

A novel mechanism is shown by which TRPV5 and Ca2+ reabsorption by the kidney is regulated. This supports the idea that histidine phosphorylation plays other, yet-uncovered roles in mammalian biology.
Abstract
The kidney, together with bone and intestine, plays a crucial role in maintaining whole-body calcium (Ca2+) homoeostasis, which is primarily mediated by altering the reabsorption of Ca2+ filtered by the glomerulus. The transient receptor potential-vanilloid-5 (TRPV5) channel protein forms a six- transmembrane Ca2+-permeable channel that regulates urinary Ca2+ excretion by mediating active Ca2+ reabsorption in the distal convoluted tubule of the kidney. Here we show that the histidine kinase, nucleoside diphosphate kinase B (NDPK-B), activates TRPV5 channel activity and Ca2+ flux, and this activation requires histidine 711 in the carboxy-terminal tail of TRPV5. In addition, the histidine phosphatase, protein histidine phosphatase 1, inhibits NDPK-B–activated TRPV5 in inside/out patch experiments. This is physiologically relevant to Ca2+ reabsorption in vivo, as short hairpin RNA knockdown of NDPK-B leads to decreased TRPV5 channel activity, and urinary Ca2+ excretion is increased in NDPK-B−/− mice fed a high-Ca2+ diet. Thus these findings identify a novel mechanism by which TRPV5 and Ca2+ reabsorption is regulated by the kidney and support the idea that histidine phosphorylation plays other, yet-uncovered roles in mammalian biology.
INTRODUCTION
The kidney plays an important role in the control of Ca2+ homeostasis and secretion. Of the filtered Ca2+, only ∼1–2% of the Ca2+ filtered at the glomerulus is excreted in the urine (Hoenderop et al., 2005). Although the majority of Ca2+ is reabsorbed in the proximal tubule and thick ascending limb of the loop of Henle via paracellular pathways (Biner et al., 2002), between 3 and 7% of the filtered Ca2+ is reabsorbed in the distal convoluted tubule and collecting duct via the transient receptor potential-vanilloid-5 (TRPV5) channel (Boros et al., 2009). TRPV5 in the apical plasma membrane provides a channel for Ca2+ entry into the cell and its subsequent reabsorption across the basolateral membrane via the Na+/Ca2+ exchanger NCX1 and Ca-ATPase PMCA1b (Boros et al., 2009; Dimke et al., 2011). Although TRPV5 is responsible for reabsorption of a relatively small portion of filtered Ca2+, it plays a critical role in maintaining whole-body Ca2+ homeostasis. This is supported by the finding that TRPV5−/− mice exhibit urinary Ca2+ wasting accompanied by secondary hyperparathyroidism (Hoenderop et al., 2003).
TRPV5 is regulated by a number of mechanisms, which include transcriptional activation, intracellular trafficking, and posttranslational modification (Boros et al., 2009; Dimke et al., 2011). For example, parathyroid hormone (PTH), 1,25(OH)2 vitamin D3, and estrogen increase TRPV5 expression via transcriptional activation of the TRPV5 gene (Hoenderop et al., 2002; Boros et al., 2009). PTH also increases TRPV5-mediated Ca2+ influx by inhibiting endocytosis of TRPV5 from the plasma membrane (PM) by activating a protein kinase C pathway and activating TRPV5 channels in the PM by protein kinase A–mediated phosphorylation of threonine 709 in the carboxy-terminus (CT) of TRPV5 (Cha et al., 2008; Boros et al., 2009; de Groot et al., 2009). Other receptors also regulate TRPV5 trafficking and TRPV5 channel activity via activation of downstream serine/threonine kinases. For example, the Ca2+-sensing receptor activates TRPV5 by stimulating phosphorylation of TRPV5 on serines 299 and 654 (Topala et al., 2009), and tissue kallikrein, by activating the bradykinin receptor, increases TRPV5 activity by inhibiting endocytosis of TRPV5 from the PM in response to phosphorylation of the same serine residues (Gkika et al., 2006).
We previously found that the K+ channel KCa3.1 is activated by direct phosphorylation of histidine358 in the CT of KCa3.1 by nucleoside diphosphate kinase-B (NDPK-B) and is inhibited by the histidine phosphatase, protein histidine phosphatase 1 (PHPT1), which mediates dephosphorylation of the same histidine residue (Srivastava et al., 2006, 2008). PHPT1 is an evolutionarily conserved, 14-kDa enzyme that dephosphorylates phosphohistidine, although it bears no resemblance to either serine/threonine or tyrosine phosphatases (Klumpp and Krieglstein, 2009). In searching the literature for other channels that resembled KCa3.1 and therefore might be candidates for being regulated in a similar manner, we identified TRPV5. Both channels are tetramers, bind calmodulin in the CT, and are regulated via binding of calcium to calmodulin (Maylie et al., 2004; de Groot et al., 2011). In addition, data demonstrate that mutation of histidine 711 (H711) in the CT of human TRPV5 leads to increase in TRPV5 channel activity that is mimicked by substitution of aspartic acid, an acidic amino for the same histidine (de Groot et al., 2010), suggesting that phosphorylation of H711 could also activate TRPV5. An important role for H711 is also supported by the finding that H711 is evolutionarily conserved in TRPV5 and not in the related TRPV6 channel (Figure 1).
FIGURE 1:

(A) Alignment of amino acid sequence surrounding H711 of human TRPV5 with the corresponding sequences of TRPV5 and TRPV6 from a number of different species. (B) Schematic representation of TRPV5: the six transmembrane domains (S), the CT calmodulin-binding domain, and H711.
We show here that TRPV5 is regulated in a similar manner to KCa3.1. TRPV5 is activated by the histidine kinase NDPK-B and inhibited by the histidine phosphatase, PHPT1. Consistent with the role for activation via direct histidine phosphorylation, NDPK-B activates TRPV5 inside/out patch experiments, and the increase in TRPV5 channel activity stimulated by NDPK-B is reversed by addition of PHPT1 or mutation of H711 on TRPV5 to alanine. Moreover, NDPK-B−/– mice exhibit increased renal Ca2+ loss on a high-Ca2+ diet. Thus, in addition to providing evidence for a new mechanism for regulation of TRPV5 and Ca2+ homeostasis in vivo, these findings reinforce the importance of histidine phosphorylation in mammalian biology.
RESULTS
NDPK-B wild type, but not kinase-dead NDPK-B, activates TRPV5
To assess whether NDPK-B activates TRPV5, we transfected HEK293 cells with TRPV5 either alone or together with NDPK-B(wild type [WT]) or a kinase-dead NDPK-B (NDPK-B(KD)) and assessed TRPV5 activity by whole-cell patch. HEK293 cells transfected with TRPV5 exhibited a characteristic inward rectifying Na+ current that was absent in vector control–transfected cells (Figure 2A, i and ii). Cotransfection of NDPK-B(WT) with TRPV5 resulted in a marked increase in Na+ current at −80 mV from 290 to 470 pA/pF (Figure 2A, i and ii). The increase in channel activity required NDPK-B kinase activity because the increase in Na+ current was not seen in cells cotransfected with NDPK-B(KD) (Figure 2A, i and ii).
FIGURE 2:

NDPK-B activates TRPV5 channels. HEK293 cells were transfected with TRPV5(WT) or TRPV5(H711N) alone or together with NDPK-B(WT) or NDPK-B(KD), and (A) Na+ and (B) Ca2+ currents were determined by whole-cell patch clamp. (Ai) Representative I/V tracing in response to a voltage ramp. (Bi) Representative current-time graph. (Aii, Bii) Bar graph summaries of average Na+ (at −80 mV) and Ca2+ current, respectively. Also shown in Bii is the inability of NDPK-A to activate TRPV5. Data are from n = 10–15 cells. *p < 0.05 as compared with the current from TRPV5 WT–transfected cells analyzed using a one-way analysis of variance. (C) Cells transfected as in A were loaded with Fura-2-AM, and calcium influx was determined as described in Materials and Methods. Calcium flux experiments were done 10–15 times, and each time the data were obtained by averaging 30 cells.
The increase in channel activity observed by cotransfection with NDPK-B(WT), but not NDPK-B(KD), suggested that NDPK-B activates TRPV5 by directly phosphorylating H711 in the CT of TRPV5. H711 was mutated to asparagine (TRPV5-H711N) and channel activity assessed in cells transfected with NDPK-B(WT) or NDPK-B(KD). Whereas basal activity of TRPV5(H711N) was similar to that of wild-type TRPV5 (TRPV5-WT) when transfected with a vector control, cotransfection with NDPK-B(WT) did not result in an increase in TRPV5(H711N) channel activity (Figure 2Aii).
NDPK-B(WT), but not NDPK-B(KD), also significantly increased TRPV5-mediated inward Ca2+ current in HEK293 cells and decreased the feedback inhibition of TRPV5 mediated by Ca2+ (Figure 2B, i and ii). Activation by NDPK-B(WT) is specific because we found that NDPK-A(WT), which is 88% identical to NDPK-B(WT), did not activate TRPV5 (Figure 2Bii). Consistent with the increase in Ca2+ current, NDPK-B(WT), but not NDPK-B(KD), also led to an increase in TRPV5-mediated influx of Ca2+ into whole cells as assessed with the Ca2+-sensitive dye fura-2-aceotxymethyl ester (Fura-2-AM; Figure 2C). Thus, NDPK-B(WT) stimulates Ca2+ influx into cells by TRPV5 by both increasing inward Ca2+ current and decreasing the feedback inhibition mediated by Ca2+-induced association of calmodulin with CT of TRPV5 (de Groot et al., 2011).
NDPK-B activates TRPV5 channel activity at the plasma membrane
To assess whether NDPK-B activated TRPV5 by increasing the number of TRPV5 channels in the PM, we transfected HEK293 cells with green fluorescent protein (GFP)–TRPV5 alone or together with mCherry-FLAG-NPDK-B and determined TRPV5 channels inserted in the PM after surface labeling of cells with biotin. These studies demonstrated that the amount of TRPV5 labeled with biotin was not increased in cells overexpressing NDPK-B (Figure 3A), indicating that NDPK-B does not increase TRPV5 channel activity by increasing the number of TRPV5 channels inserted into the PM.
FIGURE 3:
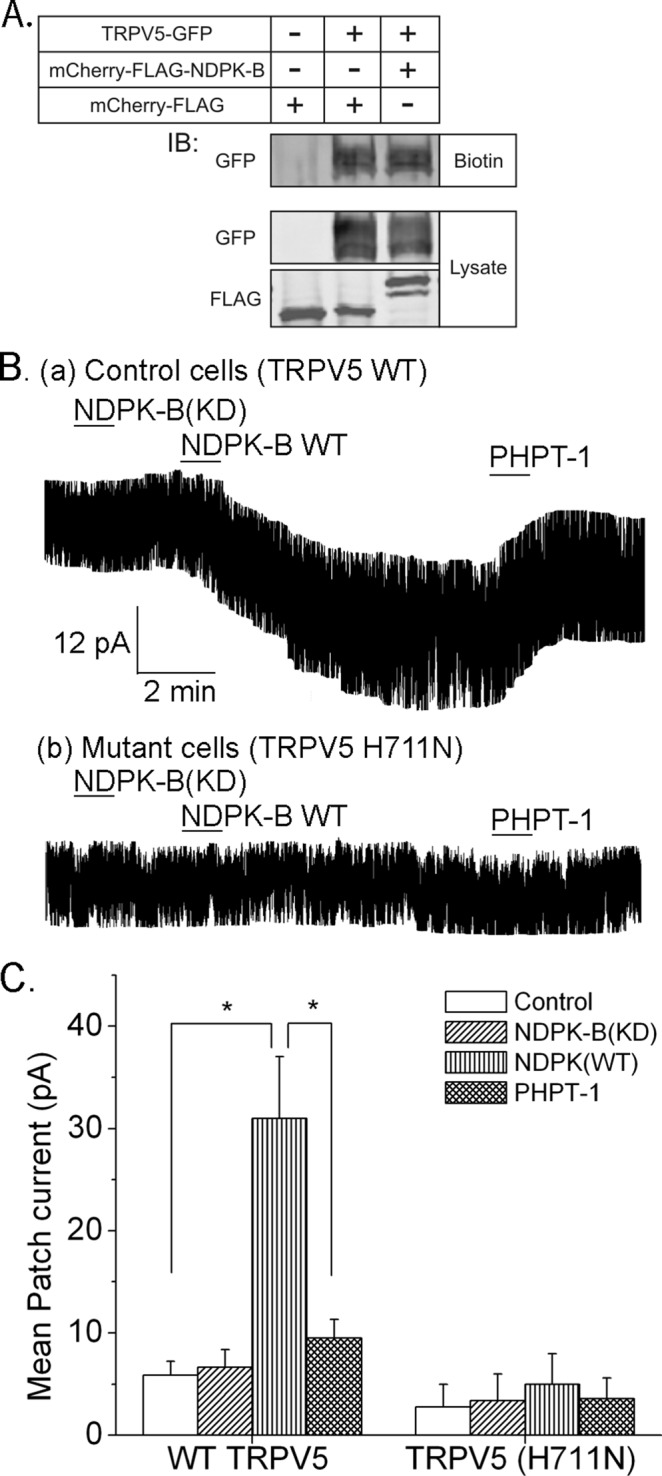
NDPK-B activates and PHPT1 inhibits TRPV5 in inside/out patch experiments. (A) HEK293 cells were transfected with GFP-TRPV5 together with mCherry-FLAG-NDPK-B or mCherry FLAG vector control. After surface labeling with biotin, cells were lysed, and biotin-labeled proteins were concentrated with High Capacity NeutrAvidin in Agarose Resin. Surface labeled GFP-TRPV5 was determined by immunoblotting precipitates with anti-GFP antibodies (top, biotin). To control for transfection efficiency, lysates were immunoblotted with anti-GFP or anti-FLAG antibodies as shown. n = 4 experiments. (B) Inside/out patch experiments on patches derived from HEK293 cells transfected with TRPV5(WT) or TRPV5(H711N). Na+ current was measured before and after addition of proteins as indicated. (C) Summary of mean patch current from TRPV5(WT)- or TRPV5(H711N)-transfected cells. n = 10–12 cells. *p < 0.05.
We next tested whether NDPK-B directly increased the activity of TRPV5 channels in the PM in inside/out (I/O) patch experiments performed on patches obtained from HEK293 cells overexpressing TRPV5(WT) or TRPV5(H711N). The addition NDPK-B(WT) to I/O patches containing TRPV5(WT) led to a marked increase in TRPV5 channel activity, which was not observed after addition of NDPK-B(KD) (Figure 3, Ba and C). Consistent with the idea that NDPK-B directly activates TRPV5 by phosphorylating H711, the addition of the histidine phosphatase, PHPT1, reversed the increase in TRPV5 channel activity stimulated by NDPK-B (Figure 3, Ba and C). Moreover, NDPK-B(WT) failed to activate TRPV5(H711N) in similar I/O patch experiments (Figure 3, Bb and C).
Endogenous NDPK-B is also required for full activation of TRPV5
To assess whether endogenous NDPK-B regulates TRPV5, we assessed TRPV5 channel activity after short hairpin RNA (shRNA) knockdown of NDPK-B in HEK293 cells. shRNA knockdown of NDPK-B led to decreased TRPV5-mediated Na+ (Figure 4A) and Ca2+ (Figure 4B) currents compared with cells transfected with an shRNA control. shRNA knockdown of NDPK-B also led to decreased TRPV5-mediated Ca2+ influx into cells (Figure 4C).
FIGURE 4:

shRNA knockdown of NDPK-B in HEK293 cells inhibits TRVP5 channel activity. NDPK-B was silenced by infecting HEK293 cells with an shRNA lentivirus to NDPK-B or a control shRNA, and (A) Na+ and (B) Ca2+ currents were measured as described in Figure 2. (C) Ca2+ flux measured in the same cells as described in Figure 1C. (D) Lysates from shRNA-infected cells were immunoblotted with antibodies to NDPK-B or β-actin, which served as loading control. n = 10–15 cells. p < 0.05 as compared with the WT.
Madin–Darby canine kidney (MDCK) cells constitute a renal epithelial cell line derived from the distal/tubule collecting duct and are more physiologically relevant cells in which to study TRPV5 than HEK293 cells. Both Na+ current (Figure 5A) and TRPV5-mediated Ca2+ influx (Figure 5B) into MDCK cells decreased after knockdown of NDPK-B compared with cells transfected with shRNA control. Moreover, the decrease in Na+ current in shRNA-transfected cells could be rescued by overexpressing NDPK-B containing point mutations that abrogate binding of the shRNA, indicating that the shRNA inhibits by specifically silencing NDPK-B.
FIGURE 5:

shRNA knockdown of NDPK-B in MDCK cells inhibits TRPV5 channel activity. NDPK-B was silenced in MDCK cells overexpressing GFP-TRPV5 as described in Figure 4, and (A) Na+ current and (B) Ca2+ flux were determined as described in Figures 2 and 4. (Aii) Bar graph summary of average Na+ current at −80 mV. The decrease in Na+ current after knockdown of NDPK-B was specific, as the Na+ current in NDPK-B shRNA–infected cells could be rescued by expressing a mutant NDPK-B that did not bind the shRNA (A). n = 10–15 cells for each experiment. *p < 0.05 as compared with the WT or as shown in the figure. (C) Lysates from shRNA- or control-transfected cells were immunoblotted with anti–NDPK-B or anti-actin antibodies.
NDPK-B–knockout mice have increased urinary Ca2+ excretion on a high-Ca2+ diet
NDPK-B–knockout mice are viable and appear phenotypically normal (Di et al., 2010b). To assess whether NDPK-B contributes to Ca2+ reabsorption in vivo, we determined urinary Ca2+ excretion in NDPK-B−/− mice subjected to high- and low-Ca2+ diets. Serum electrolytes, including Ca2+, Na+, K+, HCO3−, blood urea nitrogen, creatinine, and phosphorus, were similar between NDPK-B+/+ and NDPK-B−/− mice on both the low- and high-Ca2+ diets. However, whereas excretion of urinary Ca2+ was similar between NDPK-B+/+ and NDPK-B−/− mice on a low-Ca2+ diet (Figure 6Ai), Ca2+ excretion on a high-Ca2+ diet (Figure 6Aii), when normalized against urinary creatinine, was significantly increased in NDPK-B−/− mice. The increase in urinary Ca2+ was specific because under the same conditions the excretion of both Na+ and K+ was similar between NDPK-B+/+ and NDPK-B−/− mice (Figure 6B and C).
FIGURE 6.

NDPK-B−/− mice have increased urinary excretion of Ca2+. NDPK-B+/+ or NDPK-B−/− mice were placed on low- or high-Ca2+ diet for 2 wk, after which they were placed in metabolic cages and 24-h urine was collected. Shown is the amount of Ca2+ in the urine on a low-Ca2+ (Ai) or high-Ca2+ (Aii) diet normalized to urinary creatinine. As a control, there was no difference in the amount of Na+ (B) or K+ (C) excreted in the urine when normalized to urine creatinine on the same samples from mice on a high-Ca2+diets. n = 10 mice in each group. Statistics was done in Prism (GraphPad, La Jolla, CA), and t test was used for the significance. *p < 0.05 as compared with the WT.
DISCUSSION
Histidine phosphorylation is the predominant mechanism for regulating signaling pathways in prokaryotes and some fungi and plants (Attwood et al., 2007; Klumpp and Krieglstein, 2009; Kee and Muir, 2012). However, whereas for >40 years histidine phosphorylation has been proposed to play critical roles in regulating biological processes in mammals, until recently there has been little evidence that histidine phosphorylation also plays an essential role in mammalian biology (Attwood et al., 2007, 2010; Klumpp and Krieglstein, 2009). Recent biochemical and genetic experiments demonstrating that NDPK-B phosphorylates and activates the K+ channel KCa3.1 and the β-subunit of heterotrimeric G proteins has now provided definitive proof that histidine phosphorylation is also relevant to mammals (Cuello et al., 2003; Srivastava et al., 2006; Hippe et al., 2009; Di et al., 2010a, b). We now demonstrate that NDPK-B activates and PHPT1 reverses NDPK-B activation of TRPV5 channel activity, suggesting that TRPV5 is also regulated by reversible histidine phosphorylation.
NDPKs are encoded by the NME genes, which comprise 10 genes divided phylogenetically into two distinct groups, of which NDPK-A and -B (also known as Nm23-H1 and H2, respectively) are the best studied (Boissan et al., 2009; Boissan and Lacombe, 2011). In addition, NDPK-A and -B have been shown to function as histidine kinases in mammalian cells and are the only histidine kinases molecularly identified in mammals (Klumpp and Krieglstein, 2009). For nucleoside diphosphate and histidine kinase activities, NDPK-A and -B autophosphorylate on histidine 118 to generate a high-energy phosphate intermediate, which is then transferred to a nucleoside diphosphate to generate a nucleoside triphosphate or to a histidine residue on another protein to generate phosphohistidine (Klumpp and Krieglstein, 2009). Whereas NDPK-A and -B are 88% homologous and share some redundant functions, NDPK-B has been shown to histidine phosphorylate specific targets and regulate specific biologically processes that are not regulated by NDPK-A. For example, NDPK-B, but not NDPK-A, has been shown to histidine phosphorylate KCa3.1 and the β-subunit of heterotrimeric G proteins (Hippe et al., 2003; Srivastava et al., 2006). We now show that NDPK-B, but not NDPK-A, also specifically activates TRPV5, and this activation requires both the NDPK-B histidine kinase activity and histidine 711 in the C-terminus of TRPV5.
We provide several lines of evidence that NDPK-B and PHPT1 directly regulate TRPV5 channels at the PM by reversible histidine phosphorylation and that this regulation is physiologically relevant. This is supported by the finding that NDPK-B wild type, but not a kinase-dead NDPK-B, activates TRPV5 in whole-cell patch experiments, and activation of TRPV5 by NDPK-B requires H711 in the CT of TRPV5. Surface labeling experiments demonstrate that NDPK-B does not affect the amount of TRPV5 at the PM. Instead, I/O patch experiments demonstrate that NDPK-B directly activates TRPV5 channels in the PM, and this activation is reversed after addition of the histidine phosphatase, PHPT1, or by mutating H711 on TRPV5 to alanine. Furthermore, the finding that shRNA knockdown of NDPK-B results in decreased TRPV5 channel activity in two cell lines, including MDCK cells, which are derived from renal distal convoluted tubule/collecting duct cells where TRPV5 functions, coupled with the increase in urinary calcium excretion in NDPK-B−/− mice, indicates that NDPK-B activation of TRPV5 is biologically relevant to calcium homeostasis in vivo. We have been unable to demonstrate direct phosphorylation of TRPV5 by NDPK-B in reconstitution experiments. We believe that this is likely due to technical problems with our assays, as well as reagents that are available to detect histidine phosphorylated proteins. With regard to NPDK-B, it was reported that NDPK-B requires an unidentified cofactor in order to act as a histidine kinase, which is likely why we have been unsuccessful in our reconstitution experiments (Cuello et al., 2003).
Given the critical role TRPV5 plays in regulating whole-body calcium homeostasis (Hoenderop et al., 2003), it is not surprising that TRVP5 is subject to tight regulation by a variety of factors. For example, PTH, CaSR, tissue kallikrein, and bradykinin have all been shown to regulate TRPV5 by either affecting the trafficking of TRPV5 to and from the PM or directly regulating TRPV5 channel activity (Boros et al., 2009; Dimke et al., 2011). The signaling pathways that are activated downstream of these pathways that subsequently regulate TRPV5 activity are poorly defined, although PKC and PKA have been the predominant kinases shown to regulate TRPV5. Of interest, PTH-stimulated phosphorylation of threonine 709 in the CT of TRPV5 by PKA has been shown to increase the open probability of TRPV5 by diminishing the amount of Ca2+-induced binding of calmodulin to TRPV5 (de Groot et al., 2011); the rapid influx of Ca2+ leads to TRPV5 inactivation by promoting binding of calmodulin to TRPV5. Thus we speculate that NDPK-B phosphorylation of H711, which is contained within the amino acid sequence that mediates calmodulin binding to TRPV5, functions in a similar manner to increase TRPV5 channel activity.
We have only limited understanding of how NDPK-B is regulated in mammalian cells. Previous studies showed that generation of phosphatidylinositol 3-phosphate at the PM by the class II phosphatidylinositol 3-kinase (PI3-kinase) C2β is required for NPDK-B to phosphorylate and activate KCa3.1 in CD4 T-cells and mast cells (Srivastava et al., 2009, 2012). Future studies will address whether PI3-kinase C2β is also required for NDPK-B activation of TRPV5 and whether any of the factors known to regulate TRPV5 channel activity mediate their effects by signaling via PI3-kinase C2β and/or NDPK-B. de Groot et al. (2010) reported that mutation of histidine 711 to aspartic acid (TRPV5(H711D)) led to increased TRPV5 channel activity. However, in contrast to our results, they found that the increase in TRPV5 channel activity of TRPV5(H711D) was predominantly due to an increase in total plasma membrane TRPV5 due to decreased endocytosis of the mutant channel. We do not have an explanation for these differences. One possibility is that NDPK-B activates TRPV5 via H711-dependent and -independent pathways. It is also possible that subtle differences in experimental conditions account for the different results. For example, under our experimental conditions, we found that both human and rabbit TRPV5(H711D) behave like wild-type TRPV5 when transfected into HEK293 or MDCK cells.
Protein phosphorylation is the most common and arguably best-studied mechanism for posttranslational modification. An overwhelming number of studies in mammalian systems have focused on serine/threonine and tyrosine phosphorylation, with relatively few studies addressing the role of histidine phosphorylation in mammalian biology. In addition to providing a new mechanism for the regulation of TRPV5 and Ca2+ homeostasis in vivo, our findings provide further justification to search for additional roles for histidine kinases and phosphatases in mammalian biology. It is anticipated that identification of signaling pathways regulated by histidine phosphorylation will uncover new insights into mammalian biology and disease mechanisms and result in new therapeutic targets to treat disease.
MATERIALS AND METHODS
DNA constructs, shRNA constructs, and cell culture
The human TRPV5 cDNA was kindly provided by Ji-Bin Peng (University of Alabama at Birmingham, Birmingham, AL; Zhang et al., 2008) and was cloned into the pEGFP-N1 vector. Kinase-dead NDPK-B was generated by mutating histidine 118 on NDPK-B as previously described (Srivastava et al., 2006). FLAG-NDPK-B was generated by cloning human NDPK-B into the mCherry-C1 vector. shRNA against NDPK-B were purchased from Sigma-Aldrich, St. Louis, MO (clone ID: TRCN0000054518), and lentiviruses were generated and used to infect cells. The shRNA-resistant NDPK-B clone contained the sequence CAGGTAGGACGAAACATC. The mutated nucleotides are underlined and did not alter the amino acid sequence. TRPV5H711N was generated using the following primer in the coding region (mutation underlined): ACCCTGGGGAACTTGAATCTT. cDNA constructs were transiently expressed in HEK293 cells by using Lipofectamine (Life Technologies, Grand Island, NY). Purified bacterial-expressed glutathione S-transferase (GST) fusions of full-length NDPK-B(WT), NDPK-B(KD), and histidine (His)-tagged PHPT1 have been described (Srivastava et al., 2006, 2008). NDPK-B (NME2) antibody is a mouse monoclonal (MO8) purchased from Abgent.
Electrophysiology
Whole-cell patch clamp.
Patch-clamp experiments were performed on single cells in the whole-cell configuration 24–48 h after transfection. Cells were plated on a 15-mm coverslip and positioned in a recording chamber (RC-25F; Warner Instruments), and patch clamping was performed on an IX51 Olympus microscope. An Axopatch 200B amplifier was used along with the Clampex10 software to record the currents. Membrane currents were filtered (−3 dB at 1 kHz) and digitized at 10 kHz (pClamp10 with Digidata1440A interface; Axon Instruments). AgCl was used a reference electrode. A voltage ramp of 450 ms was applied ranging from +100 to −100 mV every 5 s from a holding potential of +20 mV as previously described, with some modifications (de Groot et al., 2011). To measure Na+ current, the internal pipette solution contained 140 mM KCl, 10 mM ethylene glycol tetraacetic acid, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.2), and the external solution contained 140 mM NaCl, 5 mM EDTA, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH 7.2). To measure Ca2+ currents, 10 mM Ca2+ (CaCl2) was added in the bath solution, and currents were measured for 3s at −100 mV, stepping from a holding potential of +70 mV. Currents were not corrected for junction potential change.
Inside/out patch.
TRPV5(WT) or TRPV5(H711N) was transfected into 293 cells. At 24–48 h posttransfection, TRPV5 channel activity was determined using I/O patch experiments as previously described (Cha et al., 2007). Briefly, the internal pipette solution contained 140 mM NaAsp, 10 mM NaCl, 10 mM EDTA, and 10 mM HEPES, and membrane patches were excised in a bath solution containing 140 mM NaAsp, 10 mM NaCl, 1 mM EDTA, and 10 mM HEPES. Once a stable current was obtained, GST-NDPK-B(WT) or NDPK-B(KD) purified from bacteria (Srivastava et al., 2006) was added to the I/O patch and channel activity recorded. To assess whether PHPT1 could reverse activation by of TRPV5, bacterial-expressed His-PHPT1 (Srivastava et al., 2008) was added to I/O patch after addition of NDPK-B(WT).
Cell surface biotinylation
HEK293 cells were transfected with GFP-TRPV5 with or without NDPK-B. At 24 h after transfection, cells were labeled with EZ Link Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, IL) as previously described (van de Graaf et al., 2008). After labeling, unreacted biotin was quenched with 100 mM glycine and 5% bovine serum albumin in phosphate-buffered saline, and cells were then lysed in lysis buffer containing 1% NP40. Biotinylated proteins were then precipitated using High Capacity NeutrAvidinAgarose Resin (Thermo Scientific Pierce). Cell surface TRPV5 was assessed by immunoblotting the avidin precipitates with anti-GFP antibodies.
Calcium measurements
Cells were trypsinized and plated on 15-mm, round coverslips, and 24 h after transfection, cells were loaded with 5 μM Fura-2-AM (Molecular Probes) in DMEM for 30 min at room temperature. Cells were washed and then resuspended in DMEM. Ca2+ imaging was done with an IX81 epifluorescence microscope (Olympus) and OpenLab imaging software (Improvision). For single-cell analysis, 340/380 nm Fura-2-AM emission ratios of >50 cells/experiment were analyzed. Background fluorescence obtained from regions containing no cells was digitally subtracted from each image. Data are represented as the ratio 340/380 nm after background subtraction.
NDPK-B−/− mice and urinary Ca2+ determination
NDPK-B−/−mice that had been backcrossed 10 generations into C57Bl/6 mice have been described (Di et al., 2010b). Littermate NDPK-B−/- or NDPK-B+/+ mice were placed on standard (0.5%) or high-Ca2+ (2%) diet from Lab Diet for 2 wk. The mice were then placed into metabolic cages, and 24-h urine samples were collected. Urine Ca2+, K+, Na+, and creatinine concentrations were measured by Antech GLP (Morrisville, NC). All animal experiments were approved under Institutional Animal Care and Use Committee Protocol 110114.
Abbreviations used:
- KD
kinase dead
- MDCK
Madin–Darby canine kidney
- NDPK-B
nucleoside diphosphate kinase B
- PHPT1
protein histidine phosphatase 1
- shRNA
short hairpin RNA
- TRPV5
transient receptor potential-vanilloid-5
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-04-0180) on February 12, 2014.
REFERENCES
- Attwood PV, Ludwig K, Bergander K, Besant PG, Adina-Zada A, Krieglstein J, Klumpp S. Chemical phosphorylation of histidine-containing peptides based on the sequence of histone H4 and their dephosphorylation by protein histidine phosphatase. Biochim Biophys Acta. 2010;1804:199–205. doi: 10.1016/j.bbapap.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Attwood PV, Piggott MJ, Zu XL, Besant PG. Focus on phosphohistidine. Amino Acids. 2007;32:145–156. doi: 10.1007/s00726-006-0443-6. [DOI] [PubMed] [Google Scholar]
- Biner HL, Arpin-Bott MP, Loffing J, Wang X, Knepper M, Hebert SC, Kaissling B. Human cortical distal nephron: distribution of electrolyte and water transport pathways. J Am Soc Nephrol. 2002;13:836–847. doi: 10.1681/ASN.V134836. [DOI] [PubMed] [Google Scholar]
- Boissan M, Dabernat S, Peuchant E, Schlattner U, Lascu I, Lacombe ML. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol Cell Biochem. 2009;329:51–62. doi: 10.1007/s11010-009-0120-7. [DOI] [PubMed] [Google Scholar]
- Boissan M, Lacombe ML. Learning about the functions of NME/NM23: lessons from knockout mice to silencing strategies. Naunyn Schmiedebergs Arch Pharmacol. 2011;384:421–431. doi: 10.1007/s00210-011-0649-3. [DOI] [PubMed] [Google Scholar]
- Boros S, Bindels RJ, Hoenderop JG. Active Ca(2+) reabsorption in the connecting tubule. Pflugers Arch. 2009;458:99–109. doi: 10.1007/s00424-008-0602-6. [DOI] [PubMed] [Google Scholar]
- Cha SK, Jabbar W, Xie J, Huang CL. Regulation of TRPV5 single-channel activity by intracellular pH. J Membr Biol. 2007;220:79–85. doi: 10.1007/s00232-007-9076-2. [DOI] [PubMed] [Google Scholar]
- Cha SK, Wu T, Huang CL. Protein kinase C inhibits caveolae-mediated endocytosis of TRPV5. Am J Physiol Renal Physiol. 2008;294:F1212–F1221. doi: 10.1152/ajprenal.00007.2008. [DOI] [PubMed] [Google Scholar]
- Cuello F, Schulze RA, Heemeyer F, Meyer HE, Lutz S, Jakobs KH, Niroomand F, Wieland T. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 IN Gbeta. J Biol Chem. 2003;278:7220–7226. doi: 10.1074/jbc.M210304200. [DOI] [PubMed] [Google Scholar]
- de Groot T, Kovalevskaya NV, Verkaart S, Schilderink N, Felici M, van der Hagen EA, Bindels RJ, Vuister GW, Hoenderop JG. Molecular mechanisms of calmodulin action on TRPV5 and modulation by parathyroid hormone. Mol Cell Biol. 2011;31:2845–2853. doi: 10.1128/MCB.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot T, Lee K, Langeslag M, Xi Q, Jalink K, Bindels RJ, Hoenderop JG. Parathyroid hormone activates TRPV5 via PKA-dependent phosphorylation. J Am Soc Nephrol. 2009;20:1693–1704. doi: 10.1681/ASN.2008080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot T, Verkaart S, Xi Q, Bindels RJ, Hoenderop JG. The identification of histidine 712 as a critical residue for constitutive TRPV5 internalization. J Biol Chem. 2010;285:28481–28487. doi: 10.1074/jbc.M110.117143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Srivastava S, Zhdanova O, Ding Y, Li Z, Wulff H, Lafaille M, Skolnik EY. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proc Natl Acad Sci USA. 2010a;107:1541–1546. doi: 10.1073/pnas.0910133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L, Srivastava S, Zhdanova O, Sun Y, Li Z, Skolnik EY. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J Biol Chem. 2010b;285:38765–38771. doi: 10.1074/jbc.M110.168070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimke H, Hoenderop JG, Bindels RJ. Molecular basis of epithelial Ca2+ and Mg2+ transport: insights from the TRP channel family. J Physiol. 2011;589:1535–1542. doi: 10.1113/jphysiol.2010.199869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkika D, Topala CN, Chang Q, Picard N, Thebault S, Houillier P, Hoenderop JG, Bindels RJ. Tissue kallikrein stimulates Ca(2+) reabsorption via PKC-dependent plasma membrane accumulation of TRPV5. EMBO J. 2006;25:4707–4716. doi: 10.1038/sj.emboj.7601357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippe HJ, Lutz S, Cuello F, Knorr K, Vogt A, Jakobs KH, Wieland T, Niroomand F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B.Gbetagamma complex in H10 cells. J Biol Chem. 2003;278:7227–7233. doi: 10.1074/jbc.M210305200. [DOI] [PubMed] [Google Scholar]
- Hippe HJ, et al. The interaction of nucleoside diphosphate kinase B with Gbetagamma dimers controls heterotrimeric G protein function. Proc Natl Acad Sci USA. 2009;106:16269–16274. doi: 10.1073/pnas.0901679106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenderop JG, Dardenne O, Van Abel M, Van Der Kemp AW, Van Os CH, St-Arnaud R, Bindels RJ. Modulation of renal Ca2 +transport protein genes by dietary Ca2+ and 1,25-dihydroxyvitamin D3 in 25-hydroxyvitamin D3-1alpha-hydroxylase knockout mice. FASEB J. 2002;16:1398–1406. doi: 10.1096/fj.02-0225com. [DOI] [PubMed] [Google Scholar]
- Hoenderop JG, Nilius B, Bindels RJ. Calcium absorption across epithelia. Physiol Rev. 2005;85:373–422. doi: 10.1152/physrev.00003.2004. [DOI] [PubMed] [Google Scholar]
- Hoenderop JG, et al. Renal Ca2+ wasting, hyperabsorption, and reduced bone thickness in mice lacking TRPV5. J Clin Invest. 2003;112:1906–1914. doi: 10.1172/JCI19826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee JM, Muir TW. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem Biol. 2012;7:44–51. doi: 10.1021/cb200445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J. Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci Signal. 2009;2:pe13. doi: 10.1126/scisignal.261pe13. [DOI] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol. 2004;554:255–261. doi: 10.1113/jphysiol.2003.049072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Cai X, Li Z, Sun Y, Skolnik EY. Phosphatidylinositol-3-kinase C2beta and TRIM27 function to positively and negatively regulate IgE receptor activation of mast cells. Mol Cell Biol. 2012;32:3132–3139. doi: 10.1128/MCB.00019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Mol Cell. 2006;24:665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Srivastava S, et al. The class II phosphatidylinositol 3 kinase C2beta is required for the activation of the K+ channel KCa3.1 and CD4 T-cells. Mol Biol Cell. 2009;20:3783–3791. doi: 10.1091/mbc.E09-05-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, Skolnik EY. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proc Natl Acad Sci USA. 2008;105:14442–14446. doi: 10.1073/pnas.0803678105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topala CN, Schoeber JP, Searchfield LE, Riccardi D, Hoenderop JG, Bindels RJ. Activation of the Ca2+-sensing receptor stimulates the activity of the epithelial Ca2+ channel TRPV5. Cell Calcium. 2009;45:331–339. doi: 10.1016/j.ceca.2008.12.003. [DOI] [PubMed] [Google Scholar]
- van de Graaf SF, Rescher U, Hoenderop JG, Verkaart S, Bindels RJ, Gerke V. TRPV5 is internalized via clathrin-dependent endocytosis to enter a Ca2+-controlled recycling pathway. J Biol Chem. 2008;283:4077–4086. doi: 10.1074/jbc.M706959200. [DOI] [PubMed] [Google Scholar]
- Zhang W, Na T, Peng JB. WNK3 positively regulates epithelial calcium channels TRPV5 and TRPV6 via a kinase-dependent pathway. Am J Physiol Renal Physiol. 2008;295:F1472–F1484. doi: 10.1152/ajprenal.90229.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]