

Abstract.
The application of time domain (TD) fluorescence lifetime multiplexing for the detection of fluorescent proteins (FPs) in whole animals, in the presence of a strong background tissue autofluorescence and excitation light leakage is discussed. Tissue autofluorescence (AF) exhibits a nonexponential temporal response, distinct from the mono-exponential decay of FPs. This allows a direct separation of FP fluorescence from AF using a dual basis function approach. We establish the detection limits of this approach using in vitro and in vivo measurements. We also demonstrate, using an experimental model of lymph node metastasis, that FP-AF lifetime multiplexing provides a greater than 30-fold improvement in contrast-to-background ratio compared with continuous wave data. In addition, we show that TD detection can simultaneously discriminate between up to three red shifted FPs placed under the skin of a nude mouse based on their distinct fluorescence lifetimes.
Keywords: fluorescent proteins, lifetime multiplexing, autofluorescence, metastasis
1. Introduction
Whole body imaging of genetically encoded optical reporters in small animals has proven to be a powerful tool for studying the progression of disease and response to therapy.1,2 These reporters can be classified as either bioluminescent enzymes,3 or fluorescent proteins (FPs).4 Both bioluminescence and fluorescence are well suited for superficial imaging and can be employed successfully for imaging deeper () in tissue at longer wavelengths (). An advantage of fluorescence over bioluminescence is the availability of a wide variety of probes with spectral and lifetime tunability.5 In addition, the direct extension of lifetime multiplexing for tomographic imaging6 can enable whole body localization and tracking of multiple-labeled disease components. However, there are two major confounds for in vivo fluorescence imaging in small animals: the presence of background tissue autofluorescence (AF)7 and the small wavelength separation between the excitation and emission peaks (Stokes shift) of commonly used fluorophores, coupled with the poor rejection of excitation light by typical emission filters.8 Filter sets for fluorescence imaging function well when there is adequate fluorescence yield (product of quantum yield, extinction coefficient, and fluorophore concentration) or minimal tissue attenuation. However, when imaging deep seated tumors, small metastatic foci or cells with low FP expression or low-quantum yields, the relatively high-excitation intensities, and extended acquisition times required, can lead to significant contributions from both tissue AF and excitation light bleed-through.3,9 Both multispectral imaging9–11 and time domain (TD) imaging12,13 have been previously employed to alleviate background AF. Multispectral imaging attempts to eliminate tissue AF using multiple excitation and emission wavelength pairs along with spectral unmixing techniques10,14 but the broad and overlapping spectra of fluorophores remains a major confound for spectral unmixing. TD imaging, on the other hand, enables the exploitation of the distinct nanosecond-timescale fluorescence decay profiles [fluorescence lifetime (FL)] of both endogenous and extrinsic fluorescence. A multiexponential analysis of the asymptotic (decay) portion of the measured fluorescence decay from a diffuse medium directly leads to independent fluorescence datasets corresponding to all fluorophores present in tissue6 (Note that this separation is possible in multispectral imaging only with fluorophores that exhibit minimal spectral overlap, such as quantum dots15). Practically, the separation of fluorescence signals into multiple lifetime components results in increased specificity and sensitivity, reducing the demand for elevated excitation powers and long acquisition times. Furthermore, the decay amplitudes resulting from the multiexponential analysis can be used in efficient tomographic algorithms for localizing multiple fluorophores in vivo.6,16 TD imaging offers an additional feature, in that it can allow the rejection of excitation light leakage based on the distinct temporal responses of excitation and fluorescence decays.17
Previously, we have demonstrated that TD imaging can provide a 10-fold improvement in contrast compared with continuous wave (CW) imaging for distinguishing FP fluorescence from tissue AF12 in large breast tumors of living mice. Here, we build on this earlier work and present quantitative measurements of varying FP concentrations in vitro, in vivo, and in an experimental model of lymph node metastasis in nude mice. The results demonstrate that TD lifetime multiplexing of FPs and tissue AF can enable the in vivo detection of metastatic lesions at earlier stages than CW imaging alone, without the need for cumbersome background subtraction techniques that can depend on experimental scaling parameters.9,18 We also demonstrate that TD FL multiplexing allows for the accurate discrimination of up to three subcutaneously placed, spectrally overlapping red shifted FPs in mice, purely based on their distinct lifetimes. The techniques presented here have the potential to positively impact studies necessitating in vivo visualization of multiple genetically encoded tags.19
2. Methods
2.1. Cell Lines
MDA-MB-231-GFP, mouse LM2, and mouse 4T1(-tdTomato, -dsRed, -mCherry) cells were maintained in DMEM with 7% FCS and 1% Penicillin streptomycin at 37°C under a humidified 5% CO2 atmosphere. For in vitro experiments, cells were harvested by trypsin, diluted in culture medium followed by two cycles of centrifugation and resuspension in phosphate buffered saline (PBS). Cell dilutions were then transferred to clear, nonfluorescent PCR tubes (Thermo scientific S98752, Waltham, Massachusetts) followed by centrifugation and imaging.
2.2. Animals
All procedures were performed in accordance with the Massachusetts General Hospital animal welfare guidelines. Female Nu/Nu mice were obtained from the Cox-7 defined-flora animal facility at MGH. Prior to experimentation mice were either anesthetized or euthanized by inhaled Isoflurane (2% or 5% respectively). For subcutaneous injections, MDA-MB-231-GFP were diluted in PBS, and 30 μL were injected in anesthetized mice via 30 gauge needle followed immediately by imaging. For direct injection of the superficial cervical lymph nodes, a skin flap was performed by cutting below the sternum and along both midaxillary lines followed by careful, blunt dissection to expose the cervical lymph nodes. Approximately 10 μL of MDA-MB-231-GFP were injected via 30 gauge needle into one of the superficial cervical lymph nodes, followed by replacement of the skin flap and imaging.
2.3. Imaging System
Time resolved images were acquired with a custom built imaging system described in detail previously.20 Briefly, fluorescence was excited with the pulsed broadband (480 to 850 nm) output of a titanium sapphire driven (800 nm @ 80-MHz repletion rate, Spectraphysics, Santa Clara, California) photonic crystal fiber (Thorlabs NL-PM-750, Newton, New Jersey) filtered through either or filter. The resulting fluorescence emission was detected with either a filter, or a 560-nm long pass filter coupled to an intensified CCD camera (PicostarHR, LAVision, Gmbh; 500-ps gate width, 600-V gain, 150-ps steps, hardware binning). Excitation powers were at both 495 and 543 nm. Camera integration times ranged from 100 ms to 3 s.
2.4. Statistics
Variance between groups was first assessed via one way analysis of variation (ANOVA). Differences in means between two groups were assessed via a two tailed -test, significance was determined at a value of 0.05.
3. Results/Discussion
3.1. TD FL Multiplexing of FP Fluorescence and Tissue AF
We first validated the ability of TD FL multiplexing to detect small numbers of GFP expressing cells in the presence of high AF background and excitation leakage with in vitro measurements. 1 to 12 K MDA-MB-231-GFP cells were mixed and pelleted with LM2 cells that serve as the background AF component. Fluorescence was excited at and detected using a filter. In order to minimize the influence of excitation leakage through the emission filter, the fluorescence temporal point spread function (TPSF) was analyzed between 2.3 and 11 ns after the excitation peak. The TPSF from a tube containing only GFP expressing cells can be fit with a single exponential decay corresponding to the GFP FL of 2.6 ns while the AF from LM2 cells is best fit with a biexponential decay with lifetimes of 0.6 and 2.1 ns.
In Fig. 1(a), we show that the recorded TPSF from the mixed cell pellets approaches the TPSF from pellets containing only GFP expressing cells as the number of GFP cells in the mixture is increased. Figure 1(b) shows the CW fluorescence intensity (grayscale), calculated by integrating the TPSF for all time points and the FL maps (color) obtained from single exponential fitting of the TPSF at each pixel. The presence of GFP fluorescence is evident from increases in both CW signal and FL. However, the FLs in the images do not accurately represent the known GFP lifetime of 2.6 ns since the observed TPSF is a mixture of the lifetimes of the tissue AF and the FP. In order to assess the contribution of each lifetime component, we fit the decay of the TPSF at each image pixel according to
| (1) |
where is the net measured TPSF, is a systemic constant that accounts for experimental scaling factors such as CCD integration time and laser power, and are the decay amplitudes of the FP and AF components, is the FL of GFP and is a biexponential basis function representing the tissue AF decay, determined experimentally by performing a biexponential fit on the TPSF from either LM2 cells alone (in vitro) or on a region of skin not containing FP (in vivo). The amplitudes, and , resulting from the fits are displayed for each tube in Fig. 1(c). For visualization purposes the amplitudes of each lifetime component are normalized to the maximum value in the series, and assigned to the red (for AF) or green (for GFP) channels of an RGB image. It is clear that while the AF component does not fluctuate greatly (no significant difference in the mean ), the GFP contribution increases significantly with the number of included GFP cells. We note that in this controlled experiment, the CW increase is also statistically significant and that simply subtracting the value of the AF (determined from the tube containing only non FP expressing cells) can provide a reliable estimate of the GFP contribution to each tube. However, the accuracy of this estimation is subject to errors introduced with fluctuations in laser power, or camera integration times and is not realistic when the control AF measurement is not available. On the other hand, in TD images, these fluctuations are incorporated into the systemic constant , and equally impact both the FP and AF contributions. We can compute the GFP AF amplitude ratio and eliminate these variations, since the basis function analysis of Eq. (1) directly measures and separates the AF and FP signal in each sample. As a result, we observe a statistically significant, linear relationship between the increase in ratio and the number of GFP cells in the tube [Fig. 1(d)].
Fig. 1.
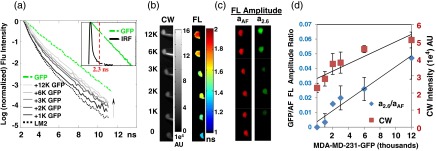
Time domain (TD) fluorescence lifetime (FL) multiplexing enables in vitro detection of GFP expressing cells in the presence of high autofluorescence and excitation leakage. MDA-MB 231-GFP cells were mixed with 2.6 million LM2 cells, pelleted and imaged. (a) TD measurements were made from 2.3 to 11 ns after the excitation peak to avoid excitation light leakage. Inset shows the instrument response function (black) obtained using a white paper placed on the imaging stage. Red dashed line shows the start of data acquisition (2.3 ns), avoiding the excitation signal. The autofluorescence temporal point spread function (TPSF) from LM2 cells is best fit with a biexponential decay with lifetimes 0.6 and 2.1 ns (dotted black line), while the shape of the normalized fluorescence TPSF from measured cell pellets approached the single exponential decay of GFP (lifetime 2.6 ns, green) with increasing number of GFP expressing cells, indicated by direction of the arrow. (b) Similarly, continuous wave (CW) fluorescence intensity and FL increase with the addition of GFP cells. (c) A dual basis function analysis [Eq. (1) in the text] of each TPSF reveals a constant AF amplitude (, red) and an increasing GFP amplitude (, green) with increasing GFP cell number. (d) The CW fluorescence intensity (red square, right axis) shows a large background due to the AF from LM2 cells and increases further with the addition of GFP cells. The GFP/AF amplitude ratio, (blue diamond, left axis) eliminates systemic variations between measurements (such as fluctuations in illumination power or integration time) and shows a linear dependence on the true GFP concentration, approaching zero for the case of pure AF cells.
We reiterate that in the above measurements, we have exploited an important advantage of TD imaging, namely the ability to exploit the distinct time scales of the fluorescence excitation and emission processes. This is illustrated in the inset of Fig. 1(a): the excitation laser pulse (black line) is rapidly diminished by 2.3 ns after the peak whereas the GFP fluorescence decay extends to an additional 8.7 ns. Thus, TD techniques allow the use of filter sets with small spectral band separations under conditions where excitation light bleed-through becomes a significant factor, such as low-probe concentrations or cells seated deeply within tissue.
Next, we performed in vivo experiments to ascertain the suitability of TD FL multiplexing for detecting small numbers of GFP expressing cells under the skin in nude mice. We injected 1 K to 15 k GFP cells subcutaneously in 30-μL saline solution followed immediately by imaging. As in our in vitro model, skin AF (excited at 495 nm) exhibits a biexponential decay, and changes in the measured TPSF are observed following the addition of GFP expressing cells [Fig. 2(a)]. From the relatively minor impact on the shape of the TPSF from 15 K subcutaneous GFP cells, it is apparent that the mouse skin AF contributes significantly more to the total fluorescence than did the AF cells in the in vitro measurements (Fig. 1). Indeed, the location of the injected cells is not clear in the CW images below 10-K GFP cells [In Fig. 2(b), black spots indicate boundary of injected area]. However, the subcutaneous location of the injected GFP cells can be observed in lifetime maps as areas of increased lifetime, compared with the background skin AF [Fig. 2(b)], well before these are visible in the CW images. As shown in Fig. 2(c), we once again observe a statistically significant linear relationship between the GFP:AF amplitude ratio and the number of implanted GFP cells. In addition, we can conclude that we have an approximate detection limit of 1.5-K subcutaneous GFP cells [Fig. 2(c)].
Fig. 2.
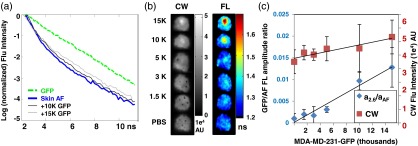
TD FL multiplexing enables in vivo detection of subcutaneous GFP expressing cells at lower levels than CW imaging. MDA-MB-231-GFP cells in 30 μL of phosphate buffered saline (PBS) were injected subcutaneously in a nude mouse. (a) The fluorescence TPSF measured from 10 and 15 K subcutaneous GFP cells (gray lines) is a combination of the biexponential decay of mouse skin (blue solid line) and the single exponential decay of a tube of GFP expressing cells (green dashed line). (b) Subcutaneous GFP cells in CW intensity images (grayscale) are only visible for cells (black spots indicate injection site boundaries) due to the strong background AF. In FL images (color), the location of GFP cells is revealed as a localized increase in FL, down to 1.5 K injected cells. (c) Both CW fluorescence intensity (red squares) and the amplitude ratio increase with the number of subcutaneous GFP cells. Note that the CW intensity is dominated by the AF background.
To demonstrate the applicability of TD FL multiplexing of FP and AF in a realistic animal disease model, we employed a model of experimental lymph node metastasis. Approximately 100 K MDA-MB-231-GFP cells were directly injected into the right-central superficial cervical lymph node of a freshly sacrificed mouse. Comparison of the CW images from before and after injection [Figs. 3(a) and 3(d), respectively] reveal a minor increase in intensity directly over the injected lymph node, with the predominant signal arising from tissue AF. In the single-exponential lifetime maps, the area directly above the injected lymph node shows a longer lifetime than the surrounding tissue [Fig. 3(e)]. Figures 3(c) and 3(f) show the amplitude maps generated using the dual basis function approach outlined above [Eq. (1)]; the basis functions consisted of a single exponential for the GFP fluorescence (2.6-ns lifetime) and a biexponential basis function for AF (lifetimes of 0.6 and 2.5 ns), determined from the TPSF of the mouse skin from the contralateral side. The contrast-to-background ratio (CBR) was estimated12 for the CW and TD approaches as and , where and are the average CW intensity and FP decay amplitude within the tumor region and and are the corresponding values outside the tumor region. The injected node is easily detectable in the FL amplitude map [Fig. 3(f)], with a much higher CBR (37.5) than the CW image (1.2). Images acquired after removing the skin [Figs. 3(g)–3(i)] and dissection of the superficial cervical lymph nodes [Figs. 3(j)–3(l)] confirm that the GFP cells were indeed located only in one superficial lymph node. These results demonstrate that lifetime multiplexing dramatically improves the contrast for detecting FPs in intact mice, potentially enabling early stage detection of metastasis in shallow organs such the mammary fat pad or lymph nodes. However, for imaging deeper organs, red shifted FPs18 are more appropriate.
Fig. 3.
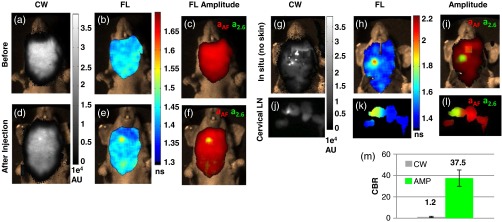
TD imaging enables detection of experimental lymph node metastasis with a high contrast-to-background ratio (CBR). The superficial cervical lymph nodes were exposed by performing a skin flap procedure in a freshly sacrificed nude mouse. Images were taken before (a–c) and after (d–f) injection of the right cervical node with of a MDA-MB-231-GFP cell solution in PBS. The corresponding FL amplitudes, , resulting from a multiexponential fit of the fluorescence TPSF with basis functions [Eq. (1)] for mouse skin AF and GFP, are shown in (c) and (f) as red and green components, respectively, of a composite RGB image (Thus, yellow indicates complete overlap of the two signals). Prior to injection, there were no detectable regions of increase in (a) CW intensity, (b) FL, or (c) FL amplitude corresponding to GFP (, green). After injection of the GFP cells, the location of the injected node can be determined from a localized increase FL directly over the injected node (E), but is barely visible in the CW fluorescence intensity (d). The decay amplitude images (f) clearly reveals the location of the injected node under the mouse skin with a (m) increase in CBR compared with the CW image. Imaging performed in situ (g–i) and of dissected cervical lymph nodes (j–l) confirm that only the right superficial cervical lymph node was injected.
3.2. TD FL Multiplexing Enables Simultaneous Imaging of Multiple Spectrally Overlapping FPs
In addition to the separation of FP and tissue AF signals, TD measurements enable multiplexed imaging of spectrally similar FPs based on their distinct lifetimes.12,21 This is important, since the large overlap in the excitation and emission spectra of FPs poses a limit on the number of FPs that can efficiently be used simultaneously using spectral unmixing alone. Multiplexed imaging of several FPs can allow the simultaneous in vivo visualization of multiple genetically encoded tags with high specificity in a living animal. Figure 4(a) shows the distinct FLs of three red shifted FPs and mouse skin AF excited at 543 nm. As we observed with GFP, the FPs exhibit single exponential decay behavior, while the mouse skin AF for 543-nm excitation is best fit with a biexponential decay (lifetimes of 0.27 and 2.1 ns). In measurements of tubes containing cells expressing dsRed, mCherry, or tdTomato, we find FLs of , , or , respectively [Fig. 4(c)], while the CW images do not distinguish the three FPs [Fig. 4(b)].
Fig. 4.

TD FL multiplexing enables the separation of three red shifted fluorescent proteins (RFPs) from mouse skin autofluorescence. Measurements of pelleted RFPs were made in vitro before and after placement under the skin of a sacrificed nude mouse. (a) tdTomato, dsRed, and mCherry have distinct, single exponential fluorescence decays with corresponding lifetimes of 3, 2.4, and 1.5 ns, respectively, while mouse skin AF (543-nm excitation) is best fit by a biexponential decay (lifetimes 0.27 and 2.1 ns). (b) CW intensity provides no identifying contrast between the tubes containing the three RFPs. (c) A single exponential fit of the fluorescence TPSF correctly reveals the lifetime of the FP expressed in each tube. (d) CW image cannot distinguish tubes containing the three RFPs placed under the skin of a nude mouse. (e) Lifetime map of the tubes under the mouse skin from a single exponential fit to each pixel. The effect of tissue AF leads to incorrect lifetimes for the tubes. (f) The contribution of each source of fluorescence can be determined by performing a multiexponential fit of the total fluorescence TPSF with four basis functions: one corresponding to the biexponential decay of the skin AF, and three single exponential decays corresponding the known FL of each RFP. For visualization, the resulting FL amplitudes: , , and are shown in red, green, and blue, respectively, of a single RGB image. Here, the FL amplitude corresponding to each FP correctly highlights and identifies the contents of each tube. The tissue AF amplitude distribution is shown in grayscale.
Figure 4(d) shows the CW image of the three FPs placed under the skin of a sacrificed nude mouse. Despite the relatively high concentration of the FPs, tissue AF is as bright as the tdTomato CW intensity. In the TD, the tissue AF contribution is also manifest in the relatively muted FL image of these subcutaneous tubes [Fig. 4(e)]. It should be noted especially that the longer lifetime tdTomato and dsRed are more strongly affected by the tissue AF, effectively reducing their observed lifetimes. Thus, single exponential fits lead to erroneous lifetimes and quantitation when analyzing in vivo data.6 However, the basis function approach [Eq. (1)] allows the correct identification and quantitation of the different FPs based on the corresponding decay amplitudes and [Fig. 4(f)]. The ratio of the recovered amplitudes was for the FPs under the skin and was similar to the in vitro amplitude ratio of , indicating the ability of the method to quantitate multiple FPs simultaneously present in an animal.
4. Conclusion
FPs have revolutionized biological imaging by enabling the visualization of biochemical processes at the cellular level. The application of FPs for whole animal imaging has enabled the tracking of disease progression in intact physiological conditions. However, many practical considerations have limited the application of FPs in living mice to large tumors or in superficial organs.22–24 Most important of the limitations is the presence of strong background tissue AF. Although CW fluorescence imaging has been applied to detect FPs in whole mice,9 the presence of tissue AF limits the minimum detectable levels of fluorescence, thereby limiting the sensitivity of the technique for whole body imaging.3 In addition, these measurements necessitate very high laser powers [ (Refs. 18 and 25)], long acquisition times (approximately minutes) and background referencing schemes9 that may not be possible in realistic in vivo measurements. In this article, we have demonstrated the unique advantages of TD FL multiplexing for the separation of tissue AF and FP fluorescence. Our results indicate that lifetime contrast can dramatically improve the contrast for FP detection in vivo, requiring lower laser powers ( in this study) or faster acquisition (seconds). These results suggest the potential of the TD lifetime multiplexing for detecting metastasis in whole mice at earlier stages than currently possible using optical imaging, thereby impacting studies that monitor cancer progression and therapeutic intervention in small animal models.
We have also demonstrated the application of TD FL multiplexing for the separation of multiple spectrally similar far red-FPs in whole mice. Indeed, a unique advantage of fluorescence over bioluminescence is the ability to multiplex based on spectrum and lifetime. This feature is a consequence of the availability of a wide range of fluorescent compounds with spectral and lifetime tunability. Our studies show the advantage of a basis function based approach over single exponential fits for recovering FP fluorescence from tissue AF. The basis function approach relies on a prior knowledge of the FP and AF component lifetimes, which can be readily obtained using in vitro and in vivo measurements with control mice. The use of a linear unmixing algorithm with fixed lifetimes reduces a complex nonlinear problem to a linear inversion for the decay amplitudes. The amplitudes provide a direct readout of the relative strength of each fluorophore with distinct lifetime. The decay amplitudes are also readily amenable for tomographic inversion using rigorous mathematical models for lifetime multiplexing,20,26 to recover quantitative in vivo yield distributions. Our ongoing work is focused on tomographic imaging of deep tissue metastasis in organs such as the lungs using far-red FP-labeled cells. In future work, we will also apply FP-AF lifetime multiplexing for simultaneously following the changes and therapy response of multiple fluorescently labeled tissue components.
Acknowledgments
This work was supported by the National Institutes of Health grant: NIH R01 EB015325.
Biographies
William L. Rice is a research fellow in the Department of Radiology at the Athinoula A. Martinos Center for Biomedical Imaging, Massachusetts General Hospital. He received his PhD in biomedical engineering from Tufts University in 2010. His current research is focused on developing fluorescence lifetime based methods for small animal imaging.
Anand T. N. Kumar is an assistant professor at the Athinoula A Martinos Center for Biomedical Imaging in the Department of Radiology, Massachusetts General Hospital. He received his MSc and PhD degrees in physics from the Indian Institute of Technology, Chennai, India, and Northeastern University, Boston, respectively. His current research focus is on diffuse optical tomography and optical molecular imaging.
References
- 1.Zhang J., et al. , “Creating new fluorescent probes for cell biology,” Nat. Rev. Mol. Cell Biol. 3(12), 906–918 (2002). 10.1038/nrm976 [DOI] [PubMed] [Google Scholar]
- 2.Tsien R. Y., “Fluorescence readouts of biochemistry in live cells and organisms,” in Molecular Imaging: Principles and Practice, Weissleder R., et al., Eds., pp. 808–828, PMPH, USA: (2010). [Google Scholar]
- 3.Troy T., et al. , “Quantitative comparison of the sensitivity of detection of fluorescent and bioluminescent reporters in animal models,” Mol. Imaging 3(1), 9–23 (2004). 10.1162/153535004773861688 [DOI] [PubMed] [Google Scholar]
- 4.Shaner N. C., Steinbach P. A., Tsien R. Y., “A guide to choosing fluorescent proteins,” Nat. Methods 2, 905–909 (2005). 10.1038/NMETH819 [DOI] [PubMed] [Google Scholar]
- 5.Berezin M. Y., Achilefu S., “Fluorescence lifetime measurements and biological imaging,” Chem. Rev. 110(5), 2641–2684 (2010). 10.1021/cr900343z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond S. B., et al. , “Lifetime-based tomographic multiplexing,” J. Biomed. Opt. 15(4), 046011 (2010). 10.1117/1.3469797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garofalakis A., et al. , “Three-dimensional in vivo imaging of green fluorescent protein – expressing T cells in mice with noncontact fluorescence molecular tomography,” Mol. Imaging 6(2), 96–107 (2007). 10.2310/7290.2007.00007 [DOI] [PubMed] [Google Scholar]
- 8.Zhu B., et al. , “Reduction of excitation light leakage to improve near-infrared fluorescence imaging for tissue surface and deep tissue imaging,” Med. Phys. 37(11), 5961–5970 (2010). 10.1118/1.3497153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deliolanis N. C., et al. , “In vivo tomographic imaging of red-shifted fluorescent proteins,” Biomed. Opt. Express 2(4), 887–900 (2011). 10.1364/BOE.2.000887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mansfield J. R., et al. , “Autofluorescence removal, multiplexing, and automated analysis methods for in-vivo fluorescence imaging,” J. Biomed. Opt. 10(4), 41207–41209 (2005). 10.1117/1.2032458 [DOI] [PubMed] [Google Scholar]
- 11.Tam J. M., et al. , “Improved in vivo whole-animal detection limits of green fluorescent protein-expressing tumor lines by spectral fluorescence imaging,” Mol. Imaging 6(4), 269–276 (2007). [PubMed] [Google Scholar]
- 12.Kumar A. T. N., et al. , “Feasibility of in vivo imaging of fluorescent proteins using lifetime contrast,” Opt. Lett. 34(13), 2066–2068 (2009). 10.1364/OL.34.002066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCormack E., et al. , “Nitroreductase, a near-infrared reporter platform for in vivo time-domain optical imaging of metastatic cancer,” Cancer Res. 73(4), 1276–1286 (2013). 10.1158/0008-5472.CAN-12-2649 [DOI] [PubMed] [Google Scholar]
- 14.Gao X., et al. , “In vivo cancer targeting and imaging with semiconductor quantum dots,” Nat. Biotechnol. 22(8), 969–976 (2004). 10.1038/nbt994 [DOI] [PubMed] [Google Scholar]
- 15.Stroh M., et al. , “Quantum dots spectrally distinguish multiple species within the tumor milieu in vivo,” Nat. Med. 11(6), 678–682 (2005). 10.1038/nm1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rice W. L., Hou S., Kumar A. T. N., “Resolution below the point spread function for diffuse optical imaging using fluorescence lifetime multiplexing,” Opt. Lett. 38(12), 2038–2040 (2013). 10.1364/OL.38.002038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diamandis E. P., “Immunoassays with time-resolved fluorescence spectroscopy: principles and applications,” Clin. Biochem. 21(3), 139–150 (1988). 10.1016/0009-9120(88)90001-X [DOI] [PubMed] [Google Scholar]
- 18.Deliolanis N. C., et al. , “Performance of the red-shifted fluorescent proteins in deep-tissue molecular imaging applications,” J. Biomed. Opt. 13(4), 044008 (2008). 10.1117/1.2967184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winkler F., et al. , “Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis,” Glia 57(12), 1306–1315 (2009). 10.1002/glia.v57:12 [DOI] [PubMed] [Google Scholar]
- 20.Kumar A. T. N., et al. , “A time domain fluorescence tomography system for small animal imaging,” IEEE Trans. Med. Imaging 27(8), 1152–1163 (2008). 10.1109/TMI.2008.918341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pepperkok R., et al. , “Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy,” Curr. Biol. 9(5), 269–272 (1999). 10.1016/S0960-9822(99)80117-1 [DOI] [PubMed] [Google Scholar]
- 22.Hoffman R. M., “Green fluorescent protein imaging of tumour growth, metastasis, and angiogenesis in mouse models,” Lancet Oncol. 3(9), 546–556 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Yang M., et al. , “Transgenic nude mouse with ubiquitous green fluorescent protein expression as a host for human tumors,” Cancer Res. 64(23), 8651–8656 (2004). 10.1158/0008-5472.CAN-04-3118 [DOI] [PubMed] [Google Scholar]
- 24.Yang M., et al. , “Whole-body and intravital optical imaging of angiogenesis in orthotopically implanted tumors,” Proc. Natl. Acad. Sci. U. S. A. 98(5), 2616–2621 (2001). 10.1073/pnas.051626698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zacharakis G., et al. , “Fluorescent protein tomography scanner for small animal imaging,” IEEE Trans. Med. Imaging 24(7), 878–885 (2005). 10.1109/TMI.2004.843254 [DOI] [PubMed] [Google Scholar]
- 26.Hou S. S., et al. , “Tomographic lifetime imaging using combined early- and late-arriving photons,” Opt. Lett. 39(5), 3–7 (2014). 10.1364/OL.39.001165 [DOI] [PMC free article] [PubMed] [Google Scholar]