

Abstract
Maternal diabetes during pregnancy is responsible for the occurrence of diabetic embryopathy, a spectrum of birth defects that includes heart abnormalities, neural tube defects, and caudal dysgenesis syndromes. Here, we report that mice transgenic for the homeodomain transcription factor Isl-1 develop profound caudal growth defects that resemble human sacral/caudal agenesis. Isl-1 is normally expressed in the pancreas and is required for pancreas development and endocrine cell differentiation. Aberrant regulation of this pancreatic transcription factor causes increased mesodermal cell death, and the severity of defects is dependent on transgene dosage. Together with the finding that mutation of the pancreatic transcription factor HLXB9 causes sacral agenesis, our results implicate pancreatic transcription factors in the pathogenesis of birth defects associated with diabetes.
Keywords: maternal diabetes, pancreas, embryopathy
Diabetes of the mother during pregnancy is a major risk factor for congenital defects, collectively termed diabetic embryopathies (1, 2). The most common abnormalities in diabetic embryopathy are neural tube and heart defects (3), with caudal regression syndrome particularly characteristic (4). This syndrome is now recognized as caudal dysgenesis (5) and comprises a spectrum of growth anomalies, malformations of interior organs, and deformities or lack of skeletal structures, in particular, defects of variable portions of the lumbar spine and absence of the sacrum.
It has remained unclear how systemic metabolic changes result in developmental defects that affect specific tissues and developmental processes (6). One hypothesis (7) is that maternal diabetes alters the expression of developmental control genes that play important roles for the morphogenesis of tissues affected in diabetic embryopathy. Consistent with this proposition is the finding that Pax3 expression is reduced in mouse embryos from diabetic mothers (8). As Pax3 is well known to be required for normal neural tube development (9-13), its reduction would explain the occurrence of neural tube defects in diabetic embryopathy. Genes that play a role in the etiology of other abnormalities in diabetic embryopathy, however, have not been identified to date. We here present evidence from a mouse model that invokes a second hypothesis for developmental anomalies in diabetic embryopathy, namely that maternal diabetes leads to dysregulation of pancreatic transcription factors. Pancreatic transcription factors are known to respond to metabolic status (14-20), and therefore would be prime candidates for dysregulation by altered metabolic state. As some pancreatic transcription factors are also expressed in non-pancreatic tissues (21), or could be ectopically activated in such tissues by metabolic disturbances, the tissue-specificity of diabetic embryopathies would be explained by the particular expression patterns in normal or ectopic locations. This hypothesis is consistent with our mouse model, in which experimental ectopic expression of the pancreatic transcription factor Islet-1 leads to developmental defects that resemble caudal growth defects in diabetic embryopathy.
MATERIALS AND METHODS
Generation of transgenic mice
To generate Islet-1 (abbreviated Isl-1) transgenic mice, we used the VP-16-based binary transgenic system (22, 23) (Fig. 1A) because initial experiments showed that animals with a conventional Hoxc-8-Isl-1 transgene die shortly after birth (24). The transactivator (TA) transgenic lines express the viral transactivator VP16 under control of the Hoxc-8 promoter (25). The transresponder (TR) transgenic lines carry the cDNA from the rat Isl-1 gene linked to the 360 bp fragment from the immediate early (IE) gene promoter of the ICP4 gene of herpes simplex virus (26). Mice double transgenic for the Hoxc-8-VP16 and IE-Isl-1 transgenes were obtained by crossing the two parental transgenic lines. The transgenic mouse strains have been published previously (22, 25, 26), and genotyping was performed according to protocols in those publications. Some double transgenic mice were viable and could be further bred to generate mice homozygous for one or both transgene loci. To detect homozygosity at either transgene locus, we performed semi-quantitative PCR by using DNA isolated from yolk sacs of individual embryos (22, 26).
Figure 1. Caudal growth defects in Isl-1 transgenic mice.
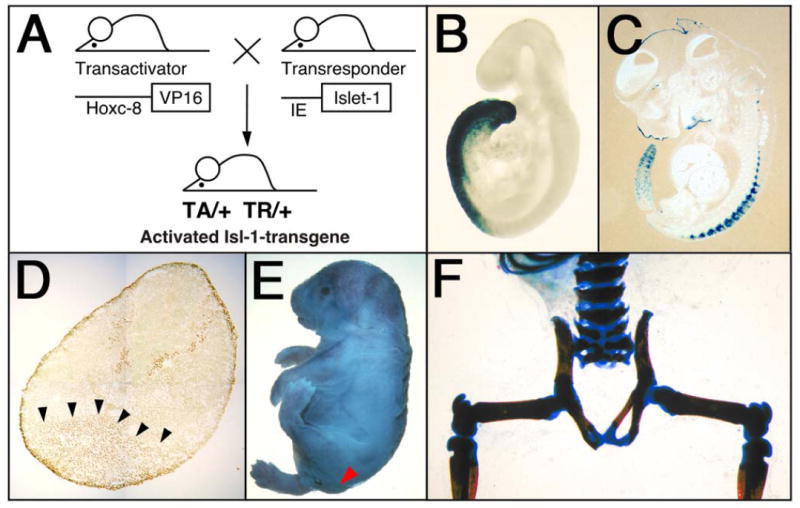
The VP-16-based binary transgenic system (A) was used to overcome perinatal lethality, and to consistently generate Isl-1 transgenics from stable parental transgenic mouse lines (22, 23). The transactivator (TA) lines express the viral transactivator VP16 under control of the Hoxc-8 promoter (25), and transresponder (TR) lines carry the cDNA from the rat Isl-1 gene linked to the VP16-responsive immediate early (IE) gene promoter from herpes simplex virus (26). Mice double transgenic for both the TA and TR transgenes were obtained by crossing the two parental transgenic lines. Some double transgenic mice were viable and were further bred to generate mice homozygous for one or both transgene loci. B, C) The domain of VP16 expression under control of the Hoxc-8 promoter is revealed by crossing the Hoxc-8-VP16 transactivator to IE-LacZ transresponder mice. Blue staining for β-galactosidase activity indicates the region of transgene activation in the posterior region at E9.5 B) in mesoderm and neural tube, and at E12.5 (C), in the tail and mesodermal derivatives. D) Ectopic Isl-1 protein (arrows) in posterior mesoderm of an Isl-1 transgenic embryo at E11.5. As expected for a transcription factor, Isl-1 protein is localized to nuclei. This picture was composed from photographs taken at 1000× magnification. E) Isl-1 transgenic newborn mouse with open eyes (open arrow) and absence of tail (closed arrow). F) Skeletal preparation of an Isl-1 transgenic newborn mouse shows absence of the tail, of caudal and sacral vertebrae, and unclosed neural arches of sacral vertebrae.
Skeletal preparations
Preparations of newborn skeletons were processed as described before (22), for embryos at younger developmental stages, the same protocol was followed, except that embryos were first cooled in PBS on ice for 10 min to stop circulation and heart beat.
Histology, in situ hybridization, and immunohistochemistry
Histological sectioning was done as described before (22, 27), typically at 10 μm thickness for paraffin-embedded tissues, and at up to 30 μm thickness for frozen tissue (24). In situ hybridizations were performed exactly as described (24), and immunohistochemistry followed established protocols (22) by using the secondary anti-mouse immunoglobulin reagents provided with the M.O.M. (Vector, Burlingame, CA) and DAB substrate (Pierce, Rockford, IL) kits. Primary monoclonal antibodies specific for Pax-7, Isl-1, Nkx-2.2, HNF3-β, and Shh were obtained from the Developmental Studies Hybridoma Bank (University of Iowa), and the Nestin-specific antibody was purchased from PharMingen (Palo Alto, CA). Where indicated, sections were photographed at 1000× magnification and images were composed by using Photoshop 5.5 (Adobe Systems, San Jose, CA).
TUNEL assays were performed using the ApopTag fluorescein direct in situ apoptosis detection kit (Intergen, Purchase, NY). Normal and transgenic mouse embryos were isolated at E10.5, E11.5, and E12.5; fixed in 4% paraformaldehyde; and embedded in paraffin. Sections of 10 μm thickness were cut, deparaffinized, and treated with Proteinase K. DNA fragments were tailed with fluorescein-nucleotide in a reaction containing TdT enzyme for 1 h at 37°C. The signals for apoptotic cells were confirmed to be specific by using an independent Peroxidase-based in situ apoptosis detection kit (Intergen).
Measurements of glucose in serum
Serum was obtained from the tail vein of mice as described previously (28). Glucose was measured by Elite XL glucometer (Bayer). Results were statistically evaluated using two-sided t-tests.
RESULTS and DISCUSSION
We found caudal growth deficiencies in mice transgenic for the pancreatic homeodomain transcription factor Islet-1 (Isl-1). These mice were generated by using the VP16-based binary system (23) (Fig. 1A). Isl-1 transgene expression is under control of the Hoxc-8 promoter, which directs gene expression to the posterior region of the early developing embryo (Fig. 1B) with restriction to mesodermal tissues at later stages (Fig. 1C). Expression of the Isl-1 transgene in the posterior region of Isl-1 transgenic embryos could be detected by RT-PCR (26) and by immunohistochemistry (Fig. 1D). Isl-1 transgenic newborn mice were born with open eyes, and had either a short tail or no tail; those without tail often died shortly after birth (Fig. 1E). Staining of newborn skeletons revealed absence of caudal structures as well as some sacral vertebrae (Fig. 1F). This phenotype was specific for Isl-1 because i) it was also produced with a conventional Hoxc-8-Isl-1 transgene, resulting in posterior truncation, hindlimb paralysis and postnatal death (data not shown); and ii) it was found with multiple TA strains and TR mouse lines when the binary system was used (Table 1). Furthermore, in this system, Hoxc-8 and Hoxd-4 TR transgenes did not affect posterior development. Developmental defects arose already with moderate activation of the Isl-1 transgene and, congruently, the severity of the Isl-1 induced phenotype was found to be dependent on transgene dosage (Fig. 2). In the binary system, if animals hemizygous for the TA and TR transgenes survive and are fertile, crossing of such parents will result in offspring with homozygosity for either the TA or the TR transgene, respectively, or, at very low frequency (see Fig. 2), homozygosity for both loci. Shortened tails were present in animals hemizygous for both TA and TR (TA/+ TR/+; Fig. 3B, E). Compound transgenic animals with transgene loci in excess of hemizygosity (TA/TA TR/+ or TA/+TR/TR or TA/TA TR/TR) exhibited postnatal lethality and, in comparison to their littermates, more severe caudal truncations of their skeletons (Fig. 3C, F). The caudal skeletal defects became evident in developing embryos and, in some cases, they were associated with a pre-sacral mass detectable at 13.5 days of development (Fig. 4). The extraneous tissue consisted of neuroepithelial cells (Fig. 4D), suggesting that neural development itself was only secondarily affected. Reproducibly, the posterior defects in Isl-1 transgenic mice were characterized by the absence of caudal and sacral vertebrae, although the precise level of truncation was variable (cf. Figs. 1F, 3C, and 3F). Taken together, the Isl-1 transgene induced developmental defects phenocopy human caudal regression syndrome.
Table 1.
Caudal growth defects are Isl-1 transgene specific
| Crosses of transactivator (TA) to transresponder (TR) strain | Phenotype | |
|---|---|---|
| TA-9 | TR-Isl-1.5 | viable: short, kinked, short and kinked tail |
| TA-9 | TR-Isl-1.81 | viable: short tail |
| TA-239 | TR-Isl-1.16 | lethal, no tail |
| TA-239 | TR-Isl-1.50 | viable and lethal: short to absent tail |
| TA-239 | TR-Isl-1.73 | normal |
| TA-239 | TR-Isl-1.81 | viable: short tail |
| TA-239 | TR-Hoxc-8 | tail normal, rib cartilage defect (Ref. 22) |
| TA-239 | TR-Hoxd-4 | tail normal, rib cartilage defect (Ref. 22) |
Isl-1 transgenic mice were generated by crossing mice containing a transactivator (TA) transgene and a transresponder (TR) transgene, respectively. The TA strains express the VP16 transactivator protein in the caudal region of developing embryos (25). The relative transactivation capacity of the two independent TA lines is 1.69 and 5.7, respectively (26); both TA lines produced comparable phenotypes. Phenotypes of Hoxc-8 and Hoxd-4 transgenic animals were reported previously (22). Here, five independent Isl-1 TR strains were used, four produced caudal growth defects. The pictures in Fig. 1 E,F show animals generated with the Isl-1.16 TR line, all other studies reported here used the Isl-1.50 TR strain.
Figure 2. Dosage dependency of caudal growth defects in Isl-1 transgenic mice.
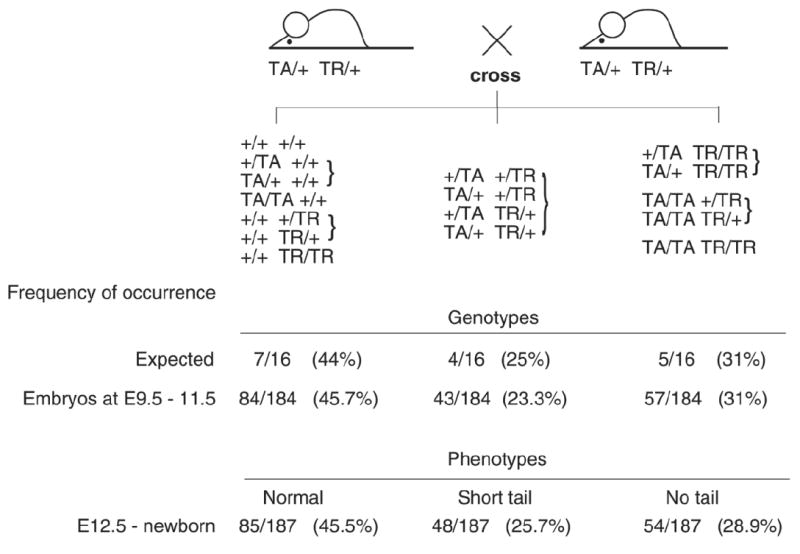
Occurrence of phenotypes/genotypes after crossing mice double hemizygous for the TA and the TR transgenes (TA/+ TR/+ × TA/+ TR/+). Embryos were isolated at various days of gestation. From day E12.5 on, phenotypes were visibly discernible, while for E9.5 to E11.5, genotypes were determined as described (25-27). The expected frequencies and the observed frequencies for occurrence of all genotypes correlate, as do the ratios of a given genotype with expected phenotype. Thus, while severity is dependent on transgene dosage, the phenotype is fully penetrant.
Figure 3. Severity of growth defects in Isl-1 transgenic mice correlates with gene dosage.
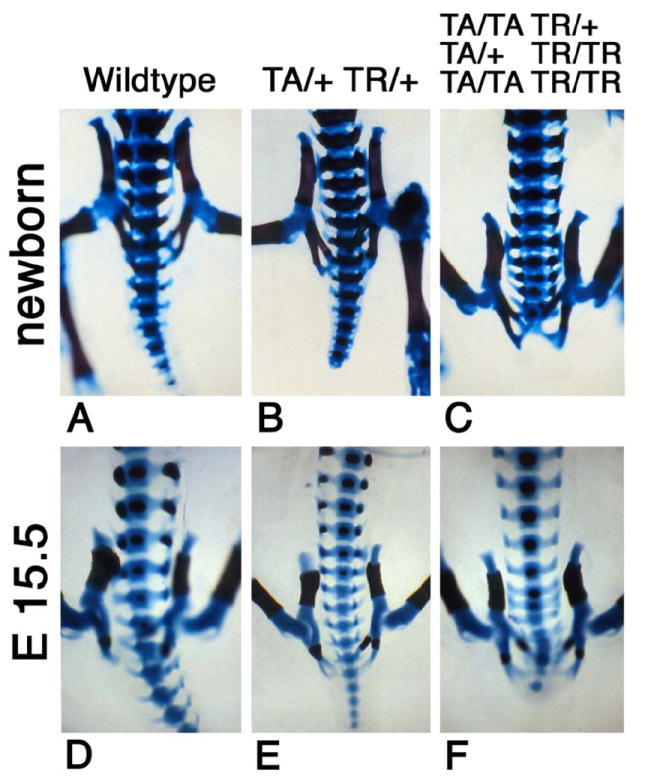
Skeletons were prepared from wildtype FVB (A) and Isl-1 transgenic newborns (B, C) and E15.5 embryos (D–F). The skeletons from Isl-1 transgenic animals show normal pelvic bone structure but partial or complete absence of caudal and sacral vertebrae. Severity of the defects increases with transgene dosage: shortened tails are observed in double hemizygous animals (TA/+TR/+), complete absence of the tail is found in animals with genotypes in excess of double hemizygosity (TA/TA TR/+ or TA/+ TR/TR or TA/TA TR/TR). Wild type skeletons exhibit normal posterior development.
Figure 4. Pre-sacral mass in Isl-1 transgenic embryos.
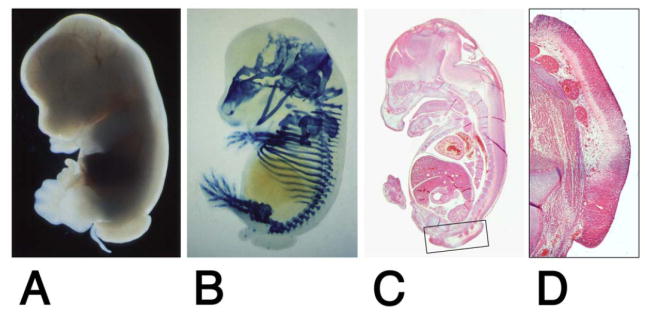
An Isl-1 transgenic embryo isolated at E13.5 shows absence of the tail and presence of pre-sacral mass (A). E14.5 Isl-1 transgenic embryo with a pre-sacral mass. Skeletal cartilage was stained with Alcian blue. The truncation of the vertebral column is obvious (B). Sagittal section through an embryo at 13.5 days (C), and magnification of the area of the pre-sacral mass (D). The pre-sacral mass consists of neuroepithelium in continuity with the neural tube.
It has been suggested that malformations seen in caudal regression result from mesodermal insult (29). Caudal regression was also found in mouse embryos exposed to retinoic acid, which causes excessive cell death in the primitive streak, leading to absence of the tail (30). Vascular insufficiency in the caudal region has been suggested as a cause (31), as well as inadequate separation of gut endoderm from the caudal eminence (32). Lynch et al. (33) proposed that dominant inherited human sacral agenesis might be caused by incomplete notochord development and defective signaling with impaired induction of sclerotomal cell migration and failure in formation of caudal skeletal elements. To discern these possible scenarios for the pathogenesis of posterior defects in our Isl-1 transgenic mice, we performed histology and immunohistochemical studies.
During development, Isl-1 transgenic embryos exhibited no histological abnormalities before day E11.5 (Fig. 5B–E). The absence of tail development was observed as early as E12.5. In the caudal region of Isl-1 transgenic embryos isolated at E12.5 and E13.5, development of the neural tube and mesoderm was incomplete. The neural tube was either reduced in size (Fig. 5G) or failed to close (Fig. 5H). The presence of the notochord in the Isl-1 transgenic embryos was clearly evident at E10.5 (C), E11.5 (E), and E12.5 (H). While these data argue against the first part of the proposition by Lynch et al. (33), they leave open the possibility that signaling from the notochord could be impaired. This prompted us to investigate the expression of markers for notochord signaling.
Figure 5. Histological changes in Isl-1 transgenic mice during embryonic development.
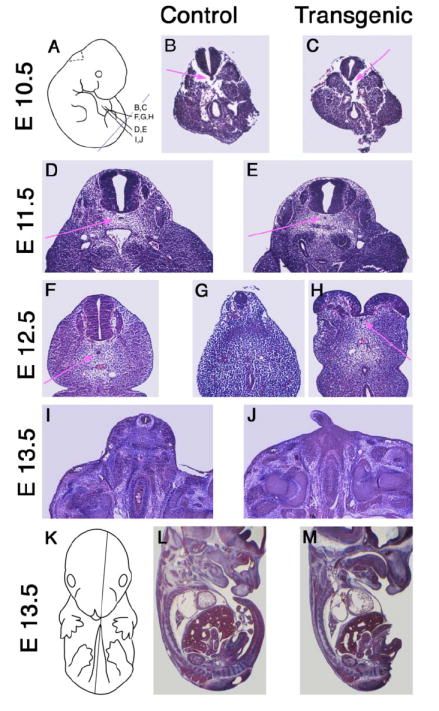
Sections from non-transgenic control (B, D, F, I, L) and Isl-1 transgenic (C, E, G, H, J, M) embryos were stained with Hematoxylin/Eosin. Sections at 10 μm thickness from embryos at E10.5 (B, C), E11.5 (D, E), E12.5 (F–H) and E13.5 (I, J) were taken at corresponding transverse levels as shown in (A). For L (E13.5, normal) and M (E13.5, transgenic), para-sagittal sections were taken as shown in (K). The phenotype of impaired posterior growth is evident by day E12.5.
Nestin was used as a marker for neuronal and muscle precursor cells (34) (Fig. 6A, H). Pax-7 was used as a neuronal marker for the dorsal neural tube (35) (Fig. 6B, I), Isl-1 as a marker for dorsal root ganglia and ventral cells (36) (Fig. 6C, J), and Nkx2.2 as a marker for a subset of motorneurons (37, 38) (Fig. 6D, K). HNF3-β (Fig. 6E, L), and Shh (Fig. 6F, M) were used as markers for the notochord and floor plate (39-41). Overall, there were no differences in expression of these markers between control and transgenic mice at E11.5. Most notably, Shh expression was found in notochord and floor plate of Isl-1 transgenic embryos, demonstrating the presence of the notochord and one of its important signaling molecules. Thus, with reference to marker genes whose expression is known to be exquisitely sensitive to Shh signaling (42), these results argue against impairment of notochord signaling in our Isl-1 transgenic mice.
Figure 6. Expression of markers for neural tube patterning in normal and Isl-1 transgenic mice.
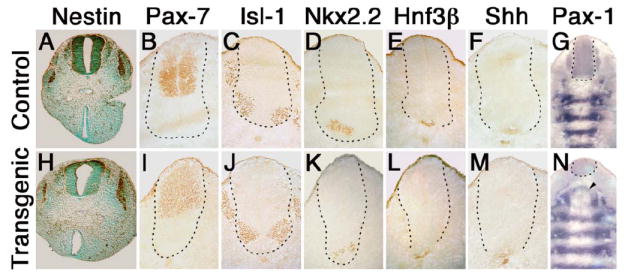
Immunohistochemistry was performed on 10 μm transverse sections taken at E11.5 at the level shown for B in Fig. 5A for deparaffinized (A, B), and at level shown for I in Fig. 5A for frozen sections (B–F, I–M). Sections from control (A–G) and Isl-1 transgenic (H–N) embryos were stained using primary antibodies specific for neuron and muscle precursor cell marker Nestin (A, H), Pax-7 (B, I), Isl-1 (C, J), Nkx-2.2 (D, K), HNF3-β (E, L) and Shh (F, M). Sections were very lightly counterstained with methylgreen. All markers were also tested in E10.5 embryos and did not reveal differences between normal and transgenic mice (data not shown). In situ hybridizations for Pax-1 (G, N) were performed on frozen sections with the direction of sectioning indicated by the light purple line in Fig. 5A, and images composed from photographs taken at 1000× magnification. The broken line provides an outline of the neural tube.
Since Isl-1 transgene expression affects posterior growth particularly of skeletal structures, we examined Pax-1 expression as a marker for skeletogenic cells (43, 44). In situ hybridization revealed normal expression of Pax-1 in developing prevertebrae (Fig. 6G, N), except for areas close to the site of truncation, where the pattern of cell condensations was perturbed (Fig. 6N, arrowhead). This suggested that the caudal defect in our transgenic mice resulted from an insult to sclerotomal cells, or their mesodermal precursors. We performed “TUNEL” assays on sections of normal and transgenic embryos to determine whether cell death could be responsible for the growth defects. Control and transgenic embryos isolated at E10.5 showed no staining difference (not shown). At the stage of E11.5, some apoptotic cells were detected in the mesoderm, but there was little difference between control (Fig. 7B) and transgenic mice (Fig. 7C). By E12.5, however, a striking increase of cell death was observed in Isl-1 transgenic mice (Fig. 7E, F) as compared with normal embryos (Fig. 7D). The cell death occurred predominantly in mesodermal cells, and only to a lesser extent in the neural tube, consistent with the subsequent absence of skeletal development in the posterior region.
Figure 7. Excessive cell death in mesodermal tissues of Isl-1 transgenic mice.
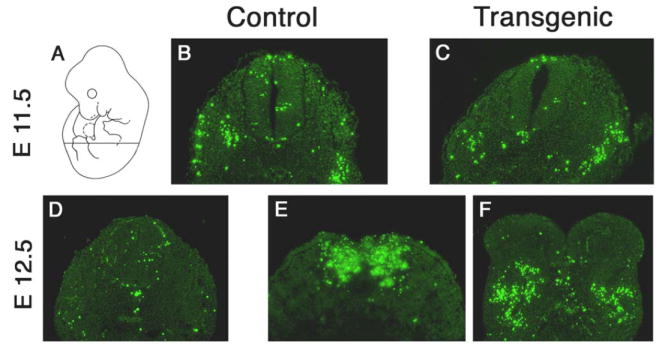
TUNEL assays on sections from normal and Isl-1 transgenic embryos at E11.5 (B, C) and E12.5 (D–F). The signals for apoptotic cells were confirmed to be specific by independent colorimetric detection on adjacent slides (data not shown). The transverse sections were taken as shown in (A).
Isl-1 is normally expressed in motor neurons and dorsal root ganglia during development (36, 45). In our transgenic mice, the Hoxc-8 promoter directs expression of Isl-1 not only to motor neurons and dorsal root ganglia, but also ectopically to neuroectodermal and mesodermal tissues in the posterior region (25, 46) (see Fig. 1). Isl-1 transgene expression was detectable by RT-PCR (26) and, albeit at low level, by immunohistochemistry in mesodermal cells (see Fig. 1). Normally, these cells are proliferating and express proliferating cell nuclear antigen (PCNA, not shown). In developing motorneurons and pancreatic cells, Isl-1 expression is correlated with exit from the cell cycle (42, 47). The possibility that Isl-1 expression in proliferating cells may actively inhibit cell division is consistent with the Isl-1 transgene induced phenotype. With the activation of Isl-1 transgene expression in posterior tissues of the developing embryo, caudal growth defects were observed with high penetrance and reproducibility. Growth inhibitory effects of transgenic Isl-1 expression were particularly evident in mesodermal derivatives and are consistent with earlier reports (29, 30) that implicate mesodermal insult in the caudal region of developing embryos as the cause for malformations seen in syndromes of caudal dysgenesis. Our results provide strong evidence that phenocopies of human caudal regression can be caused by aberrant Isl-1 expression in the posterior mesoderm of developing embryos.
Caudal regression, neural tube defects, and heart abnormalities are hallmarks of diabetic embryopathy in humans (48). These congenital abnormalities are significantly more frequent in infants born to diabetic mothers and are thought to arise from yet unidentified insults during early gestation (4, 49). It has been suggested that glucose toxicity and the accumulation of metabolites (50), or depletion of important nutrients (51), could be responsible for the malformations, possibly through oxidative stress (52). But it has remained unclear how systemic changes in metabolism become translated into specific developmental anomalies (6). The finding of caudal growth defects our Isl-1 transgenic mouse model prompts the hypothesis that the Isl-1 gene may be a specific target of metabolic deregulation. This proposition implies that altered metabolic state can aberrantly regulate or activate Isl-1 gene expression through specific DNA regulatory elements. A potential substrate for activation in the posterior region of developing embryos could be the posterior region-specific enhancer that we have identified in the Isl-1 gene (Salbaum and Kappen, unpublished results). Studies are currently underway to determine in which way this element responds to metabolic imbalance in diabetic embryopathy.
Recently, it has been suggested that predisposition to diabetes may be caused by exposure to a diabetic environment during pregnancy (49, 53-56), such that there is an association of low birth weight and diabetes (57, 58). Interestingly, our double transgenic Isl-1 mice are born smaller than their single transgenic littermates. Although there is no indication that the transgene would be activated in the pancreas (25, 46, 59), we nevertheless investigated whether those Isl-1 transgenic mice that survive after birth showed signs of metabolic perturbations. Glucose levels were measured in serum starting from 3 weeks to 6 month of age. Fig. 8 displays the results, which indicate that any differences are not statistically significant. Thus, we do not detect abnormal glucose levels in the Isl-1 transgenic mice, which is consistent with the fact that at the age of 2 weeks, internal organs such as pancreas and kidney of surviving transgenic mice showed no obvious histological differences to control mice (data not shown). Indeed, the surviving Isl-1 transgenic mice eventually grow to a size indistinguishable from their control littermates by 8–10 weeks. However, these results do not exclude the possibility that the Isl-1 transgenic mice with higher transgene dosage, which die shortly after birth, may have metabolic defects. We are currently investigating whether metabolic aberrations could be involved in postnatal lethality of Isl-1 transgenic mice.
Figure 8. Measurement of glucose levels in Isl-1 transgenic mice.
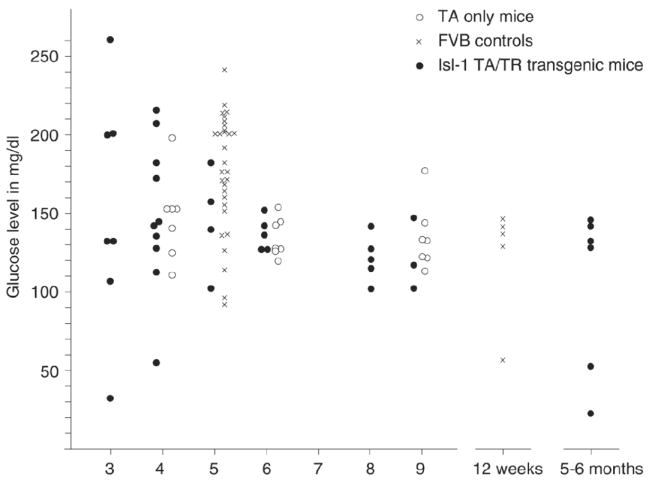
Glucose measurements were performed on serum from Isl-1 transgenic mice at various time points. Each Isl-1 transgenic individual is represented by a filled circle, with mice transgenic for transactivator only (open circles) and normal FVB mice (X) serving as controls. While younger animals display greater variance than adults, statistical evaluation reveals no significant differences between transgenic and control groups at comparable ages, indicating normal glucose levels in isl-1 transgenic mice.
Intriguingly, in human sacral agenesis, another pancreatic homeobox gene has been implicated: HLXB9 was found to be mutated in Currarino triad (33, 60), a form of dominant inherited sacral agenesis that consists of anorectal malformations, agenesis of sacral vertebrae and presence of a pre-sacral mass (anterior meningocoele or teratoma; 61, 62). It is intriguing that pre-sacral mass (in form of extruded neuroepithelial tissue) is also found in our Isl-1 transgenic mice (Fig. 4), indicating that both Isl-1 and HLXB9 can be involved in pathogenesis of similar phenotypes. Like Isl-1 (45, 47), Hlxb9 is essential for proper pancreatic development (63, 64) and for differentiation of motorneurons in the spinal cord (65, 66). Hlxb9 in the mouse acts, as has been shown for in differentiating motorneurons and pancreatic endocrine cells, in the same pathway as Isl-1 (21, 66), affecting subpopulations of Isl-1 expressing cells at later stages of differentiation. This places Hlxb9 downstream of Isl-1 in differentiated cells. Mice with null mutations in Isl-1 or Hlxb9 do not exhibit sacral agenesis, and it is currently not known how mutated HLXB9 causes sacral agenesis in humans (61). In analogy to the Isl-1 induced phenotype in mice, ectopic expression or dominant-negative interference of Hlxb9 with cell growth would provide a plausible explanation for the dominant inheritance of the human sacral agenesis phenotype. The parallel phenotypes induced by HLXB9 mutation in humans and Isl-1 misexpression in mice indicate that transcription factors from common cellular pathways can have comparable pathogenic roles.
Interestingly, when Isl-1 was overexpressed during neural tube development, we found neural tube defects (Yaworsky, Salbaum and Kappen, manuscript in preparation), indicating that Isl-1 can also induce another developmental abnormality characteristic for diabetic embryopathy. With regard to developmental defects, our results advance the hypothesis that metabolic imbalance during diabetic pregnancy might lead to deregulation of pancreatic transcription factors, which, in turn, induce phenotypes of diabetic embryopathy.
Supplementary Material
Acknowledgments
We thank Rudi Balling for the Pax-1 probe plasmid, MiMi P.-W. Macias for making the IE-Isl-1 plasmid, the transgenic facility at Mayo Clinic Scottsdale for generating, and Per Jambeck, Kristin Smith and Diane Costanzo for genotyping the transgenic mice, Diane Costanzo for the serum measurements, M. Anita Jennings for expert histology services, Jörg Rahnenführer for advice on statistics, and Melanie Schrack and Saralyn Fisher for assistance with manuscript preparation. We are grateful to Frank H. Ruddle for encouragement and to David P. Gardner for his contributions during the early phase of this project. This work was funded in parts by The Neurosciences Support Corporation (to J.M.S.), by Mayo Foundation for Medical Education and Research (to C.K.), by a grant from the Nebraska Research Initiative (to C.K.), and by NIH grant R01-HD-37804 (to C.K.).
References
- 1.Kucera J. Rate and type of congenital anomalies among offspring of diabetic women. J Reprod Med. 1971;7:73–82. [PubMed] [Google Scholar]
- 2.Goto MP, Goldman AS. Diabetic embryopathy. Curr Opin Pediatr. 1994;6:486–491. doi: 10.1097/00008480-199408000-00023. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Frias ML. Epidemiological analysis of outcomes of pregnancy in diabetic mothers: identification of the most characteristic and most frequent congenital anomalies. Am J Med Genet. 1994;51:108–113. doi: 10.1002/ajmg.1320510206. [DOI] [PubMed] [Google Scholar]
- 4.Mills JL. Malformations in infants of diabetic mothers. Teratology. 1982;25:385–394. doi: 10.1002/tera.1420250316. [DOI] [PubMed] [Google Scholar]
- 5.Bohring A, Lewin SO, Reynolds JF, Voigtlander T, Rittinger O, Carey JC, Kopernik M, Smith R, Zackai EH, Leonard NJ, et al. Polytopic anomalies with agenesis of the lower vertebral column. Am J Med Genet. 1999;87:99–114. [PubMed] [Google Scholar]
- 6.Greene MF. Diabetic embryopathy 2001: moving beyond the “diabetic milieu”. Teratology. 2001;63:116–118. doi: 10.1002/tera.1021. [DOI] [PubMed] [Google Scholar]
- 7.Chang TI, Loeken MR. Genotoxicity and diabetic embryopathy: impaired expression of developmental control genes as a cause of defective morphogenesis. Semin Reprod Endocrinol. 1999;17:153–165. doi: 10.1055/s-2007-1016222. [DOI] [PubMed] [Google Scholar]
- 8.Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46:1189–1197. doi: 10.2337/diab.46.7.1189. [DOI] [PubMed] [Google Scholar]
- 9.Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell. 1991;67:767–774. doi: 10.1016/0092-8674(91)90071-6. [DOI] [PubMed] [Google Scholar]
- 10.Goulding M, Sterrer S, Fleming J, Balling R, Nadeau J, Moore KJ, Brown SD, Steel KP, Gruss P. Analysis of the Pax-3 gene in the mouse mutant splotch. Genomics. 1993;17:355–363. doi: 10.1006/geno.1993.1332. [DOI] [PubMed] [Google Scholar]
- 11.Mansouri A, Stoykova A, Gruss P. Pax genes in development. J Cell Sci Suppl. 1994;18:35–42. doi: 10.1242/jcs.1994.supplement_18.5. [DOI] [PubMed] [Google Scholar]
- 12.Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT. Mutations in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome (WS-III) as well as Waardenburg syndrome type I (WS-I) Am J Hum Genet. 1993;52:455–462. [PMC free article] [PubMed] [Google Scholar]
- 13.Tassabehji M, Read AP, Newton VE, Patton M, Gruss P, Harris R, Strachan T. Mutations in the PAX3 gene causing Waardenburg syndrome type 1 and type 2. Nat Genet. 1993;3:26–30. doi: 10.1038/ng0193-26. [DOI] [PubMed] [Google Scholar]
- 14.Habener JF, Stoffers DA. A newly discovered role of transcription factors involved in pancreas development and the pathogenesis of diabetes mellitus. Proc Assn Am Physicians. 1998;110:12–21. [PubMed] [Google Scholar]
- 15.Marshak S, Totary H, Cerasi E, Melloul D. Purification of the beta-cell glucose-sensitive factor that transactivates the insulin gene differentially in normal and transformed islet cells. Proc Natl Acad Sci USA. 1996;93:15057–15062. doi: 10.1073/pnas.93.26.15057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson LK, Qian J, Poitout V. Glucose rapidly and reversibly decreases Ins-1 cell insulin gene transcription via decrements in stf-1 and C1 activator transcription factor activity. Mol Endocrinol. 1998;12:207–219. doi: 10.1210/mend.12.2.0066. [DOI] [PubMed] [Google Scholar]
- 17.Roduit R, Morin J, Masse F, Segall L, Roche E, Newgard CB, Assimacopoulos-Jeannet F, Prentki M. Glucose down-regulates the expression of the peroxisome proliferator- activated receptor-alpha gene in the pancreatic beta -cell. J Biol Chem. 2000;275:35799–35806. doi: 10.1074/jbc.M006001200. [DOI] [PubMed] [Google Scholar]
- 18.Sharma A, Olson LK, Robertson RP, Stein R. The reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to high glucose concentration is associated with the loss of RIPE3b1 and STF-1 transcription factor expression. Mol Endocrinol. 1995;9:1127–1134. doi: 10.1210/mend.9.9.7491105. [DOI] [PubMed] [Google Scholar]
- 19.Weir GC, Sharma A, Zangen DH, Bonner-Weir S. Transcription factor abnormalities as a cause of beta cell dysfunction in diabetes: A hypothesis. Acta Diabetol. 1997;34:177–184. doi: 10.1007/s005920050071. [DOI] [PubMed] [Google Scholar]
- 20.Edlund H. Transcribing pancreas. Diabetes. 1998;47:1817–1823. doi: 10.2337/diabetes.47.12.1817. [DOI] [PubMed] [Google Scholar]
- 21.Edlund H. Developmental biology of the pancreas. Diabetes. 2001;50(Suppl.1):S5–S9. doi: 10.2337/diabetes.50.2007.s5. [DOI] [PubMed] [Google Scholar]
- 22.Yueh YG, Gardner DP, Kappen C. Evidence for regulation of cartilage differentiation by the homeobox gene Hoxc-8. Proc Natl Acad Sci USA. 1998;95:9956–9961. doi: 10.1073/pnas.95.17.9956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kappen C. The VP16-dependent binary system for inducible gene expression in transgenic mice. In: Accili D, editor. Genetic manipulation of receptor expression and function. John Wiley & Sons; 1999. pp. 69–92. [Google Scholar]
- 24.Salbaum JM. Punc, a novel mouse gene of the immunoglobulin superfamily, is expressed predominantly in the developing nervous system. Mech Dev. 1998;71:201–204. doi: 10.1016/s0925-4773(98)00005-7. [DOI] [PubMed] [Google Scholar]
- 25.Gardner DP, Byrne GW, Ruddle FH, Kappen C. Spatial and temporal regulation of a LacZ reporter transgene in a binary transgenic mouse system. Transgenic Res. 1996;5:37–48. doi: 10.1007/BF01979920. [DOI] [PubMed] [Google Scholar]
- 26.Rundle CH, Macias MP, Gardner DP, Yueh YG, Kappen C. Transactivation of Hox Gene Expression in a VP16-Dependent Binary Transgenic Mouse System. Biochim Biophys Acta. 1998;1398:164–178. doi: 10.1016/s0167-4781(98)00046-3. [DOI] [PubMed] [Google Scholar]
- 27.Yaworsky PJ, Gardner DP, Kappen C. Transgenic analyses reveal neuron and muscle specific elements in the murine neurofilament light chain gene promoter. J Biol Chem. 1997;272:25112–25120. doi: 10.1074/jbc.272.40.25112. [DOI] [PubMed] [Google Scholar]
- 28.Gause A, Yoshida N, Kappen C, Rajewsky K. In vivo generation and function of B cells in the presence of monoclonal anti-IgM antibody: Implications for B cell tolerance. Eur J Immunol. 1987;17:981–990. doi: 10.1002/eji.1830170714. [DOI] [PubMed] [Google Scholar]
- 29.Duhamel B. From mermaid to anal imperforation: The syndrome of caudal regression. Arch Dis Child. 1961;36:152–155. doi: 10.1136/adc.36.186.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alles AJ, Sulik KK. A review of caudal dysgenesis and its pathogenesis as illustrated in an animal model. Birth Defects Orig Artic Ser. 1993;29:83–102. [PubMed] [Google Scholar]
- 31.Jones KL. Smith’s recognizable patterns of human malformations. Philadelphia: WB Saunders Co; 1988. [Google Scholar]
- 32.Dias MS, Azizkhan RG. A novel embryogenetic mechanism for Currarino’s triad: inadequate dorsoventral separation of the caudal eminence from hindgut endoderm. Pediatr Neurosurg. 1998;28:223–229. doi: 10.1159/000028655. [DOI] [PubMed] [Google Scholar]
- 33.Lynch SA, Bond PM, Copp AJ, Kirwan WO, Nour S, Balling R, Mariman E, Burn J, Strachan T. A gene for autosomal dominant sacral agenesis maps to the holoprosencephaly region at 7q36. Nat Genet. 1995;11:93–95. doi: 10.1038/ng0995-93. [DOI] [PubMed] [Google Scholar]
- 34.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 35.Jostes B, Walther C, Gruss P. The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech Dev. 1990;33:27–38. doi: 10.1016/0925-4773(90)90132-6. [DOI] [PubMed] [Google Scholar]
- 36.Ericson J, Thor S, Edlund T, Jessell TM, Yamada T. Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science. 1992;256:1555–1560. doi: 10.1126/science.1350865. [DOI] [PubMed] [Google Scholar]
- 37.Briscoe J, Ericson J. The specification of neuronal identity by graded Sonic Hedgehog signalling. Semin Cell Dev Biol. 1999;10:353–362. doi: 10.1006/scdb.1999.0295. [DOI] [PubMed] [Google Scholar]
- 38.Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- 39.Ericson J, Morton S, Kawakami A, Roelink H, Jessell TM. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87:661–673. doi: 10.1016/s0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- 40.Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 41.Roelink H, Augsburger A, Heemskerk J, Korzh V, Norlin S, Ruiz i Altaba A, Tanabe Y, Placzek M, Edlund T, Jessell TM, et al. Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell. 1994;76:761–775. doi: 10.1016/0092-8674(94)90514-2. [DOI] [PubMed] [Google Scholar]
- 42.Tanabe Y, Jessell TM. Diversity and pattern in the developing spinal cord. Science. 1996;274:1115–1123. doi: 10.1126/science.274.5290.1115. [DOI] [PubMed] [Google Scholar]
- 43.Wallin J, Wilting J, Koseki H, Fritsch R, Christ B, Balling R. The role of pax-1 in axial skeleton development. Development. 1994;120:1109–1121. doi: 10.1242/dev.120.5.1109. [DOI] [PubMed] [Google Scholar]
- 44.Balling R, Deutsch U, Gruss P. undulated, a mutation affecting the development of the mouse skeleton, has a point mutation in the paired box of Pax 1. Cell. 1988;55:531–535. doi: 10.1016/0092-8674(88)90039-6. [DOI] [PubMed] [Google Scholar]
- 45.Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM. Requirement for LIM homeobox gene Isl-1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84:309–320. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 46.Shashikant CS, Bieberich CJ, Belting H-G, Wang JCH, Borbely MA, Ruddle FH. Regulation of Hoxc-8 during mouse embryonic development: identification and characterization of critical elements involved in early neural tube expression. Development. 1995;121:4339–4347. doi: 10.1242/dev.121.12.4339. [DOI] [PubMed] [Google Scholar]
- 47.Ahlgren U, Pfaff SL, Jessell TM, Edlund T, Edlund H. Independent requirement for ISL1 in formation of pancreatic mesenchyme and islet cells. Nature. 1997;385:257–260. doi: 10.1038/385257a0. [DOI] [PubMed] [Google Scholar]
- 48.National Diabetes Data Group. Diabetes in America. National Institutes of Health; 1995. NIH Publication No. 95-1468. [Google Scholar]
- 49.Buchanan TA, Kitzmiller JL. Metabolic interactions of diabetes and pregnancy. Annu Rev Med. 1994;45:245–260. doi: 10.1146/annurev.med.45.1.245. [DOI] [PubMed] [Google Scholar]
- 50.Baker L, Piddington R. Diabetic embryopathy: a selective review of recent trends. J Diabetes Complications. 1993;7:204–212. doi: 10.1016/1056-8727(93)90046-2. [DOI] [PubMed] [Google Scholar]
- 51.Freinkel N. Diabetic embryopathy and fuel-mediated organ teratogenesis: Lessons from animal models. Horm Metabol Res. 1988;20:463–475. doi: 10.1055/s-2007-1010861. [DOI] [PubMed] [Google Scholar]
- 52.Cederberg J, Siman CM, Eriksson UJ. Combined treatment with vitamin E and vitamin C decreases oxidative stress and improves fetal outcome in experimental diabetic pregnancy. Pediatr Res. 2001;49:755–762. doi: 10.1203/00006450-200106000-00007. [DOI] [PubMed] [Google Scholar]
- 53.Van Assche FA, Holemans K, Aerts L. Fetal growth and consequences for later life. J Perinat Med. 1998;26:337–346. doi: 10.1515/jpme.1998.26.5.337. [DOI] [PubMed] [Google Scholar]
- 54.Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet. 1999;353:1789–1792. doi: 10.1016/S0140-6736(98)07546-1. [DOI] [PubMed] [Google Scholar]
- 55.Meigs JB, Cupples LA, Wilson PW. Parental transmission of type 2 diabetes: the Framingham Offspring Study. Diabetes. 2000;49:2201–2207. doi: 10.2337/diabetes.49.12.2201. [DOI] [PubMed] [Google Scholar]
- 56.Dabelea D, Pettitt DJ. Intrauterine diabetic environment confers risks for type 2 diabetes mellitus and obesity in the offspring, in addition to genetic susceptibility. J Pediatr Endocrinol Metab. 2001;14:1085–1091. doi: 10.1515/jpem-2001-0803. [DOI] [PubMed] [Google Scholar]
- 57.Casson IF, Clarke CA, Howard CV, McKendrick O, Pennycook S, Pharoah PO, Platt MJ, Stanisstreet M, van Velszen D, Walkinshaw S. Outcomes of pregnancy in insulin dependent diabetic women: results of a five year population cohort study. B M J. 1997;315:275–278. doi: 10.1136/bmj.315.7103.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Terauchi Y, Kubota N, Tamemoto H, Sakura H, Nagai R, Akanuma Y, Kimura S, Kadowaki T. Insulin effect during embryogenesis determines fetal growth: a possible molecular link between birth weight and susceptibility to type 2 diabetes. Diabetes. 2000;49:82–86. doi: 10.2337/diabetes.49.1.82. [DOI] [PubMed] [Google Scholar]
- 59.Bieberich CJ, Utset MF, Awgulewitsch A, Ruddle FH. Evidence for positive and negative regulation of the Hox-3.1 gene. Proc Natl Acad Sci USA. 1990;87:8462–8466. doi: 10.1073/pnas.87.21.8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ross AJ, Ruiz-Perez V, Wang Y, Hagan DM, Scherer S, Lynch SA, Lindsay S, Custard E, Belloni E, Wilson DI, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998;20:358–361. doi: 10.1038/3828. [DOI] [PubMed] [Google Scholar]
- 61.Hagan DM, Ross AJ, Strachan T, Lynch SA, Ruiz-Perez V, Wang YM, Scambler P, Custar E, Reardon W, Hassan S, et al. Mutation analysis and embryonic expression of the HLXB9 Currarino syndrome gene. Am J Hum Genet. 2000;66:1504–1515. doi: 10.1086/302899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lynch SA, Wang Y, Strachan T, Burn J, Lindsay S. Autosomal dominant sacral agenesis: Currarino syndrome. J Med Genet. 2000;37:561–566. doi: 10.1136/jmg.37.8.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23:71–75. doi: 10.1038/12674. [DOI] [PubMed] [Google Scholar]
- 64.Li H, Arber S, Jessell TM, Edlund H. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23:67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- 65.Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- 66.Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/s0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.