

Abstract
Myeloproliferative neoplasms are uncommon disorders in children, for which we have limited understanding of the pathogenesis and optimal management. JAK2 and MPL mutations, while common drivers of myeloproliferative neoplasms in adult patients, are not clearly linked to pediatric disease. Management and clinical outcomes in adults have been well delineated with defined recommendations for risk stratification and treatment. This is not the case for pediatric patients, for whom there is neither a standard approach to workup nor any consensus regarding management. This review will discuss thrombocytosis in children, including causes of thrombocytosis in children, the limited knowledge we have regarding pediatric primary thrombocytosis, and our thoughts on potential risk stratification and management, and future questions to be answered by laboratory research and collaborative clinical study.
Introduction
Thrombocytosis is now a common finding on the complete blood count (CBC) of children. It is often transient, and occurs secondary to various underlying medical, usually inflammatory, disorders because an increase in the platelet count is one aspect of the acute phase reaction. This occurs more frequently in younger children, either because of the immaturity of their innate and/or adaptive immunity, or because they have more frequent infections.
Primary thrombocytosis is substantially less common in children than it is in adults. Essential thrombocytosis or thrombocythemia (ET) in adults is well known as a member of the family of myeloproliferative neoplasms (MPN), also including polycythemia vera (PV) and primary myelofibrosis (PMF). These disorders share several features, such as splenomegaly, growth factor independent hematopoiesis, and the potential to transform to acute myeloid leukemia (AML). Since identification of the JAK2 V617F mutation in 2005, and subsequently of mutations in relevant JAK-STAT pathway genes, these MPN are now relatively well-understood entities in adults. Hereditary thrombocytosis is also a defined entity, much like ET, reported in a number of families, with causative mutations identified in the thrombopoietin (TPO) gene, the TPO receptor gene cMPL (MPL), and most recently in JAK2 (see further discussion of molecular findings).
The same cannot be said for primary thrombocytosis in children. The pathogenesis of the disease in the pediatric population is less clear, with a far smaller percentage of children than adults having mutations in JAK2 or MPL. This review will discuss the causes of thrombocytosis in children, the pathogenesis of primary thrombocytosis, and our current understanding of this entity in the pediatric population, and will consider important questions that still remain unanswered.
Thrombocytosis defined
Thrombocytosis is generally considered a platelet count more than 450×109/L. One definition considers a count from 450–700×109/L as mild thrombocytosis, 700–900×109/L as moderate, and more than 900×109/L as severe.1 Counts over 1000×109/L are considered extreme thrombocytosis.1
Thrombopoietin and its role in thrombocytosis
Megakaryopoiesis involves interplay between various growth factors and cytokines, such as interleukin-3 (IL-3) and stem cell factor (SCF), with megakaryocyte progenitors. By far the most essential is thrombopoietin (TPO) which is produced predominantly in the liver, but is also generated within marrow stroma and the kidney.2,3 TPO is required both for stem cell differentiation and for all stages of megakaryocyte maturation.4,5 TPO also interacts synergistically with a number of other growth factors, such as IL-11 and erythropoietin, to enhance megakaryocyte colony growth.6
The receptor for TPO was identified as the protein MPL, produced by the cMPL gene. cMPL was recognized as a growth factor receptor by virtue of its structure, and identification of its ligand is how TPO was discovered.7,8 The most direct evidence for the critical role of cMPL, and by extension thrombopoietin, in thrombopoiesis and stem cell differentiation, is that absence of cMPL expression has been shown to be responsible for congenital amegakaryocytic thrombocytopenia. The resulting TPO insensitivity often results in aplastic anemia, as TPO promotes progenitor survival.9 Conversely, excess production of TPO (beyond what is needed based on the platelet count) has been found in familial thrombocytosis secondary to a gain of function mutation.10,11 In addition to platelets, endothelial cells have been shown to display c-MPL receptors, but these are not thought to participate in TPO regulation.12
In steady state, there is constitutive TPO production and thus the TPO level depends upon its rate of clearance and not generally on control at the transcriptional level.13 This rate depends upon TPO binding to its receptor, which in turn depends upon how many TPO-R bearing cells are accessible, as well as how many receptors they express.14 Low platelet counts will lead to decreased TPO clearance (less receptors in the circulation) and therefore increased levels of TPO; the reverse occurs with higher platelet counts, i.e. thrombocytosis. There are variations to this basic principal, for example in the setting of idiopathic thrombocytopenia (thrombocytopenia due to destruction). In these cases, the number of megakaryocytes and megakaryocyte mass may also influence TPO regulation and circulating levels.15,16
Causes of reactive thrombocytosis in children
Secondary, or reactive, thrombocytosis is a common occurrence in children. It has been reported to occur in 6–15% of hospitalized children, with variations based on age. Most of these children had thrombocytosis that could be characterized as mild, but others transiently reached levels over 900,000.17–20
Causes of secondary thrombocytosis are many and varied (Table 1). Infection is the most common, including viral and bacterial pathogens, and both acute and chronic infections. Especially in children under one year of age, any infection seems capable of triggering a high platelet count. Inflammatory diseases, e.g. Kawasaki disease, rheumatoid arthritis, and inflammatory bowel disease, are commonly associated with reactive thrombocytosis, as are hypoxia, trauma, blood loss, and malignancy.19,21,22 Iron deficiency also seems to be a frequent cause of secondary thrombocytosis.20
Table 1.
Reactive (secondary thrombocytosis).

It is believed that underlying mechanisms of secondary thrombocytosis can be explained by upregulation of TPO expression and resultant increased TPO levels. Hepatic TPO mRNA expression is increased with inflammation.23 Interleukin-6 (IL-6) is increased in various inflammatory states, and has been associated with elevation of plasma TPO levels and TPO mRNA expression. TPO levels correlate with C-reactive protein, another inflammatory marker.24 It is appropriate to note that “viral suppression” of thrombopoiesis is also common, and thus in the setting of infection the platelet count may go up or down depending on the relative effects on platelet production and (possibly) on platelet destruction. Furthermore, in the setting of viral suppression resulting in thrombocytopenia, there may be an “overshoot” during recovery with transient thrombocytosis.
There is significant homology between erythropoietin (EPO) and thrombopoietin. In settings of anemia in which there is thrombocytosis, some authors have proposed that increased levels of EPO might bind MPL and lead to a TPO-like effect to increase platelet counts.25 However, other authors have demonstrated lack of competition between EPO and TPO.26,27 The relationship between platelets and iron deficiency is likely to be multifaceted and the causative mechanism of thrombocytosis in iron-deficiency anemia has not been established.
Overall, in children, transient reactive thrombocytosis is much more common than primary thrombocytosis. However, primary thrombocytosis is recognized more commonly than in the past, likely due to the increase in the number of blood counts being drawn on children. Also, following a patient with presumed reactive thrombocytosis to see if the blood count normalizes can lead to the diagnosis of primary thrombocytosis (Figure 1). Thus the asymptomatic child with primary thrombocytosis is likely to be identified earlier.
Figure 1.
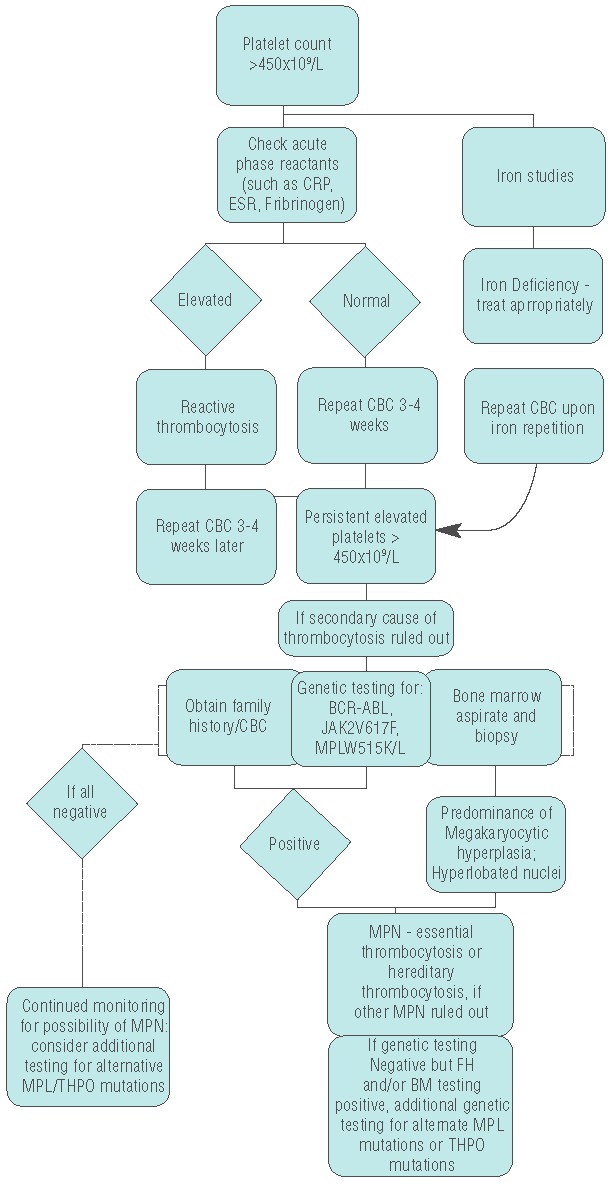
Diagnostic algorithm for persistent elevated platelets. Figure 1 shows our groups’ proposed diagnostic algorithm for approaching patients with elevated platelets. Evaluation for secondary causes, such as iron deficiency or inflammatory or infectious disorders is conducted. Iron deficiency and other underlying causes are treated. If no secondary cause is found, or treatment of the underlying disorder does not remedy the platelet count, further evaluation is done to look for signs of essential thrombocytosis. Bone marrow evaluation is recommended at this point. Also, genetic studies are recommended, and if there is clinical suspicion then BCR-ABL testing should be done. If it is not tested for, or if it is negative, testing for JAK2V617F or MPLW5151K/L is done. Positive JAK or MPL testing, and/or diagnostic bone marrow findings, contribute to the diagnosis of an MPN. Specific criteria for ET (or if needed, PV or PMF) are evaluated to identify the correct MPN. Concurrently, a detailed family history should be obtained to evaluate the possibility of hereditary thrombocytosis. Testing for alternative THPO or MPL mutations is recommended in the setting of presumed hereditary thrombocytosis. Absence of the more common genetic mutation does not rule out diagnosis of an MPN, and if no other cause is identified genetic testing for additional MPL or THPO mutations should be considered and patients should be monitored for the continued possibility of MPN.
MPN and primary thrombocytosis/thrombocythemia
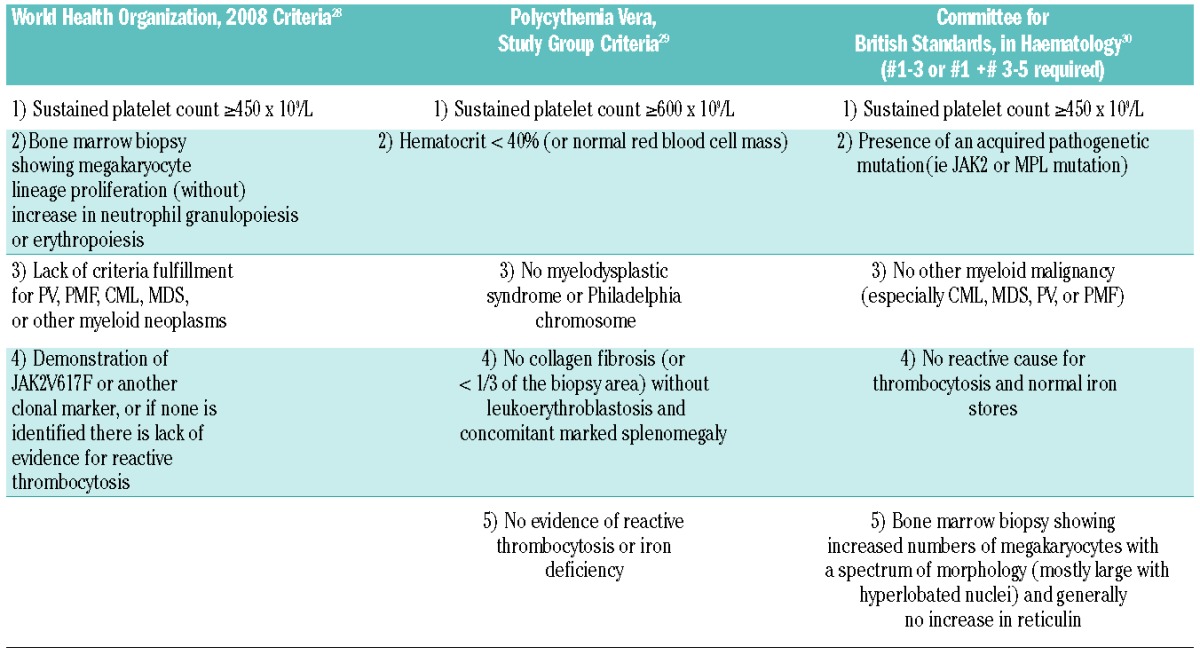
As stated above, ET is a disorder of elevated platelet number that is a type of MPN, i.e. clonal hematologic disorders that stem from specific genetic alterations. One of the classical MPN is BCR-ABL-positive chronic myeloid leukemia (CML), which occasionally presents with thrombocytosis. BCR-ABL-negative MPN include polycythemia vera (PV), primary myelofibrosis (PMF), and ET. A number of different diagnostic criteria are used for ET and these are summarized in Table 2.28–30 ET in adults leads to several constitutional symptoms, the most dangerous of which are thrombosis, myelofibrosis, and leukemic transformation.31
Table 2.
Criteria for diagnosis of essential thrombocytosis.
Hereditary, or familial, thrombocytosis is clinically similar to essential thrombocytosis. Genetically, it has Mendelian inheritance and is polyclonal. It typically only affects platelet lineage. While it was previously thought to be a benign entity, it is now recognized that patients with the hereditary form of primary thrombocytosis may be at risk for thrombosis or bleeding, as well as splenomegaly, bone marrow fibrosis, and leukemic transformation.32–34
Molecular derangements in primary thrombocytosis
In 1951, Dameshek first suggested interrelatedness of the various MPN.35 In 2005, a mutation in the JAK2 gene, a member of the Janus Kinase family of non-receptor tyrosine kinases, was identified in a significant proportion of patients with MPN.36,37 JAK2V617F is a somatically acquired, constitutive activating mutation in the JAK2 pseudokinase domain that turns on the JAK/STAT pathway and promotes continuous signal transduction and proliferation. Activation of downstream mediators such as STAT-5 and Bcl-xL can promote erythroid proliferation and growth in the absence of cytokines (such as erythropoietin, EPO).38 In adults, this mutation is present in approximately 95% of patients with PV, and approximately 50% of patients with ET or PMF.39,40
Additional mutations in JAK2 in exon 12 have been identified in a number of patients with PV and idiopathic erythrocytosis as well.41,42 In JAK2V617F-negative MPN patients, mutations in the cMPL gene were discovered; the MPLW515L/K mutations have been identified in both ET and PMF patients.40,43 These activating mutations also turn on JAK/STAT signaling pathways and can lead to cellular proliferation. Overexpression of the PRV-1 gene (Polycythemia rubra vera-1), which is involved in TPO-induced proliferation and cytokine signaling pathways, was identified in numerous patients with PV and ET.44,45 It is not, however, generally used in the diagnosis of MPN. Most recently, mutations in the CALR gene were identified by two different groups in a significant proportion of JAK2 and MPL wild-type MPN patients.46,47
Much work has been done to understand how particular mutations can produce these clinically distinct, yet related, phenotypes. Allele burden, loss of heterozygosity, and uniparental disomy all may affect the clinical phenotype in MPN patients.48–50 Looking at both a mouse model and human samples, Tiedt et al. showed there was relatively more JAK2V617F mRNA expressed in PV patients and relatively more wild-type JAK2 mRNA expressed in ET patients.50 Findings of low-penetrance inherited alleles and certain single nucleotide polymorphisms (SNPs) may also contribute to the resulting phenotype. Pardanani and colleagues reported that some JAK2 SNPs were more likely to be associated with PV or ET.51 Cytogenetic abnormalities have been linked to MPN phenotypes as well, in both JAK2 mutant and wild-type patients. Both deletion of 20q and 13q, and trisomy 9, have all been identified.52
Hereditary, or familial thrombocytosis has been identified in several families of varying ethnic origin. A number of different mutations in the thrombopoietin gene (THPO) have been identified which affect the upstream open reading frame 7 (uORF 7), which normally has an inhibitory effect on TPO translation. Additional cases with various mutations of the cMPL gene have also been reported; the mutations identified lead either to constitutive MPL activation or secondary increases in TPO due to decreased MPL/TPO binding.10,11,34,53 More recently, a mutation of JAK2, JAK2V617I, has been found in a family with hereditary thrombocytosis.54
Pediatric primary thrombocytosis
There is evidence to clarify the mechanisms of primary thrombocytosis and other MPN in adult patients, as MPN are significantly more common in adults than in pediatric patients. Dame and Sutor reported that ET is over 60 times more common in adults than in children.18 Case series of pediatric ET patients are usually small (Table 3). This rarity, combined with a lower rate of JAK2V617F mutation, means that gaining a clear understanding of the pathogenic mechanisms and diagnosis of ET in children remains a challenge. In addition, reports of sequelae from ET in children are anecdotal and treatment has not been standardized.
Table 3.
Case series of pediatric primary thrombocytosis.
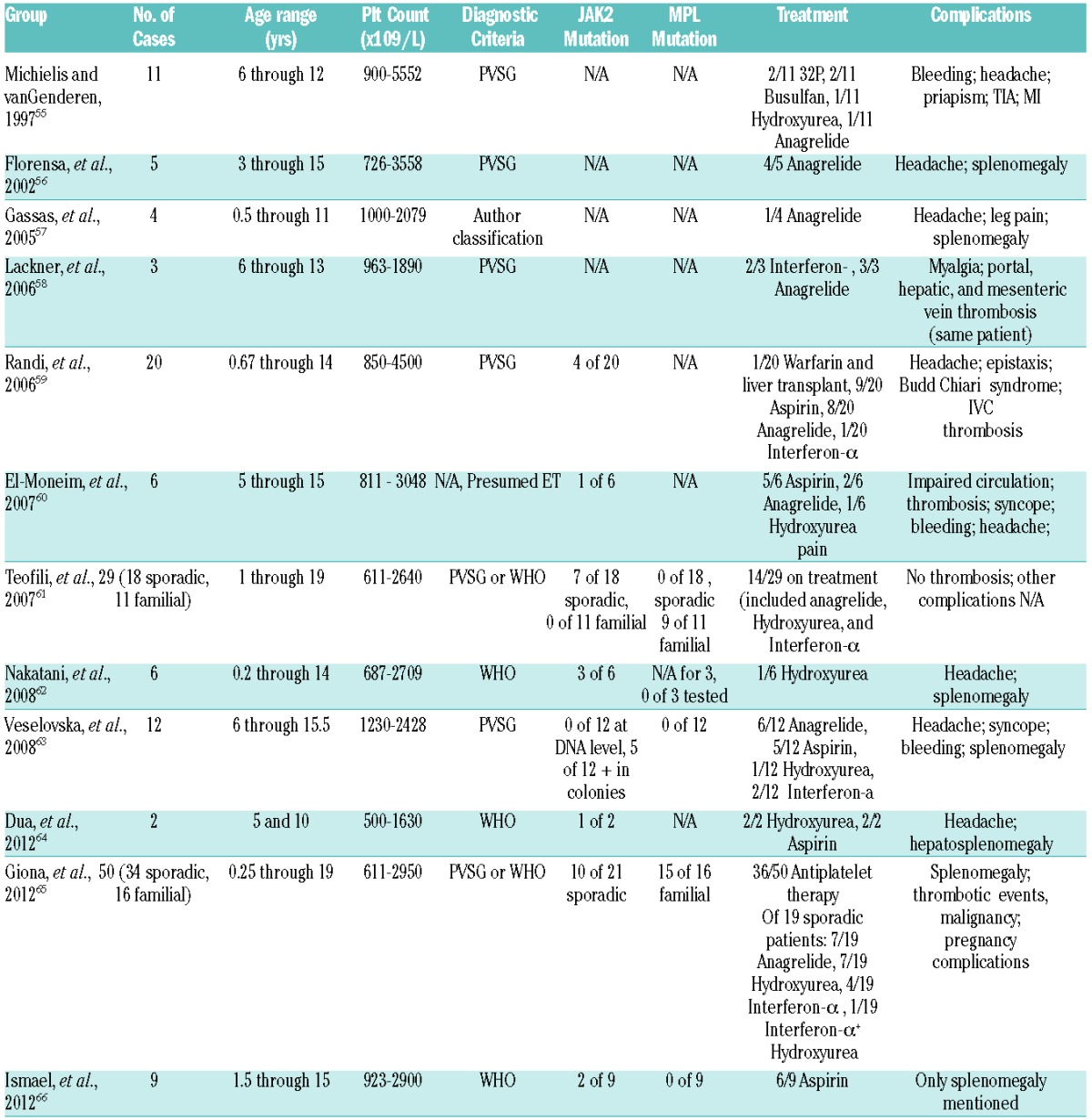
While primary thrombocytosis in children may seem superficially similar to the disease in adults, the symptoms and sequelae of primary thrombocytosis are more benign in the pediatric population. In adults with ET, vascular events, both venous and arterial, were the most common clinical outcome. Girodon and colleagues looked at a number of case series and also at 311 patients of their own; thrombosis occurred in approximately 10–30%.67 A recent series showed similar rates.67,68 In 2012, a retrospective analysis by Giona and colleagues of 34 pediatric ET patients showed that only one experienced a thrombotic event while only 2 out of 16 patients with hereditary thrombocytosis had a thrombotic event.65 All 3 of these patients had concomitant infections at the time.65
There have been additional case reports of pediatric ET patients developing thrombosis. A teenage girl with ET was found to have a portal vein thrombus in the setting of a newly diagnosed urinary tract infection (UTI),69 and a boy developed a cerebral venous sinus thrombus, despite an otherwise negative thrombophilia workup.70 Overall though, the general risk of thrombosis in pediatric ET patients appears to be lower than in the adult population.
The molecular pathogenesis of ET in childhood is not fully aligned with its adult counterpart. Fewer children than adults with ET are JAK2-mutant positive. One small study showed that 3 of 6 pediatric ET patients harbored a JAK2V617F mutation, but all had increased PRV-1 expression.62 A larger study showed a significantly decreased frequency of JAK2V617F mutation in pediatric patients compared to adults. It also showed a significantly lower degree of clonality in the pediatric patients.59 Another study in childhood ET showed rare JAK2V617F+ erythroid colonies with no detectable mutation in peripheral blood cells implying rare clones in these patients, making the role of JAK2V617F unclear.63
In view of the significant number of JAK2V617F-negative children with ET, a recent study out of Japan looked for alternative causative mutations in pediatric patients.66 TET2, ASXL1, IDH1, and IDH2 have recently been identified as mutated in adult MPN patients.71–73 Another gene, CBL, is mutated in many patients with juvenile myelomonocytic leukemia, a form of MPN unique to children.74 Ismael and colleagues performed direct sequencing on a small gene panel, which included JAK2, cMPL, TET2, ASXL1, IDH1, IDH2, and CBL in 13 patients including 9 with ET and 4 with PV.66 Two patients with ET were found to have JAK2V617F, and one was found to have a mutation in ASXL1 that was also found in some controls. The majority of patients did not have a clearly pathogenic mutation identified in the screen.66 A recent report showed a novel cMPL mutation, MPLY252H, in a young child with essential thrombocytosis.75
ET and hereditary forms of thrombocytosis seem to differ in their pathogenesis as well in pediatric patients. Among a group of 29 pediatric primary thrombocytosis patients, those with a hereditary form of thrombocytosis showed a lower percentage of JAK2V617F mutation, and PRV-1 expression.61 Alternatively, these children had more MPL mutations compared to the ET cohort.
Management of thrombocytosis
The bulk of available data on the management of primary thrombocytosis comes from treatment of adult patients with ET. The hallmark of treatment for ET in adults is based on risk stratification for thrombosis. Classically, high-risk features are age over 60 years and having had a prior thrombus. Conversely, low-risk patients are those under 60 years of age who have no thrombosis history. Extreme thrombocytosis also affects treatment decisions.76 Most treatment strategies utilize these risk groups. In 2012, a study was published demonstrating a new international prognostic score that was developed for ET patients (WHO-defined). This score included low-, intermediate-, and high-risk groups. The study utilized risk factors such as age and previous thrombosis history, as well as cardiovascular risk factors and JAK2V617F status, and may play a role in future treatment algorithms.77
Patients without extreme thrombocytosis who are low-risk are generally treated with low-dose aspirin therapy. One concern in patients with extreme thrombocytosis is acquired von Willebrand disease (vWD).76 One study suggested that anti-platelet therapy is not needed in all low-risk patients but may be best for JAK2-positive patients or those with associated cardiovascular risk factors.78 Testing for ristocetin co-factor activity is important in this group, as their risk of bleeding with acquired vWD when on aspirin can be significant.78
In addition to low-dose aspirin, cytoreductive therapy is indicated in high-risk patients. Hydroxyurea is often the first-line agent chosen.76,30,79 A randomized control trial comparing hydroxyurea to no cytoreductive treatment showed significantly fewer patients experiencing thrombosis in the hydroxyurea group.80 Concerns regarding leukemic transformation in patients on hydroxyurea have not been substantiated, and reports are conflicting.
Interferon-α (IFN-α) also significantly reduces the platelet count and has no increased risk of leukemogenesis or teratogenicity. It does, however, have a number of persistent, common side effects that limit its use. Younger high-risk patients or pregnant women may benefit from IFN-α use. Low-dose busulfan has been shown to improve hematologic parameters and is sometimes used as a second-line therapy in elderly patients.76,79 Anagrelide, a potent anti-platelet drug, can successfully lower platelet counts and is still used by some as second-line therapy for those who cannot tolerate hydroxyurea.30 A large study using the PVSG criteria for ET showed higher rates of arterial thrombosis, hemorrhage, and transformation to myelofibrosis in patients on anagrelide with aspirin, compared to those on hydroxyurea with aspirin. Only rates of venous thromboembolism were lower.81 Due to these more frequent hematologic side effects, anagrelide is not favored by some practitioners,79 or its use may lead to additional monitoring.81 A recent study in adults looking specifically at anagrelide versus hydroxyurea in ET patients diagnosed using the WHO criteria showed non-inferiority of anagrelide when compared with hydroxyurea.82 This may lead to additional study of anagrelide in the future.
In the past several years, targeted therapy with JAK2 inhibitors has attracted great interest. Ruxolitinib, a JAK1/2 inhibitor, is the most advanced compound, and has been looked at in various phases of study in patients with MF, as well as with ET and PV; it is FDA-approved for certain patients with myelofibrosis. Symptomatic improvement was seen; however, long-term benefit was less clear. Other JAK inhibitors are also being tested, but their roles in treatment, along with safety aspects, are still not clear.76,79,83,84
Treatment approaches adopted for primary thrombocytosis in children have been reported but there are no consensus guidelines. A recent article by Barbui described his recommendations for treatment of children and young adults with ET or PV. He appropriately points out that there is little information on management in children and it is challenging to plan a treatment protocol for children with ET.85 The European Leukemia Network (ELN) recommended using cytoreductive therapy as a last resort and expressed caution regarding use of aspirin in children under 12 years of age.86 Giona and colleagues made recommendations for children with primary thrombocytosis, suggesting cytoreductive therapy when ET patients fail aspirin therapy or have worsening organomegaly, and less aggressive management for children with hereditary thrombocytosis.65
Our suggested algorithm, which requires validation, selects treatments based on clinical symptoms and risk of thrombotic complications, and tailors therapy as appropriate (Figure 2). While these should be taken with caution, the rarity of Reye syndrome has allowed aspirin to be successfully used in younger children.
Figure 2.
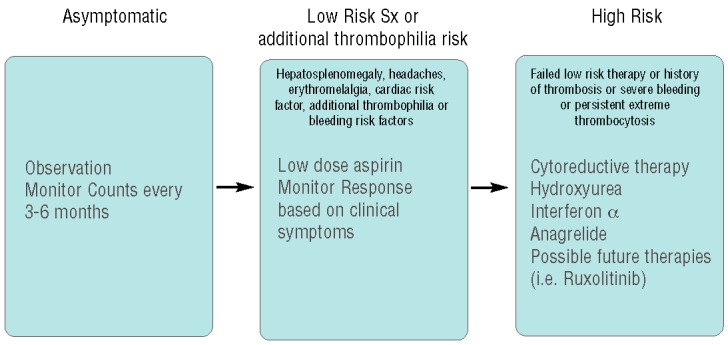
Proposed treatment stratification for pediatric ET showing details of our group’s proposed treatment stratification for children with ET. Asymptomatic patients are observed with monitoring of blood counts. Patients with lower-risk symptoms such as organomegaly, erythromelalgia, or headache, or those with additional cardiovascular or thrombophilia risk factors (such as elevated cholesterol or Factor V Leiden mutation) are treated with aspirin and are monitored for change in symptoms. High-risk patients, including those with thrombosis or severe bleeding, those who failed aspirin therapy, or those who have persistent extreme thrombocytosis, are treated with cytoreductive therapy. Our current first line is hydroxyurea, followed by Interferon-alpha or anagrelide in certain patients. Ruxolitinib is not currently first-line therapy but is being studied in clinical trial for children with MPN and targeted inhibitors may some day become an important component of therapy for children with MPN.
Unanswered questions that require evaluation
We know that children with persistently elevated platelet counts may turn out to have primary thrombocytosis. As described above, the diagnosis of ET in adults is made after demonstration of a persistently elevated platelet count, in combination with having associated bone marrow findings, failure to meet diagnostic criteria for other MPN, and presence of a clonal marker such as JAK2V617F (or absence of a cause for reactive thrombocytosis.) Is this the same criteria we should utilize in diagnosing children with primary thrombocytosis? Given the lower frequency of JAK2 and MPL mutations in children, this set of diagnostic criteria probably needs to be modified for childhood ET and this concept has been supported by other groups.87 Similarly, absence of a causative mutation in a child with a positive family history does not preclude the diagnosis of a hereditary thrombocytosis.
Since children have rapid growth and hormonal changes, more frequent marrow evaluations may be needed. Alternatively, if primary thrombocytosis in children is discovered to be generally more benign, in addition to the lower risk of thrombosis, with decreased risk of fibrotic or leukemic transformation, then perhaps marrows could be evaluated less often.
The underlying pathogenesis is not nearly as clear in children. If children with ET do not present with JAK2 or MPL mutation, can they develop JAK2V61F or MPLW515L at some point during the course of their illness? If so, does it change the clinical course of their disease¿ JAK2-mutant adults with MPN may transform to JAK2-WT leukemias.89 Can reversion to normal occur spontaneously in certain children if they are/become JAK2-mutant? While in JAK2 and MPL negative pediatric ET patients it is likely that there is another causative mutation, the genetic landscape of ET in children is unknown. Are there situations in which children may have persistently elevated platelet counts that are not reactive, but also not truly ET¿ Absence of JAK or MPL mutations does not eliminate upregulation of the JAK/STAT or other relevant pathways as the causative mechanism of disease. To find the driver mutation in these patients, should further genetic evaluation be carried out in additional genes of the relevant pathways, as was done by Ismael and colleagues? We believe the answer is “yes”, and we are currently initiating a broader, targeted analysis in our patients.
The clinical course for adults with ET has established thrombosis, bleeding, and the possibility of progression to myelofibrosis or AML as being the major end points of concern. More is being learned about clinical outcomes of patients with hereditary thrombocytosis as well. Since we do not know enough about the natural course of primary thrombocytosis in childhood, is it possible that a number of young adult patients developed disease earlier and went undetected for many years¿ Thrombocytosis on its own, if platelet function is not normal, may lead to thrombosis even without progression of disease. The only way to answer these questions concerning disease course is to systematically follow well-characterized young ET patients so we can learn the natural history of the disease in children.
Treatment of primary thrombocytosis in children is also unclear. These patients should be at lower risk of vascular damage and thrombosis, due to decreased likelihood of associated risk factors (such as diabetes and atherosclerosis) and a healthier vasculature. Alternatively, diagnosis of primary thrombocytosis in childhood may mean they will be thrombocythemic for 50 or 60 years instead of 20 or 30 years. Does a more prolonged exposure to endothelial damage and activation and/or platelet dysfunction worsen the later risk of long-term complications¿
Should episodes of thrombosis or hemorrhage be used as treatment criteria in pediatric patients or should the focus be on prevention of long-term vascular and organ damage? A number of patients are treated with low-dose aspirin, as for low-risk adults, and this is generally well tolerated. While there is concern of development of Reye syndrome in younger children on aspirin, fortunately this is very rare and aspirin can be stopped when an infection occurs or patients can be temporarily changed to ibuprofen. One report recommends aspirin in young adult patients with erythromelalgia or cardiovascular risk factors.85 In addition, one study in adults suggested that aspirin should be given twice daily in these patients because of enhanced platelet turnover.90
A case report of hydroxyurea use in 2 children with ET and platelet counts over 1,000×109/L has shown a good response64 and it is recommended by Barbui in young ET patients with major thrombotic events.85 Since there is good evidence for the safety of hydroxyurea in even young children with sickle cell disease,91,92 is this something that should be considered first line for pediatric patients with ET? And should cytoreductive therapy be considered for children with HT who show more progressive signs of disease¿
Currently a phase I Children’s Oncology Group study of ruxolitinib is underway for pediatric patients with MPN and certain malignancies. This will likely provide valuable treatment-related data for children with ET even if they are not JAK2-mutated. It may be of particular interest in the future to see the role JAK inhibition plays in JAK2V617F-negative patients, especially as more insight is gained regarding new driver mutations, extent of involvement of the JAK/STAT pathway, and pathogenic mechanisms. This highlights the need for cooperative trials for pediatric primary thrombocytosis, so that biological understanding of the disease can be expanded, treatment strategies standardized, and counseling families of affected pediatric patients improved.
Footnotes
Funding
NK receives support from the Weill Cornell Medical College CTSC grant KL2-TR-000458.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Dame C. Thrombocytosis. In: Arceci RJ, Hahn IM, Smith OP, eds. Pediatric Hematology. 3rd Ed. Oxford, UK: Blackwell Publishing Ltd; 2006, p.548–561 [Google Scholar]
- 2.Qian S, Fu F, Li W, Chen Q, de Sauvage FJ. Primary Role of the Liver in Thrombopoietin Production Shown by Tissue-Specific Knockout. Blood. 1998;92(6):2189–91 [PubMed] [Google Scholar]
- 3.Sungaran R, Markovic B, Chong BH. Localization and regulation of thrombopoietin mRNa expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood. 1997;89(1):101–7 [PubMed] [Google Scholar]
- 4.Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369(6481):568–71 [DOI] [PubMed] [Google Scholar]
- 5.Zeigler FC, de Sauvage F, Widmer HR, Keller GA, Donahue C, Schreiber RD, et al. In vitro megakaryocytopoietic and thrombopoietic activity of c-mpl ligand (TPO) on purified murine hematopoietic stem cells. Blood. 1994;84(12):4045–52 [PubMed] [Google Scholar]
- 6.Broudy VC, Lin NL, Kaushansky K. Thrombopoietin (c-mpl ligand) Acts Synergistically With Erythropoietin, Stem Cell Factor, and Interleukin-11 to enhance Murine Megakaryocyte Colony Growth and Increases Megakaryocyte Ploidy In Vitro. Blood. 1995;85(7):1719–26 [PubMed] [Google Scholar]
- 7.Vignon I, Mornon JP, Cocault L, Mitjavila MT, Tambourin P, Gisselbrecht S, et al. Molecular cloning and characterization of MPL, the human homolog of the v-mpl oncogene: Identification of a member of the hematopoietic growth factor receptor superfamily. Proc Natl Acad Sci USA. 1992;89:5640–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wendling F, Maraskovsky E, Debili N, et al. c-Mpl ligand is a humoral regulator of megakaryocytopoiesis. Nature. 1994;369(6481):571–4 [DOI] [PubMed] [Google Scholar]
- 9.Ballmaier M, Germeshausen M, Schulze H, Cherkaoui K, Lang S, Gaudig A, et al. c-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood. 2001;97(1):139–46 [DOI] [PubMed] [Google Scholar]
- 10.Ghilardi N, Wiestner A, Kikuchi M, Ohsaka A, Skoda RC. Hereditary thrombocythaemia in a Japanese family is caused by a novel point mutation in the thrombopoietin gene. Br J Haematol. 1999;107(2):310–6 [DOI] [PubMed] [Google Scholar]
- 11.Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet. 1998;18(1):49–52 [DOI] [PubMed] [Google Scholar]
- 12.Geddis AE, Fox NE, Kaushansky K. The Mpl receptor expressed on endothelial cells does not contribute significantly to the regulation of circulating thrombopoietin levels. Exp Hematol. 2006;34:82–6 [DOI] [PubMed] [Google Scholar]
- 13.Stoffel R, Wiestner A, Skoda RC. Thrombopoietin in Thrombocytopenic Mice: Evidence Against Regulation at the mRNA Level and for a Direct Regulatory Role of Platelets. Blood. 1996;87(2):567–73 [PubMed] [Google Scholar]
- 14.Fielder PJ, Gurney AL, Stefanich E, Marian M, Moore MW, Carver-Moore K, et al. Regulation of thrombopoietin levels by c-mpl mediated binding to platelets. Blood. 1996;87(6):2154–61 [PubMed] [Google Scholar]
- 15.Nagasawa T, Hasegawa Y, Shimizu S, Kawashima Y, Nishimura S, Suzukawa K, et al. Serum thrombopoietin level is mainly regulated by megakarycotye mass rather than platelet mass in human subjects. Br J Haematol. 1998;101:242–4 [DOI] [PubMed] [Google Scholar]
- 16.Kaushansky K. The molecular mechanisms that control thorombopoiesis. J Clin Invest. 2005;115(2):3339–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan KW, Kaikov Y, Wadsworth LD. Thrombocytosis in childhood: a survey of 94 patients. Pediatrics. 1989;84(6):1064–7 [PubMed] [Google Scholar]
- 18.Dame C, Sutor AH. Primary and secondary thombocytosis in childhood. Br J Haematol. 2005;129(2):165–77 [DOI] [PubMed] [Google Scholar]
- 19.Heath HW, Pearson HA. Thrombocytosis in pediatric outpatients. J Pediatr. 1989;114(5):805–7 [DOI] [PubMed] [Google Scholar]
- 20.Matsubara K, Fukaya T, Nigami H, Harigaya H, Hirata T, Nozaki H, et al. Age-dependent changes in the incidence and etiology of childhood thrombocytosis. Acta Haematol. 2004;111(3):132–7 [DOI] [PubMed] [Google Scholar]
- 21.Dror Y, Blanchette VS. Essential thrombocythaemia in children. Br J Haematol. 1999;107(4):691–8 [DOI] [PubMed] [Google Scholar]
- 22.Vora AJ, Lilleyman JS. Secondary thrombocytosis. Arch Dis Child. 1993;68(1):88–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolber EM, Fandrey J, Frackowski U, Jelkmann W. Hepatic thrombopoietin mRNA is increased in acute inflammation. Thromb Haemost. 2001;86(6):1421–4 [PubMed] [Google Scholar]
- 24.Ishiguro A, Suzuki Y, Mito M, Shimbo T, Matsubara K, Kato T, et al. Elevation of serum thrombopoietin precedes thrombocytosis in acute infections. Br J Haematol. 2002;116(3):612–8 [DOI] [PubMed] [Google Scholar]
- 25.Bilic E, Bilic E. Amino acid sequence homology of thrombopoietin and erythropoietin may explain thrombocytosis in children with iron deficiency anemia. J Pediatr Hematol Oncol. 2003;25(8):675–6 [DOI] [PubMed] [Google Scholar]
- 26.Broudy VC, Lin NL, Sabath DF, Papayannopoulou T, Kaushansky K. Human platelets display high-affinity receptors for thrombopoietin. Blood. 1997;89:1896–904 [PubMed] [Google Scholar]
- 27.Geddis AE, Kaushansky K. Cross-reactivity between erythropoietin and thrombopoietin at the level of Mpl does not account for the thrombocytosis seen in iron deficiency. J Pediatr Hematol Oncol. 2003;25(11):919–20 [DOI] [PubMed] [Google Scholar]
- 28.Thiele J, Kvasnicka HM. The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis. Curr Hematol Malig Rep. 2009;4(1):33–40 [DOI] [PubMed] [Google Scholar]
- 29.Murphy S, Peterson P, Iland H, Laszlo J. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol. 1997;34(1):29–39 [PubMed] [Google Scholar]
- 30.Harrison CN, Bareford D, Butt N, Campbell P, Conneally E, Drummond M, et al. Guideline for the investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol. 2010;149(3):352–75 [DOI] [PubMed] [Google Scholar]
- 31.Passamonti F, Rumi E, Pungolino E, Malabarba L, Bertazzoni P, Valentini M. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–61 [DOI] [PubMed] [Google Scholar]
- 32.Posthuma HL, Skoda RC, Jacob FA, van der Maas AP, Valik PJ, Posthuma EF. Hereditary thrombocytosis not as innocent as thought? Development into acute leukemia and myelofibrosis. Blood. 2010;116(17):3375–6 [DOI] [PubMed] [Google Scholar]
- 33.Teofili L, Giona F, Martin M, Torti L, Cenci T, Foà R, et al. Thrombopoietin receptor activation, thrombopoietin mimetic drugs, and hereditary thrombocytosis: remarks on bone marrow fibrosis. J Clin Oncol. 2010;28(19):e317–8 [DOI] [PubMed] [Google Scholar]
- 34.Teofili L, Larocca LM. Advances in understanding of the pathogenesis of famililal thrombocythaemia. Br J Haematol. 2011;152(6):701–12 [DOI] [PubMed] [Google Scholar]
- 35.Dameshek W. Some speculations on the Myeloproliferative syndromes. Blood. 1951;6(4):372–5 [PubMed] [Google Scholar]
- 36.Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR, et al. A gain-of-function mutation of JAK2 in myeloproliferative neoplasms. N Engl J Med. 2005;352(17):1779–90 [DOI] [PubMed] [Google Scholar]
- 37.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–97 [DOI] [PubMed] [Google Scholar]
- 38.Garçon L, Rivat C, James C, Lacout C, Camara-Clayette V, Ugo V, et al. Constitutive activation of STAT5 and Bcl-xL overexpression can induce endogenous erythroid colony formation in human primary cells. Blood. 2006;108(5):1551–4 [DOI] [PubMed] [Google Scholar]
- 39.Hoffman R, Prchal JT, Samuelson S, Ciurea O, Rondelli D. Philadelphia chromosomenegative myeloproliferative disorders: biology and treatment. Biol Blood Marrow Transplant. 2007;13(1 Suppl 1):64–72 [DOI] [PubMed] [Google Scholar]
- 40.Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3(7):e270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pietra D, Li S, Brisci A, Passamonti F, Rumi E, Theocharides A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111(3):1686–9 [DOI] [PubMed] [Google Scholar]
- 43.Pardanani AD, Levine RL, Lasho T, Pikman Y, Mesa RA, Wadleigh M, et al. MPLW515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108(10):3472–6 [DOI] [PubMed] [Google Scholar]
- 44.Temerinac S, Klippel S, Strunck E, Röder S, Lübbert M, Lange W, et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood. 2000;95(8):2569–76 [PubMed] [Google Scholar]
- 45.Martini M, Teofili L, Larocca LM. Overexpression of PRV-1 gene in polycythemia rubra vera and essential thrombocytosis. Methods Mol Med. 2006;125:265–73 [DOI] [PubMed] [Google Scholar]
- 46.Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klampfl T, Gisslinger H, Harutyunyan AS, Nivrathi H, Rumie E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90 [DOI] [PubMed] [Google Scholar]
- 48.Antonioli E, Guglielmelli P, Poli G, Bogani C, Pancrazzi A, Longo G, et al. Influence of JAK2V617F burden on phenotype in essential thrombocythemia. Haematologica. 2008;93(1):41–8 [DOI] [PubMed] [Google Scholar]
- 49.Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30(3):229–36 [DOI] [PubMed] [Google Scholar]
- 50.Tiedt R, Hao-Shen H, Sobas MA, Looser R, Dirnhofer S, Schwaller J, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MOD phenotype in transgenic mice. Blood. 2008;111(8):3931–40 [DOI] [PubMed] [Google Scholar]
- 51.Pardanani A, Fridley BL, Lasho TL, Gilliland DG, Tefferi A. Host genetic variation contributes to phenotypic diversity in myeloproliferative disorders. Blood. 2008;111(5):2785–9 [DOI] [PubMed] [Google Scholar]
- 52.Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJ, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108(10):3548–55 [DOI] [PubMed] [Google Scholar]
- 53.Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythaemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood. 2004;103(11):4198–200 [DOI] [PubMed] [Google Scholar]
- 54.Mead AJ, Rugless MJ, Jacobsen SE, Schuh A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med. 2012;366(10):967–9 [DOI] [PubMed] [Google Scholar]
- 55.Michiels JJ, Van Genderen PJ. Essential Thrombocythemia in Childhood. Semin Thromb Hemost. 1997;23(3):295–301 [DOI] [PubMed] [Google Scholar]
- 56.Florensa L, Zamora L, Besses C, Ortega JJ, Bastida P, Toll T, et al. Cultures of myeloid progenitor cells in pediatric essential thrombocythemia. Leukemia. 2002;16(9):1876–7 [DOI] [PubMed] [Google Scholar]
- 57.Gassas A, Doyle JJ, Weitzman S, Freedman MH, Hitzler JK, Sharathkumar A, et al. A basic classification and a comprehensive examination of pediatric myeloproliferative syndromes. J Pediatr Hematol Oncol. 2005;27(4):192–6 [DOI] [PubMed] [Google Scholar]
- 58.Lackner H, Urban C, Benesch M, Moser A, Sovinz P, Schwinger W, et al. Long-term use of anagrelide in the treatment of children with essential thrombocythemia. Eur J Haematol. 2006;77(4):358–9 [DOI] [PubMed] [Google Scholar]
- 59.Randi ML, Putti MC, Scapin M, Pacquola E, Tucci F, Micalizzi C, et al. Pediatric patients with essential thrombocythemia are mostly polyclonal and V617FJAK2 negative. Blood. 2006;108(10):3600–2 [DOI] [PubMed] [Google Scholar]
- 60.El-Moneim AA, Kratz CP, Böll S, Rister M, Pahl HL, Niemeyer CM. Essential versus reactive thrombocythemia in children: retrospective analyses of 12 cases. Pediatr Blood Cancer. 2007;49(1):52–5 [DOI] [PubMed] [Google Scholar]
- 61.Teofili L, Giona F, Martini M, Cenci T, Guidi F, Torti L, et al. Markers of myeloproliferative diseases in childhood polycythemia vera and essential thrombocythemia. J Clin Oncol. 2007;25(9):1048–53 [DOI] [PubMed] [Google Scholar]
- 62.Nakatani T, Imamura T, Ishida H, Wakaizumi K, Yamamoto T, Otabe O, et al. Frequency and clinical features of the JAK2 V617F mutation in pediatric patients with sporadic essential thrombocythemia. Pediatr Blood Cancer. 2008;51(6):802–5 [DOI] [PubMed] [Google Scholar]
- 63.Veselovska J, Pospisilova D, Pekova S, Horvathova M, Solna R, Cmejlova J, et al. Most pediatric patients with essential thrombocythemia show hypersensitivity to erythropoietin in vitro, with rare JAK2 V617F-positive erythroid colonies. Leuk Res. 2008;32(3):369–77 [DOI] [PubMed] [Google Scholar]
- 64.Dua V, Yadav SP, Kumar V, Saxena R, Sachdeva A. Two cases of pediatric essential thrombocythemia managed effectively with hydroxyurea. Int J Hematol. 2012;96(6):810–3 [DOI] [PubMed] [Google Scholar]
- 65.Giona F, Teofili L, Moleti ML, Martini M, Palumbo G, Amendola A, et al. Thrombocythemia and polycythemia in patients younger than 20 years at diagnosis: clinical and biologic features, treatment, and long-term outcome. Blood. 2012;119(10):2219–27 [DOI] [PubMed] [Google Scholar]
- 66.Ismael O, Shimada A, Hama A, Sakaguchi H, Doisaki S, Muramatsu H, et al. Mutations profile of polyctyhemia vera and essential thrombocythemia among Japanese children. Pediatr Blood Cancer. 2012;59(3):530–5 [DOI] [PubMed] [Google Scholar]
- 67.Girodon F, Dutrillaux F, Broséus J, Mounier M, Goussot V, Bardonnaud P, et al. Leukocytosis is associated with poor survival but not with increased risk of thrombosis in essential thrombocythaemia: a population-based study of 311 patients. Leukemia. 2010;24:900–3 [DOI] [PubMed] [Google Scholar]
- 68.Stein BL, Rademaker A, Spivak JL, Moliterno AR. Gender and vascular complications in the JAK2 V617F-positive myeloproliferative neoplasms. Thrombosis. 2001;2011:8741–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mossier C, Kerbl R, Wagner T, Muntean W, Sorantin E, Urban CE. Portal vein thrombosis in a 17-yo female adolescent with essential thrombocytosis. Pediatr Hematol Oncol. 1997;14(5):457–62 [DOI] [PubMed] [Google Scholar]
- 70.Jensen AW, Tefferi A, Arndt CA. Cerebral venous sinus thrombosis associated with essential thrombocytosis in a pediatric patient. J Pediatr Hematol Oncol. 2007;29(3):156–9 [DOI] [PubMed] [Google Scholar]
- 71.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adélaïde J, Rey J, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23(11):2183–6 [DOI] [PubMed] [Google Scholar]
- 73.Tefferi A, Lasho TL, Abdel-Wahab O, Guglielmelli P, Patel J, Caramazza D, et al. IDH1 and IDH2 mutation studies in 1473 paients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114(9):1859–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lambert MP, Jiang J, Batra V, Wu C, Tong W. A novel mutation in MPL (Y252H) results in increased thrombopoietin sensitivity in essential thrombocythemia. Am J Hematol. 2012;87(5):532–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barbui T, Finazzi MC, Finazzi G. Front-line therapy in polycythemia vera and essential thrombocythemia. Blood Rev. 2012;26(5):205–11 [DOI] [PubMed] [Google Scholar]
- 77.Barbui T, Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, et al. Development and Validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):5128–33 [DOI] [PubMed] [Google Scholar]
- 78.Alvarez-Larrán A, Cervantes F, Pereira A, Arellano-Rodrigo E, Pérez-Andreu V, Hernández-Boluda JC, et al. Observation versus antiplatelet therapy as primary prophylaxis for thrombosis in low-risk essential thrombocythemia. Blood. 2010;116(8):1205–10 [DOI] [PubMed] [Google Scholar]
- 79.Tefferi A. Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol. 2012;87(3):285–93 [DOI] [PubMed] [Google Scholar]
- 80.Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332(17):1132–6 [DOI] [PubMed] [Google Scholar]
- 81.Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. :. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med. 2005;353(1):33–45 [DOI] [PubMed] [Google Scholar]
- 82.Gisslinger H, Gotic M, Holwiecki J, Penka M, Thiele J, Kvasnicka HM, et al. Anagrelide compared to hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013;121(10):1720–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kucine N, Levine RL. JAK inhibitors and other novel agents in myeloproliferative neoplasms: are we hitting the target? Ther Adv Hematol 2011;2(4):203–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012;119(12):2721–30 [DOI] [PubMed] [Google Scholar]
- 85.Barbui T. How to manage children and young adults with myeloproliferative neoplasms. Leukemia. 2012;26(7):1452–7 [DOI] [PubMed] [Google Scholar]
- 86.Barbui T, Barosi G, Birgegard G, Cervantes F, Finazzi G, Griesshammer M, et al. Philadelphia-Negative Classical Myeloproliferative Neoplasms: Critical Concepts and Management Recommendations From European LeukemiaNet. J Clin Oncol. 2011;29(6):761–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Teofili L, Giona F, Martini M, Cenci T, Guidi F, Torti L, et al. The revised WHO diagnostic criteria for Ph-negative myeloproliferative diseases are not appropriate for the diagnostic screening of childhood polycythemia vera and essential thrombocythemia. Blood. 2007;110(9):3384–6 [DOI] [PubMed] [Google Scholar]
- 89.Beer PA, Delhommeau F, LeCouédic JP, Dawson MA, Chen E, Bareford D, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115(14):2891–900 [DOI] [PubMed] [Google Scholar]
- 90.Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. 2012;119(15):3595–603 [DOI] [PubMed] [Google Scholar]
- 91.Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010;115(26):5300–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thornburg CD, Files BA, Luo Z, Miller ST, Kalpatthi R, Iyer R, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood. 2012;120(22):4304–10 [DOI] [PMC free article] [PubMed] [Google Scholar]