

Abstract
Background
In the pancreas, poorly differentiated neuroendocrine carcinomas include small cell carcinoma and large cell neuroendocrine carcinoma and are rare; data regarding their pathologic and clinical features are very limited.
Design
One hundred and seven pancreatic resections originally diagnosed as poorly differentiated neuroendocrine carcinomas were reassessed using the classification and grading (mitotic rate/Ki67 index) criteria put forth by the WHO in 2010 for the gastroenteropancreatic system. Immunohistochemical labeling for neuroendocrine and acinar differentiation markers was performed. Sixty-three cases were reclassified, mostly as well differentiated neuroendocrine tumor or acinar cell carcinoma, and eliminated. The clinicopathologic features and survival of the remaining 44 poorly differentiated neuroendocrine carcinomas were further assessed.
Results
The mean patient age was 59 years (range, 21–82) and the male/female ratio was 1.4. Twenty-seven tumors were located in the head of the pancreas, 3 in the body and 11 in the tail. The median tumor size was 4 cm (range, 2–18). Twenty-seven tumors were large cell neuroendocrine carcinomas and 17 were small cell carcinomas (mean mitotic rate, 37/10 HPF and 51/10 HPF; mean Ki67 index, 66% and 75%, respectively). Eight tumors had combined components, mostly adenocarcinomas. In addition, 2 tumors had components of well differentiated neuroendocrine tumor. Eighty-eight percent of the patients had nodal or distant metastatic disease at presentation and an additional 7% developed metastases subsequently. Follow-up information was available for 43 patients; 33 died of disease with a median survival of 11 months (range, 0–104); 8 were alive with disease, with a median follow-up of 19.5 months (range, 0–71). The 2-year and 5-year survival rates were 22.5% and 16.1%, respectively.
Conclusion
Poorly differentiated neuroendocrine carcinoma of the pancreas is a highly aggressive neoplasm, with frequent metastases and poor survival. Most patients die within less than a year. Most (61%) are large cell neuroendocrine carcinomas. Well differentiated neuroendocrine tumor and acinar cell carcinoma are often misdiagnosed as poorly differentiated neuroendocrine carcinoma, emphasizing that diagnostic criteria need to be clearly followed to ensure accurate diagnosis.
Keywords: Pancreas, neuroendocrine, carcinoma, small cell, large cell, high-grade, poorly differentiated
BACKGROUND
With the description of large cell neuroendocrine carcinomas, best characterized in the lung and subsequently recognized in the tubular gastrointestinal tract, the spectrum of poorly differentiated neuroendocrine carcinomas has been extended to include both small cell carcinoma and large cell neuroendocrine carcinoma [1]. Based on evidence from both pulmonary and extrapulmonary sites, it appears that these poorly differentiated neuroendocrine carcinomas are highly aggressive neoplasms with a propensity for early metastases and poor outcomes, whether presenting as a pure histologic pattern or as a component of a combined carcinoma [1–11]. Poorly differentiated neuroendocrine carcinomas, especially the small cell subtype, also typically have a favorable but short-lived response to platinum-based chemotherapy [1–11].
However, the literature on poorly differentiated neuroendocrine carcinomas primary in the pancreas is very limited, probably due to the rarity of this disease. Our current understanding of this neoplasm is mainly based on individual case reports, analyses of small series of cases, or opinions presented in textbooks [1, 11–19]. The diagnostic criteria have also been poorly defined, leading to inconsistent pathologic classification that has impacted clinical management [20]. In the current (2010) WHO classification [1], all pancreatic “high-grade neuroendocrine carcinomas”, along with their tubular gastrointestinal tract counterparts, are now included under the Grade-3 category, defined by a mitotic rate >20/10 high power fields or a Ki67 labeling index >20%. However, preliminary studies have shown that the Grade-3 category includes two distinct entities: 1) a more highly proliferative group of well differentiated neuroendocrine tumors (NETs); and 2) the true poorly differentiated neuroendocrine carcinomas, small cell carcinoma and large cell neuroendocrine carcinoma [21]. The first group appears to have a significantly more protracted clinical course [21] but less dramatic response to platinum-based chemotherapy [11] compared to poorly differentiated neuroendocrine carcinomas, and poorly differentiated neuroendocrine carcinomas have distinctive genetic alterations such as inactivation of TP53 and RB [22]. It is also the impression of many pathologists that true poorly differentiated neuroendocrine carcinomas usually have a very high Ki67 labeling index (typically above 50%), well above the 20% threshold in the current WHO classification [1] for a neuroendocrine neoplasm to be high-grade.
Here, we present one of the largest clinicopathologic studies of pancreatic poorly differentiated neuroendocrine carcinoma, with the aim of further defining the histologic features and clinical outcome in the era of the current WHO classification. Diagnostic issues revealed by our study are also explored.
MATERIALS AND METHODS
Surgical pathology databases of the authors’ 14 institutions were searched for cases with a diagnosis of pancreatic “poorly differentiated neuroendocrine carcinoma” “high-grade neuroendocrine carcinoma” “small cell carcinoma” or “large cell neuroendocrine carcinoma” from 1988 to 2012. One hundred and seven resections for which the slides were available were identified. Medical records including radiology and pathology reports were reviewed to: 1) obtain clinical data including age, gender, associated genetic syndromes, functionality, distant metastases and treatment modalities and 2) exclude the possibility of a metastasis from another organ or direct invasion from a contiguous site, particularly the ampulla of Vater [23]. A representative formalin-fixed paraffin-embedded tissue section of each case was immunolabeled using the standard avidin-biotin peroxidase method with antibodies against the neuroendocrine markers chromogranin A (DakoCytomation, Carpinteria, CA) and synaptophysin (DakoCytomation, Carpinteria, CA), the acinar differentiation markers trypsin (Biodesign, Memphis, TN) and chymotrypsin (Biodesign, Memphis, TN), and Ki67/MIB1 (DakoCytomation, Carpinteria, CA). Five cases revealed sheets of small, round, monotonous cells with a syncytial appearance and small pseudorosettes; these cases were also immunolabeled for CD99 (DakoCytomation, Carpinteria, CA) to explore the alternative diagnosis of primitive neuroectodermal tumor (PNET). The slides were then reassessed using the diagnostic and grading criteria put forth in the current (2010) WHO classification of neuroendocrine neoplasms of the digestive system [1]. Specifically, small cell carcinomas were characterized by sheets or nests of relatively small cells with a high nucleus-to-cytoplasm ratio, hyperchromatic and finely granular chromatin, inconspicuous nucleoli, and nuclear molding. Large cell neuroendocrine carcinomas displayed neuroendocrine architectural patterns (i.e., organoid or nested structures, trabeculae, peripheral palisading, or rosettes) and were composed of monotonous, round to polygonal cells with moderate amounts of cytoplasm. The nuclei were round with vesicular chromatin or prominent nucleoli. For large cell neuroendocrine carcinomas, diagnostic criteria included labeling with at least one neuroendocrine marker (chromogranin or synaptophysin, see below). Mitotic rate was determined by counting 50 high power fields (at 400X on an Olympus microscope = 0.45mm2) and averaged to 10 high power fields. The Ki67 labeling index was determined by manual counting of 1000 cells using captured images of the proliferation hot-spots, as described previously [24].
Four tumors that did not label with any of the neuroendocrine or acinar markers and did not have small cell carcinoma morphology were excluded for further analysis. Seventeen tumors immunolabeled with trypsin and/or chymotrypsin in greater than 25% of the neoplastic cells and were thus reclassified as mixed acinar-neuroendocrine carcinoma (12 tumors) or pure acinar cell carcinoma (5 tumors) [25]. Of the remaining cases, forty-two were excluded due to a well differentiated NET morphology or a low mitotic rate (≤ 20 per 10 high power fields) and low Ki67 labeling index (≤ 20%) (Table 1).
Table 1.
Case Selection
 |
For the remaining 44 cases, the following histopathologic information was recorded: tumor size; cell type; growth pattern; necrosis; lymphovascular and perineural invasion; margin status; extension into the peripancreatic soft tissue, duodenal wall, or other adjacent organs; and number of involved lymph nodes. Also tabulated was the presence of non-neuroendocrine components such as ductal adenocarcinoma or squamous cell carcinoma. The carcinomas were staged following the criteria of the 7th edition American Joint Cancer Committee (AJCC) cancer staging manual [26]. Overall survival data were obtained from hospital records or the United States Social Security Death Index. Survival analysis was performed using the Kaplan-Meier method and log-rank (Mantel-Cox) comparison, which was performed with StatView software version 5.0 (SAS Institute, Cary, NC) and/or MedCalc V12.7.5 (MedCalc Software bvba, Ostend, Belgium).
RESULTS
We identified a total of 44 cases (43 surgical specimens, 1 autopsy) that met the criteria for poorly differentiated neuroendocrine carcinoma outlined above.
Clinical Features
The mean patient age was 59 years (range, 21 to 82) with a male to female ratio of 1.4. The age and sex of the patients with large cell neuroendocrine carcinoma (mean age, 60.4; male to female ratio = 1.9) were similar to those of patients with small cell carcinoma (mean age, 57.5; male to female ratio = 0.9; p=0.57 [student’s t test] and p=0.34 [Fisher’s exact test], respectively (Table 2). None of the patients had a known genetic syndrome except for one patient who carried a germline BRCA1 mutation. One patient had a history of breast carcinoma and melanoma, and another had a gastric adenocarcinoma. Of 35 patients with information available, only one had an increased serum hormone level (hyperinsulinism). Thirty patients underwent pancreatoduodenectomy and 11 underwent distal pancreatectomy and splenectomy. In 3 patients, the type of surgery was not known. Adjuvant therapy information was available for only 14 patients and was variable, although the majority of recent patients received cisplatin-based chemotherapy and/or radiation therapy.
Table 2.
Clinicopathologic Features of Small Cell and Large Cell Subtypes
| Small Cell (n=17, 39%) |
Large Cell (n=27, 61%) |
|
|---|---|---|
| Mean Age (range) |
57.5 (21–82) |
60.4 (29–79) |
| Male:Female | 0.9 | 1.9 |
| Tumor Location | ||
| Head | 12 | 15 |
| Body | 1 | 2 |
| Tail | 3 | 8 |
| Median Tumor Size (cm) (range) | 3.5 (2.3–18) | 4 (2–14.5) |
| Mitoses/10HPF*(range) | 51 (21–92) | 37 (21–83) |
| Ki67 Labeling Index (range) | 75% (50–98%) | 66% (40–95%) |
| T Stage | ||
| T1 | 0 | 1 |
| T2 | 1 | 1 |
| T3 | 12 | 20 |
| Only Lymph Node Metastasis at Presentation | 10 | 16 |
| Any Metastasis at Presentation | ||
| Present | 14 | 23 |
| Absent | 2 | 3 |
| Median Survival (Months) (95% CI) | 6 (5, 20) | 16 (11, 18) |
| 2-year DSS** | 24.2% ± 11.7% | 22.2% ± 8.7% |
| 5-year DSS** | 16.2% ± 10.2% | 16.6% ± 8.1% |
HPF High power field
DSS disease-specific survival (expressed as mean ± standard error)
Pathologic Features
Twenty-seven tumors were located in the head of the pancreas, 3 in the body and 11 in the tail. Tumor location information was not available for 3 patients. Tumor size varied from 2 – 18 cm (median, 4 cm). Grossly, the tumors were tan-red or yellowish, solid, and were generally described as “vaguely nodular”, “lobulated” or “relatively circumscribed” (Figure 1). Hemorrhage was common and necrosis was occasionally noted.
Figure 1.

This large, tan-yellow, vaguely nodular, fleshy mass with areas of necrosis suggests that the lesion is not a ductal adenocarcinoma, and the differential diagnoses include poorly differentiated neuroendocrine carcinoma as well as acinar cell carcinoma.
Microscopically, twenty-seven carcinomas were large cell neuroendocrine carcinoma, in which the growth pattern was more variable. Diffuse, organoid/nested, trabecular (Figure 2A) and peripheral palisading growth patterns, often intermingled in various proportions, were seen in most of these carcinomas. In some of the large cell neuroendocrine carcinomas, pseudopapillae, composed of viable tumor cells surrounding fibrovascular cores, were seen at the periphery of necrotic areas. Rarely, tumors displayed glandular formations (Figure 2B). Apoptotic cells and mitotic figures were abundant, but mitotic figures in the large cell neuroendocrine carcinomas were not as numerous as in the small cell carcinomas, averaging 37 per 10 high power fields (range, 21–83). The average Ki67 labeling index in the large cell neuroendocrine carcinomas was 66 % (range, 40–95%). The remaining 17 cases were small cell carcinomas displaying predominantly a diffuse, sheet-like growth pattern with confluent areas of necrosis and entrapment of pancreatic parenchyma. Scattered tumor giant cells with hyperchromatic, bizarre nuclei (Figure 3) or rosettes were also noted in some tumors. Extensive apoptosis was present in all small cell carcinomas, and mitoses averaged 51 per 10 high power fields (range, 21–92). The average Ki67 labeling index was 75% (range, 50–98%).
Figure 2.
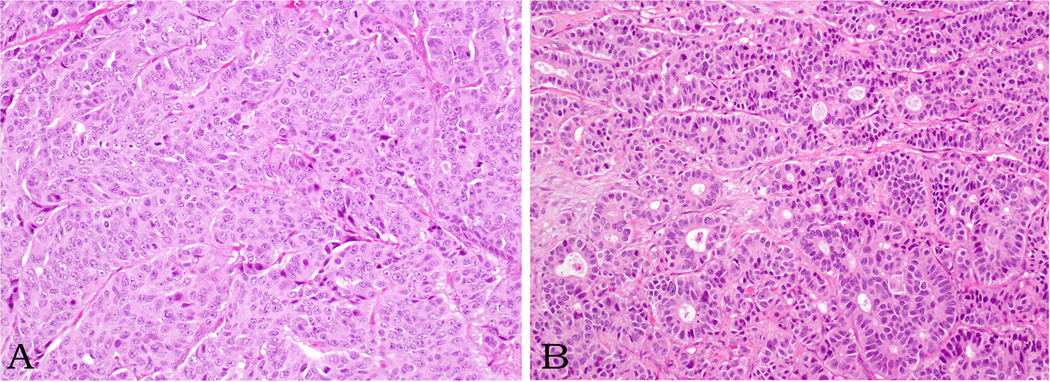
Large cell neuroendocrine carcinomas reveal various growth patterns (trabecular-A- and glandular-B- patterns are depicted here). The cells are often round to polygonal and the nuclei have either vesicular chromatin or prominent nucleoli. Note multiple mitotic figures.
Figure 3.

Small cell carcinoma. The majority of the tumor cells are relatively small with a high nucleus-to-cytoplasm ratio, hyperchromatic nuclei, and nuclear molding. However, scattered giant tumor cells with bizarre, large nuclei are also common.
Although the majority (36 of the 44) of the poorly differentiated neuroendocrine carcinomas was pure, 8 had combined components. An associated conventional ductal adenocarcinoma was present in 6 cases. The poorly differentiated neuroendocrine carcinoma was large cell subtype in 5 of these cases and small cell subtype in one. While the poorly differentiated neuroendocrine carcinoma and ductal adenocarcinoma components were sharply segregated in two cases, they merged intimately in four (Figure 4). Large cell neuroendocrine carcinoma arose in association with an intraductal papillary mucinous neoplasm (IPMN), pancreatobiliary type, with high-grade dysplasia in one case. Another case revealed sheets of small cell carcinoma admixed with squamous cell carcinoma (Figure 4).
Figure 4.

Combined neuroendocrine carcinomas: In 4 cases, individual ductal adenocarcinoma glands are intimately admixed with nests of neuroendocrine tumor cells (A). The ductal adenocarcinoma glands express glycoprotein markers (CEA is shown here, B). Conventional ductal adenocarcinoma and neuroendocrine carcinoma components are sharply segregated in 2 cases (C). One small cell carcinoma exhibits widespread squamous differentiation (D).
In addition, two cases had regions demonstrating features of well differentiated NET, composed of cells with moderate amounts of cytoplasm and round, regular nuclei with stippled chromatin (Figure 5A). In those sharply segregated well differentiated foci, the mitotic rate was less than 1 per 10 high power fields and the Ki67 labeling index was 1% and 5%, respectively, as opposed to frequent mitotic figures, necrosis and Ki67 labeling indices of 50% and 55%, respectively, in the associated poorly differentiated neuroendocrine carcinoma components, both of which were large cell neuroendocrine carcinomas (Figure 5B).
Figure 5.
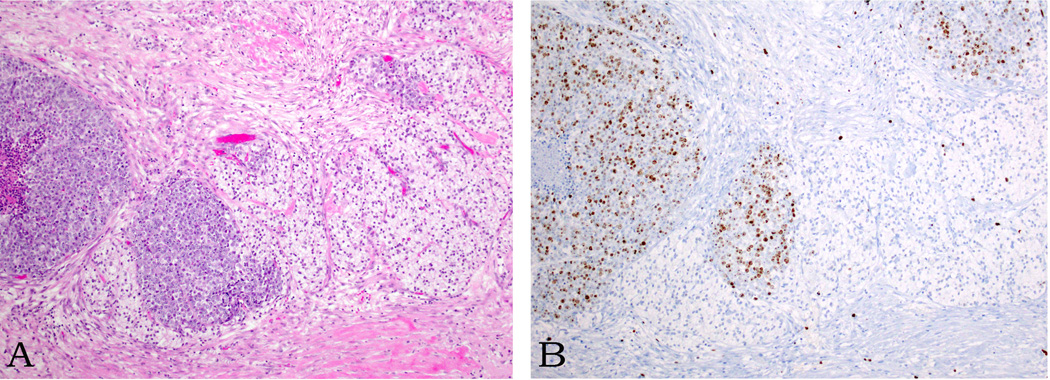
Sharply segregated well differentiated neuroendocrine tumor regions, composed of cells with clear cytoplasm and monotonous, round nuclei, exhibit a nested growth pattern (A). A Ki67 stain shows a labeling index of 1% in this component (as opposed to 50% in the poorly differentiated neuroendocrine carcinoma component, B).
Perineural invasion and angiolymphatic invasion were identified in the majority of the carcinomas (71% and 79%, respectively). All but 3 carcinomas extended beyond the pancreas (pT3), usually into the peripancreatic soft tissue but also into the duodenum in 5, portal vein in 3, splenic artery and vein in one and spleen in one. In addition, 17 of 30 cases had one or more positive surgical margins: of those with information available, the posterior-inferior (uncinate) margin was involved in 9 cases, the pancreatic neck (duct) margin in 3 cases, and the proximal margin in 1 case.
Although high-grade pancreatic intraepithelial neoplasia (PanIN-3) was identified in all 6 cases with an associated adenocarcinoma component, pathologic changes in the adjacent pancreatic parenchyma, when present, were largely limited to foci of chronic pancreatitis. It should be noted, however, that in many cases little uninvolved pancreas was available for study.
Metastases to regional lymph nodes and liver were common at presentation. Twenty-six patients had metastases to regional lymph nodes only; 8 had metastases to both regional lymph nodes and to the liver; and 3 had metastases to the liver only. Fifteen patients, including 3 patients without metastases at the time of diagnosis, developed (additional) metastases as the disease progressed. The majority of subsequent metastases were to the liver; however, brain, lung, mediastinum, adrenal gland and kidney were involved in rare cases.
Immunohistochemistry
Immunohistochemically, all of the poorly differentiated neuroendocrine carcinomas labeled with at least one neuroendocrine marker (chromogranin and/or synaptophysin) in >10% of the tumor cells. There were 7 cases that were positive for only one neuroendocrine marker, emphasizing the need for both stains. Synaptophysin was more sensitive and was expressed in 95% of the cases, and chromogranin was expressed in 84%. In most cases, 30% to 75% of the tumor cells were immunolabeled with neuroendocrine marker(s) moderately to strongly. The labeling was often patchy.
The acinar markers were either negative or labeled rare, scattered cells in the poorly differentiated neuroendocrine carcinomas. None of the cases tested labeled with CD99.
Clinical Course
Follow-up information was available for 43 of the 44 patients. The follow-up period ranged from 0 to 104 months (median 9 months), and 35 patients were followed to the time of their death. At the last follow-up, 33 patients had died of disease with a median survival of 11 months (range, 0–104 months). One patient died of surgical complications and another patient with disease died in an accident at 8 months. Eight patients were alive with disease at a median follow-up of 19.5 months (range, 0–71 months). The 2-year and 5-year disease-specific survival rates were 22.5%±6.9% and 16.1%±6.3% (mean±standard error), respectively (Figure 6).
Figure 6.

The Kaplan-Meier analysis of the overall disease-specific survival of all poorly differentiated neuroendocrine carcinomas.
The median survival of the patients with large cell neuroendocrine carcinoma (n=27) was longer than that of the patients with small cell carcinoma (n=17) (16 vs. 6 months) with overlapping 95% confidence interval; however, 2-year and 5-year disease-specific survival rates (24.2% vs. 22.2% and 16.2% vs. 16.6%, respectively) were equivalent (Table 2). Thus, the difference in survival between these 2 groups was not statistically significant (Log rank p=0.3653). Likewise, the survival difference between pure and combined poorly differentiated neuroendocrine carcinomas (median survival, 12 vs. 20 months; 2-year and 5-year disease-specific survival rates, 21.8% vs. 25.4% and 9.1% vs. 0%, respectively) was not statistically significant (Log rank p=0.722).
By Kaplan-Meier survival analysis, none of the prognostic factors tested (age, gender, tumor location, tumor size, lymphovascular and perineural invasion, margin status, T stage, lymph node metastasis), was significantly associated with survival (Table 3). In particular, the patients with a Ki67 labeling index <55% (n = 12, mean index = 43%) did not live longer than those with a Ki67 labeling index ≥55% (n = 26, mean index = 79%).
Table 3.
Survival Analysis of Prognostic Factors
| Number of Cases |
Median Survival (Months) (95% CI) |
p value (Log rank) |
||
|---|---|---|---|---|
| Age | ||||
| <40 | 6 | 15.5 (5, 104) | 0.0944 | |
| 40–65 | 18 | 20 (9, 50) | ||
| >65 | 19 | 11 (5, 17) | ||
| Gender | ||||
| M | 25 | 16 (11, 24) | 0.24 | |
| F | 18 | 9 (5, 20) | ||
| Tumor Location | ||||
| Head | 27 | 12 (5, 18) | 0.858 | |
| Body | 3 | 20 (5, -) | ||
| Tail | 11 | 13 (9, 50) | ||
| Tumor Size | ||||
| <4 cm | 18 | 13.5 (6, 20) | 0.9974 | |
| 4–10 cm | 19 | 11 (5, 45) | ||
| >10 cm | 3 | 13 (6, 50) | ||
| Cell Subtype | ||||
| Small cell | 17 | 6 (5, 20) | 0.3653 | |
| Large cell | 27 | 16 (11, 18) | ||
| Tumor Type | ||||
| Pure | 34 | 12 (6, 16) | 0.722 | |
| Combined | 10 | 20 (5, 50) | ||
| Mitoses/10HPF* | ||||
| >20–≤40 | 18 | 11 (5, 18) | 0.9087 | |
| >40–≤60 | 8 | 16 (15, 17) | ||
| >60 | 10 | 9 (6, 20) | ||
| Ki67 Labeling Index | ||||
| <55% | 12 | 13 (6, 20) | 0.4756 | |
| ≥55% | 26 | 16 (6, 24) | ||
| Lymphovascular Invasion | ||||
| Present | 23 | 11 (6, 24) | 0.5343 | |
| Absent | 6 | 15 (6, 20) | ||
| Perineural Invasion | ||||
| Present | 20 | 11 (6, 24) | 0.8463 | |
| Absent | 8 | 11 (6, 20) | ||
| Margin | ||||
| Positive | 17 | 11 (5, 20) | 0.8344 | |
| Negative | 13 | 15 (9, 20) | ||
| T stage | ||||
| T1 | 1 | 0.7504 | ||
| T2 | 2 | 16 (6, 20) | ||
| T3 | 32 | 13 (6, 18) | ||
| Any Metastasis at Presentation | ||||
| Yes | 37 | 13 (6, 18) | 0.1519 | |
| No | 5 | 20 (6, 104) | ||
| AJCC Stage (2010) at Presentation | ||||
| II | 28 | 17 (9, 20) | 0.1348 | |
| IV | 15 | 11 (5, 16) | ||
HPF High power field
DISCUSSION
The current WHO classification of gastroenteropancreatic neuroendocrine neoplasms, which is based on the 2007 proposals from the European Neuroendocrine Tumor Society [27], recommends the use of proliferative rate-based grading to separate the neoplasms into three grades, with the highest grade (Grade-3) stated to correspond to poorly differentiated neuroendocrine carcinomas (small cell carcinoma and large cell neuroendocrine carcinoma). Although several recent studies have verified the prognostic significance of the WHO system [28, 29], the threshold of 20 mitoses per 10 high power fields or 20% Ki67 labeling index used to separate Grade-2 (i.e., well differentiated NETs) from Grade-3 has been questioned. Anecdotal experience suggests some NETs that are histologically well differentiated may have a proliferative rate somewhat in excess of these cut-points [30]. The current study was conducted to specifically evaluate those pancreatic neuroendocrine neoplasms that have both histologic features of a poorly differentiated carcinoma and a high proliferative rate (both mitotic rate and Ki67 index above the threshold) – the “true” poorly differentiated neuroendocrine carcinomas. Cases with a well differentiated morphology but an elevated proliferative rate, which fell in the Grade-3 category solely on the basis of the Ki67 index, were therefore excluded. The term “poorly differentiated” neuroendocrine carcinoma is used deliberately to specify this group, in distinction to “high-grade” neuroendocrine carcinoma, which includes all neoplasms currently within the WHO Grade-3 category.
This study constitutes the largest series of histologically confirmed; primary pancreatic poorly differentiated neuroendocrine carcinoma, a neoplasm whose pathologic features have not been analyzed in detail. A larger collection of Grade-3 primary pancreatic neuroendocrine neoplasms was reported by Sorbye et al. (“The Nordic neuroendocrine carcinoma study”) [11]; however, their study mainly focused on treatment aspects without a detailed description of the pathologic characteristics. Also, some of their findings, such as only 30% of their pancreatic high-grade neuroendocrine carcinomas had a Ki67 index ≥ 55% (vs. 60% in our study), suggest that some of their cases were likely well differentiated NETs with an elevated proliferative rate, rather than poorly differentiated neuroendocrine carcinomas as defined in our study.
Most of the patients in this series were in their late 50s and there was a slight male predilection (male/female ratio of 1.4). In contrast to well differentiated NETs, the carcinomas were not associated with hereditary syndromes and were clinically non-functional. Most arose in the head of the pancreas and presented as a large (median tumor size of 4 cm), relatively circumscribed, solid mass. The small cell subtype was less common than the large cell subtype (39% vs. 61%).
Review of these cases culled from the archives of many institutions clearly demonstrates that the accurate diagnosis of pancreatic poorly differentiated neuroendocrine carcinoma can be challenging. Entities prominent in the differential diagnosis include well differentiated NETs, acinar cell carcinoma, mixed acinar-neuroendocrine carcinomas [25, 31, 32], and primitive neuroectodermal tumor [33]. The histologic features of these entities can overlap significantly, but immunohistochemical staining can usually establish the correct diagnosis if all possibilities are considered. Table 4 includes some of the distinguishing histologic findings; along with the results of targeted immunohistochemical labeling that can be used to correctly classify these neoplasms.
Table 4.
Differential Diagnosis of Pancreatic Poorly Differentiated Neuroendocrine Carcinoma
| Poorly Differentiated Neuroendocrine Carcinoma |
Well Differentiated Neuroendocrine Tumor |
Acinar Cell Carcinoma* | Primitive Neuroectodermal Tumor |
|
|---|---|---|---|---|
| Histology | • Markedly cellular • Solid and nesting growth patterns • Acinar (glandular) units or pseudorosettes may be present. • Substantial necrosis |
• Less cellular • Various organoid growth patterns • Limited or no necrosis |
• Markedly cellular • Solid and acinar growth patterns • Necrosis may be extensive. • Eosinophilic granular cytoplasm |
• Markedly cellular • Diffuse growth pattern • Pseudorosettes may be present. |
| Nuclei | • Hyperchromatic nuclei and inconspicuous nucleoli (small cell) • Vesicular chromatin or prominent nucleoli (large cell) |
• Monotonous, round nuclei with vesicular chromatin • Nucleoli may be seen. |
• Monotonous nuclei • Solitary large nucleoli |
• Small, round monotonous nuclei • Inconspicuous nucleoli |
| Mitoses/10 HPF** | High (by definition > 20 [1]; usually > 30) |
Low to intermediate (by definition ≤ 20 [1]) |
Variable but usually high (> 20) |
Variable but usually high (> 20) |
| Ki67 Labeling Index | High (by definition > 20%; usually > 40%) | High (by definition ≤ 20%) | Variable but usually high (> 20%) | Variable but usually high (> 20%) |
| Immunohistochemistry | ||||
| Keratins | Positive | Positive | Positive | Usually positive [33] |
| Trypsin/Chymotrypsin | Rare, scattered cells may be positive. | Rare, scattered cells may be positive. | Positive | Negative |
| Chromogranin | Positive, may be weak or focal | Positive, usually strongly | Rare, scattered cells may be positive. | Usually negative [33] |
| Synaptophysin | Positive, may be weak or focal | Positive, usually strongly | Rare, scattered cells may be positive. | Usually negative [33] |
| CD99 | Negative | May be positive (45% of cases) [38]. | Negative | Positive (diffuse, membranous) |
Mixed acinar-neuroendocrine carcinomas exhibit both acinar and neuroendocrine features.
HPF High power field
In our study, the potential for poorly differentiated neuroendocrine carcinomas to be misdiagnosed as well differentiated NETs was not assessed, since our search parameters targeted only cases originally diagnosed as poorly differentiated or high grade. However, in our experience, this scenario is not common, since the necrosis, nuclear atypia and high proliferative rate of poorly differentiated neuroendocrine carcinomas are usually readily recognizable. The converse diagnostic confusion is much more common; indeed, in our study, forty-two cases originally diagnosed as poorly differentiated neuroendocrine carcinomas proved to be well differentiated NETs on our review. Well differentiated NETs can show nuclear pleomorphism, sometimes marked [34], and features such as a diffuse or markedly infiltrative growth pattern and necrosis can also be identified [19]. On casual examination, these falsely suggest a poorly differentiated neoplasm but it is the proliferative rate that defines a neuroendocrine neoplasm to be poorly differentiated [1]. If careful mitotic count and immunolabeling for Ki67 are not performed, misclassification can occur, which can have profound therapeutic consequences [35]. Recently, it has become clear that some well differentiated NETs may have a proliferative rate in excess of the threshold established by the WHO for high-grade (Grade-3) neuroendocrine carcinomas [21, 30]. Although detailed pathological studies of this group are still underway, the available evidence suggests that these NETs are genetically and biologically more closely related to low- and intermediate-grade (Grade-1 and -2) NETs than to poorly differentiated neuroendocrine carcinomas [36]. The “Nordic neuroendocrine carcinoma study” has also shown that “high-grade” neuroendocrine carcinomas with a Ki67 labeling index less than 55% did not respond to platinum-based chemotherapy, in contrast to those with Ki67 labeling index greater than 55% [11], supporting the concept that the tumors that are at the lower end of the Grade-3 range are in fact well differentiated NETs with an elevated proliferative rate (or “high-grade well differentiated neuroendocrine tumors”). Of note, there was no survival difference between poorly differentiated neuroendocrine carcinomas with Ki67 labeling index < 55% vs. ≥ 55% in our study (Table 3), as we excluded the highly proliferative well differentiated NETs that likely impacted the outcome and therapeutic responsiveness of the < 55% group in that study. Furthermore, our group has recently demonstrated that the outcome of well differentiated NETs with an elevated proliferative rate is worse than that of intermediate-grade well differentiated NETs, but not a poor as the poorly differentiated neuroendocrine carcinomas (5-year disease-specific survival rates of 22% vs. 60.5% vs. 17% months, respectively) [21]. Obviously, careful assessment of proliferative rate is necessary to separate these tumor types, and recognition of a more organoid growth pattern, more limited necrosis, and more classic neuroendocrine nuclear features would favor a diagnosis of well differentiated NET.
Much has been written about the distinction of acinar cell carcinomas and the related mixed acinar carcinomas (mixed acinar-neuroendocrine carcinoma in particular) from well differentiated NETs [19, 37]. The presence of acinar formations, basal nuclear polarization, granular eosinophilic cytoplasm, and solitary large nucleoli all favor a diagnosis of an acinar neoplasm. Confirmation of acinar differentiation with immunohistochemical stains for trypsin and chymotrypsin is important to establish the diagnosis. Another feature more typical of acinar cell carcinoma and mixed acinar carcinomas, relative to well differentiated NETs, is a high proliferative rate, since these acinar neoplasms commonly (but not always) have a mitotic rate greater than 20 per 10 high power fields and a Ki67 labeling index of 30–60%. As our study demonstrates; however, the distinction of acinar cell carcinoma and mixed acinar carcinomas from poorly differentiated neuroendocrine carcinomas is even more problematic. Both entities usually have a high proliferative rate, acinar cell carcinomas can have a diffuse growth pattern and can lack the distinguishing features mentioned above, and large cell type poorly differentiated neuroendocrine carcinomas can have prominent nucleoli. In our study, seventeen cases originally diagnosed as poorly differentiated neuroendocrine carcinomas proved to be acinar cell carcinomas or mixed acinar-neuroendocrine carcinomas on our review. Although cytologic features suggesting acinar differentiation were found in some cases, there were also cases in which no distinguishing characteristics could be recognized, and the finding of immunolabeling for chromogranin or synaptophysin in some cases wrongly suggested a pure poorly differentiated neuroendocrine carcinoma. In all of these cases, immunohistochemical staining for trypsin and chymotrypsin had not been performed, and the correct diagnosis was readily established once these stains were completed. Given the rarity of primary pancreatic poorly differentiated neuroendocrine carcinomas, relative to acinar neoplasms, it is thus recommended that a diagnosis of poorly differentiated neuroendocrine carcinoma should not be rendered unless acinar differentiation has been excluded immunohistochemically.
A final diagnostic consideration is primitive neuroectodermal tumor, especially in younger patients. Primitive neuroectodermal tumors generally have small, round, monotonous nuclei with inconspicuous nucleoli and scant cytoplasm, although pancreatic examples can be more epithelioid and can express keratin strongly [33]. The proliferative rate is variable, but can overlap with that of poorly differentiated neuroendocrine carcinoma. In cases with suggestive histologic features (or in younger patients), immunolabeling for CD99 can be helpful, since most primitive neuroectodermal tumors show strong, diffuse membranous staining. Weak or focal staining may be seen in other tumors [38], but in the cases of poorly differentiated neuroendocrine carcinoma with suggestive morphology in the current study, CD99 stains were consistently negative. In questionable cases, molecular studies can be performed to further explore this diagnosis.
Consistent with previous reports, the pancreatic poorly differentiated neuroendocrine carcinomas in this study behaved aggressively, commonly featuring vascular and perineural invasion and lymph node metastases, with dismal survival. The overall median survival was only 12 months and seventy-seven percent of the patients died of disease after a mean follow-up of less than one year, despite presenting with resectable disease. Similar to the recent European multicenter study [11], there was no difference in survival among the morphologic subtypes of poorly differentiated neuroendocrine carcinoma (small cell carcinoma vs. large cell neuroendocrine carcinoma).
Eight (18%) of our cases contained neoplastic elements of non-neuroendocrine lineage, usually ductal adenocarcinomas or a precursor thereof (intraductal papillary mucinous neoplasm). The presence of an associated adenocarcinoma, IPMN, or squamous cell carcinoma component also was not associated with different survival. In most cases these elements were mixed throughout the tumor, and although molecular studies to prove their relationship have yet to be performed, we suspect the glandular or squamous components of these combined carcinomas share a common histogenesis. Similar combinations exist in poorly differentiated neuroendocrine carcinomas of other organs, such as the lung [39] and large bowel [8], where non-neuroendocrine elements are found in over 40% of carefully examined cases. Insufficient treatment response data exist to determine whether the biology of the poorly differentiated neuroendocrine carcinoma component should dictate the choice of chemotherapy in the pancreas, but in other sites, these combined carcinomas are generally managed as neuroendocrine carcinomas.
Since any survival differences between the different morphologic types of pancreatic poorly differentiated neuroendocrine carcinoma are minimal, an obvious question is whether there is a need to separate small cell carcinoma from large cell neuroendocrine carcinoma for clinical management. Currently, we believe that the evidence is still insufficient to justify combining the subtypes into a single entity, despite the admitted difficulty of distinguishing them in cases with borderline nuclear features. Although the management of large cell neuroendocrine carcinomas of several anatomic sites has been extrapolated from that of small cell carcinoma, the evidence that platinum-based chemotherapy is the optimal choice for large cell neuroendocrine carcinoma is not compelling [40]. The characteristic dramatic responses seen in small cell carcinomas are not common in large cell neuroendocrine carcinomas, and randomized clinical trials to prove the benefit of platinum-based chemotherapy have yet to be conducted.
Another goal of this study was to reassess the mitotic rate and Ki67 index thresholds used to separate Grade-2 well differentiated NETs from Grade-3 neuroendocrine carcinomas, since the cut-points of 20 mitoses per 10 high power fields and Ki67 of 20% have been questioned. Our results demonstrated that when only the pancreatic neuroendocrine neoplasms with poorly differentiated morphologic features are considered, both the mitotic rate (average, 51 and 37 per 10 high power fields for small cell neuroendocrine carcinoma and large cell neuroendocrine carcinoma, respectively) and Ki67 index (average, 75% and 66% for small cell neuroendocrine carcinoma and large cell neuroendocrine carcinoma, respectively) are much higher than the WHO-recommended thresholds for the Grade-3 category. Thus, if the intent of the Grade-3 category is to capture only the poorly differentiated neuroendocrine neoplasm, the cut-points for this grade could be raised. Whether the well differentiated NETs that currently fall in the Grade-3 category should continue to be separated from Grade-2 well differentiated NETs requires further study.
Finally, the potential relationship of poorly differentiated neuroendocrine carcinomas to well differentiated NETs has been a subject of debate. Although it is rare to encounter a poorly differentiated neuroendocrine carcinoma arising in association with a well differentiated NET, neoplastic progression of well differentiated NETs has been proposed as the origin of some poorly differentiated neuroendocrine carcinomas of the pancreas [19, 41]. However, it has been shown recently that pancreatic small cell carcinoma and large cell neuroendocrine carcinoma are genetically related entities and that the genetic changes frequently seen in these neoplasms, such as co-inactivation of the TP53 and the Rb/p16 cell cycle pathways, are infrequently observed in pancreatic well differentiated NETs [22]. Conversely, inactivating mutations in DAXX and ATRX and mutations in MEN1 are exclusively found in pancreatic well differentiated NETs but not in small cell carcinoma or large cell neuroendocrine carcinoma of the pancreas [22]. While these findings suggest that poorly differentiated neuroendocrine carcinoma is a genetically distinct entity from well differentiated NETs, it does not fully rule out the possibility that pancreatic well differentiated NETs may rarely transform genetically into poorly differentiated neuroendocrine carcinomas [19]. In fact, in our study, 5% of the poorly differentiated neuroendocrine carcinomas arose in a background of well differentiated (WHO Grade-1 and -2) NETs. Although the frequency with which pancreatic well differentiated NETs may transform to true poorly differentiated neuroendocrine carcinomas is unknown, most observations to date suggest it is an uncommon pathway for the development of pancreatic poorly differentiated neuroendocrine carcinoma. Progression of Grade-1 well differentiated NETs to higher grade well differentiated NETs does occur with greater frequency; however, and this topic is the subject of ongoing research [36].
In summary, this study illustrates the pathologic characteristics of diagnostically challenging primary pancreatic poorly differentiated neuroendocrine carcinomas. These are uncommon, highly aggressive neoplasms that are often locally advanced at the time of diagnosis and have a dismal prognosis. Distinction of these carcinomas from their mimics and application of current grading parameters will allow more consistent treatment decisions to help better understand their biology and optimal management.
Acknowledgement
The authors thank Allyne Manzo and Lorraine Biedrzycki for assistance with the figures.
Footnotes
Ralph H. Hruban and N. Volkan Adsay have a grant from National Institutes of Health (#5P50 CA62924); Zhaohai Yang has received honoraria from Up To Date, Inc; Wendy L. Frankel has been an alliance for Clinical Trials in Oncology and received honoraria from Frozen Section Library: Pancreas: Springer; David S. Klimstra was on the speaker’s bureau for Novartis and has received honoraria from Up To Date. For the remaining authors none were declared.
This study was presented in part at the 102nd annual meeting of the United States and Canadian Academy of Pathology in Baltimore, MD, in March 2013, and supported in part by the National Cancer Institute Specialized Program in Research Excellence (SPORE) CA62924.
REFERENCES
- 1.Bosman FT, et al. Neuroendocrine tumors of the GI tract. In: Bosman FT, et al., editors. WHO classification of tumors of Digestive System. Lyon: WHO Press; 2010. [Google Scholar]
- 2.Bernick PE, et al. Neuroendocrine carcinomas of the colon and rectum. Dis Colon Rectum. 2004;47(2):163–169. doi: 10.1007/s10350-003-0038-1. [DOI] [PubMed] [Google Scholar]
- 3.Brenner B, et al. Small-cell carcinomas of the gastrointestinal tract: a review. J Clin Oncol. 2004;22(13):2730–2739. doi: 10.1200/JCO.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 4.Travis WD, et al. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. World Health Organization Classification of Tumours 2004: IARC Publications [Google Scholar]
- 5.Lee SS, et al. Extrapulmonary small cell carcinoma: single center experience with 61 patients. Acta Oncol. 2007;46(6):846–851. doi: 10.1080/02841860601071893. [DOI] [PubMed] [Google Scholar]
- 6.Faggiano A, et al. Pulmonary and extrapulmonary poorly differentiated large cell neuroendocrine carcinomas: diagnostic and prognostic features. Cancer. 2007;110(2):265–274. doi: 10.1002/cncr.22791. [DOI] [PubMed] [Google Scholar]
- 7.Kang H, et al. Rare tumors of the colon and rectum: a national review. Int J Colorectal Dis. 2007;22(2):183–189. doi: 10.1007/s00384-006-0145-2. [DOI] [PubMed] [Google Scholar]
- 8.Shia J, et al. Is nonsmall cell type high-grade neuroendocrine carcinoma of the tubular gastrointestinal tract a distinct disease entity? Am J Surg Pathol. 2008;32(5):719–731. doi: 10.1097/PAS.0b013e318159371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walenkamp AM, Sonke GS, Sleijfer DT. Clinical and therapeutic aspects of extrapulmonary small cell carcinoma. Cancer Treat Rev. 2009;35(3):228–236. doi: 10.1016/j.ctrv.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Carbonero R, et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): results from the National Cancer Registry of Spain (RGETNE) Ann Oncol. 2010;21(9):1794–1803. doi: 10.1093/annonc/mdq022. [DOI] [PubMed] [Google Scholar]
- 11.Sorbye H, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann Oncol. 2013;24(1):152–160. doi: 10.1093/annonc/mds276. [DOI] [PubMed] [Google Scholar]
- 12.Corrin B, et al. Oat cell carcinoma of the pancreas with ectopic ACTH secretion. Cancer. 1973;31(6):1523–1527. doi: 10.1002/1097-0142(197306)31:6<1523::aid-cncr2820310633>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 13.Reyes CV, Wang T. Undifferentiated small cell carcinoma of the pancreas: a report of five cases. Cancer. 1981;47(10):2500–2502. doi: 10.1002/1097-0142(19810515)47:10<2500::aid-cncr2820471032>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 14.Hobbs RD, et al. Hypercalcemia in small cell carcinoma of the pancreas. Cancer. 1984;53(7):1552–1554. doi: 10.1002/1097-0142(19840401)53:7<1552::aid-cncr2820530722>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 15.Morant R, Bruckner HW. Complete remission of refractory small cell carcinoma of the pancreas with cisplatin and etoposide. Cancer. 1989;64(10):2007–2009. doi: 10.1002/1097-0142(19891115)64:10<2007::aid-cncr2820641006>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 16.O'Connor TP, et al. Small cell undifferentiated carcinoma of the pancreas. Report of a patient with tumor marker studies. Cancer. 1992;70(6):1514–1519. doi: 10.1002/1097-0142(19920915)70:6<1514::aid-cncr2820700612>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 17.Chetty R, Clark SP, Pitson GA. Primary small cell carcinoma of the pancreas. Pathology. 1993;25(3):240–242. doi: 10.3109/00313029309066580. [DOI] [PubMed] [Google Scholar]
- 18.Ordonez NG, Cleary KR, Mackay B. Small cell undifferentiated carcinoma of the pancreas. Ultrastruct Pathol. 1997;21(5):467–474. doi: 10.3109/01913129709021947. [DOI] [PubMed] [Google Scholar]
- 19.Hruban R, Pitman MB, Klimstra DS. AFIP Atlas of Tumor Pathology. Washington, DC: American Registry of Pathology; 2007. Tumors of the Pancreas. [Google Scholar]
- 20.Iwasa S, et al. Cisplatin and etoposide as first-line chemotherapy for poorly differentiated neuroendocrine carcinoma of the hepatobiliary tract and pancreas. Jpn J Clin Oncol. 2010;40(4):313–318. doi: 10.1093/jjco/hyp173. [DOI] [PubMed] [Google Scholar]
- 21.Basturk O, et al. Increased (>20%) Ki67 Proliferation Index in Morphologically Well Differentiated Pancreatic Neuroendocrine Tumors (PanNETs) Correlates with Decreased Overall Survival (Abstract) Modern Pathology. 2013;26(2S):423A. [Google Scholar]
- 22.Yachida S, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36(2):173–184. doi: 10.1097/PAS.0b013e3182417d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nassar H, Albores-Saavedra J, Klimstra DS. High-grade neuroendocrine carcinoma of the ampulla of vater: a clinicopathologic and immunohistochemical analysis of 14 cases. Am J Surg Pathol. 2005;29(5):588–594. doi: 10.1097/01.pas.0000157974.05397.4f. [DOI] [PubMed] [Google Scholar]
- 24.Bagci P, et al. Comparative Analysis of Different Counting Methodologies for Ki-67 in Pancreatic Neuroendocrine Tumors (Abstract) Modern Pathology. 2012;25(2):441A. [Google Scholar]
- 25.Stelow EB, et al. Pancreatic acinar cell carcinomas with prominent ductal differentiation: Mixed acinar ductal carcinoma and mixed acinar endocrine ductal carcinoma. Am J Surg Pathol. 2010;34(4):510–518. doi: 10.1097/PAS.0b013e3181cfcac7. [DOI] [PubMed] [Google Scholar]
- 26.AJCC Cancer Staging Manual. 7ed. New York: Springer; 2010. [Google Scholar]
- 27.Couvelard A, Scoazec JY. [A TNM classification for digestive endocrine tumors of midgut and hindgut: proposals from the European Neuroendocrine Tumor Society (ENETS)] Ann Pathol. 2007;27(6):426–432. doi: 10.1016/S0242-6498(07)71414-1. [DOI] [PubMed] [Google Scholar]
- 28.Wang DS, et al. Prognostic factors and survival in patients with neuroendocrine tumors of the pancreas. Tumour Biol. 2011;32(4):697–705. doi: 10.1007/s13277-011-0170-9. [DOI] [PubMed] [Google Scholar]
- 29.Araujo PB, et al. Evaluation of the WHO 2010 grading and AJCC/UICC staging systems in prognostic behavior of intestinal neuroendocrine tumors. PLoS One. 2013;8(4):e61538. doi: 10.1371/journal.pone.0061538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Tang LH, Klimstra DS. Gastroenteropancreatic neuroendocrine neoplasms: Historical context and current issues. Semin Diagn Pathol. 2013;30(3):186–196. doi: 10.1053/j.semdp.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Klimstra DS, et al. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol. 1992;16(9):815–837. doi: 10.1097/00000478-199209000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Klimstra DS, Rosai J, Heffess CS. Mixed acinar-endocrine carcinomas of the pancreas. Am J Surg Pathol. 1994;18(8):765–778. doi: 10.1097/00000478-199408000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Movahedi-Lankarani S, et al. Primitive neuroectodermal tumors of the pancreas: a report of seven cases of a rare neoplasm. Am J Surg Pathol. 2002;26(8):1040–1047. doi: 10.1097/00000478-200208000-00009. [DOI] [PubMed] [Google Scholar]
- 34.Zee SY, et al. Pleomorphic pancreatic endocrine neoplasms: a variant commonly confused with adenocarcinoma. Am J Surg Pathol. 2005;29(9):1194–1200. doi: 10.1097/01.pas.0000164370.81132.25. [DOI] [PubMed] [Google Scholar]
- 35.Strosberg JR, et al. The NANETS consensus guidelines for the diagnosis and management of poorly differentiated (high-grade) extrapulmonary neuroendocrine carcinomas. Pancreas. 2010;39(6):799–800. doi: 10.1097/MPA.0b013e3181ebb56f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang L, et al. High grade transformation of differentiated neuroendocrineneoplasms (NENs) of the enteropancreatic system - a unique entity distinct from de-novo high grade neuroendocrine carcinoma (HGNECa) in pathogenesis and clinical behavior. Mod Pathol. 2008;21(S1):137A. [Abstract #622]. [Google Scholar]
- 37.Basturk O, Klimstra DS. Acinar cell carcinoma of the pancreas and related neoplasms: a review. Diagnostic Histopathology. 2012;18(1):8–16. [Google Scholar]
- 38.Hochwald SN, et al. Prognostic factors in pancreatic endocrine neoplasms: an analysis of 136 cases with a proposal for low-grade and intermediate-grade groups. J Clin Oncol. 2002;20(11):2633–2642. doi: 10.1200/JCO.2002.10.030. [DOI] [PubMed] [Google Scholar]
- 39.Rusch VW, Klimstra DS, Venkatraman ES. Molecular markers help characterize neuroendocrine lung tumors. Ann Thorac Surg. 1996;62(3):798–809. doi: 10.1016/s0003-4975(96)00435-3. discussion 809-10. [DOI] [PubMed] [Google Scholar]
- 40.Moertel CG, et al. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in the anaplastic variants of these neoplasms. Cancer. 1991;68(2):227–232. doi: 10.1002/1097-0142(19910715)68:2<227::aid-cncr2820680202>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 41.Motojima K, et al. Small cell carcinoma of the pancreas and biliary tract. J Surg Oncol. 1990;45(3):164–168. doi: 10.1002/jso.2930450306. [DOI] [PubMed] [Google Scholar]