

Abstract
Background
Optimization of cerebral oxygenation after pediatric cardiac arrest (CA) may reduce neurological damage associated with the post-CA syndrome. We hypothesized that important alterations in regional partial pressure of brain tissue oxygen (PbO2) occur after resuscitation from CA and that clinically relevant interventions such as hyperoxia and blood pressure augmentation would influence PbO2.
Methods
Cortical and thalamic PbO2 were monitored in immature rats subjected to asphyxial CA (9 or 12 min asphyxia) and sham-operated rats using oxygen sensors.
Results
Thalamus and cortex showed similar baseline PbO2. Post-resuscitation there was early and sustained cortical hypoxia in an insult-duration fashion. In contrast, thalamic PbO2 initially increased four-fold, and afterwards returned to baseline values. PbO2 was FiO2-dependent, and the response to oxygen was more pronounced after a 9 min vs. 12 min CA. After a 12 min CA, PbO2 was modestly affected by blood pressure augmentation using epinephrine in the thalamus but not cortex.
Conclusion
After asphyxial pediatric CA, there is marked regional variability of cerebral oxygenation. Cortical hypoxia is pronounced and appears early, while thalamic hyperoxia is followed by normoxia. Compromised PbO2 in the cortex may represent a relevant and clinically measurable therapeutic target aimed at improving neurological outcome after pediatric CA.
Introduction
Hypoxic ischemic brain injury affects 53-76% of children successfully resuscitated from cardiac arrest (CA) (1). Brain tissue hypoxia, cerebral hypoperfusion and impaired autoregulation in the early post-CA syndrome have potential implications for secondary brain injury in patients successfully resuscitated from CA. The International Liaison Committee on Resuscitation emphasized in a recent statement that good neurological outcome is contingent upon optimal post-CA care, and identified important knowledge gaps in optimal oxygen delivery during the initial stages of reperfusion, and optimal cerebral monitoring after CA (2). Early optimization of cerebral oxygen delivery to meet metabolic demands may reduce the neurological damage associated with post-CA brain injury.
After experimental pediatric asphyxial CA, cortical regions have decreased perfusion, while thalamic regions are hyperemic (3). Cortical hypoperfusion observed in our model may be associated with cortical hypoxia. Serial and regional partial pressure of brain tissue oxygen (PbO2) after experimental asphyxial CA has not been characterized. Multiple studies have demonstrated that after traumatic brain injury (TBI) direct monitoring of PbO2 is safe and some studies have suggested that targeting specific thresholds for PbO2 has been associated with improved outcomes compared with historical controls (4-8). Although brain tissue monitoring was included in the treatment guidelines for adult victims of TBI in 2007, PbO2 monitoring after pediatric CA is not used routinely.
Based on our previous studies of post-resuscitation cerebral perfusion, we hypothesized that important differences in regional PbO2 would be observed during early post-CA syndrome after experimental asphyxial CA and that clinically relevant interventions such as hyperoxia and blood pressure augmentation would influence PbO2.
Results
Regional PbO2 during and after asphyxial CA
Table 1 presents physiological data for the groups of rats that underwent CA. Mean arterial pressure (MAP) increased at 5-10 min post resuscitation, then decreased and was lower than baseline after 30 min post resuscitation. The baseline MAP was lower in the 12 min asphyxial CA groups that underwent thalamic PbO2 measurements vs. 9 min asphyxial CA that underwent cortical or thalamic PbO2 measurements (55±2 vs. 67±2 and 72±4 mmHg, respectively, p<0.05). PaCO2 was maintained in the normal range at all time points except for an increase to 46±1.6 mm Hg only at 60 min post resuscitation in the 9 min thalamic group. Hemoglobin decreased vs. baseline at 30, 60, and 120 min after 9 and 12 min CA, with no difference between groups at each time point.
Table 1.
MAP, pH, PaO2, PaCO2, and hemoglobin (Hb) at baseline (immediately before asphyxia) and after asphyxial CA
| Baseline | 10 min | 30 min | 60 min | 120 min | |||
|---|---|---|---|---|---|---|---|
| Cortex | 9 min | MAP | 67±2 | 78±2* | 56±2* | 52±2* | 52±2* |
| pH | 7.40±0.01 | 7.29±0.01* | 7.46±0.02* | 7.47±0.01* | 7.44±0.03 | ||
| PaO2 | 207±11 | 402±12* | 412±10* | 183±15* | 235±6 | ||
| PaCO2 | 37±1 | 41±1 | 36±1 | 37±1 | 36±4 | ||
| Hb | 9.0±0.2 | 8.9±0.2 | 8.2±0.2* | 8.2±0.5* | 8.1±0.3* | ||
| 12 min | MAP | 65±2 | 86±3* | 55±5* | 49±3* | 47±3* | |
| pH | 7.39±0.01 | 7.27±0.02* | 7.40±0.01 | 7.44±0.01 | 7.39±0.01 | ||
| PaO2 | 212±3 | 411±20* | 403±1* | 266±38 | 208±5 | ||
| PaCO2 | 36±1 | 39±3 | 37±2 | 37±1 | 38±1 | ||
| Hb | 8.7±0.3 | 8.9±0.3 | 8.4±0.2* | 7.9±0.3* | 7.7±0.3* | ||
| Thalamus | 9 min | MAP | 72±4 | 73±1 | 58±2* | 49±3* | 55±2* |
| pH | 7.36±0.10 | 7.32±0.10* | 7.37±0.10* | 7.34±0.10* | 7.42±0.10* | ||
| PaO2 | 210±7 | 302±17* | 360±7* | 204±3 | 228±7 | ||
| PaCO2 | 37±0.8 | 39±2.5 | 39±2.0 | 46±1.6* | 33±2.5 | ||
| Hb | 10±0.2 | 9.6±0.7 | 8.9±0.2 | 8.0±0.1* | 8.5±0.1 | ||
| 12 min | MAP | 55±2 | 72±4* | 42±5* | 41±2* | 40±1* | |
| pH | 7.35±0.01 | 7.25±0.02 | 7.36±0.01 | 7.39±0.01 | 7.34±0.01 | ||
| PaO2 | 192±11 | 385±36* | 375±14* | 184±6 | 207±5 | ||
| PaCO2 | 41±2 | 40±4 | 38±1 | 42±1 | 44±1 | ||
| Hb | 9.1±0.1 | 9.7±0.2* | 8.9±0.2 | 8.1±0.2* | 7.7±0.2* |
(p<0.05 vs. baseline)
Baseline PbO2 was similar for cortical and thalamic areas. After the onset of asphyxia, PbO2 decreased to less than 5 mm Hg within 60 s in both regions and all rats. After CPR, we observed a rapid rise in PbO2 in the thalamus and a more gradual rise in PbO2 in the cortex.
Figure 1a illustrates cortical PbO2 after resuscitation from 9 and 12 min asphyxial CA. After 9 min asphyxial CA, mean cortical PbO2 was similar to baseline at 5, 10, and 15 min (64±22, 39±8, and 33±6 mm Hg at 5, 10 and 15 min, respectively), and then lower than baseline from 30 min (19±4 mm Hg) to 120 min (25±5 mm Hg) post-resuscitation. After 12 min asphyxial CA, cortical PbO2 was similar to baseline at 5-15 min (43±18, 25±7 and 25±6 mm Hg), and then lower than baseline from 30 min (15±4 mm Hg) to 120 min (9±2 mm Hg) post-resuscitation (Figure 1a). Cortical PbO2 was lower in rats subjected to 12 min asphyxial CA vs. 9 min asphyxial CA at 120 min post-resuscitation (p<0.05).
Figure 1.
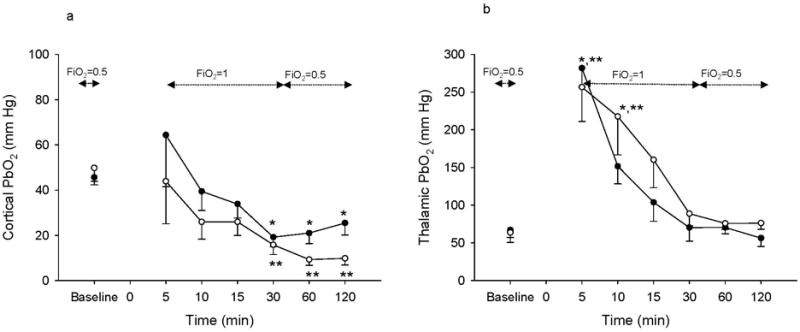
Cortical (a) and thalamic (b) PbO2 at baseline and after resuscitation from 9 and 12 min of asphyxia. Time 0 represents the time of CA. Black circles represent 9 min CA group, and white circles represent 12 min CA group. n= 6/group for thalamic PbO2 measurements and n=12/group for cortical PbO2 measurements. (*p<0.05 vs. baseline for 9 min insult, **p<0.05 vs. baseline for 12 min insult).
Figure 1b illustrates thalamic PbO2 after resuscitation from 9 and 12 min asphyxial CA. After both 9 and 12 min asphyxial CA, thalamic PbO2 was markedly increased vs. baseline at 5 min (281±25 vs. 66±16.3, and 256±45 vs. 63±6 mm Hg, p<0.05 after 9 and 12 min asphyxia, respectively) and remained elevated at 10 min after resuscitation (151±23 and 217±50 mm Hg, p<0.05 vs. baseline after 9 and 12 min asphyxia, respectively). After the initial increase, thalamic PbO2 decreased towards baseline for the duration of monitoring, and remained well above 20 mm Hg.
Post-CA response of PbO2 to supplemental oxygen
The response to supplemental oxygen was evaluated at 120 min after resuscitation from 9 or 12 min CA. At 120 min after resuscitation the rats receive a fraction of inspired oxygen (FiO2) =0.5 in our model. At that time point we decreased the FiO2 to 0.21 and gradually increased the FiO2 to 1.0. Decreasing the FiO2 to 0.21 at 120 min after CA resulted in arterial hypoxemia (Transcutaneous oxygen saturation, SatO2=83±3%, PaO2=46.2±4.2 mmHg). At an FiO2 of 0.21, cortical PbO2 was 16±2 and 5±2 mm Hg after 9 and 12 min asphyxial CA, respectively, while thalamic PbO2 was 19±5 and 30±3 after 9 and 12 min asphyxial CA, respectively (Figure 2).
Figure 2.
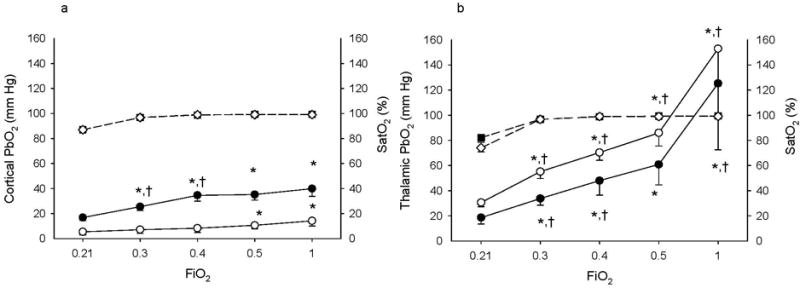
Cortical (a) and thalamic (b) PbO2 and Sat O2 response to increase in FiO2 from 0.21 to 0.3, 0.4, 0.5, and 1.0 after 9 and 12 min asphyxia. Black circles represent PbO2 for the 9 min CA group, white circles represent PbO2 for the 12 min CA group, black squares represent SatO2 for the 9 min CA group, and white diamonds represent SatO2 for the 12 min CA group. n= 6/group/region. (*p<0.05 vs. FiO2=0.21, †p<0.05 vs. previous FiO2).
Cortical and thalamic PbO2 and SatO2 values for individual FiO2 concentrations are shown in Figure 2. Increasing FiO2 from 0.21 to 0.3 resolved the observed hypoxemia and increased the SatO2 above 95% in all rats. Further increases in FiO2 increased SatO2 to 99-100%. Under these conditions, cortical and thalamic PbO2 had different responses to increased FiO2. Cortical PbO2 after 9 min of asphyxia increased progressively with supplemental oxygen from FiO2=0.21 to FiO2=0.4 (p<0.05). Further increase in FiO2 to 0.5 and 1 did not result in further increase in cortical PbO2 after 9 min of asphyxia. After 12 min of asphyxia, cortical PbO2 values were 7.1±3 and 8.3±3 mm Hg at FiO2=0.3 and 0.4, respectively (p=0.3 and 0.2, FiO2=0.3 and 0.4 vs. FiO2=0.21, respectively). PbO2 was slightly increased at FiO2=0.5 and 1 (PbO2=10.5±8 mm Hg, p<0.05 for FiO2=0.5 vs. FiO2=0.21).
Thalamic PbO2 gradually increased with increases in supplemental oxygen after both 9 and 12 min of asphyxia (Figure 2).
Post-CA response of PbO2 to epinephrine infusion
To determine whether low cortical PbO2 levels after 12 min asphyxial CA were related to low MAP, we administered epinephrine to increase MAP at 120 min after CA. A 2 mcg kg-1 min-1 epinephrine infusion increased MAP from 47±3.7 to 66.6±6.8 mm Hg (baseline MAP), and further infusion of epinephrine to 20 mcg kg-1 min-1 increased MAP to 82.3±5.8 mmHg (above baseline MAP). Cortical PbO2 did not increase with epinephrine infusion (Figure 3). Conversely, thalamic PbO2 did increase during high dose epinephrine infusion (p<0.05, epinephrine 20 mcg kg-1 min-1 vs. before epinephrine infusion, Figure 3). In sham rats treated with epinephrine infusion we observed a similar pattern of PbO2 response: no increase in cortical PbO2 and increase in thalamic PbO2 at 4, 10, and 20 mcg kg-1 min-1 (data not shown).
Figure 3.
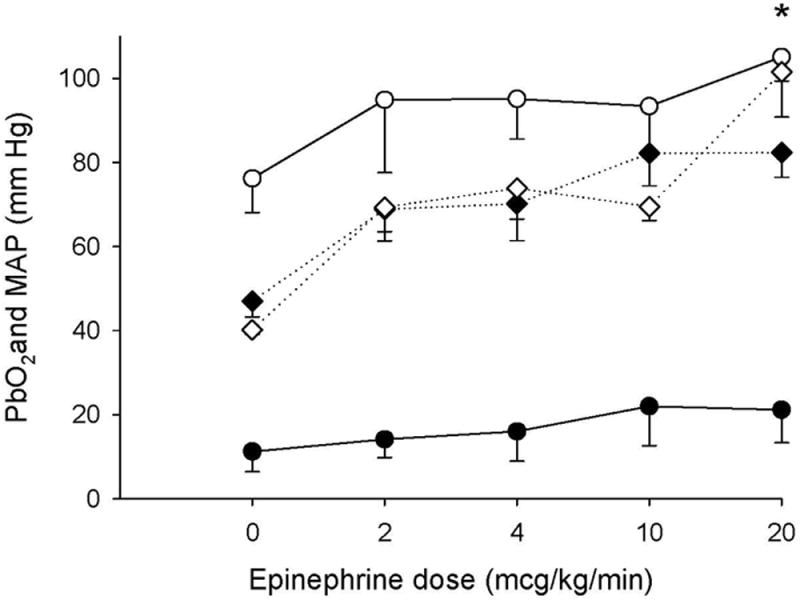
Cortical PbO2 at 120 min after resuscitation from CA (12 min asphyxia), before epinephrine (epinephrine dose=0), and after infusion of epinephrine at doses of 2, 4, 10, and 20 mcg kg-1 min-1. Black circles represent cortical PbO2, white circles represent thalamic PbO2, black diamonds represent MAP for the cortical group, and white diamonds represent MAP for the thalamic group. n= 6/group. (*p<0.05 vs. before epinephrine).
Discussion
To our knowledge, this is the first study reporting direct measurements of PbO2 in a model of pediatric asphyxial CA. Our data suggest important region- and insult duration-dependent alterations in PbO2 after CA. Cortical PbO2 was reduced in the first two hours after resuscitation, especially after longer insults, reaching thresholds consistent with cerebral ischemia (9). In distinct contrast, thalamic PbO2 was markedly increased initially after CA, and then returned to baseline. PbO2 response to supplemental oxygen was also insult duration and region dependent.
PbO2 reflects the local oxygen concentration in brain tissue, and serves as a marker of the balance between regional oxygen supply and consumption. It is influenced by cerebral blood flow (CBF), MAP, PaO2, hemoglobin concentration, and factors affecting diffusion of oxygen in brain (10). In clinical studies, PbO2 values less than 10 mm Hg are associated with higher risk for ischemic brain injury (11, 12) and with unfavorable outcome after TBI (8). Similar threshold values have not been reported in either experimental or clinical CA.
Post-CA cortical hypoxia in this study reached levels that are below the accepted PbO2 ischemic thresholds of 20 (5) or even 10 mm Hg in humans (8, 11). Human data demonstrated ongoing ischemia and anaerobic metabolism in the cortex during the first 10 h and up to 40 h after CA (13, 14). Similar to our study, in an experimental model of global cerebral ischemia produced by neck tourniquet in adult primates, PbO2 was region dependent with cortical hypoxia starting at 1 h post ischemia (15). Conversely, upon resuscitation from neonatal hypoxia with brief (1 min) CA, Linner et al. observed initial cortical hyperoxia followed by return to baseline levels in newborn piglets (16). This is consistent with the hypothesis that PbO2 changes are insult-duration dependent, with shorter durations of CA producing less hypoxia, similar to our previously described insult-duration dependency of CBF post CA (3). Age-related differences or species differences could also be involved. If this observation translates to children resuscitated from out-of-hospital CA, where the duration of no-flow is most often unknown, PbO2 could have potential utility in insult-duration stratification and prognostication.
Our data suggest that PbO2 parallels CBF in our model. Post-CA cortical hypoxia mirrors the cortical hypoperfusion we previously observed, while thalamic hyperoxia parallels thalamic hyperemia (3). Isoflurane, shown to preferentially increase subcortical CBF (17-20), increased thalamic PbO2 in our sham rats while having no effect on cortical PbO2 (pilot data, not shown). The association between PbO2 and CBF was also reported in non-injured rats (21) and models of global ischemia (22). These observations suggest that continuous PbO2 monitoring may be used as a surrogate marker for cerebral perfusion early after CA.
Supplemental oxygen increased thalamic and cortical PbO2, with a more robust effect after 9 vs. 12 min CA. It was previously reported that the response of PbO2 to hyperoxia is diminished in pathological compared with normal tissue (23-25), and increased supplemental oxygen does not correspond to an improvement in brain oxygen delivery in areas with severely reduced CBF (26). We previously reported that CBF is decreased in cortex, more pronounced after prolonged insults, and increased in thalamus after CA in our model (3). Our findings are in accordance with the hypothesis that raising FiO2 in the injured brain yields an increase in PbO2 but is highly dependent on adequate CBF. These observations suggest that interventions targeted to increase PbO2 after long durations of CA should primarily focus on increasing CBF. Further studies of strategies to increase cortical PbO2 via blood flow promoting vasodilatatory agents are warranted. Possible CBF promoting strategies include: endothelin receptor antagonists (27), nitrite (28), or inhibition of 20-HETE (29), among other therapies. In our study, conditions of mild arterial hypoxemia, on the contrary, resulted in lower PbO2 in shams and post-CA, suggesting that arterial hypoxemia should be avoided post-CA.
Increasing MAP using epinephrine infusion after long insults did not significantly increase cortical PbO2, although it increased thalamic PbO2 at high doses only. This suggests that nonspecific interventions such as MAP augmentation may not be sufficient to increase PbO2 in areas of cerebral hypoxia after CA. Our results also indicate that epinephrine could compromise cerebral microcirculatory flow after CA, similar to its effects in a piglet model of CA where despite increase in cerebral perfusion pressure after epinephrine infusion, microcirculatory blood flow decreased (30). As epinephrine is frequently used clinically post resuscitation from CA to stabilize hemodynamic parameters, it would be important to determine if other strategies to raise MAP might be able to improve PbO2 and microcirculatory flow. While MAP was measured during the epinephrine infusion, measurement of the intracranial pressure to assess the cerebral perfusion pressure could have given us additional information regarding the cerebrovascular effect of epinephrine.
We chose to study the effect on PbO2 of two clinically relevant and frequently used therapies during post-CA syndrome: oxygen and epinephrine. Other clinical strategies to augment PbO2, such as blood transfusion, administration of pentobarbital or controlled hypoventilation, not studied here, might have some utility.
We found some degree of variability of PbO2 in our study, both in shams and injured rats. This variability was observed in other studies (15) and is consistent with data showing that cortical PbO2 varies with as much as 20 mm Hg depending on the cortical area and layers of the somatosensory cortex (31). Likewise, hippocampal PbO2 varies within different areas (32). Our data suggest, however, that marked reductions of PbO2 in the cortex are observed after CA and represent a potential therapeutic target for post-resuscitation approaches designed to raise PbO2.
The ratio of PbO2/PaO2 followed the same pattern as PbO2 (data not shown), confirming the region specific variability to PbO2 in response to CA is not a function of changes in FiO2. We found some variability in our physiological data. The thalamic group with lower MAP (12 min asphyxial CA) had a similar PbO2 tracing with the 9 min thalamic group, and thus MAP values were unlikely to have affected our results. We also observed a decrease in hemoglobin of 0.9-1.5 mg/dl over time in all groups, with no difference between groups. In our model we observe a similar decrease in hemoglobin in sham rats with no change in PbO2, suggesting that this degree of decrease in hemoglobin does not affect PbO2 in sham or injured rats.
Relevant to the current study, in previous studies we did not observe sex-based differences in post-arrest CBF (3), although we did observe a sex-based difference in response to a specific treatment (polynitroxyl albumin) (33). As one aspect of the current study was to evaluate PbO2 as a potential surrogate for continuous CBF measurements after CA, we chose to limit the experiments to males. Future studies utilizing PbO2, particularly those evaluating treatments, should include males and females.
In this model of pediatric asphyxial CA selective neuronal death in cortex, thalamus, hippocampus and cerebellum is seen, along with behavioral deficits (34, 35). However, the relationship between cortical PbO2 and histopathological and functional outcome in our model remains to be determined. The threshold for cortical hypoxia that produces neuronal injury may even be lower than 10 mm Hg and remains uncertain. Age-related differences in the PbO2 threshold for hypoxia after CA also remain undefined. An intriguing hypothesis is that post-ischemic thalamic hyperoxia might also be detrimental. Further experiments in our model assessing the PbO2 threshold and whether goal directed PbO2 therapy improves outcome are warranted.
In conclusion, PbO2 after asphyxial CA in immature rats is region-dependent: there is cortical hypoxia but early thalamic hyperoxia. These findings mimic the pattern of CBF in this model, suggesting that PbO2 is representative of oxygen delivery and CBF. Increasing supplemental oxygen improves cortical hypoxia. If our model translates to the human condition, these data suggest a possible utility for PbO2 monitoring after CA in children. Further studies in this model are warranted to assess if goal directed therapy targeting cerebral oxygenation improves outcome after experimental pediatric asphyxial CA.
Materials and methods
Studies were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh and the care and handling of the animals were in accord with the National Institutes of Health (Bethesda, MD) guidelines. We used male postnatal day 16-18 Sprague-Dawley rats (30-46 g, n=56). We measured PbO2 after CA in cortex and thalamus in separate groups of rats after asphyxial CA and the response of PbO2 to hyperoxia or MAP augmentation after CA.
Anesthesia and surgery
Rats were initially anesthetized with 3% isoflurane/50% N2O/balance O2 in a Plexiglas chamber until unconscious and then their tracheas were intubated with an 18-gauge catheter. Mechanical ventilation was started and ventilatory rates and tidal volumes were adjusted to maintain PaCO2 at 35-45 mm Hg. Femoral arterial and venous catheters (PE 10) were inserted to monitor MAP and infuse medications. During surgery, anesthesia was maintained with 1.5% isoflurane/50% N2O/balance O2. Isoflurane was discontinued after the placement of arterial and venous catheters and anesthesia was maintained with fentanyl as described below. MAP and heart rate were continuously monitored. Rectal temperature was maintained at 37°C via a heated water blanket.
PbO2 electrode placement
The head was stabilized in a stereotaxic instrument using ear bars. A small burr hole (2 mm) was drilled in the skull 2 mm lateral and 3.3 mm posterior to bregma. PbO2 was measured continuously using a Clark type tissue electrode (Ox-50, Unisense, Denmark) inserted at either a depth of 1 mm for cortical PbO2 measurements or at a depth of 6 mm for thalamic PbO2 measurements. We chose to measure PbO2 in the cortex and thalamus because these are brain regions with the lowest or highest post-resuscitation CBF, respectively, in our previous report (3). The location of the electrode in cortex or thalamus was verified in brain sections after injection of Evans Blue at the conclusion of the experiment. Brain temperature was continuously monitored with an intraparenchymal sensor. Three groups (cortical 9 and 12 min, thalamic 9 min groups) had mean brain temperatures between 36.2 and 36.9. Due to an experimental oversight, the thalamic 12 min group did not have brain temperature monitored; however the body temperature, which parallels brain temperature in our model, was maintained constant in this group. Baseline parameters for MAP, PaO2, pH, PaCO2 and PbO2 were obtained immediately before the asphyxial insult.
PbO2 measurements in shams under different anesthetic conditions
In pilot experiments we determined the effects of two anesthetics: isoflurane and fentanyl on cortical and thalamic PbO2 in sham rats. Cortical PbO2 was measured in one group, and thalamic PbO2 in the other (n=8, 4/group). Each group initially was anesthetized with isoflurane and PbO2 was measured after a 10 min stabilization period. Isoflurane was then discontinued and a fentanyl infusion at 50 μg kg-1 h-1 was started. The rats were observed for 30 min prior to measuring the PbO2 to assure wash out of isoflurane. Cortical PbO2 values were similar using isoflurane or fentanyl anesthesia. Thalamic PbO2 were higher using isoflurane vs. fentanyl anesthesia (109±32% increase in thalamic PbO2 using isoflurane vs. fentanyl, p<0.05). Therefore, in all CA experiments, anesthesia was maintained with fentanyl infusion to minimize effects of anesthesia on PbO2.
Asphyxial cardiac arrest
We used an established asphyxial CA protocol (3). Rats received intravenous fentanyl infusion at 50 μg kg-1h-1 to provide anesthesia and vecuronium infusion at 5 mg kg-1h-1 to induce neuromuscular blockade. We used fentanyl as the anesthetic agent during the CA experiments because it is clinically relevant and unlike the inhaled anesthetics, it does not affect CBF (19). The FiO2 was reduced from 0.5 to 0.21 for 1 min before asphyxia to avoid preinsult hyperoxygenation. The tracheal tube was disconnected from the ventilator for 9 or 12 min. Resuscitation was started by reconnecting the ventilator and reinstituting mechanical ventilation at an FiO2 of 1.0. Epinephrine (0.005 mg kg-1) and sodium bicarbonate (1 mEq kg-1) were administered intravenously, followed by manual chest compressions until return of spontaneous circulation. The fentanyl infusion was restarted 30 min after resuscitation at the pre-arrest infusion rate. At 30 min after resuscitation, FiO2 was decreased to 0.5. This oxygenation sequence during CPR and post-resuscitation is consistent with common clinical practice at our institution. Arterial blood gas measurements were obtained at the time of arterial catheter insertion, at 30 and 60 min after CA and at the end of the experiment, and the ventilatory rates and tidal volumes were adjusted to a target of PaCO2 of 35-45 mm Hg. Transcutaneous oxygen saturation (SatO2) was measured continuously with a pulse oximeter (MouseOx, Starr Life Sciences).
PbO2 measurements during and after CA
In separate groups of rats, cortical or thalamic PbO2 were measured continuously before, during, and after 9 or 12 min asphyxial CA (n= 6/group for thalamic PbO2 measurements and n=12/group for cortical PbO2 measurements). PbO2 was recorded for analysis at baseline, and at the following time points post-resuscitation: 5, 10, 15, 30, 60, and 120 min. At the completion of 120 min post- resuscitation period, these rats underwent PbO2 measurements to assess the response to supplemental oxygen or epinephrine as described below.
Post-CA response of PbO2 to supplemental oxygen
We assessed cortical and thalamic PbO2 responses to a gradual increase in FiO2 from 0.21 to 1.0 (FiO2= 0.21, 0.3, 0.4, 0.5, and 1.0) at 120 min after resuscitation from 9 or 12 min asphyxial CA (n=6/group/region). According to our asphyxial CA protocol, rats receive FiO2= 1 at resuscitation and during the first 30 min post-resuscitation. From 30-120 min after resuscitation the rats receive an FiO2=0.5. At 120 min after resuscitation we decreased the FiO2 to 0.21, and then gradually increased the FiO2 as above. At each FiO2 we recorded the transcutaneous oxygen saturation (SatO2), PbO2, and MAP after a 10 min stabilization period for PbO2.
Post-CA response of PbO2 to epinephrine
We assessed the post-CA cortical and thalamic PbO2 response to an increase in MAP initially to baseline values, followed by MAP increase above baseline (n=6/group). At 120 min after 12 min asphyxia, we increased MAP via infusion of epinephrine. We started at a dose of 2 mcg kg-1 min-1 to achieve MAP equal to baseline, and then increased the infusion to 4, 10, and 20 mcg kg-1 min-1 (3, 36) while the FiO2 was maintained at 0.5. We administered the same infusion of epinephrine to sham rats (n=6/group) and measured PbO2.
Statistical Analysis
Data were analyzed with the statistical software Systat, Sigmastat 11.2 (Systat Software, Inc., Chicago, IL). Data were expressed as mean ± SEM. A p < 0.05 was considered significant. We used repeated measures ANOVA with Student-Newman Keuls post-hoc test to compare MAP, PaCO2, PaO2, pH, and PbO2 values at each time point and within each group over time. For data that failed equal variance and normality we ranked the data and afterwards performed repeated measures ANOVA.
Acknowledgments
Statement of financial support: National Institutes of Health (NIH; Bethesda, MD) grant K08 HD058798 (to MDM), NIH grant R01 HD075760 (to MDM), American Heart Association grant 10BGIA3580040 (to MDM), Children’s Hospital of Pittsburgh Research Grant, Pittsburgh, PA (to MDM), NIH grant R01 HD045968 (to RSBC), and NIH grant K23 NS065132 (to ELF).
Footnotes
DISCLOSURE: none
References
- 1.Moler FW, Meert K, Donaldson AE, et al. In-hospital versus out-of-hospital pediatric cardiac arrest: a multicenter cohort study. Crit Care Med. 2009;37:2259–2267. doi: 10.1097/CCM.0b013e3181a00a6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nolan JP, Neumar RW, Adrie C, et al. Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation. 2008;79:350–379. doi: 10.1016/j.resuscitation.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 3.Manole MD, Foley LM, Hitchens TK, et al. Magnetic resonance imaging assessment of regional cerebral blood flow after asphyxial cardiac arrest in immature rats. J Cereb Blood Flow Metab. 2009;29:197–205. doi: 10.1038/jcbfm.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stiefel MF, Udoetuk JD, Storm PB, et al. Brain tissue oxygen monitoring in pediatric patients with severe traumatic brain injury. J Neurosurg. 2006;105:281–286. doi: 10.3171/ped.2006.105.4.281. [DOI] [PubMed] [Google Scholar]
- 5.Narotam PK, Morrison JF, Nathoo N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111:672–682. doi: 10.3171/2009.4.JNS081150. [DOI] [PubMed] [Google Scholar]
- 6.McCarthy MC, Moncrief H, Sands JM, et al. Neurologic outcomes with cerebral oxygen monitoring in traumatic brain injury. Surgery. 2009;146:585–590. doi: 10.1016/j.surg.2009.06.059. discussion 590-581. [DOI] [PubMed] [Google Scholar]
- 7.Lang EW, Mulvey JM, Mudaliar Y, Dorsch NW. Direct cerebral oxygenation monitoring--a systematic review of recent publications. Neurosurg Rev. 2007;30:99–106. doi: 10.1007/s10143-006-0062-4. discussion 106-107. [DOI] [PubMed] [Google Scholar]
- 8.Maloney-Wilensky E, Gracias V, Itkin A, et al. Brain tissue oxygen and outcome after severe traumatic brain injury: a systematic review. Crit Care Med. 2009;37:2057–2063. doi: 10.1097/CCM.0b013e3181a009f8. [DOI] [PubMed] [Google Scholar]
- 9.Vespa PM. The implications of cerebral ischemia and metabolic dysfunction for treatment strategies in neurointensive care. Curr Opin Crit Care. 2006;12:119–123. doi: 10.1097/01.ccx.0000216577.57180.bd. [DOI] [PubMed] [Google Scholar]
- 10.Maloney-Wilensky E, Le Roux P. The physiology behind direct brain oxygen monitors and practical aspects of their use. Childs Nerv Syst. 26:419–430. doi: 10.1007/s00381-009-1037-x. [DOI] [PubMed] [Google Scholar]
- 11.Goodman JC, Valadka AB, Gopinath SP, Uzura M, Robertson CS. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit Care Med. 1999;27:1965–1973. doi: 10.1097/00003246-199909000-00041. [DOI] [PubMed] [Google Scholar]
- 12.Kiening KL, Unterberg AW, Bardt TF, Schneider GH, Lanksch WR. Monitoring of cerebral oxygenation in patients with severe head injuries: brain tissue PO2 versus jugular vein oxygen saturation. J Neurosurg. 1996;85:751–757. doi: 10.3171/jns.1996.85.5.0751. [DOI] [PubMed] [Google Scholar]
- 13.Nordmark J, Rubertsson S, Mortberg E, Nilsson P, Enblad P. Intracerebral monitoring in comatose patients treated with hypothermia after a cardiac arrest. Acta Anaesthesiol Scand. 2009;53:289–298. doi: 10.1111/j.1399-6576.2008.01885.x. [DOI] [PubMed] [Google Scholar]
- 14.Bauer R, Gabl M, Obwegeser A, et al. Neurochemical monitoring using intracerebral microdialysis during cardiac resuscitation. Intensive Care Med. 2004;30:159–161. doi: 10.1007/s00134-003-2015-5. [DOI] [PubMed] [Google Scholar]
- 15.Nemoto EM, Erdmann W, Strong E, Rao GR, Moossy J. Regional brain PO2 after global ischemia in monkeys: evidence for regional differences in critical perfusion pressures. Stroke. 1979;10:44–52. doi: 10.1161/01.str.10.1.44. [DOI] [PubMed] [Google Scholar]
- 16.Linner R, Werner O, Perez-de-Sa V, Cunha-Goncalves D. Circulatory recovery is as fast with air ventilation as with 100% oxygen after asphyxia-induced cardiac arrest in piglets. Pediatr Res. 2009;66:391–394. doi: 10.1203/PDR.0b013e3181b3b110. [DOI] [PubMed] [Google Scholar]
- 17.Farber NE, Harkin CP, Niedfeldt J, et al. Region-specific and agent-specific dilation of intracerebral microvessels by volatile anesthetics in rat brain slices. Anesthesiology. 1997;87:1191–1198. doi: 10.1097/00000542-199711000-00024. [DOI] [PubMed] [Google Scholar]
- 18.Hansen TD, Warner DS, Todd MM, Vust LJ, Trawick DC. Distribution of cerebral blood flow during halothane versus isoflurane anesthesia in rats. Anesthesiology. 1988;69:332–337. doi: 10.1097/00000542-198809000-00008. [DOI] [PubMed] [Google Scholar]
- 19.Hendrich KS, Kochanek PM, Melick JA, et al. Cerebral perfusion during anesthesia with fentanyl, isoflurane, or pentobarbital in normal rats studied by arterial spin-labeled MRI. Magn Reson Med. 2001;46:202–206. doi: 10.1002/mrm.1178. [DOI] [PubMed] [Google Scholar]
- 20.Loeb AL, Raj NR, Longnecker DE. Cerebellar nitric oxide is increased during isoflurane anesthesia compared to halothane anesthesia: a microdialysis study in rats. Anesthesiology. 1998;89:723–730. doi: 10.1097/00000542-199809000-00024. [DOI] [PubMed] [Google Scholar]
- 21.Lowry JP, Boutelle MG, Fillenz M. Measurement of brain tissue oxygen at a carbon past electrode can serve as an index of increases in regional cerebral blood flow. J Neurosci Methods. 1997;71:177–182. doi: 10.1016/s0165-0270(96)00140-9. [DOI] [PubMed] [Google Scholar]
- 22.Rossi S, Balestreri M, Spagnoli D, et al. Oxygen delivery and oxygen tension in cerebral tissue during global cerebral ischaemia: a swine model. Acta Neurochir Suppl. 2000;76:199–202. doi: 10.1007/978-3-7091-6346-7_40. [DOI] [PubMed] [Google Scholar]
- 23.Longhi L, Valeriani V, Rossi S, et al. Effects of hyperoxia on brain tissue oxygen tension in cerebral focal lesions. Acta Neurochir Suppl. 2002;81:315–317. doi: 10.1007/978-3-7091-6738-0_80. [DOI] [PubMed] [Google Scholar]
- 24.Hlatky R, Valadka AB, Gopinath SP, Robertson CS. Brain tissue oxygen tension response to induced hyperoxia reduced in hypoperfused brain. J Neurosurg. 2008;108:53–58. doi: 10.3171/JNS/2008/108/01/0053. [DOI] [PubMed] [Google Scholar]
- 25.Meixensberger J, Dings J, Kuhnigk H, Roosen K. Studies of tissue PO2 in normal and pathological human brain cortex. Acta Neurochir Suppl (Wien) 1993;59:58–63. doi: 10.1007/978-3-7091-9302-0_10. [DOI] [PubMed] [Google Scholar]
- 26.Rossi S, Longhi L, Balestreri M, et al. Brain oxygen tension during hyperoxia in a swine model of cerebral ischaemia. Acta Neurochir Suppl. 2000;76:243–245. doi: 10.1007/978-3-7091-6346-7_49. [DOI] [PubMed] [Google Scholar]
- 27.Armstead WM. Age dependent endothelin contribution to NOC/oFQ induced impairment of NMDA cerebrovasodilation after brain injury. Peptides. 2001;22:39–46. doi: 10.1016/s0196-9781(00)00354-5. [DOI] [PubMed] [Google Scholar]
- 28.Jung KH, Chu K, Ko SY, et al. Early intravenous infusion of sodium nitrite protects brain against in vivo ischemia-reperfusion injury. Stroke. 2006;37:2744–2750. doi: 10.1161/01.STR.0000245116.40163.1c. [DOI] [PubMed] [Google Scholar]
- 29.Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab. 2006;26:1551–1561. doi: 10.1038/sj.jcbfm.9600309. [DOI] [PubMed] [Google Scholar]
- 30.Ristagno G, Tang W, Huang L, et al. Epinephrine reduces cerebral perfusion during cardiopulmonary resuscitation. Crit Care Med. 2009;37:1408–1415. doi: 10.1097/CCM.0b013e31819cedc9. [DOI] [PubMed] [Google Scholar]
- 31.Masamoto K, Takizawa N, Kobayashi H, Oka K, Tanishita K. Dual responses of tissue partial pressure of oxygen after functional stimulation in rat somatosensory cortex. Brain Res. 2003;979:104–113. doi: 10.1016/s0006-8993(03)02882-8. [DOI] [PubMed] [Google Scholar]
- 32.Andreasen A, Danscher G, Juhl S, et al. Distinct differences in partial oxygen pressure at micrometer ranges in the rat hippocampal region. J Neurosci Methods. 1997;72:15–21. doi: 10.1016/s0165-0270(96)00149-5. [DOI] [PubMed] [Google Scholar]
- 33.Manole MD, Kochanek PM, Foley LM, et al. Polynitroxyl albumin and albumin therapy after pediatric asphyxial cardiac arrest: effects on cerebral blood flow and neurologic outcome. J Cereb Blood Flow Metab. doi: 10.1038/jcbfm.2011.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fink EL, Alexander H, Marco CD, et al. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatr Crit Care Med. 2004;5:139–144. doi: 10.1097/01.pcc.0000112376.29903.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoykhet M, Simons DJ, Kochanek PM, Clark RSB. Anatomic evidence of injury to thalamocortical structures in a rodent model of pediatric asphyxia cardiac arrest. Critical Care Med. 2011;38(Suppl):A152. [Google Scholar]
- 36.Dewachter P, Raeth-Fries I, Jouan-Hureaux V, et al. A comparison of epinephrine only, arginine vasopressin only, and epinephrine followed by arginine vasopressin on the survival rate in a rat model of anaphylactic shock. Anesthesiology. 2007;106:977–983. doi: 10.1097/01.anes.0000265157.09438.1f. [DOI] [PubMed] [Google Scholar]