

Abstract
Transposable elements (TEs) are the major component of plant genomes where they contribute significantly to the >1,000-fold genome size variation. To understand the dynamics of TE-mediated genome expansion, we have undertaken a comparative analysis of the TEs in two related organisms: the weed Arabidopsis thaliana (125 megabases) and Brassica oleracea (≈600 megabases), a species with many crop plants. Comparison of the whole genome sequence of A. thaliana with a partial draft of B. oleracea has permitted an estimation of the patterns of TE amplification, diversification, and loss that has occurred in related species since their divergence from a common ancestor. Although we find that nearly all TE lineages are shared, the number of elements in each lineage is almost always greater in B. oleracea. Class 1 (retro) elements are the most abundant TE class in both species with LTR and non-LTR elements comprising the largest fraction of each genome. However, several families of class 2 (DNA) elements have amplified to very high copy number in B. oleracea where they have contributed significantly to genome expansion. Taken together, the results of this analysis indicate that amplification of both class 1 and class 2 TEs is responsible, in part, for B. oleracea genome expansion since divergence from a common ancestor with A. thaliana. In addition, the observation that B. oleracea and A. thaliana share virtually all TE lineages makes it unlikely that wholesale removal of TEs is responsible for the compact genome of A. thaliana.
Arabidopsis thaliana and Brassica oleracea are closely related species (they belong to the same taxonomic family, Brassicaceae) that diverged from a common ancestor ≈15–20 million years ago and now share ≈85% nucleotide sequence identity in their protein coding regions (1). A. thaliana is a weed, whereas several B. oleracea cultivars, such as cabbage, kale, broccoli, cauliflower, and brussel sprouts, are of worldwide economical importance. Whereas the sequence of the A. thaliana genome has been available for >3 years (2), a shotgun sequence of B. oleracea was recently generated by The Institute for Genomic Research (TIGR) to assist in its annotation. At this time, the B. oleracea database encompasses approximately one-third of the genome (≈220 megabases, Mb, of ≈600 Mb) and consists of ≈350,000 short sequence reads (average length ≈650 bp).
The availability of these sequence databases offers an unprecedented opportunity for detailed genomic comparisons in closely related organisms. Although the major reason for sequencing B. oleracea genomic DNA was to identify the coding exons of cellular genes, in reality the vast majority of the shared sequences are expected to be proteins encoded by transposable elements (TEs). This reflects the fact that TEs make up the largest fraction of the genomes of most multicellular organisms, especially higher plants, and most encode proteins required for their mobility (3–5).
In eukaryotes, TEs have been divided into two classes based on their transposition intermediate (6). Class 1 (RNA) elements transpose via an RNA intermediate and either have long terminal repeats (LTR retrotransposons) or terminate at one end with a poly(A) tract [long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs)] (7, 8). LTR retrotransposons have been further classified as either Ty1/copia-like or Ty3/gypsy-like elements based on the order of their encoded proteins that include a reverse transcriptase (RT) and integrase required for reverse transcription and integration (7). Class 2 (DNA) elements transpose via a DNA intermediate, have terminal inverted repeats, and have been grouped into superfamilies [e.g., Tc1/mariner, hAT, CACTA, Mutator-like elements (MULEs), and PIF/Pong] based on the similarity of transposases, the element-encoded protein that catalyzes transposition and integration (9–19).
TEs are major components of plant genomes and contribute significantly to the >1,000-fold genome size variation. A series of recent studies has demonstrated that differential amplification of one element type, LTR retrotransposons, largely accounts for the C-value paradox among the agronomically important members of the grass clade. The C-value paradox is the observed lack of correlation between increases in DNA content and an organism's complexity (20). For the members of the grass clade examined, the fraction of the genome contributed by LTR retrotransposons increases with genome size from rice, the smallest characterized grass genome (≈14% of its 430-Mb genome consists of LTR retrotransposons) (21), through maize (≈2,500 Mb, 50–60% retrotransposons) (4, 22), to barley (≈4,800 Mb, >70% retrotransposons) (23).
A. thaliana harbors all of the TE types found in larger plant genomes; however, copy number is generally low, with all TEs accounting for only ≈10% of the A. thaliana genome (2). Although A. thaliana and B. oleracea diverged from a common ancestor ≈15–20 million years ago (1), the B. oleracea genome at ≈600 Mb is almost 5-fold larger (24). Recent studies indicate that the B. oleracea genome has expanded through triplication since its divergence from Arabidopsis (25–27). However, genome triplication cannot fully explain the genome size difference between the two species. Because the proliferation of TEs has been implicated in the expansion of grass genomes (4, 23), it is possible that they are also involved in the recent genome size expansion of B. oleracea. Alternatively, if the last common ancestor of A. thaliana and B. oleracea had a large genome, preferential loss of sequences in the lineage leading to A. thaliana may help to explain its compact genome (28, 29).
As reported in this study, analysis of the TEs in the two genomes has permitted an estimation of the patterns of amplification, diversification, and loss that has occurred since divergence from their last common ancestor. This was made possible by devising strategies to compare TEs with significant coding capacity in the complete A. thaliana genomic sequence and the fragmentary B. oleracea database. Nearly all TE lineages are shared in both species, but the number of elements in each lineage is usually greater in B. oleracea than in A. thaliana. Although we find that class 1 elements are the most abundant TE class in both species, several families of class 2 elements have amplified to very high copy number in B. oleracea, where they have contributed significantly to genome expansion. Taken together, the results of this analysis indicate that amplification of both RNA and DNA elements is responsible, in part, for B. oleracea genome expansion since divergence from a common ancestor with A. thaliana. In addition, the observation that the two species share virtually all TE lineages makes it unlikely that wholesale removal of TEs is responsible for the compact genome of A. thaliana.
Materials and Methods
Database Search Strategies and Sequence and Phylogenetic Analysis. The following procedure was used to identify TE encoding sequences from A. thaliana and B. oleracea. For each type of TE, the most conserved coding region was first identified by comparing previously described A. thaliana elements (see text for specific examples). The amino acid sequences of these regions were used as queries in tblastn searches against the Arabidopsis database [AT H1_bacs.seq, available at TIGR (http://tigrblast.tigr.org/er-blast/index.cgi?project=ath1)] to identify all A. thaliana homologs. Next, these homologs were compared by clustalw multiple alignments and resolved into lineages by generating phylogenetic trees (not shown). Sequences from major A. thaliana lineages were then used as queries in tblastn searches against the TIGR B. oleracea database (brassica prelim sequences; http://tigrblast.tigr.org/euk-blast/index.cgi?project=bog1). Hits from B. oleracea that contained the entire query region and their A. thaliana homologs were pooled, compared by clustalw multiple alignments, and used to generate the phylogenetic trees described in the text. Preliminary B. oleracea sequence data were obtained from the TIGR web site at www.tigr.org. TE sequences from both species are available upon request. Multiple sequence alignments were performed with the clustalw server available at European Bioinformatics Institute (www.ebi.ac.uk/clustalw) with default parameters. Phylogenetic trees were generated based on the neighbor-joining method, using paup* version 4.0b8 with default parameters (30).
B. oleracea TE Copy Number Estimate. The number of hits from B. oleracea could not be directly converted into TE copy number because the TIGR B. oleracea database consists of short reads (on average ≈650 bp) and, for example, two hits from two different reads could represent different regions of the same element. For this reason, the following equation was derived to estimate TE copy number based on effective query length (Leq), average length of database reads (Ldr), and the number of hits (Nhits):
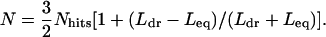 |
[1] |
Detailed descriptions of the derivation and feasibility tests of this equation are provided in Supporting Text and Fig. 6, which are published as supporting information on the PNAS web site.
Results
TE Abundance. The abundance of TE families in A. thaliana and B. oleracea was determined as described in Materials and Methods. For this comparison, the absolute values obtained from the complete A. thaliana sequence were compared with the partial and fragmented B. oleracea sequence. For the latter species, an equation was derived that converts database characteristics and search output into an estimate of TE copy number. In both species, class 1 elements are more abundant than class 2 elements (Fig. 1A), with LTR-retrotransposons as the predominant TE type. The most abundant class 2 superfamily in B. oleracea is CACTA, whereas MULEs have the most copies in A. thaliana.
Fig. 1.

Comparison of the abundance (A) and the density (copies per Mb) (B) of different types of TEs in A. thaliana and B. oleracea. Values from A. thaliana are shown in green, and those from B. oleracea are shown in red.
This comparison of copy number revealed that all TE types are more numerous in B. oleracea (Fig. 1A). This is not surprising because the genome of B. oleracea is about five times larger than that of A. thaliana. For this reason, TE densities (copy number per Mb) were calculated to identify TEs with copy numbers higher than expected for the proportional increase in genome size. As shown in Fig. 1B, the densities of hAT, MULEs, and PIF-like elements are similar in both genomes, whereas the densities of Pong-like, CACTA-like, and all class 1 elements are significantly higher in B. oleracea.
Comparative Phylogenetic Analyses of TEs. In addition to taking an inventory of element types and their approximate copy number, the availability of a large amount of genomic sequence from related plant species provided an unprecedented opportunity to understand how TEs evolve after divergence of their hosts from a common ancestor. To do this, phylogenic trees were generated for all major types of TE by comparing sequences from both genomes using clustalw multiple alignments. The results of this analysis for select TE types are summarized below.
Pong-Like and PIF-Like Elements. PIF/Pong is a recently discovered superfamily of eukaryotic transposons (9, 10, 15, 19). Members are particularly widespread and abundant in plants, where they group into two clades named PIF-like and Pong-like (15). Both clades encode two ORFs: a putative DNA-binding protein (called ORF1) and a transposase (9, 10).
Pong-like elements in A. thaliana and B. oleracea were identified by tblastn searches using as query the catalytic domain of the Pong transposase. The evolution of this superfamily was of immediate interest because Pong-like elements were found to be present at much higher density in B. oleracea (1.6 copies per Mb) than in A. thaliana (0.2 copy per Mb) (Fig. 1B). A phylogenetic tree generated from 27 A. thaliana and 139 B. oleracea entries containing the query sequence resolved three lineages corresponding to three previously described dicot Pong lineages (O1, P1, and P2; Fig. 2A) (9). The three lineages were found in both species, suggesting that they were present in the last common ancestor. Of the three lineages, P1 includes the majority of sequences from A. thaliana (18 of 27) and nearly all from B. oleracea (137 of 139). Within P1, B. oleracea sequences clustered into four large, species-specific groups with short branch lengths (P1b–P1e), suggesting that several lineages of Pong-like elements have undergone extensive amplification since their last common ancestor.
Fig. 2.

Phylogeny of Pong-like TPases (A), Pong-like ORF1s (B), and PIF-like TPases (C) in A. thaliana (green) and B. oleracea (red). These phylogenetic trees were generated by using the neighbor-joining method and rooted with the TPase and ORF1 of the rice Pong element and the TPase of the maize PIF element, respectively. Bootstrap values were calculated from 500 replicates.
In addition to the catalytic domain, ≈1,000 B. oleracea reads were homologous to other regions of the Pong transposase. It is unlikely that this large number is an artifact of database bias (possibly caused by sequencing a few elements many times) because only four pairs of sequences used to generate the phylogenetic tree in Fig. 2A were identical. Additional evidence for the abundance and explosive amplification of Pong-like elements was furnished when a similar analysis using, as query, the rice ORF1 (340 aa), the second Pong-like ORF, identified >700 ORF1 homologs in B. oleracea (E value < e-10), and only 21 (E value < e-5) from A. thaliana. Furthermore, a phylogenetic tree generated by using ORF1 from A. thaliana and B. oleracea was found to be highly consistent with the transposase tree from these species (Fig. 2B).
In contrast to Pong-like elements, the density of PIF-like elements is not significantly different in A. thaliana and B. oleracea. tblastn searches using the catalytic domain of the maize PIF transposase (120 aa) identified 35 hits from A. thaliana and 21 from B. oleracea that contained the entire query region. Their phylogenies in these species (shown in Fig. 2C) reveal that PIF-like transposases are significantly more divergent than Pong-like transposases (Fig. 2C, note the longer branches vs. Fig. 2A) and cluster into three lineages (A3, B, and C), with each lineage containing sequences from both species and no lineage containing smaller, species-specific clusters.
CACTA-Like Elements. Prior studies of TEs in A. thaliana identified four families of CACTA-like elements (named Atenspm1-4), of which one (Atenspm1) was shown to be transpositionally active (31, 32). Comparison of the transposases of Atenspm1 (889 aa) with a CACTA-like element (703 aa) isolated from another brassica species, Brassica rapa (33), served to identify the most conserved region as a 100-aa segment (77% identical) corresponding to positions 272–371 in Atenspm1. tblastn searches using this region as query identified 121 and 541 hits containing the entire query region in A. thaliana and B. oleracea, respectively. A phylogenetic tree generated from a clustalw multiple alignment of these sequences (Fig. 3) indicates that all elements from both species cluster into two major clades, called A and B. Clade A contains 40 of the 121 A. thaliana sequences and only two of the 540 B. oleracea sequences. Clade B consists of three lineages (B1–B3), all of which are present in both A. thaliana and B. oleracea. Lineage B1 is present at low copy number in both species, whereas lineage B2 resembles clade A in that it is relatively abundant in A. thaliana (50 sequences) but scarce in B. oleracea (three sequences). The vast majority of B. oleracea sequences (n = 500) comprise the B3 lineage and cluster into three large, species-specific groups. Two groups, named BoC1 and BoC2, include many highly similar sequences. Forty two of the 118 BoC1 sequences were identical, as were 46 of the 181 BoC2 sequences. High intrafamily sequence identity, such as this, is indicative of families that have very recently amplified.
Fig. 3.

Phylogeny of CACTA-like elements in A. thaliana (green) and B. oleracea (red). This phylogenetic tree was generated by using the neighbor-joining method and rooted with the TPase of the maize Doppia element. Bootstrap values were calculated from 250 replicates.
MULEs and hAT-Like Elements. Previous analysis of a subset of genomic sequence led to the identification of several dozen MULEs and hAT-like elements in A. thaliana (16, 17). Additional MULEs and hAT-like elements were identified by tblastn searches using, as queries, the most conserved coding regions of previously described elements (the catalytic domain for MULEs and the dimerization domain for hAT-like elements) (16). The phylogenies of these two superfamilies of transposons were determined and provided in Figs. 7 and 8, which are published as supporting information on the PNAS web site. Overall, both MULEs and hAT-like elements are represented by multiple small lineages (each containing few elements) that diverged before the divergence of A. thaliana and B. oleracea.
Copia-Like LTR-Transposons. The A. thaliana genome harbors ≈300 copia-like elements (34). A previous study of a subset of 25 elements resolved six lineages (Copia I–VI) (34). tblastn searches using a 156-aa segment from the RT domain (the most conserved region among described A. thaliana elements) as query identified 268 and 638 hits containing the entire query region from A. thaliana and B. oleracea, respectively. A phylogenetic tree generated from the clustalw multiple alignment of these hits resolved two major clades (A and B, see Fig. 4). Clade A includes all six previously reported lineages as well as five lineages identified in this study. Clade B, which does not include previously described copia-like elements from either species, can be divided into two lineages. Of the 13 copia-like lineages, one (Copia XIII) was only found in B. oleracea. The remaining 12 are present in both species and all at a higher density in B. oleracea. Two lineages, Copia IX and XI, have the largest difference in density at ≈2 copies per Mb (≈1,200–1,400 genomic copies) in B. oleracea, but each with only ≈0.08 copy per Mb in A. thaliana (<10 copies genome wide). B. oleracea sequences in these two lineages clustered into species-specific groups with short branch lengths (Fig. 4); an indication of their amplification since divergence of the two species. Three small clusters of B. oleracea sequences (≈25 sequences per cluster) are highly similar (>98% identical), suggesting recent activity (named BoCP_ IXa, -b, and -c; arrows in Fig. 4).
Fig. 4.

Phylogeny of copia-like LTR retrotransposons in A. thaliana (green) and B. oleracea (red). This phylogenetic tree was generated by using the neighbor-joining method and rooted with the corresponding RT from the yeast Ty1 element. Bootstrap values were calculated from 250 replicates.
Gypsy-Like LTR-Retrotransposons. Approximately 60 gypsy-like elements in A. thaliana were previously described and grouped into eight lineages (35). tblastn searches using, as query, the most conserved coding sequence (a 156-aa segment of the RT domain) identified 286 and 714 sequences with the entire query region from A. thaliana and B. oleracea, respectively. A phylogenetic tree generated from a clustalw multiple alignment of these sequences resolved two clades, E and M, consisting of 11 and 12 lineages, respectively (Fig. 5). Nine of the 23 gypsy-like lineages are in both species, of which one (Tat) has similar copy number in both species (≈15 copies), whereas eight have significantly more elements in B. oleracea (≈10- to ≈600-fold). Of the remaining 14 lineages, five included only A. thaliana sequences, whereas nine are specific to B. oleracea. Significantly, all 14 species-specific lineages have shorter branch lengths than the nine shared lineages, indicating that they emerged after the divergence of the two species.
Fig. 5.

Phylogeny of gypsy-like LTR retrotransposons in A. thaliana (green) and B. oleracea (red). This phylogenetic tree was generated by using the neighbor-joining method and rooted with the corresponding RT from the yeast Ty3 element. Bootstrap values were calculated from 250 replicates.
LINEs. A recent survey of A. thaliana LINEs identified 219 elements, and phylogenetic analysis of a subset of 62 elements defined two clades (I and II) (36). tblastn searches using the most conserved coding sequence in these LINEs (a 151-aa region in their RT domain) as query identified 238 and 498 sequences with the entire query region from A. thaliana and B. oleracea, respectively. The phylogeny of LINEs in A. thaliana and B. oleracea was determined based on these sequences and is provided in Fig. 9, which is published as supporting information on the PNAS web site. Eighteen lineages were resolved, of which 15 are present in both species, whereas three include only sequences from A. thaliana. In general, LINE lineages are characterized by very long branches, suggesting that the LINEs in both species are old.
Discussion
Here we report the use of large quantities of genomic data from two related plant species to estimate the TE landscape in their last common ancestor. By exploiting the genomic resources of A. thaliana and B. oleracea, we were able to infer the number of lineages present in their last common ancestor and determine the success of different lineages since species divergence.
The A. thaliana genome is compact in large part because of its low TE content. Although the biological basis for its compact genome remains unresolved, two hypotheses have been entertained. The first proposes that low TE content results from an inability to amplify to high copy numbers. This could be due, for example, to stringent host control or to high gene density. The second hypothesis is that TEs have successfully proliferated in the lineage leading to A. thaliana but have been lost by deletion (28). Results from this study support the former view and provide little evidence for the latter. Overall, nearly all lineages of each type of TE are present in both A. thaliana and B. oleracea. For example, the two species share all three Pong-like lineages (Fig. 2), all four CACTA-like lineages (Fig. 3), 12 of the 13 copia-like lineages (Fig. 4), and 15 of the 18 LINE lineages (Fig. 9), suggesting that these lineages were in their last common ancestor. In a few cases, some lineages are in only one species. For example, one copia-like lineage (Copia XII, Fig. 4) was only in B. oleracea, whereas three LINE lineages (II-a, -f, and -h in Fig. 9) only included A. thaliana sequences. The species-specific lineages have shorter branch lengths compared to lineages that are in both species, suggesting that they emerged after divergence from a common ancestor. A lineage found in only one species may have evolved from a related element, been lost from one species, or become established after horizontal transfer. Despite these few exceptions, the fact that the vast majority of TE lineages are retained in both species suggests that A. thaliana has not lost substantial amounts of TEs since it diverged from the last common ancestor, making it unlikely that A. thaliana has a history of genome-wide elimination of protein-coding TEs.
Data presented in this study support the view that the low TE content of A. thaliana is largely due to the lack of significant amplification of any TE type. First, ancestral lineages retained in both species almost always have much lower copy number in A. thaliana than in B. oleracea. For example, all 12 shared copia-like lineages are at much lower density (from 2-fold to >70-fold) in A. thaliana. Second, few lineages of any TE have amplified in A. thaliana and, for those that have, the extent of amplification is modest. For example, the only lineages to attain significant copy number in A. thaliana are Pong-like lineage P1a (19 sequences) and CACTA-like lineages A (40 sequences) and B2 (50 sequences). However, even for these lineages, the extent of amplification is much less extensive than that in B. oleracea, where Pong-like lineages P1b–P1e and CACTA-like lineage B3 have amplified to ≈1,000 and ≈2,300 copies, respectively (extrapolated to the whole genome).
In contrast, many lineages, including both class 1 and class 2 elements, have amplified in B. oleracea since its divergence from A. thaliana. The total length of TEs in B. oleracea (≈120 Mb or ≈20% of its genome) is ≈15 times more than that in A. thaliana (≈8 Mb or 6% of its genome). This difference results from the relatively recent amplification of both class 1 and class 2 elements, such as copia-like (lineages IX and XI, Fig. 4), Pong-like (lineages P1b-e, Fig. 2 A and B) and CACTA-like (lineage B3, Fig. 3) elements. Class 1 elements, including both LTR and non-LTR retrotransposons, account for ≈78 Mb of nuclear DNA in B. oleracea (≈14% of its genome) but only 5–6 Mb in A. thaliana (≈4% of its genome). Class 2 elements also make up a larger fraction of nuclear DNA in B. oleracea (≈37 Mb or 6% of its genome) than in A. thaliana (≈3 Mb or 2–3% of its genome).
As more is learned about the TE component of diverse plant genomes, the more it becomes apparent that each is unique. For example, analyses of large monocot genomes have revealed that the amplification of a few families of LTR retrotransposons is largely responsible for genome size variations. Four LTR-retrotransposon families (Ji, Opie, Huck, and Zeon-1) account for 32% of the ≈2,500-Mb maize genome (22), and one family (IRRE) accounts for ≈10% of the ≈10,000-Mb genome of Iris brevicaulis (37). Even the relatively small sorghum genome (≈700 Mb) contains a high copy number retrotransposon family (Retrosor6, ≈6,000–7,000 copies) that accounts for ≈6% of the genome (38). In contrast, massive amplification of one or more retrotransposon family has not occurred in B. oleracea; the most abundant family contains only ≈140 copies (BoCP_IXc). Thus, although class 1 elements comprise the largest fraction of the B. oleracea genome at 14%, this is due to the amplification of numerous class 1 element families to relatively low copy number. In this regard, B. oleracea is reminiscent of rice with its relatively small genome (≈450 Mb, 14% retrotransposons), that also harbors only small families of LTR retrotransposons (N. Jiang and S.R.W., unpublished data).
Although class 1 elements have long been known to predominate in plant genomes (3), the ability of class 2 elements to attain very high copy numbers has only recently become apparent. In this study we find that DNA elements account for ≈6% of the B. oleracea genome. Most notably, two families of CACTA-like elements (BoC1 and BoC2, Fig. 3) have amplified to >500 and >800 copies, respectively, in B. oleracea and together account for >2% of the total genomic DNA. High copy number CACTA-like families were recently reported in a few species of grasses with large genomes. For example, there are ≈5,000 Tpo1 elements in ryegrass (Lolium perenne, 5,000 Mb) (39) and ≈3,000 Caspar elements in wheat (Triticum monococcum, ≈5,000 Mb) (40).
In summary, the comparative analyses presented in this study indicate that A. thaliana and B. oleracea inherited and retained largely the same collection of TE lineages from their last common ancestor ≈15–20 million years ago. However, since diverging from that ancestor, TEs have been able to attain much higher copy numbers in the lineage leading to B. oleracea. It should be noted that the differential accumulation of TEs in the modern genomes of A. thaliana and B. oleracea must take into account the combined effects of TE amplification and elimination. Although the retention of nearly all ancestral TE lineages in A. thaliana suggests that large-scale TE elimination is highly unlikely, this type of data alone is not sufficient to completely rule out this possibility. Higher resolution studies might involve, for example, systematic comparisons of regions of the A. thaliana genome to a close relative as well as to an appropriate outgroup. With regard to the low TE content of A. thaliana or any other organism with high gene density, what is apparent is that such organisms cannot tolerate large-scale TE amplification because of the mutagenic effects of TE insertions. In support of this view is the finding that a high percentage (≈48%) of T-DNA insertions in A. thaliana are knockouts (41). On the other hand, the B. oleracea genome was recently reported to be the product of a triplication event after its divergence from A. thaliana (25–27). Such an event would have produced numerous safe havens for TE insertions because of functional redundancy. This may be particularly relevant with regard to the amplification of DNA transposons, such as Pong-like elements, which are known to target genic regions (10).
Supplementary Material
Acknowledgments
Preliminary sequence data were obtained from TIGR web site at www.tigr.org. Sequencing of B. oleracea was funded by the National Science Foundation. This work was supported by grants from the National Institutes of Health and National Science Foundation Plant Genome Initiative (to S.R.W.).
Abbreviations: TIGR, The Institute for Genomic Research; Mb, megabase; TE, transposable elements; LINE, long interspersed nuclear element; MULE, Mutator-like elements; RT, reverse transcriptase.
References
- 1.Yang, Y. W., Lai, K. N., Tai, P. Y. & Li, W. H. (1999) J. Mol. Evol. 48, 597–604. [DOI] [PubMed] [Google Scholar]
- 2.The Arabidopsis Genome Initiative (2000) Nature 408, 796–815. [DOI] [PubMed] [Google Scholar]
- 3.Kumar, A. & Bennetzen, J. L. (1999) Annu. Rev. Genet. 33, 479–532. [DOI] [PubMed] [Google Scholar]
- 4.SanMiguel, P. & Bennetzen, J. L. (1998) Ann. Bot. 81, 37–44. [Google Scholar]
- 5.SanMiguel, P., Tikhonov, A., Jin, Y.-K., Motchoulskaia, N., Zakharov, D., Melake-Berhan, A., Springer, P. S., Edwards, K. J., Lee, M., Avramova, Z., et al. (1996) Science 274, 765–768. [DOI] [PubMed] [Google Scholar]
- 6.Capy, P., Bazin, C., Higuet, D. & Langin, T. (1998) Dynamics and Evolution of Transposable Elements (Landes, Austin, TX).
- 7.Xiong, Y. & Eickbush, T. H. (1990) EMBO J. 9, 3353–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doolittle, R. F., Feng, D.-F., Johnson, M. S. & McClure, M. A. (1989) Q. Rev. Biol. 64, 1–30. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, X., Jiang, N., Feschotte, C. & Wessler, S. (2004) Genetics 166, 971–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang, N., Bao, Z., Zhang, X., Hirochika, H., Eddy, S. R., McCouch, S. R. & Wessler, S. R. (2003) Nature 421, 163–167. [DOI] [PubMed] [Google Scholar]
- 11.Plasterk, R. H. A. & van Luenen, H. G. (2002) in Mobile DNA II, eds. Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M. (Am. Soc. Microbiol. Press, Washington, DC), pp. 519–532.
- 12.Feschotte, C. & Wessler, S. R. (2002) Proc. Natl. Acad. Sci. USA 99, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lisch, D. (2002) Trends Plant Sci. 7, 498–504. [DOI] [PubMed] [Google Scholar]
- 14.Kunze, R. & Weil, C. F. (2002) in Mobile DNA II, eds. Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M. (Am. Soc. Microbiol. Press, Washington, DC), pp. 565–610.
- 15.Zhang, X., Feschotte, C., Zhang, Q., Jiang, N., Eggleston, W. B. & Wessler, S. R. (2001) Proc. Natl. Acad. Sci. USA 98, 12572–12577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubin, E., Lithwick, G. & Levy, A. A. (2001) Genetics 158, 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu, Z., Wright, S. I. & Bureau, T. E. (2000) Genetics 156, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mao, L., Wood, T. C., Yu, Y., Budiman, M. A., Tomkins, J., Woo, S., Sasinowski, M., Presting, G., Frisch, D., Goff, S., et al. (2000) Genome Res. 10, 982–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker, E. L., Eggleston, W. B., Demopulos, D., Kermicle, J. & Dellaporta, S. L. (1997) Genetics 146, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas, C. A. (1971) Annu. Rev. Genet. 5, 237–256. [DOI] [PubMed] [Google Scholar]
- 21.Jiang, N. & Wessler, S. R. (2001) Plant Cell 13, 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyers, B. C., Tingey, S. V. & Morgante, M. (2001) Genome Res. 11, 1660–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vicient, C. M., Suoniemi, A., Anamthawat-Jonsson, K., Tanskanen, J., Beharav, A., Nevo, E. & Schulman, A. H. (1999) Plant Cell 11, 1769–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arumuganathan, K. & Earle, E. D. (1991) Plant. Mol. Biol. Rep. 9, 208–218. [Google Scholar]
- 25.Lan, T. H., DelMonte, T. A., Reischmann, K. P., Hyman, J., Kowalski, S. P., McFerson, J., Kresovich, S. & Paterson, A. H. (2000) Genome Res. 10, 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavell, A. C., Lydiate, D. J., Parkin, I. A., Dean, C. & Trick, M. (1998) Genome 41, 62–69. [PubMed] [Google Scholar]
- 27.Lagercrantz, U. (1998) Genetics 150, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devos, K. M., Brown, J. K. & Bennetzen, J. L. (2002) Genome Res. 12, 1075–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennetzen, J. L. & Kellogg, E. A. (1997) Plant Cell 9, 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swofford, D. L. (1999) PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) (Sinauer, Sunderland, MA).
- 31.Miura, A., Yonebayashi, S., Watanabe, K., Toyama, T., Shimada, H. & Kakutani, T. (2001) Nature 411, 212–214. [DOI] [PubMed] [Google Scholar]
- 32.Kapitonov, V. V. & Jurka, J. (1999) Genetica 107, 27–37. [PubMed] [Google Scholar]
- 33.Suzuki, G., Kai, N., Hirose, T., Fukui, K., Nishio, T., Takayama, S., Isogai, A., Watanabe, M. & Hinata, K. (1999) Genetics 153, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Terol, J., Castillo, M. C., Bargues, M., Perez-Alonso, M. & de Frutos, R. (2001) Mol. Biol. Evol. 18, 882–892. [DOI] [PubMed] [Google Scholar]
- 35.Marin, I. & Llorens, C. (2000) Mol. Biol. Evol. 17, 1040–1049. [DOI] [PubMed] [Google Scholar]
- 36.Noma, K., Ohtsubo, H. & Ohtsubo, E. (2000) DNA Res. 7, 291–303. [DOI] [PubMed] [Google Scholar]
- 37.Kentner, E. K., Arnold, M. L. & Wessler, S. R. (2003) Genetics 164, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson, D. G., Schulze, S. R., Sciara, E. B., Lee, S. A., Bowers, J. E., Nagel, A., Jiang, N., Tibbitts, D. C., Wessler, S. R. & Paterson, A. H. (2002) Genome Res. 12, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Langdon, T., Jenkins, G., Hasterok, R., Jones, R. N. & King, I. P. (2003) Genetics 163, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wicker, T., Guyot, R., Yahiaoui, N. & Keller, B. (2003) Plant Physiol. 132, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szabados, L., Kovacs, I., Oberschall, A., Abraham, E., Kerekes, I., Zsigmond, L., Nagy, R., Alvarado, M., Krasovskaja, I., Gal, M., et al. (2002) Plant J. 32, 233–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.