2-terminal protein kinase signaling, or blocking cell cycle by inhibition of NF-κB signaling), contrasting cancer progression. Similarly, i6A has been reported to inhibit cell cycle progression [11,14,15] and induce apoptosis through the inhibition of NF-κB signaling [14]. It is interesting to note that i6A and its analogs, in addition to their activation of the NRF2-mediated antioxidant response, also modulated genes in the p53 signaling pathway, although at lower statistical significance. Since the p53 pathway is a master regulator of the cell cycle and of apoptosis [36], these observations provide some molecular insight into the antiproliferative actions of these modified nucleosides.
A recent paper showed that i6A specifically binds to the A3 adenosine receptor [19]. The four known adenosine receptors, A(1)AR, A(2A)AR, A(2B)AR and A(3)AR, play important roles in a large number of biological pathways and are also involved in different physiological and pathological conditions, such as cancer and inflammatory disease [37]. Through A(3)AR, adenosine exerts an anti-inflammatory effect on neutrophils by inhibiting superoxide production and chemotaxis [38]. Various A(3)AR agonists have been investigated for their anti-inflammatory effects in preclinical and clinical studies (reviewed in [39]). Therefore, we hypothesize that the antioxidant effects of i6A observed in our in vitro experiments and its activation of the NRF2 pathway, are also mediated by A(3)AR. The same may also be true for benzyl6A which behaved similarly to i6A in our experiments, while the divergent effects of ally6A and butyl6A may be mediated by other receptors. Indeed, our transcriptome analysis suggested that i6A and its analogs also modulated several genes belonging to the “glucocorticoid receptor signaling” pathway which is known to exert anti-inflammatory effects [40], thus suggesting another possible molecular mechanism of action of these chemicals that needs further investigation.
Possibly the most interesting result in the present study is the in vivo anti-inflammatory effect of i6A and benzyl6A when topically applied to mouse ears before TPA treatment. To the best of our knowledge, these anti-inflammatory properties have never been observed before. Further studies are needed to determine if this in vivo activity is mediated by the NRF2 pathway. Some insight on this possibility comes from studies using Nrf2-null mice, which developed a lupus-like autoimmune syndrome characterized by multiorgan inflammatory lesions [41]. Comparison of wild-type and Nrf2-null mice showed that Nrf2 protects the liver from oxidative stress, DNA damage and steatohepatitis induced by the tumor promoter 2,3,7,8-tetrachlorodibenzo-p-dioxin [42]. Moreover, UVB-irradiated Nrf2-null mice showed accelerated photoageing [43]. Altogether, these results highlight the importance of the Nrf2 pathway in the in vivo anti-oxidant and anti-inflammatory responses to endogenous and exogenous stimuli.
In light of the observed in vivo anti-inflammatory effects in a mouse model exerted by i6A and benzyl6A and of their strong in vitro antiproliferative effects and inhibition of ROS generation, future studies should investigate these two agents for their possible topical use in humans. In particular, they might be an attractive approach to alleviate skin inflammation and oxidative stress-induced tissue damage caused, for example, by UV radiation, or to prevent UV-related skin tumors.
Conflicts of interest
No competing financial interests exist.
Acknowledgments
We thank Dr. Valerie Matarese for scientific editing and Dr. Elena Goricioi (Department of Medicine, Surgery, and Dentistry, University of Milan, Italy) for the preparation of allyl6A and butyl6A. F.C. was recipient of a Marta Nurizzo Fellowship (http://www.martalive.org). This work was funded in part by grants from Italian Association for Cancer Research (AIRC) and the Italian Foundation for Cancer Research (FIRC) to T.A.D. and from the General Management for International Research of the Italian Ministry for Education, University and Research to E.S. The sponsors had no role in study design, in the collection, analysis and interpretation of the data, in the writing the report, or in the decision to submit the article for publication.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.redox.2014.03.001.
Appendix. Supplementary materials
Structures of i6A (A), allyl6A (B), benzyl6A (C), and butyl6A (D).
{kind=link}
Equi-effective doses of i6A and its analogs (10 µM i6A, 67 µM allyl6A, 11 µM benzyl6A, and 44 µM butyl6A) inhibited MCF7 cell growth to a similar extent (P > 0.05).
{kind=link}
Pathway diagram showing the proteins involved in the NRF2-mediated oxidative stress response and their subcellular locations, according to ingenuity pathway analysis software. Proteins encoded by genes that were differentially expressed (P < 1.0 × 10−4 and fold-change ≥1.5) between i6A-treated and untreated cells are highlighted in green when upregulated and in red when downregulated. Protein identifiers marked with an asterisk indicate that multiple identifiers in the gene list, obtained by class comparison analysis, map to a single protein.
{kind=link}
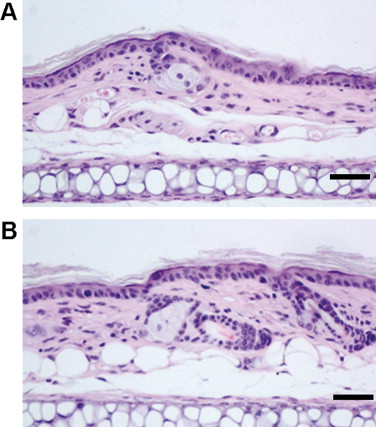
i6A pretreatment alone (without TPA application) did not induce either an inflammatory response or morphological changes in the ear dermis of Car-S mice. (A) Sample pretreated with i6A in 95% ethanol followed by treatment with acetone. (B) Sample pretreated with 95% ethanol followed by treatment with acetone. Hematoxylin–eosin staining. Scale bar: 50 µm.
{kind=link}
Primers used in quantitative PCR.
Expression levels of 10 genes in dHL-60 cells treated for 6 h with 10 µM i6A.
References
- 1.Spinola M., Galvan A., Pignatiello C., Conti B., Pastorino U., Nicander B., Paroni R., Dragani T.A. Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene. 2005;24:5502–5509. doi: 10.1038/sj.onc.1208687. 15870694 [DOI] [PubMed] [Google Scholar]
- 2.Golovko A., Hjälm G., Sitbon F., Nicander B. Cloning of a human tRNA isopentenyl transferase. Gene. 2000;258:85–93. doi: 10.1016/S0378-1119(00)00421-2. 11111046 [DOI] [PubMed] [Google Scholar]
- 3.Fradejas N., Carlson B.A., Rijntjes E., Becker N.P., Tobe R., Schweizer U. Mammalian Trit1 is a tRNA([ser]sec)-isopentenyl transferase required for full selenoprotein expression. Biochemical Journal. 2013;450:427–432. doi: 10.1042/BJ20121713. 23289710 [DOI] [PubMed] [Google Scholar]
- 4.Jenner L.B., Demeshkina N., Yusupova G., Yusupov M. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nature Structural & Molecular Biology. 2010;17:555–560. doi: 10.1038/nsmb.1790. 20400952 [DOI] [PubMed] [Google Scholar]
- 5.Bifulco M., Malfitano A.M., Proto M.C., Santoro A., Caruso M.G., Laezza C. Biological and pharmacological roles of N6-isopentenyladenosine: an emerging anticancer drug. Anti-cancer Agents in Medicinal Chemistry. 2008;8:200–204. doi: 10.2174/187152008783497028. 18288922 [DOI] [PubMed] [Google Scholar]
- 6.Vold B.S., Keith D.E., Jr., Slavik M. Urine levels of N-[9-(beta-D-ribofuranosyl)purin-6-ylcarbamoyl]-l-threonine, N6-(delta 2-isopentenyl)adenosine, and 2′-O-methylguanosine as determined by radioimmunoassay for normal subjects and cancer patients. Cancer Research. 1982;42:5265–5269. 7139629 [PubMed] [Google Scholar]
- 7.Slocum H.K., Hakala M.T. Mechanism of natural resistance to N6-(delta2-isopentenyl)adenosine in cultured cells. Cancer Research. 1975;35:423–428. 162874 [PubMed] [Google Scholar]
- 8.Jones R., Grace J.T., Mittelman A., Woodruff M.W. Human pharmacology and initial clinical trail of isopentenyl adenosine (IPA) Proceedings of the American Association for Cancer Research. 1968;9:35. [Google Scholar]
- 9.Suk D., Simpson C.L., Mihich E. Toxicological and antiproliferative effects of N6-(delta2-isopentenyl) adenosine, a natural component of mammalian transfer RNA. Cancer Research. 1970;30:1429–1436. 5426945 [PubMed] [Google Scholar]
- 10.Mittelman A., Evans J.T., Chheda G.B. Cytokinins as chemotherapeutic agents. Annals of the New York Academy of Sciences. 1975;255:225–234. doi: 10.1111/j.1749-6632.1975.tb29228.x. 1059358 [DOI] [PubMed] [Google Scholar]
- 11.Colombo F., Falvella F.S., De Cecco L., Tortoreto M., Pratesi G., Ciuffreda P., Ottria R., Santaniello E., Cicatiello L., Weisz A., Dragani T.A. Pharmacogenomics and analogues of the antitumour agent N6-isopentenyladenosine. International Journal of Cancer. Journal international Du Cancer. 2009;124:2179–2185. doi: 10.1002/ijc.24168. 19123479 [DOI] [PubMed] [Google Scholar]
- 12.Laezza C., Notarnicola M., Caruso M.G., Messa C., Macchia M., Bertini S., Minutolo F., Portella G., Fiorentino L., Stingo S., Bifulco M. N6-isopentenyladenosine arrests tumor cell proliferation by inhibiting farnesyl diphosphate synthase and protein prenylation. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2006;20:412–418. doi: 10.1096/fj.05-4044lsf. 16507758 [DOI] [PubMed] [Google Scholar]
- 13.Castiglioni S., Casati S., Ottria R., Ciuffreda P., Maier J.A. N6-isopentenyladenosine and its analogue n6-benzyladenosine induce cell cycle arrest and apoptosis in bladder carcinoma T24 cells. Anti-cancer Agents in Medicinal Chemistry. 2013;13:672–678. doi: 10.2174/1871520611313040016. 23094912 [DOI] [PubMed] [Google Scholar]
- 14.Laezza C., Malfitano A.M., Di Matola T., Ricchi P., Bifulco M. Involvement of Akt/NF-κB pathway in N6-isopentenyladenosine-induced apoptosis in human breast cancer cells. Molecular Carcinogenesis. 2010;49:892–901. doi: 10.1002/mc.20666. 20672320 [DOI] [PubMed] [Google Scholar]
- 15.Spinola M., Colombo F., Falvella F.S., Dragani T.A. N6-isopentenyladenosine: a potential therapeutic agent for a variety of epithelial cancers. International Journal of Cancer. Journal international Du Cancer. 2007;120:2744–2748. doi: 10.1002/ijc.22601. 17304507 [DOI] [PubMed] [Google Scholar]
- 16.Chheda G.B., Mittelman A. N 6-(2 -isopentenyl)adenosine metabolism in man. Biochemical Pharmacology. 1972;21:27–37. doi: 10.1016/0006-2952(72)90247-X. 5066734 [DOI] [PubMed] [Google Scholar]
- 17.Meisel H., Günther S., Martin D., Schlimme E. Apoptosis induced by modified ribonucleosides in human cell culture systems. FEBS Letters. 1998;433:265–268. doi: 10.1016/S0014-5793(98)00927-2. 9744808 [DOI] [PubMed] [Google Scholar]
- 18.Casati S., Ottria R., Baldoli E., Lopez E., Maier J.A., Ciuffreda P. Effects of cytokinins, cytokinin ribosides and their analogs on the viability of normal and neoplastic human cells. Anticancer Research. 2011;31:3401–3406. 21965753 [PubMed] [Google Scholar]
- 19.Blad C.C., von Frijtag Drabbe Künzel J.K., de Vries H., Mulder-Krieger T., Bar-Yehuda S., Fishman P., Ijzerman A.P. Putative role of the adenosine A(3) receptor in the antiproliferative action of N (6)-(2-isopentenyl)adenosine. Purinergic Signalling. 2011;7:453–462. doi: 10.1007/s11302-011-9244-9. 21720785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottria R., Casati S., Baldoli E., Maier J.A., Ciuffreda P. N-alkyladenosines: synthesis and evaluation of in vitro anticancer activity. Bioorganic and Medicinal Chemistry. 2010;18:8396–8402. doi: 10.1016/j.bmc.2010.09.030. 21035348 [DOI] [PubMed] [Google Scholar]
- 21.Teufelhofer O., Weiss R.M., Parzefall W., Schulte-Hermann R., Micksche M., Berger W., Elbling L. Promyelocytic HL60 cells express NADPH oxidase and are excellent targets in a rapid spectrophotometric microplate assay for extracellular superoxide. Toxicological Sciences: An Official Journal of the Society of Toxicology. 2003;76:376–383. doi: 10.1093/toxsci/kfg234. 14514966 [DOI] [PubMed] [Google Scholar]
- 22.Saran A., Neveu T., Covelli V., Mouton D., Pazzaglia S., Rebessi S., Doria G., Biozzi G. Genetics of chemical carcinogenesis: analysis of bidirectional selective breeding inducing maximal resistance or maximal susceptibility to 2-stage skin tumorigenesis in the mouse. International Journal of Cancer. Journal international Du Cancer. 2000;88:424–431. doi: 10.1002/1097-0215(20001101)88:3<424::AID-IJC15>3.0.CO;2-D. 11054672 [DOI] [PubMed] [Google Scholar]
- 23.NRC, National Research Council . Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals. National Academies Press; Washington, DC: 2011. [Google Scholar]
- 24.Wright G.W., Simon R.M. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–2455. doi: 10.1093/bioinformatics/btg345. 14668230 [DOI] [PubMed] [Google Scholar]
- 25.J.C. Oliveros, VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams, 2007.
- 26.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B. 1995;57:289–300. [Google Scholar]
- 27.Galvan A., Vorraro F., Cabrera W.H., Ribeiro O.G., Pazzaglia S., Mancuso M., Zolin A., Milani S., Saran A., Ibañez O.M., Dragani T.A. Genetic heterogeneity of inflammatory response and skin tumorigenesis in phenotypically selected mouse lines. Cancer Letters. 2010;295:54–58. doi: 10.1016/j.canlet.2010.02.013. 20307927 [DOI] [PubMed] [Google Scholar]
- 28.Fox J. The R commander:a basic-statistics graphical user interface to R. Journal of Statistical Software. 2005;19:1–42. [Google Scholar]
- 29.Baird L., Dinkova-Kostova A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Archives of Toxicology. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. 21365312 [DOI] [PubMed] [Google Scholar]
- 30.Jeong W.S., Jun M., Kong A.N. Nrf2: a potential molecular target for cancer chemoprevention by natural compounds. Antioxidants & Redox Signaling. 2006;8:99–106. doi: 10.1089/ars.2006.8.99. 16487042 [DOI] [PubMed] [Google Scholar]
- 31.Malhotra D., Thimmulappa R., Vij N., Navas-Acien A., Sussan T., Merali S., Zhang L., Kelsen S.G., Myers A., Wise R., Tuder R., Biswal S. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. American Journal of Respiratory and Critical Care Medicine. 2009;180:1196–1207. doi: 10.1164/rccm.200903-0324OC. 19797762 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Conklin K.A. Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integrative Cancer Therapies. 2004;3:294–300. doi: 10.1177/1534735404270335. 15523100 [DOI] [PubMed] [Google Scholar]
- 33.Chen Y., Jungsuwadee P., Vore M., Butterfield D.A., St Clair D.K. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Molecular interventions. 2007;7:147–156. doi: 10.1124/mi.7.3.6. 17609521 [DOI] [PubMed] [Google Scholar]
- 34.Tiligada E. Chemotherapy: induction of stress responses. Endocrine-related Cancer. 2006;13(Suppl. 1):S115–S124. doi: 10.1677/erc.1.01272. 17259552 [DOI] [PubMed] [Google Scholar]
- 35.Lee J.H., Khor T.O., Shu L., Su Z.Y., Fuentes F., Kong A.N. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacology & Therapeutics. 2013;137:153–171. doi: 10.1016/j.pharmthera.2012.09.008. 23041058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinhardt H.C., Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends in Genetics. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. 22265392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J.F., Eltzschig H.K., Fredholm B.B. Adenosine receptors as drug targets—what are the challenges? Nature Reviews. Drug Discovery. 2013;12:265–286. doi: 10.1038/nrd3955. 23535933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Hoeven D., Wan T.C., Auchampach J.A. Activation of the A(3) adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils. Molecular Pharmacology. 2008;74:685–696. doi: 10.1124/mol.108.048066. 18583455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fishman P., Bar-Yehuda S., Liang B.T., Jacobson K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discovery Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. 22033198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kadmiel M., Cidlowski J.A. Glucocorticoid receptor signaling in health and disease. Trends in Pharmacological Sciences. 2013;34:518–530. doi: 10.1016/j.tips.2013.07.003. 23953592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Q., Battelli L., Hubbs A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. American Journal of Pathology. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. 16723711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu H., Cui W., Klaassen C.D. Nrf2 protects against 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced oxidative injury and steatohepatitis. Toxicology and Applied Pharmacology. 2011;256:122–135. doi: 10.1016/j.taap.2011.07.019. 21846477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirota A., Kawachi Y., Yamamoto M., Koga T., Hamada K., Otsuka F. Acceleration of UVB-induced photoageing in nrf2 gene-deficient mice. Experimental Dermatology. 2011;20:664–668. doi: 10.1111/j.1600-0625.2011.01292.x. 21569103 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structures of i6A (A), allyl6A (B), benzyl6A (C), and butyl6A (D).
Equi-effective doses of i6A and its analogs (10 µM i6A, 67 µM allyl6A, 11 µM benzyl6A, and 44 µM butyl6A) inhibited MCF7 cell growth to a similar extent (P > 0.05).
Pathway diagram showing the proteins involved in the NRF2-mediated oxidative stress response and their subcellular locations, according to ingenuity pathway analysis software. Proteins encoded by genes that were differentially expressed (P < 1.0 × 10−4 and fold-change ≥1.5) between i6A-treated and untreated cells are highlighted in green when upregulated and in red when downregulated. Protein identifiers marked with an asterisk indicate that multiple identifiers in the gene list, obtained by class comparison analysis, map to a single protein.
i6A pretreatment alone (without TPA application) did not induce either an inflammatory response or morphological changes in the ear dermis of Car-S mice. (A) Sample pretreated with i6A in 95% ethanol followed by treatment with acetone. (B) Sample pretreated with 95% ethanol followed by treatment with acetone. Hematoxylin–eosin staining. Scale bar: 50 µm.
Primers used in quantitative PCR.
Expression levels of 10 genes in dHL-60 cells treated for 6 h with 10 µM i6A.