

Abstract
Huanglongbing (HLB) is a destructive citrus disease. The leading cause of HLB is Candidatus Liberibacter asiaticus. Fatty acid biosynthesis is essential for bacterial viability and has been validated as a target for the discovery of novel antibacterial agents. Enoyl−acyl carrier protein reductase (also called ENR or FabI and a product of the fabI gene) is an enzyme required in a critical step of bacterial fatty acid biosynthesis and has attracted attention as a target of novel antimicrobial agents. We determined the crystal structures of FabI from Ca. L. asiaticus in its apoform as well as in complex with b-nicotinamide adenine dinucleotide (NAD) at 1.7 and 2.7 Å resolution, respectively, to facilitate the design and screening of small molecule inhibitors of FabI. The monomeric ClFabI is highly similar to other known FabI structures as expected; however, unlike the typical tetramer, ClFabI exists as a hexamer in crystal, whereas as dimer in solution, on the other hand, the substrate binding loop which always disordered in apoform FabI structures is ordered in apo-ClFabI. Interestingly, the structure of ClFabI undergoes remarkable conformational change in the substrate-binding loop in the presence of NAD. We conclude that the signature sequence motif of FabI can be considered as Gly-(Xaa)5-Ser-(Xaa)n-Val-Tyr-(Xaa)6-Lys-(Xaa)n-Thr instead of Tyr-(Xaa)6-Lys. We have further identified isoniazid as a competitive inhibitor with NADH.
Keywords: Candidatus Liberibacter asiaticus, enoyl–acyl carrier protein reductase I, crystal structures, b-nicotinamide adenine dinucleotide, isoniazid
Introduction
Citrus Huanglongbing (HLB) is a century-old and re-emerging disease that impedes citrus production worldwide. HLB was discovered in Asian countries in the 1870s1 and has spread to the western hemisphere in recent years. HLB cases have been reported in Brazil,2 the United States, Cuba, and Mexico.3,4 HLB-associated Liberibacter species have three classes, namely, Candidatus Liberibacter asiaticus, Candidatus Liberibacter africanus, and Candidatus americanus. Ca. L. asiaticus, the most devastating species of HLB bacteria, has caused serious economic losses worldwide. The current strategy for controlling HLB mainly involves annihilation of media psyllids and eradication of infected trees. Conventional breeding and transformation approaches are also used to generate new clones that are tested for their responses to Ca. L. asiaticus infection. Other approaches, such as chemotherapeutic methods designed to eliminate or suppress Ca. L. asiaticus in infected plants, also offer promising methods for alternatives for managing this devastating disease.
Fatty acids are important components of cellular metabolic pathways5 and are essential for bacterial viability. Different from mammals which use type I fatty acid synthase, most bacteria and plants adopt type II fatty acid synthase in this pathway in which enoyl–acyl carrier protein (ACP) reductase I (ENR or FabI) catalyzes the final step in the fatty acids synthesis pathway. FabI is crucial in the completion of cycles, particularly in the elongation phase of fatty acids synthesis.6,7 Consequently, this important enzyme has recently attracted tremendous attention from researchers. Several crystal structures of FabI have been reported in other bacteria and protozoans, such as Escherichia coli,8 Staphylococcus aureus,9 Bacillus subtilis,10 Bacillus cereus,11 Helicobacter pylori,12 Francisella tularensis,13 Plasmodium falciparum,14 and Mycobacterium tuberculosis.15 These homologous structures have been used as models to understand ClFabI in HLB pathogens and to develop new antimicrobial compounds for HLB control.
As a key enzyme in fatty acids synthesis, FabI is a major target for antimicrobial compounds. FabI assays with varying concentrations of nicotinamide adenine dinucleotide (NAD+) suggest that b-nicotinamide adenine dinucleotide (NADH) oxidation is involved in the inhibition reaction; this finding indicates that NAD+ facilitates the inhibitory activity of triclosan.16 Saito et al.17 elucidated the crystal structure of the enoyl–ACP reductase FabK (isoenzyme of FabI) in Streptococcus pneumonia and the binding mode of an inhibitor. Rafi et al.6 showed that complexes are primarily stabilized by interactions with acidic residues in the helix α2 of ACP, illustrating that FabI can serve as a target for drug discovery.
Studying on the mechanism of isoniazid inhibit InhA provides significant insight for discovery antimicrobial agent. Isoniazid has been reported to form isoniazid–NAD adduct, which is a slow, tight-binding competitive inhibitor of InhA from Mycobacterium tuberculosis.15 To inhibit InhA, isoniazid requires conversion to an activated form of the drug and that a catalase peroxidase (KatG) participates in isoniazid activation, in its activated form, isoniazid forms covalent attachment with the nicotinamide ring of NAD bound within the active site of InhA.18–20 The resulting INH–NAD adducts then inhibit enzyme enoyl–ACP reductase (InhA) and thus inhibit mycolic acid biosynthesis.21 However, the activated form of isoniazid is an intermediate in the formation of isonicotinic acid, isonicotinamide, and pyridine-4-carboxaldehyde, the products of isoniazid oxidation, none of which inhibits InhA.22 The structure of INH is simple, but the complex mechanism and target of its inhibitory function remains poorly known. In this study, we attempted to understand whether ClfabI interacts with INH when ClfabI, INH, NAD+, NADH, and the substrate crotonoyl coenzyme A (CoA) are present. Studies on pre-steady-state kinetics of NADH binding have been carried out, the results showed that the limiting rate constant values for NADH dissociation from the InhA-NADH binary complexes (koff) were eleven, five, and tenfold higher for, respectively, I21V, I47T, and S94A INH-resistant mutants of InhA as compared to INH-sensitive wildtype InhA.23 The ENR inhibitor isoniazid (INH) has been used to block enoyl–ACP reductase and thus treats infections efficient of H. pylori.24 Synthesis derivatives with side chain of the fatty acid chlorides and alkyl groups as potential enoyl–ACP reductase inhibitors exhibit an available means.25 The structure of INH is simple, but the complex mechanism and target of its inhibitory function remains poorly known. In this study, we attempted to understand whether ClfabI interacts with INH when ClfabI, INH, NAD+, NADH, and the substrate crotonoyl-CoA are present.
Ca. L. asiaticus is difficult to cultivate, which makes development and discovery of effective methods for controlling the HLB pathogen challenging. In addition, cloning FabI from Ca. L. asiaticus is not straightforward. However, recent availability of the FabI sequence on completion of sequencing of the whole genome (NC_012985) of Ca. L. asiaticus26 has made this easier. FabI is a valid target for selecting antimicrobial compounds against Ca. L. asiaticus. Therefore, the crystal structure of CIFabI would facilitate structure-based strategies for virtual screening and molecular docking against libraries of potential small molecules. Taking advantage of the mechanism by which inhibitors block metabolism of fatty acids is a promising method for finding new antibacterial agents. Screening of antibacterial agents against HLB is urgently needed. At present, we hope to validate if the overexpressed protein ClFabI has vigor, and if the inhibitor INH has function to the target ClFabI, which is important to establish a foundation for new antibacterial exploitation.
In the current work, we cloned and overexpressed FabI from Ca. L. asiaticus and determined the crystal structures of FabI in its apoform and in complex with NAD+. We also examined the kinetic properties of FabI and its mode of interaction with inhibitor INH. Our findings provide a structural framework to screen and develop effective inhibitors for FabI, which are expected to lead to new solutions for the control of HLB pathogens.
Results and Discussions
Overall structure of apo-ClFabI
Apo-ClFabI exists as a hexamer in crystal with root mean square deviation (RMSD) varying from 0.19 to 0.33 Å between each two monomers. Residues Y259–N267 in each monomer and M1 of chain F are lost because of poor electron density. Each monomer adopts a classical Rossmann fold usually found in nucleotide-binding proteins with each monomer composed of a parallel β-sheet with seven strands and flanked by loops and 12 helices, including four 310 helices [η1–η4; Fig. 1(a)]. The whole structure could be considered to be prism with three dimers as its sides [Fig. 1(b)]. The accessible surface area of the monomer is ∼12,405 Å2, as calculated using Areamol in CCP4. Dimer formation buried ∼1450 Å2 with extensive hydrogen bonds at the dimer interface involving 15 residues, namely, E67, Y105, Y106, T108, R110, T125, R129, G148, S149, R151, V153, S164, S168, Y172, and D176. Two additional hydrogen bonds between K171 and M150 from each chain are found in the dimer of Chains C and D. In the dimer of Chains E and F, extra hydrogen bonds between R110 from Chain E and V66 and E126 from Chain F are included in the dimerization interaction. Aside from these dimerization interactions, some hydrogen bonds or salt bridges are formed between dimers in which more than one half is formed between AB and CD dimers. Thus, dimers AB and CD could be considered as a tetramer similar to typical ENR structures. The result of the size analysis using a chromatographic column (Sephadex G-200 10/30) indicates that ClFabI exists as a dimer in solution, and according to the protein assemblies provided by the program PISA,14 it indicated that the six monomers can be divided into three dimers: AB, CD, and EF dimers. They can be superposed on each other very well with RMSD values of about 0.2 Å [Fig. 1(b)]. The interface surface areas are all about 1600 Å2 for each dimer, and the residues involved in the dimer interactions are nearly identical. Thus, the AB dimer is chosen for comparison with homolog ENR dimers: the apo-structures of FabI from S. aureus (PDB codes 3GNS and 3GNT) and B. cereus (PDB code 3OJE). There are some disordered residues in 3GNS, 3GNT, and 3OJE; however, all the corresponding residues in apo-ClFabI are ordered, because some of these residues take part in the dimer interaction making them more stable. The interface surface areas are 1750, 1547, and 1398 Å2 for 3GNS, 3GNT, and 3OJE dimers, respectively, The corresponding values for ClFabI are similar, but the residues contained in the dimer interaction are different (Supporting Information Table S1). This means that the relative orientation of two monomers in these dimers are different, which maybe the result of crystal packing during crystallization (Supporting Information Table S2).
Figure 1.
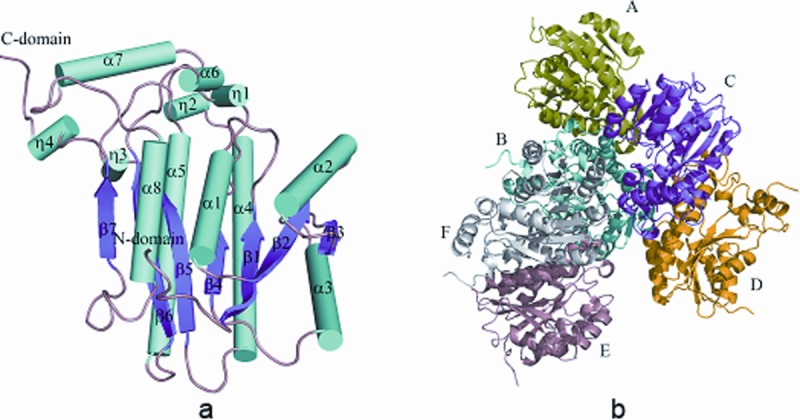
(a) Monomer structure of ClFabI in apoform with secondary structure labeled. α-Helices are shown as cylinder, 310 helices are labeled η, and β-sheets are shown as flat arrows. (b) Structure of a hexamer; Chains A–F are labeled and shown in different colors.
The theoretical molecular weight of CIFabI with 267 aa is 29263.57. Interestingly, a single peak with an elution volume corresponding to a molecular mass of ∼61.6 kDa is observed in a 120-mL gel filtration on Superdex G200 (25 mM Bis-Tris, pH 6.95, 300 mM NaCl) with respect to a ClFabI dimer (58.5 kDa). The small discrepancy is probably due to extended flexible expression tag at the amino termini of ClFabI. Therefore, we conclude that CIFabI formed stable dimers in solution (Supporting Information Fig. S1).
Comparison of apo-ClFabI and ClFabI–NAD binary complexes
There are also six monomers in the ClFabI–NAD binary complex. However, only four CIFabI molecules are incorporated into NAD; these molecules are similar to each other with RMSD varying from 0.27 to 0.30 Å. The two left monomers (Chains B and F) do not contain NAD according to electron density. Superposition of the apo-structure (Chain A) and binary complex structure in Chain B or Chain F results in an overall RMSD of 0.30 or 0.31 Å for 258 Cα atoms, suggesting that the two monomers are in apoform. Conversely, the overlay with an RMSD of 0.44 Å between Chain A in the apoform and Chain A in the binary complex indicates that monomer bound with NAD is also similar to the ones of apoform. The obvious conformational change comes from the region V192–N202, which moves toward NAD after cofactor binding, and the residues T194–S199 in α6 even deviates by more than 5 Å after NAD binding [Fig. 2(a)], and at the same time, the average B-factor of this region in ClFabI–NAD is higher than that in apo-ClFabI (71.2 in ClFabI–NAD and 40.3 in apoform).
Figure 2.
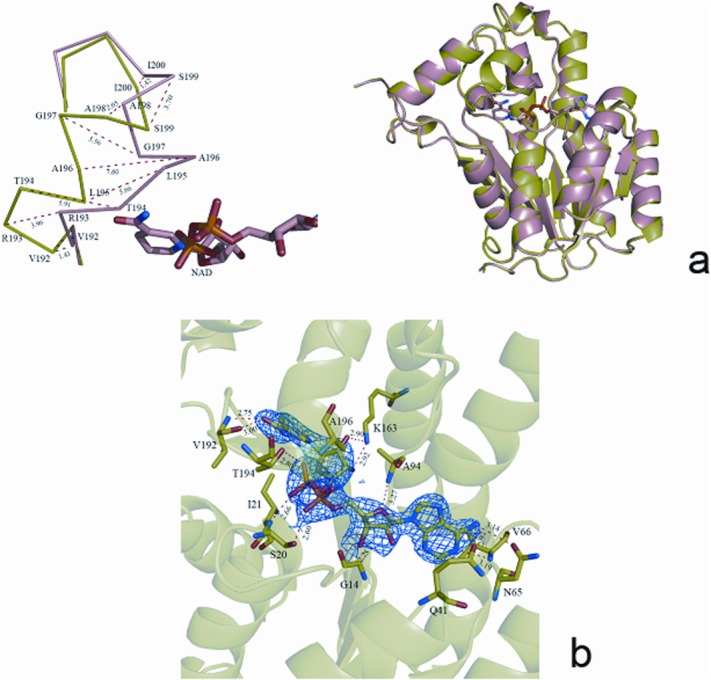
(a) Superposition of ClFabI (green) and the ClFabI–NAD binary complex (salmon). Only Chain A from each structure is shown for clarity. α6 is pulled toward NAD after cofactor binding, and the largest movement is 7.60 Å (between Cα of A196). (b) Contacts between protein and NAD mediated by hydrogen bonds are shown as dashed lines, and the numbers parallel to the lines indicate distance.
The electron density map clearly reveals the position of the cofactor without any ambiguity, and the residues S20, I21, N65, A94, K163, V192, T194, and A196 are involved in the interaction with NAD [Fig. 2(b)]. The adenosine moiety interplays with side-chain atoms of Q41 and N65 and main-chain atoms of G14 and V66, whereas the pyrophosphate moiety forms hydrogen bonds with side-chain oxygen atom of S20 and main-chain nitrogen atoms of I21 and A196. The nicotinamide ribose moiety is bounded by K163, V192, and T194. Aside from these residues, five water atoms also form hydrogen bonds with NAD. The final refinement statistics are summarized in TableI.
Table I.
ClFabI and ClFabI–NAD Complex Statistics
| Data collection | Apoform | Binary complex | |
|---|---|---|---|
| Wavelength (Å) | 0.9792 | 0.9792 | |
| Space group | P3221 | P3221 | |
| Unit-cell parameters (Å) | a = b = 203.48, c = 81.76 | a = b = 203.27, c = 82.43 | |
| Resolution (Å) | 1.65 (1.68–1.65) | 2.70 (2.75–2.70) | |
| Number of unique reflections | 230,676 (11,291) | 52,181 (2624) | |
| Completeness (%) | 99.6 (98.1) | 96.8 (97.0) | |
| Redundancy | 3.1 (3.6) | 4.6 (3.7) | |
| Mean I/σ (I) | 25.6 (2.1) | 12.9 (2.5) | |
| Molecules in asymmetric unit | 6 | 6 | |
| Rmerge (%) | 7.40 (55.0) | 13.4 (46.4) | |
| Refinement | |||
| Resolution range (Å) | 43.2–1.70 | 47.8–2.7 | |
| Rwork/Rfree (%) | 17.5/20.1 | 19.0/25.7 | |
| Number of residues/protein atoms | 1547/11,830 | 1548/11,850 | |
| Number of water atoms | 1442 | 234 | |
| Average B factor | |||
| Main chain | 19.2 | 42.1 | |
| Side chain | 22.9 | 45.6 | |
| Ligand | — | 79.8 | |
| Waters | 31.6 | 31.3 | |
| Ramachandran plot (%) | |||
| Most favored | 97.1 | 94.22 | |
| Allowed | 2.90 | 5.65 | |
| Outlier | 0.00 | 0.13 | |
| RMSD | |||
| Bond lengths (Å) | 0.007 | 0.008 | |
| Bond angles (°) | 1.177 | 1.224 | |
The values in the parenthesis indicate the corresponding parameters for the highest resolution shell.
Structural comparison of ClFabI with other ENR structures
The topology of the ClFabI monomer is similar to those of other ENRs from different species, and the results of structure-based Dali search27 showed hundreds of homologous structures. In the current study, homologous structures from the top nine species are selected to be compared with ClFabI. These species are Aquifex aeolicus VF5 (AaFabI, PDB code 2P91), Bartonella henselae (BhFabI, PDB code 4EIT), E. coli (EcFabI, PDB code 2FHS or 1DFI,6,28 B. melitensis (BmFabI, PDB code 3GRK), S. aureus29 (SaFabI, PDB code 4ALN), Anaplasma phagocytophilum (ApFabI, PDB code 3K2E or 3K31), Thermus thermophilus30 (TtFabI, PDB code 2WYU or 2WYV), B. cereus11 (BaFabI, PDB code 3OJE), and F. tularensis13 (FtFabI, PDB code 2JJY). ClFabI shows 40.1 to 54.1% amino acid sequence identities to the homologous structures (Fig. 3). Among these structures, 2P91, 4EIT, 2FHS, 3GRK, 4ALN, 3K2E, 2WYU, and 3OJE are in apoform, whereas 1DFI, 3K31, 2WYV, and 2JJY are in binary complex with NAD. When superposing apo-ClFabI to these apo-structures, the RMSD varies from 0.67 to 1.45 Å, which indicates that these structures are similar to one another, with the presence of some fragments at various locations [Fig. 4(a)]. One example of these fragments is the substrate-binding loop (V192–S214 in ClFabI). The substrate-binding loops are almost disordered without interpretable electron density in apo-FabIs, for example, among these apo-structures mentioned above, only the loops in 2FHS, 3K2E, and 2WYU are ordered in one or two monomers. However, a prominent electron density in apo-ClFabI indicates it is ordered. To our knowledge, this is the first crystal structure of apo-FabI with ordered substrate-binding loop in all monomers [Fig. 4(b)]. When binary complexes are structurally compared, the RMSD changes from 0.93 to 1.20 Å, and the difference mainly comes from the location of the substrate-binding loop. Among these four binary complexes, only the loop in 3K31 is ordered; however, the nicotinamide part of NAD is lost in this structure because of poor electron density. In addition, the average temperature factor (B-factor) of NAD is almost three times than that of the protein, suggesting that NAD is bound very loosely and may not represent the binding mechanism efficiently. Therefore, the binary complexes from Brassica napus31 (PDB code 1ENO) and M. tuberculos19 (PDB code 1ENY) are chosen to be compared with ClFabI–NAD. ClFabI–NAD, 1ENO, and 1ENY stand for the typical conformations (open, closed, and a state between them) of the substrate-binding loops when only the cofactor (NAD or NADP) is bounded in the crystal structures of ENR [Fig. 4(c)]. Although the loop occupies various positions in different species in binary complexes, it only has a closed conformation in the FabI–cofactor–inhibitor ternary complex and has lower average B-factor than in binary complexes, for example, the average B-factors of it in Thermus thermophilus FabI (TtFabI, PDB code 2WYW), E. coli FabI (EcFabI, PDB code 1C14), and Helicobacter pylori FabI (HpFabI, PDB code 2PD3) are 27.8, 43.2, and 48.6, respectively. Therefore, the loop is flexible until it is hold by hydrogen bonds with a cofactor and inhibitor or substrate, and its flexibility can be used to design new antimicrobial regent.
Figure 3.

Sequence alignment of FabI from Candidatus Liberibacter asiaticus (ClFabI), Aquifex aeolicus VF5 (AaFabI), Bartonella henselae (BhFabI), Escherichia coli (EcFabI), Brucella melitensis (BmFabI), Staphylococcus aureus (SaFabI), Anaplasma phagocytophilum (ApFabI), Thermus thermophilus (TtFabI), Bacillus cereus (BaFabI), and Francisella tularensis (FtFabI). The residues in bold red are strictly conserved, whereas the residues in light red are relatively conserved. Secondary structures of ClFabI in this study are depicted above the sequence.
Figure 4.
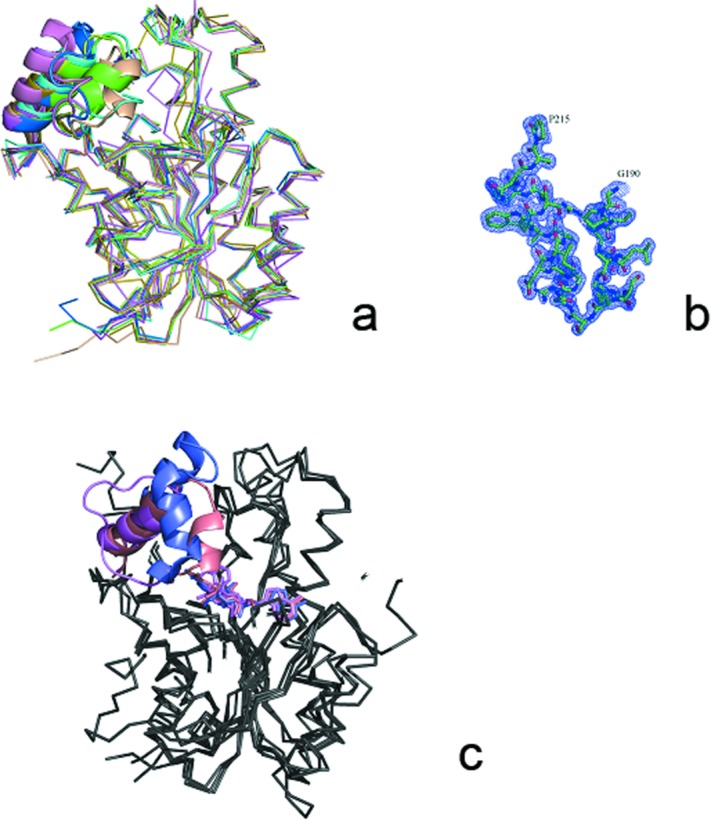
(a) Superposition of the apoform of FabI from Candidatus Liberibacter asiaticus (green), Aquifex aeolicus VF5 (cyan, 2P91), Bartonella henselae (magenta, 4EIT), Escherichia coli (pink, 2FHS), Brucella melitensis (limon, 3GRK), Staphylococcus aureus (olive, 4ALN), Anaplasma phagocytophilum (marine, 3K2E), Thermus thermophilus (wheat, 2WYU), and Bacillus cereus (violet, 3OJE). The substrate-binding loops that produced the most structural variations are indicated by an ellipse. (b) Electron density map of the substrate-binding loop of ClFabI contoured at 1.0 sigma. (c) Different conformations of the substrate-binding loops in the FabI binary complex. The loops are shown in different colors: salmon, ClFabI; violet, BnFabI (1ENO); and slate, MtFabI (1ENY). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Conversely, the average B-factor of NAD in the binary complexes was higher than that of the protein. Moreover, NAD did not occupy all monomers in the binary complexes. For instance, only one NAD molecule was bonded in the tetramer structure of 2JJY and only four NAD molecules in the current hexamer structure. However, in the ternary complex (FabI–NAD–triclosan), both NAD and triclosan had similar B-factor as the protein, and they occupied all monomers. Thus, the binary complexes were not stable until triclosan or another inhibitor was bound to FabI. The ternary complex structures (FabI–NAD–triclosan) are from PDB.9,14 In fact, in other ENR ternary complexes which include cofactor (NAD or NADP) and various inhibitors, the B-factors of cofactors and inhibitors are similar to their protein environment, indicating that the ternary structures are more stable than the binary structures.14,16,32,33 Like other ENRs, ClFabI has the highly conserved signature sequence motif Tyr-(Xaa)6-Lys. In addition, another motif dyad Gly14-(Xaa)5-Ser20 and Val66 and Thr194 in ClFabI are also highly conserved and contained in the hydrogen bond network of FabIs10,11 (Fig. 3). Therefore, the signature sequence can be considered to be Gly-(Xaa)5-Ser-(Xaa)n-Val-Tyr-(Xaa)6-Lys-(Xaa)n-Thr.
Inhibition of ClFabI enzyme activity in Ca. L. asiaticus by INH
The inhibitory mechanism of INH was investigated using the enzyme kinetic method. Lineweaver–Burk plot analysis indicated that INH acts as a competitive inhibitor with respect to NADH and as an uncompetitive inhibitor with respect to crotonoyl-CoA [Fig. 5(a,b)]. At INH concentrations of 0, 2, 4, and 8 μM, the Km values for NADH were 20.5, 85.2, 167.8, and 299.0 μM, respectively, whereas those for crotonoyl-CoA were 17.5, 15.9, 10.9, and 8.8 μM, respectively. The Ki values of INH were 0.11 and 6.8 μM with respect to NADH and crotonoyl-CoA, respectively [Fig. 5(c,d)]. The Ki value for the inhibitor INH reflects the strength of the interaction between the enzyme ClFabI and INH. A small Ki value indicates relative tight binding of INH to ClFabI at different NADH concentrations, whereas a large Ki value indicates weak binding between the enzyme and substrate [ES] complex and INH at varied crotonoyl-CoA concentrations.
Figure 5.
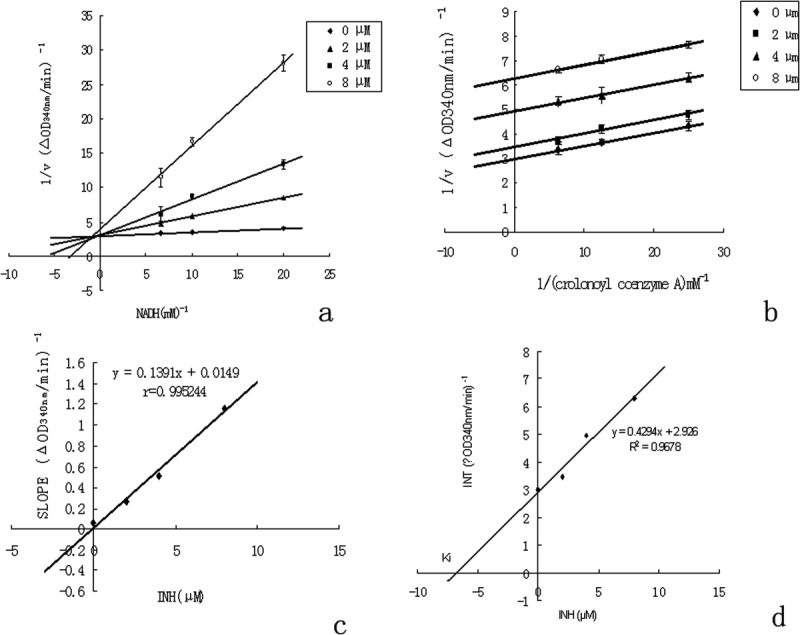
Inhibitory mechanism of ClFabI by INH. (a) Lineweaver–Burk plot showing competitive inhibition of FabI binding to NADH by INH. The enzyme was incubated in the presence of different concentrations of INH and NADH at a fixed concentration of crotonoyl-CoA (40 μM). The concentrations of INH were 0 μM (diamond), 2 μM (triangle), 4 μM (square), and 8 μM (circle). (b) Lineweaver–Burk plot showing uncompetitive inhibition of FabI binding to crotonoyl-CoA by INH. The enzyme was incubated in the presence of different concentrations of INH and crotonoyl-CoA at a fixed concentration of NADH (100 μM). The concentrations of INH were 0 μM (diamond), 2 μM (triangle), 4 μM (square), and 8 μM (circle). (c) The slope values of the lines from Figure 5(a) are plotted versus INH concentration, the secondary plot of Ki. An inhibitor constant value of 0.11 μM for NADH was determined. (d) The intercept values of the lines from Figure 5(b) are plotted versus INH concentration. A Ki value of 6.8 μM for crotonoyl-CoA was determined.
The inhibitory activity percentages corresponding to NADH were 29.7, 45.6, and 70.6%, the correlation coefficient corresponding to the inhibitor concentration was 0.9765, and the inhibitory activity percentages corresponding to crotonoyl-CoA were 10, 36.7, and 48.9%. After adding different concentrations (2, 4, and 8 μM) of the inhibitor INH, the correlation coefficient corresponding to the inhibitor concentration was 0.9581, and the inhibitory activity percentage increased with increasing inhibitor concentration (Supporting Information Table S3).
In this research, INH acted as a competitive inhibitor with respect to NADH and as an uncompetitive inhibitor with respect to crotonoyl-CoA. In competitive inhibition, Vmax was unchanged according to the statistical analysis with t-test, Km increased with increasing concentration of the inhibitor INH, and the ratio of Km/Vmax did not change. Lineweaver−Burk plot with straight lines intersected the longitudinal axis corresponding to the uninhibited and inhibited reactions. In uncompetitive inhibition, Vmax and Km decreased with increasing concentration of the inhibitor INH, and the ratio of Km/Vmax did not change. These findings resulted in a Lineweaver−Burk plot with two parallel lines corresponding to the uninhibited and inhibited reactions. This current research obtained similar results with a previous report in S. pneumonia.17
The dissociation constant was compared with previous data and this experiment. INH–NAD adduct has been shown to bind to InhA with an equilibrium dissociation constant value of 0.4 nM in Mycobacterium tuberculosis.34 INH–NAD adduct has been shown to be a slow, tight-binding competitive inhibitor, which inhibits the InhA activity with an overall inhibition constant Ki value of 0.75 (± 0.08) nM.35 Our research indicated that purified ClFabI has activity in vivo, and when ClfabI, INH, NAD+, NADH, and substrate crotonoyl-CoA were present, INH acts as a competitive inhibitor with respect to NADH; the dissociation constant Ki of INH and ClFabI is 0.11 μM, it is about 140-to 260-fold higher than that of INH–NAD adduct and InhA in Mycobacterium tuberculosis.
Ki value indicated the weak binding of INH to ClFabI at different NADH concentrations when compared with that of INH–NAD adduct to InhA. The results presume that INH without oxidation treat could bind with ClFabI at weak range.
It is possible to explain the case with structure analysis. INH adduct [(2E)-N-[(1,2-dimethyl-1H-indol-3-yl)methyl]-N-methyl-3-(7-oxo-5,6,7,8-tetrahydro-1,8-naphthyridin-3-yl)prop-2-enamide (IMJ)] has been found in the ternary crystal structures of FabI from Bacillus subtilis10 (PDB code 3OIG) and Bacillus cereus11 (PDB code 3OJF). When the ClFabI–NAD binary structure was superimposed to these ternary structures, the RMSD values were 0.68 and 0.67 Å, respectively, and the cofactor positions were nearly identical. Therefore, it is reasonable to speculate that IMJ should lie at a similar position in the ClFabI ternary structure (Fig. 4). The residues that contact INH are A98, Y148, Y158, M161, I202, and F205 in BsFabI and F96, A97, Y147, V154, Y157, P192, S197, F204, and I207 in BcFabI. Based on the sequence alignment, the possible residues in ClFabI that interact with INH are F95, S96, Y146, V153, Y156, M159, P191, A196, I200, G203, and I206. Y146 and Y156 are highly conserved and are the residues most likely to interact with INH. Serine or alanine occupies S96 in ClFabI and corresponding positions in other ENRs, and this residue makes hydrogen bonds with INH through backbone amide and/or carbonyl oxygen. Therefore, S96 is also the residue that most likely makes contacts with INH. Glycine occupies the G203 position in ClFabI; however, phenylalanine is in the corresponding position in other ENRs and makes hydrophobic interactions with INH, and thus, G203 is the least likely residue to contact INH. The remaining residues contain F95, Y146, V153, Y156, M159, P191, A196, I200, I206, Y146, and Y156 likely make hydrophobic interactions with INH.
In summary, the crystal study of CIFabI has special significance for screening of antibacterial agents against HLB in citrus with structure-based strategies. We determined the apo-ClFabI and ClFabI–NAD binary crystal structures, which are both six monomers in one asymmetric unit (ASU) in crystal. They both exist as dimer in solution, and all the disordered residues in apo-FabI crystals from other species are ordered in our apo-structure. Based on the sequence alignment and the residues involved in hydrogen bond network of FabIs, the signature sequence motif is expanded from Tyr-(Xaa)6-Lys to Gly-(Xaa)5-Ser-(Xaa)n-Val-Tyr-(Xaa)6-Lys-(Xaa)n-Thr. The resulting structure information regarding the active site pocket and details of the NAD+-binding mode could be used as bases for the rational design of more effective FabI inhibitors against Candidatus Liberibacter asiaticus.
We suggest that the crystal structure of complexes of other new inhibitors and ClfabI in Ca. L. asiaticus should be performed in the future. At present, the kinetics analysis is primary and imperfection, and thus, presteady-state kinetics analysis23 and progress cure analysis of the inhibition of FabI by inhibitor36 would be the better method, which is used for exploring the binding way of FabI–NAD and FabI–NADH and explaining apparent inhibition constant and dissociation constant for the slow, tight-binding inhibitor.
Materials and Methods
Cloning, expression, and purification
The ClFabI gene was amplified from the genomic DNA (NC_012985) of Ca. L. asiaticus and was inserted into the pET28a-SOMU1 expression vector (constructed based on pET28a by our laboratory). The construct was transformed into BL21 (DE3) for expression. Cells were grown in Luria−Bertani medium supplemented with 50 mg mL−1 kanamycin at 37°C until an OD600 value of 0.6–0.8 was reached. Isopropyl β-d-thiogalactopyranoside was added to a final concentration of 0.4 mM to induce expression at 16°C for 20 h. The cell pellet was harvested, resuspended in ice-cold lysis buffer A (25 mM Bis−Tris, pH 6.95, 400 mM NaCl, 2 mM 2-mercaptoethanol), and then subjected to French press (JN3000 PLUS). The supernatant was collected after centrifugation at 16,000 rpm (Sorvall GSA rotor) for 55 min at 4°C.
The supernatant was loaded onto a Ni2+ Hi-Trap chelating column (Amersham Biosciences), washed with 70 mM imidazole in buffer B (25 mM Bis-tris, pH 6.95, 1M NaCl), and then eluted with 500 mM imidazole in buffer B. The SUMO tag was enzymatically cleaved overnight at 4°C by SUMO protease (ULP1), and the target protein was isolated by a second Ni affinity chromatography step. After concentration, the protein sample was further purified using a Superdex™ 200 (GE Healthcare) column equilibrated with buffer C (25 mM Bis-Tris, pH 6.95, 300 mM NaCl) at 4°C in an AKTA system (Amersham Bioscience). The molecular weight of purified CLFabI protein in solution was calibrated according to a 120-mL Superdex™ 200 column in AKTA.
Crystallization and X-ray data collection
Crystallization of ClFabI was screened by the sitting-drop vapor-diffusion method at 4 and 20°C. The protein was concentrated to 15 mg mL−1 in 25 mM Bis-Tris, pH 6.95, 300 mM NaCl. Crystallization conditions were initially screened using commercially available kits from Emerald BioSystems (Bainbridge Island, WA) and Hampton Research (Aliso Viejo, CA). Microcrystals were obtained after 3–5 days using Emerald BioSystems Cryo™ I. Crystals suitable for data collection were obtained in conditions containing 0.1M CHES, pH 9.5, 200 mM NaCl, and 50% PEG 400 (v/v) after optimization. The same pathway mentioned above was adopted in the crystallization of the ClFabI–NAD complex, except that the protein solution contains 4 mM NAD, and the optimum condition containing 35% (v/v) 2-ethoxyethanol and cacodylate (0.1M, pH 6.5).
The apo-ClFabI crystal was flash-cooled directly in liquid nitrogen before mounting for data collection. Diffraction data were collected on the beamline 1W2B at Beijing Synchrotron Radiation Facility or on the beamline BL17U1 at Shanghai Synchrotron Radiation Facility. The total oscillation was 180° with 0.5° per image. The data were processed with HKL-2000.37
Structure determination and refinement
The crystal structure of ClFabI in the apoform (PDB code 4NK4) is determined by molecular replacement using Phaser.33 The crystal structure of short-chain dehydrogenase reductase glucose-ribitol dehydrogenase from Brucella melitensis (PDB code 3GRK) is used as the search model. According to the Matthews coefficient, we estimated six, seven, or eight molecules in the ASU, with solvent constants of 54.8, 47.3, or 39.7%, respectively. Thus, 3GRK contains eight molecules. The dimer (Chains B and D) is adopted, and the high value of translation function Z-score suggests that ASU contains six molecules. The binary complex structure is determined by molecular replacement using the apoform structure as the search model. Manual rebuilding is performed by operating Coot,38 and refinement is carried out by applying Phenix Refine.39 Final models are validated by adopting MolProbity,40 and all structure figures are generated using Pymol (http://www.pymol.org/).
ClFabI inhibition assays
The catalytic activity of ClFabI that reduced enoyl–ACP coupled with the concomitant oxidation of NADH was assayed based on a modified protocol.8,41 INH, NADH, crotonoyl-CoA, and dimethyl sulfoxide (DMSO) were purchased from Sigma. All other chemicals used were of analytical grade. All experiments were performed on a Spectra Max M2 Molecular Devices at 28°C. During the course of the analysis of ClFabI, it is possible for an increase in the concentration of NAD+ due to the oxidation of NADH, which is the coenzyme of ClFabI. Therefore, 50 µM NAD+ was added in all the reactions to keep the concentration of NAD+ constant and to keep it close to its steady state. Inhibition of INH against ClLFabI was assayed using crotonoyl-CoA as a substrate and monitored by decline in absorbance at 340 nm (eM 340 = 6220 M−1 cm−1). Two sets of experiments were performed to evaluate the inhibitory effect of INH on ClFabI. In the first set, the concentration of crotonoyl-CoA was held constant, whereas the concentrations of NADH and the inhibitor were varied systematically. Assays were performed for 6 min in 40 μM crotonoyl-CoA, 50 μM NAD, and 20 nM enzyme in buffer D (20 mM TrisHCl, 150 mM NaCl buffer, pH 7.5, and 1% DMSO) in a total volume of 100 µL with NADH in a concentration range of 50–150 μM and in the absence or presence of INH. After 5 h of incubation at 4°C, the reaction was started by adding crotonoyl-CoA. In the second set of experiments, the concentration of NADH was held constant, whereas the concentrations of crotonoyl-CoA and the inhibitor were varied systematically. Assays were performed for 6 min in 50 μM NAD, 100 μM NADH, and 20 nM enzyme in buffer D with crotonoyl-CoA in a concentration range of 40–160 μM in the absence or presence of the inhibitor. The Michaelis constants (Km) for NADH and crotonoyl-CoA were determined using Lineweaver–Burk plots, with initial velocities obtained by altering the concentration of one substrate while maintaining the other substrate at a fixed concentration in the absence of the inhibitor. The dissociation constant Km was obtained from the Lineweaver–Burk double-reciprocal plots and Ki in the secondary plots is inhibitor constant, it is also called as dissociation constant, and it comes from the Dixon construct method. Data are presented as means and standard errors of three replicates.
The inhibitory activity percentage (I%) was calculated using the following formula
where νi is the initial velocity in the presence of the inhibitor and ν0 is the initial velocity in the untreated control.
Glossary
- ACP
acyl carrier protein
- ASU
asymmetric unit
- CoA
coenzyme A
- DMSO
dimethyl sulfoxide
- FAS
fatty acid synthesis
- HLB
Huanglongbing
- INH
isoniazid
- NAD+
nicotinamide adenine dinucleotide
- NADH
b-nicotinamide adenine dinucleotide
Additional Supporting Information may be found in the online version of this article.
References
- 1.Islam MS, Glynn JM, Bai Y, Duan YP, Coletta-Filho HD, G Kuruba, EL Civerolo, H Lin. Multilocus microsatellite analysis of “Candidatus Liberibacter asiaticus” associated with citrus Huanglongbing worldwide. BMC Microbiology. 2012;12:39. doi: 10.1186/1471-2180-12-39. http://www.biomedcentral.com/1471-2180/12/39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coletta-Filho HD, Targon MLPN, Takita MA, Negri De JD, Jr, Pompeu JMAM. First report of the causal agent of Huanglongbing “Candidatus Liberibacter asiaticus” in Brazil. Plant Dis. 2004;88:1382. doi: 10.1094/PDIS.2004.88.12.1382C. [DOI] [PubMed] [Google Scholar]
- 3.Halbert SE, L Manjunath. Asian citrus psyllid (Stenorrhynca: Psyllidae) and greening disease of citrus: A literature review and assessment of risk in Florida. Florida Entomologist. 2004;87:330–353. [Google Scholar]
- 4.NAPPO. 2009. Detection of Huanglongbing ‘Candidatus Liberibacter asiaticus’ in the municipality of Tizimin, Yucatan, Mexico. Phytosanitary alert system. Official Pest Reports. Available from: http://www.pestalert.org/oprDetail.cfm?oprID=384. Accessed August 07 2009.
- 5.Massengo-Tiassé RP, Cronan JE. Diversity in enoyl–acyl carrier protein reductases. Cell Mol Life Sci. 2009;66:1507–1517. doi: 10.1007/s00018-009-8704-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rafi S, Novichenok P, Kolappan S, Zhang XJ, Stratton CF, Rawat R, Kisker C, Simmerling C, Tonge PJ. Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J Biol Chem. 2006;281:39285–39293. doi: 10.1074/jbc.M608758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heath RJ, Rock CO. Enoyl–acyl carrier protein reductase (FabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 8.Yao J, Zhang Q, Min J, He J, Yu Z. Novel enoyl–ACP reductase (FabI) potential inhibitors of Escherichia coli from Chinese medicine monomers. Bioorgan Med Chem Lett. 2010;20:56–59. doi: 10.1016/j.bmcl.2009.11.042. [DOI] [PubMed] [Google Scholar]
- 9.Priyadarshi A, Kim EE, Hwang KY. Structural insights into Staphylococcus aureus enoyl–ACP reductase (FabI), in complex with NADP and triclosan. Proteins. 2009;78:480–486. doi: 10.1002/prot.22581. [DOI] [PubMed] [Google Scholar]
- 10.Kim KH, Ha BH, Kim SJ, Hong SK, Hwang KY, Kim EE. Crystal structures of enoyl–ACP reductases I (FabI) and III (FabL) from B. subtilis. J Mol Biol. 2011;406:403–415. doi: 10.1016/j.jmb.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Kim SJ, Ha BH, Kim KH, Hong SK, Shin KJ, Suh SW, Kim EE. Dimeric and tetrameric forms of enoyl–acyl carrier protein reductase from Bacillus cereus. Biochem Biophys Res Commun. 2010;400:517–522. doi: 10.1016/j.bbrc.2010.08.083. [DOI] [PubMed] [Google Scholar]
- 12.Stephen PM, Sean TP, Liqun ZH, Michael JK, Craig WR, Wernimont S, McLeodc R, Ricea DW. Expression, purification and crystallization of Helicobacter pyloril-asparaginase. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:604–606. [Google Scholar]
- 13.Lu H, England K, Am Ende C, Truglio JJ, Luckner SR, Marlenee N, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem Biol. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maity K, Bhargav SP, Sankaran B, Surolia N, Surolia A, Suguna K. X-ray crystallographic analysis of the complexes of enoyl acyl carrier protein reductase of Plasmodium falciparum with triclosan variants to elucidate the importance of different functional groups in enzyme inhibition. IUBMB Life. 2010;62:467–476. doi: 10.1002/iub.327. [DOI] [PubMed] [Google Scholar]
- 15.Luckner SR, Liu N, Am Ende CW, Tonge PJ, Kisker C. A slow, tight binding inhibitor of InhA, the enoyl–acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem. 2010;285:14330–14337. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapoor M, Dar MJ, Surolia A, Surolia N. Kinetic determinants of the interaction of enoyl–ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem Biophys Res Commun. 2001;289:832–837. doi: 10.1006/bbrc.2001.6061. [DOI] [PubMed] [Google Scholar]
- 17.Saito J, Yamada M, Watanabe T, Iida M, Kitagawa H, Takahata S, Ozawa T, Takeuchi Y, Ohsawa F. Crystal structure of enoyl–acyl carrier protein reductase (FabK) from Streptococcus pneumonia reveals the binding mode of an inhibitor. Protein Sci. 2008;17:691–699. doi: 10.1110/ps.073288808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Jr, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 19.Dessen A, Quemard A, Blanchard JS, Jacobs WR, Jr, Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 20.Dias MV, Vasconcelos IB, Prado AM, Fadel V, Basso LA, Azevedo de WF, Jr, Santos DS. Crystallographic studies on the binding of isonicotinyl–NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl–ACP (CoA) reductase from Mycobacterium tuberculosis. J Struct Biol. 2007;159:369–380. doi: 10.1016/j.jsb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 21.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, Lisle de G, Jacobs WR., Jr InhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 22.Johnsson K, Schultz PG. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J Am Chem Soc. 1994;116:7425–7426. [Google Scholar]
- 23.Oliveira JS, Pereira JH, Canduri F, Rodrigues NC, Souza de ON, Azevedo de WF, Jr, Basso LA, Santos DS. Crystallographic and pre-steady-state kinetics studies on binding of NADH to wild-type and isoniazid-resistant enoyl–ACP (CoA) reductase enzymes from Mycobacterium tuberculosis. J Mol Biol. 2006;359:646–666. doi: 10.1016/j.jmb.2006.03.055. [DOI] [PubMed] [Google Scholar]
- 24.Lee HH, Moon J, Suh SW. Crystal structure of the Helicobacter pylori enoyl–acyl carrier protein reductase in complex with hydroxydiphenyl ether compounds, triclosan and diclosan. Proteins. 2007;69:691–694. doi: 10.1002/prot.21586. [DOI] [PubMed] [Google Scholar]
- 25.Kumar HSN, Parumasivam T, Ibrahim P, Asmawi MZ, Sadikun A. Synthesis of hydrophobic N-acylated isonicotinic acid hydrazide, derivatives as potential enoyl–acyl carrier protein reductase (InhA) inhibitors. Med Chem Res. DOI: 10.1007/s00044-013-0715-0 (in press) [Google Scholar]
- 26.Duan Y, Zhou L, Hall DG, Li W, Doddapaneni H, Lin H, Liu L, Vahling CM, Gabriel DW, Williams KP, Dickerman A, Sun Y, Gottwald T. Complete genome sequence of citrus Huanglongbing bacterium ‘Candidatus Liberibacter asiaticus’ obtained through metagenomics. Mol Plant Microbe Interact. 2009;22:1011–1020. doi: 10.1094/MPMI-22-8-1011. [DOI] [PubMed] [Google Scholar]
- 27.Holm L, Rosenström PD. Server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–2110. doi: 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]
- 29.Schiebel J, Chang A, Lu H, Baxter MV, Tonge PJ, Kisker C. Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure. 2012;20:802–813. doi: 10.1016/j.str.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otero JM, Noël AJ, Guardado-Calvo P, Llamas-Saiz AL, Wende W, Schierling B, Pingoud A, Raaij van MJ. Structural biology and crystallization communications. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:1139–1148. doi: 10.1107/S1744309112033982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rafferty JB, Simon JW, Baldock C, Artymiuk PJ, Baker PJ, Stuitje AR, Slabas AR, Rice DW. The enoyl–[acyl-carrier-protein] reductase (FabI) of Escherichia coli, which catalyzes a key regulatory step in fatty acid biosynthesis, accepts NADH and NADPH as cofactors and is inhibited by palmitoyl-CoA. Structure. 1995;3:927–938. [Google Scholar]
- 32.Chang A, Schiebel J, Yu W, Bommineni GR, Pan P, Baxter MV, Khanna A, Sotriffer CA, Kisker C, Tonge PJ. Rational optimization of drug-target residence time: insights from inhibitor binding to the Staphylococcus aureus FabI enzyme–product complex. Biochemistry. 2013;52:4217–4228. doi: 10.1021/bi400413c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lei B, Wei CJ, Tu SC. Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of InhA inhibitor. J Biol Chem. 2000;275:2520–2526. doi: 10.1074/jbc.275.4.2520. [DOI] [PubMed] [Google Scholar]
- 35.Rawat R, Whitty A, Tonge PJ. The isoniazid–NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu H, England K, Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee N, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem Biol. 2009;4:221–231. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergler H, Wallner P, Ebeling A, Leitinger B, Fuchsbichler S, Aschauer H, Kollenz G, Högenauer G, Turnowsky F. Protein EnvM is the NADH-dependent enoyl–ACP reductase (FabI) of Escherichia coli. J Biol Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.