
Fig.1.
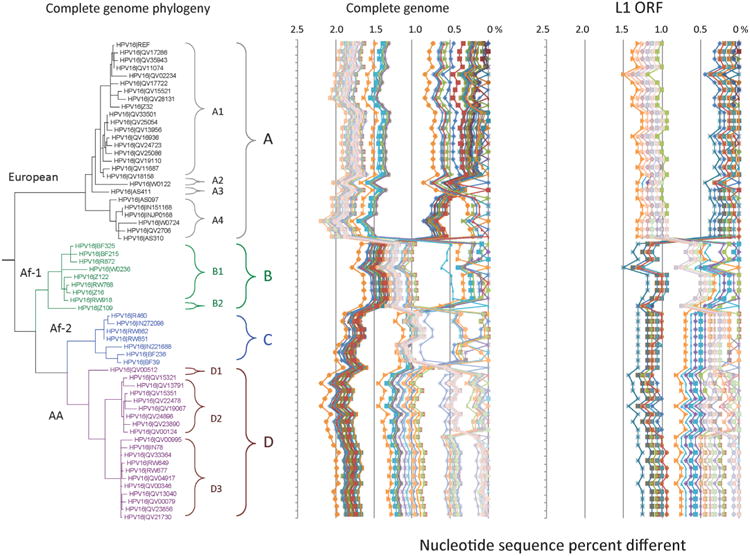
HPV16 variant tree topology and pairwise comparisons of individual complete genomes. A maximum likelihood (ML) tree was inferred from a global alignment of 62 complete genome nucleotide sequences of HPV16 using RAxML HPC v7.2.8 (Stamatakis, 2006). The 62 HPV16 genomes were previously described in Smith et al. (2011). Distinct variant lineages (i.e., termed A/B/C/D) and sublineages (e.g., termed A1/A2/A3/A4) were classified according to the topology and nucleotide sequence differences from > 1% to < 10%, and > 0.5% to < 1% ranges, respectively (Bernard et al., 2010; Chen et al., 2011). The p-distance method in MEGA5 (Tamura et al., 2011) was used to calculate pairwise nucleotide sequence differences for each isolate compared to all other isolates based on the complete genome nucleotide sequences and are shown in the middle panel, labeled “Complete genome” at the top of the figure. For comparison, the L1 ORF nucleotide sequence of each isolate was compared to that of all other isolates and is shown in the right panel, labeled “L1 ORF”. Values for each comparison of a given isolate are connected by lines and the comparison to self is indicated by the 0% difference point.