
Fig. 2.
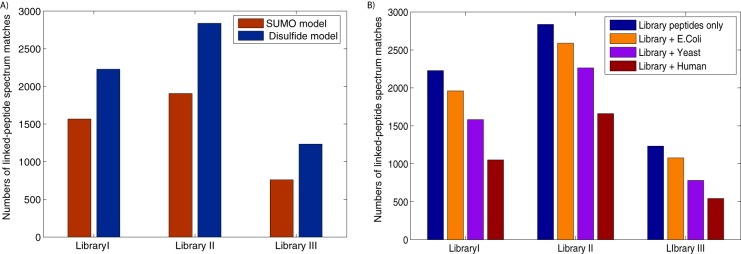
Identification of disulfide-bridged peptides from combinatorial peptide libraries. A, the initial scoring models learned from SUMOylated peptides (shown in red) were compared with the scoring models for disulfide-bridged peptides (shown in blue). The latter models improved the identification of disulfide-bridged peptides by 31% to 60%, confirming the need to model the different fragmentation patterns for different types of linked peptides. B, MXDB's ability to identify disulfide-bridged peptides against whole-proteome sequence databases was tested as follows: the library peptide sequences were concatenated with all E. coli, yeast, and human protein sequences (respectively shown in blue, orange, purple, and red) and the spectra from the peptide libraries were searched against each concatenated database. MXDB was able to identify thousands of spectra from disulfided peptides against these proteome-scale databases, and the general trend shows that as the search space of cross-linked peptides increased by ∼9-fold, the sensitivity of the identification of disulfide-bridged peptides decreased by 20% to 25%, unless a two-pass search strategy was used (see supplementary Fig. S6).