


Abstract
The RNA world hypothesis describes a stage in the early evolution of life in which RNA served as genome and as the only genome-encoded catalyst. To test whether RNA world organisms could have used cyclic trimetaphosphate as an energy source, we developed an in vitro selection strategy for isolating ribozymes that catalyze the triphosphorylation of RNA 5′-hydroxyl groups with trimetaphosphate. Several active sequences were isolated, and one ribozyme was analyzed in more detail. The ribozyme was truncated to 96 nt, while retaining full activity. It was converted to a trans-format and reacted with rates of 0.16 min−1 under optimal conditions. The secondary structure appears to contain a four-helical junction motif. This study showed that ribozymes can use trimetaphosphate to triphosphorylate RNA 5′-hydroxyl groups and suggested that RNA world organisms could have used trimetaphosphate as their energy source.
INTRODUCTION
The early evolution of life went through a stage in which RNA molecules served as genome and as genome-encoded catalyst, according to the RNA world hypothesis (1–3). This hypothesis is widely accepted due to several lines of evidence [for recent reviews, see (4,5)]. Direct fossil evidence of the RNA world, however, may never be found because the expected chemical components of these organisms would almost certainly not have survived more than three billion years until today (6). To determine how an RNA world organism could have functioned, researchers are attempting to re-create an RNA world organism in the laboratory (7–12). The replication of an RNA world organism would have relied on the synthesis of nucleotides (13–19), the generation of chemically activated nucleoside 5′-phosphates (see later in the text) and their polymerization (10). The focus of this study is the generation of chemically activated 5′-phosphates.
The polymerization of RNA is entropically unfavorable in aqueous environments. Strategies to energetically drive RNA polymerization may have been different in two phases of the RNA world. First, in the prebiotic phase and perhaps in an early phase of the RNA world, RNA polymerization may have been driven by the evaporation of water from a solution (20) and from lipid encapsulations (21), or by the activation of the nucleotide's 5′-phosphate with a wide range of leaving groups (22) such as adenine (23), cyanide (24), imidazole (25,26), or 2-methylimidazole (27). Importantly, these activation groups lead to hydrolysis in aqueous environment within hours. Second, in later phases of the RNA world, when lipid vesicles presumably encapsulated aqueous droplets of self-replicating RNA systems (9,28–30), kinetically more stable activation groups would have been important. This requirement is fulfilled by nucleoside triphosphates, the universal energy currency in all known life forms.
Nucleoside triphosphates can be generated from nucleosides and cyclic trimetaphosphate (TMP) (31). TMP is a prebiotically plausible compound because it can be generated by volcanic activity (32), by the erosion of phosphide minerals (33) and by heating of phosphate in the presence of urea (34). Additionally, TMP results from the self-reaction of the termini of linear polyphosphates in aqueous medium (35). The prebiotic availability of TMP is supported by the recent finding of abundant, reactive and reduced phosphorus species in sediments of 3.5 billion-year-old marine sediments (36). These phosphorus species can form polyphosphates including TMP if sources such as lightening provide hydroxyl radicals (33). Although TMP is the most reactive polyphosphorylating reagent of alcohols among all polyphosphates (37), the reaction between nucleoside 5′-hydroxyl groups and TMP proceeds efficiently only at pH values >12 (31). At such high pH, RNA world organisms would hydrolyze quickly. Therefore, RNA world organisms would require a catalyst to use TMP as energy source.
The triphosphorylation of nucleoside 5′-hydroxyl groups with TMP appears to be well within the range of catalysis by ribozymes. Most ribozymes are acid-base catalysts and have a particular capacity for phosphoryl transfer reactions (38–47). To identify ribozymes for new reactions, the only known route is the in vitro selection from large pools of random RNA sequences (42,48,49). Therefore, we aimed to develop an in vitro selection scheme for the isolation of ribozymes that catalyze the triphosphorylation of RNA 5′-hydroxyl groups using TMP.
Here, we describe the in vitro selection of a ribozyme that uses TMP to triphophorylate RNA 5′-hydroxyl groups. After five to eight rounds of selection, we obtained several active ribozymes, one of which was analyzed in more detail. This ribozyme was able to triphosphorylate RNAs in cis and in trans, with a kcat of up to 0.16 min−1. These results suggested that RNA world organisms could have used TMP as their energy source.
MATERIALS AND METHODS
In vitro selection
The DNA pool for the selection was generated from a 187-nt long DNA oligomer (‘ultramer', Integrated DNA Technologies (IDT)) that included 150 positions with randomized sequence (phosphoramidites were hand mixed) and primer binding sites on both termini. This 187-mer was annealed to a 79-mer, which attached the sequence of the hammerhead ribozyme and the promoter of T7 RNA polymerase to the 5′-terminus of the pool. Amplification with short primers generated double-stranded copies of the pool. The DNA pool was transcribed into the RNA pool under standard conditions, during which the 5′-terminal hammerhead ribozymes cleaved themselves from the pool, generating 5′-hydroxyl groups on the RNA pool. The sequence of the transcript was 5′-GGGCGGTCTCCTGACGAGCTAAGCGAAACTGCGGAAACGCAGTCGAGACCGAGATGTT-N150-CGCCAGTTAAGCTCCAGC-3′, where the removed hammerhead ribozyme sequence is underlined. The RNA pool was purified by denaturing polyacrylamide gel electrophoresis to remove any uncleaved pool molecules.
The 5′-hydroxylated RNA pool was incubated with TMP in a buffer containing 100 mM MgCl2, 50 mM trisodium TMP (freshly dissolved and sterile filtered) and 50 mM Tris–HCl, pH 8.3. All triphosphorylation reactions during the selection were done at pH 8.3. When the solutions with TMP and MgCl2 were combined, the pH dropped to ∼4.5. This pH was restored to the pH of the TMP solution alone (pH ∼6) before the buffer (Tris–HCl, pH 8.3) was added. After 3 h or 5 min (depending on the selection round) of incubation at room temperature, RNAs were ethanol precipitated. The large salt pellet was extracted with a small volume of cold water to remove most of the salt. The remaining pellet was dissolved in water, desalted by size exclusion chromatography (P30 spin-columns; Bio-Rad), ethanol precipitated and redissolved in water.
The recovered RNA pool molecules were heat renatured (2 min/80°C) with a 1.25-fold molar excess of the R3C ligase ribozyme (50) (whose arms were designed complementary to the RNA pool 5′-terminus and the biotinylated capture oligonucleotide) and a 1.5-fold molar excess of biotinylated capture oligonucleotide [5′-biotin-d(GAACTGAAGTGTATG)rU-3′], in 100 mM KCl and 100 mM Tris–HCl, pH 8.5. The solution was diluted to 400 nM pool RNA, 500 nM ligase ribozyme, 600 nM capture oligonucleotide and adjusted to 50 mM KCl, 25 mM MgCl2, 2 mM spermidine, 20% (w/v) PEG 8000 and 50 mM Tris–HCl, pH 8.5. After incubation for 3 h at 30°C, magnesium was chelated by an excess of Na2EDTA, and the mixture was heated (10 min/50°C) with a 10-fold excess of a DNA complementary to the ligase ribozyme to free the ligated RNAs. The biotinylated nucleic acids were captured during 30 min of agitation with streptavidin-coated magnetic beads (Promega) containing a 1.5-fold excess of biotin binding sites over biotinylated capture RNAs. Captured RNAs were washed first with 50 mM KCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)/KOH pH 7.2 and 0.01% Triton X-100, then with 20 mM NaOH and 0.01% Triton X-100. RNA pool molecules were eluted from the magnetic beads by heating (3 min/65°C) with 95% formamide/1 mM Na2EDTA. The effective complexity of the RNA pool was 1.7 × 1014 sequences, based on an initial complexity of the double-stranded DNA library of 2.4 × 1014 sequences, a total of 1.6 nmol of RNA pool molecules that entered the ligation step, losses of ∼60% in the ligation step, losses of ∼50% in the capture on streptavidin-coated beads and losses of ∼20% in further processing steps (data not shown). After ethanol precipitation, the RNAs were reverse transcribed using Superscript III reverse transcriptase (Invitrogen) and polymerase chain reaction (PCR) amplified using Taq polymerase. The 5′-PCR primer added a selective step because a part of its binding site was generated by the biotinylated capture oligonucleotide. The DNA pool for the next round of selection was regenerated by PCR amplification with the 79-nt primer that was used to attach the hammerhead ribozyme and the T7 RNA polymerase promoter. We used a previously published protocol for mutagenic PCR (51).
In pilot PCR amplifications of the reverse-transcribed products, short PCR artifacts created the dominant product after ∼30 PCR cycles. We assumed that this was because some pool molecules allowed the PCR primers to anneal within the randomized region, and shorter amplicons are known to outcompete longer amplicons during PCR (52). To avoid that problem we ‘doped' the RNA pool molecules after the incubation with TMP with 1/37 000 molar amount of pool 0 molecules that carried a 5′-triphosphate. Because these 5′-triphosphorylated molecules were ligated efficiently to the capture oligonucleotides, the doped pools required only 18–30 PCR amplification cycles and suppressed short PCR artifacts. Although this may have slowed the selection compared with a protocol without PCR problems, the 1/37 000-fold did not seem to harm the enrichment over multiple rounds of selection.
Triphosphorylation assays
For the self-triphosphorylation assay based on the R3C ligase ribozyme, cloned ribozymes were first transcribed in vitro (53) and purified by denaturing polyacrylamide gel electrophoresis (PAGE). Triphosphorylation reactions with 5 µM triphosphorylation ribozyme were incubated with 50 mM TMP, 100 mM MgCl2 (corresponding to 50 mM free Mg2+) and 50 mM Tris–HCl (final pH 8.1) for 3 h at 22°C. The reaction mixture was diluted 10-fold with a buffer such that buffer concentrations were 500 nM ribozyme, 500 nM R3C ligase ribozyme, 500 nM of an oligonucleotide (5′-[32P]-d(GAACTGAAGTGTATG)rU-3′), 100 mM KCl, 100 mM Tris–HCl, pH 8.5, and 15 mM Na2EDTA. After heat denaturation (2 min/65°C), the mixtures were diluted another 2-fold and adjusted to 20% (w/v) PEG 8000, 22 mM free Mg2+, 50 mM KCl, 2.5 mM spermidine and 100 mM Tris–HCl pH 8.5. This ligation reaction was incubated at 30°C for 2 h, stopped by ethanol precipitation and the products were separated by denaturing 10% PAGE. Gels were exposed to phosphorimager screens, scanned on a PMI phosphorimager (Bio-Rad) and signals were quantitated using the software quantity one.
The self-triphosphorylation assay based on the DNAzyme used triphosphorylation ribozymes that were internally [32P] labeled during transcription and purified by denaturing PAGE. Triphosphorylation reactions with 8 µM ribozyme were incubated as described earlier in the text. At specific time points, 2 µl aliquots of the reactions were quenched by adding them to 8 µl of a solution containing 31.25 mM Na2EDTA, 375 mM NaCl, 32 pmol of a DNAzyme that recognized the 14 nt conserved at all triphosphorylation ribozyme 5′-termini, and 32 pmol of a DNA oligonucleotide that was complementary to the first 25 nt of the ‘N150’ sequence to improve accessibility for the DNAzyme. After heat renaturation (2 min/80°C), the DNAzyme reaction was initiated by adding 10 µl of a solution containing 200 mM MgCl2, 100 mM Tris–HCl pH 8.3, and incubated for 1 h at 37°C. Two microliter of these reactions were quenched with 8 µl of PAGE loading buffer containing 16 pmol of a DNA oligonucleotide complementary to the DNAzyme, 90% (v/v) formamide and 37.5 mM Na2EDTA. Products were separated by denaturing 22.5% PAGE and analyzed as described earlier in the text.
The trans-triphosphorylation assay used substrate molecules that were internally [32P] labeled during transcription. Two hammerhead ribozymes were contained in the transcript, one at the 5′-terminus and one at the 3′-terminus. The 5′-terminal hammerhead ribozyme generated the 5′-hydroxyl group, whereas the 3′-terminal hammerhead ribozyme generated a homogeneous 3′-terminus (54). Cleaved transcripts were purified by denaturing PAGE. Reaction mixtures with 5.5 µM triphosphorylation ribozyme and substoichiometric concentrations of radiolabeled substrate were incubated as described earlier in the text. At specific time points, 1.5 µl of the reactions were added to 6.5 µl of formamide PAGE loading buffer containing 20 mM Na2EDTA, heat renatured and separated by denaturing 20% PAGE. Quantitation of the signals was as aforementioned. For the titration of the TMP concentration, the chelation of Mg2+ with TMP generated a slight reduction in the pH for the reaction with 200 mM [TMP] such that the pH of 8.1 (all other reactions) was reduced to pH 7.6.
TMP was obtained as trisodium salt from MP Biomedicals (cat # 150160; lot # 9425H). The supplier listed 0.4 mg/kg of arsenic and <4 mg/kg of lead as contamination. We tested for other polyphosphates in the preparation by electrospray-ionization Mass Spectroscopy (ESI-MS) analysis. The results suggested that the used TMP sample contained ∼80% TMP, 10% linear triphosphate and 10% of linear tetraphosphate, under the assumption that these molecules have similar ionization efficiencies under ESI-MS. Traces of orthophosphate and no detectable amounts of pyrophosphate or polyphosphates longer than tetraphosphate were found.
Generation of modified ribozyme constructs
Sequence modifications were introduced into ribozyme constructs by PCR mutagenesis or the Quickchange protocol for site-directed mutagenesis (Stratagene). All DNA sequences were confirmed by cloning into pUC19 and sequencing. The only exceptions were ribozyme constructs where mutations near the 3′-terminus could be introduced during PCR amplification of the templates for transcription. RNAs were generated by runoff transcription by T7 RNA polymerase as described (53) and purified by denaturing PAGE. The sequences in the mutated substrate recognition duplex of the trans-reaction were 5′-GAGACCGUACAUUU-3′ for the substrate and 5′-GGAUGUAC … -3′ for the ribozyme 5′-terminus. Underlined nucleotides differ from the wild-type sequence.
Mass spectrometric analysis
Substrate RNAs were prepared as described for the trans-triphosphorylation reaction, but without internal radiolabeling. A fraction of the substrate was incubated with an excess of the TPR1 ribozyme for 3 h. The product of the triphosphorylation reaction and unreacted substrate RNA were purified by denaturing PAGE, and desalted on C18 Zip-tips (Millipore). About 20 pmol of each sample were dissolved in 4 µl containing 3-hydroxypicolinic acid and diammonium hydrogen citrate, and spotted on matrix-assisted laser desorption/ionization (MALDI) targets. The mass spectra were recorded by Dr Yongxuan Su at the UCSD Molecular Mass Spectrometry Facility in negative ion mode on a Bruker Biflex IV, MALDI-time-of-flight mass spectroscopy (TOFMS).
SHAPE analysis
For SHAPE analysis, the TPR1 ribozyme was extended at its 3′-terminus by 12 nt such that a 12-nt reverse transcription primer could facilitate analysis of the complete ribozyme. The trans-reacting TPR1 ribozyme was incubated at a concentration of 1 µM with a 1.5-fold stoichiometric excess of its substrate, in 100 mM MgCl2, 50 mM trisodium TMP and 50 mM HEPES/KOH pH 8.0. To 9 µl of this solution, 1 µl of dimethyl sulfoxide (DMSO) or a 20 mM solution of 1M7 (55) in DMSO was added. After incubation for 3 min at room temperature, the reaction products were ethanol precipitated. Reverse transcription with a 5′-[32P]-radiolabeled 12-nt long reverse transcription primer was facilitated by Superscript III reverse transcriptase (Invitrogen). Reaction products were ethanol precipitated, redissolved in formamide PAGE loading buffer, heated (2 min/80°C) and separated by denaturing 10% and 20% PAGE. Signal quantitation was done as described for the triphosphorylation assays. Only primary reaction sites were observed because >80% of the reverse transcription products had full length, and signals appeared with consistent kinetics in a concentration series of 1M7 (not shown). The signals were judged as strong, or weak when their intensity was at least 30%, or 8.5% of the strongest SHAPE signal intensity, respectively. Markers were synthetic DNA molecules with 5′-[32P] label that had the exact sequence of expected reverse transcription products and a length of 25, 47 and 70 nt.
RESULTS
In vitro selection
An in vitro selection procedure was established to obtain ribozymes that use TMP to triphosphorylate RNA 5′-hydroxyl groups (Figure 1). First, a double-stranded DNA pool was generated that encoded the T7 RNA polymerase promoter, a hammerhead ribozyme and 150 randomized nucleotides flanked by two constant primer binding sites. The hammerhead ribozyme served to generate 5′-hydroxyl groups at the 5′-termini of the pool molecules (56). The hammerhead ribozyme cleaved itself efficiently off the pool under transcription conditions so that >95% of the pool was cleaved after 2 h of transcription incubation (data not shown). The gel-purified pool molecules were then incubated for 3 h with 100 mM MgCl2, 50 mM TMP and 50 mM Tris–HCl pH 8.3 to allow catalytically active pool molecules to triphosphorylate their own 5′-terminus. We found that TMP chelated an equimolar amount of Mg2+ (data not shown), therefore the triphosphorylation conditions provided 50 mM free magnesium ions. To isolate pool molecules that carried a 5′-triphosphate, the R3C ligase ribozyme was used (50). This ribozyme created 3′-5′-phosphodiester bonds between 5′-triphosphorylated pool molecules and the 3′-hydroxyl group of a 5′-biotinylated oligonucleotide. The biotin modification allowed capturing the pool on streptavidin-coated magnetic beads, and stringent washing of pool molecules that were coupled covalently to the biotin moiety. The captured pool molecules were reverse transcribed and PCR amplified. In a second PCR step, the T7 RNA polymerase promoter and the 5′-terminal hammerhead ribozyme sequence were regenerated, completing one round of the selection. In a mock selection round, we estimated that 5′-triphosphorylated RNAs were enriched at least 9000-fold in a single selection round (Supplementary Figure S1).
Figure 1.

Scheme for the in vitro selection of triphosphorylation ribozymes. (A) A double-stranded DNA library containing the promoter for T7 RNA polymerase (T7), the sequence for a hammerhead ribozyme (HhRz) and a randomized sequence (N150) was transcribed into RNA. (B) The 5′-terminal hammerhead ribozyme cleaved itself off cotranscriptionally (filled triangle), generating a 5′-terminal hydroxyl group on the RNA library. (C) The RNA library was incubated in the presence of TMP such that active ribozymes could triphosphorylate their 5′-terminus. (D) Triphosphorylated RNA molecules were reacted with the 3′-hydroxyl group of a short biotinylated RNA, by the R3C ligase ribozyme. (E) Ligated RNAs were captured via their biotin modification on streptavidin-coated magnetic beads and washed stringently. The RNAs were then (F) reverse transcribed and (G) amplified by PCR to generate the DNA pool for the next round of selection.
Based on the effective complexity of the RNA pool of ∼1.7 × 1014 sequences (see ‘Materials and Methods’ section) and the enrichment factor of ∼104-fold/selection round (Supplementary Figure S1), we anticipated that catalytically active sequences could dominate the RNA pool after four selection rounds. To follow the progress of the selection, we monitored the number of PCR cycles that were required after each reverse transcription reaction to generate clearly detectable bands on agarose gels (Supplementary Figure S2). Satisfyingly, the number of PCR cycles dropped from at least 17 cycles in rounds one to 3–8 cycles in round four. Two modifications were used for the later selection rounds. First, mutagenic PCR was introduced in the amplification steps to allow the pool molecules to explore their sequence neighbors and perhaps find more active sequences. Second, the population was split into two branches, one exposed to the same selection pressure as before (3 h of incubation with TMP), and one exposed to high selection pressure (only 5 min of incubation with TMP). In round seven, a crossover branch was added to allow clones from low selection pressure to cross into the high selection pressure branch of round eight. A total of 36 sequences were obtained from the pools after five and eight selection rounds, from low and high selection pressure branches (Supplementary Figure S3). The clones were named after their selection history: R5 or R8 denoting their round of isolation, a combination of the numbers 3 and 5 denoting the incubation times with TMP (3 h and/or 5 min) and C with the clone number.
Identification of the most active ribozyme clones
To identify ribozymes with high activity, we screened the 36 isolated clones for activity. This screen used the same procedure as during the selection, reacting the ribozyme with TMP followed by ligation to a short RNA using the R3C ligase ribozyme, but a 5′-radiolabeled oligonucleotide was used instead of a biotinylated oligonucleotide (Supplementary Figure S4). The fraction of shifted oligonucleotides in a denaturing gel reported the ligation efficiency and indirectly the triphosphorylation efficiency. The 36 ribozyme clones were sorted according to their activity, which revealed a wide distribution of activities (Figure 2). Several sequences appeared multiple times, but their multiplicity was not correlated with increased activity. The activity of several ribozyme clones in this assay was ∼2-fold higher than that of a positive control, made from pool RNA containing a 5′-triphosphate. We assumed that this was because the assay was not directly monitoring triphosphorylation activity, and the clones with peculiarly high activity worked especially well together with the R3C ligase ribozyme used in the selection procedure. To identify those ribozyme clones that were most efficient in catalyzing the triphosphorylation reaction, we chose the eight most active clones from the ligase ribozyme-based assay.
Figure 2.
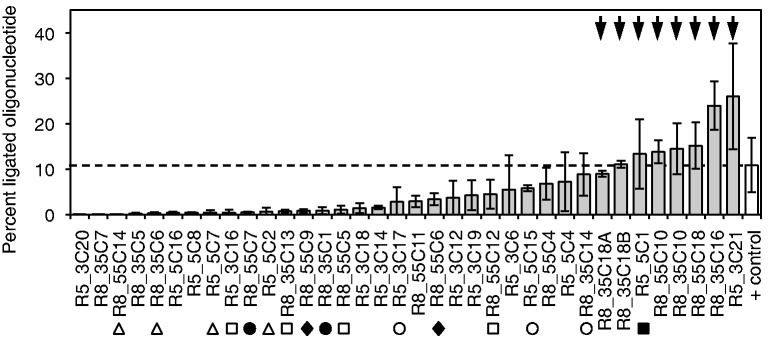
Screen of 36 in vitro selected RNAs for self-triphosphorylation activity, using an assay based on the R3C ligase ribozyme (see Supplementary Figure S4). The column heights show the percent of 5′-[32P]-radiolabeled oligonucleotide that was ligated to the individual selected RNAs by the ligase ribozyme, after the selected RNAs were incubated with TMP for 3 h to allow for self-triphosphorylation. Therefore, the percent of ligated oligonucleotide was an indirect measure of self-triphosphorylation activity. As positive control, the unselected RNA pool was ligated to the radiolabeled oligonucleotide, facilitated by a 5′-triphosphate that was incorporated during transcription (white column and horizontal dashed line). The labels on the x-axis denote the clone names (round of isolation_branch of the evolution_clone number). The clones were sorted according to the average percent of ligated oligonucleotide. The sequences of all 36 clones are shown in Supplementary Figure S3. Classes of related sequences are indicated by symbols, with empty squares (class 1), empty triangles (class 2), empty circles (class 3), filled circles (class 4), filled squares (class 5) and filled diamonds (class 6) each belonging to one class. Class 5 is represented by a single clone because the second clone had the identical sequence. No other clone showed related sequences among the 36 clones. The eight clones with the highest activity (arrows) were chosen for further analysis. Error bars are standard deviations from three independent experiments.
To directly monitor the triphosphorylation status at the ribozyme 5′-termini, a different gel shift assay was used (Figure 3). Because the gel shift of the triphosphate group was too small to detect migration differences of the 182-nt long ribozymes, we used the 8–17 DNAzyme (57) to cleave off the eight 5′-terminal nucleotides. The triphosphorylation ribozymes were internally labeled; therefore, the triphosphorylation status of the 8-nt fragment could be followed after separation on denaturing polyacrylamide gels and phosphorimaging. The 8-nt fragments of unreacted ribozymes comigrated with RNAs containing a 5′-hydroxyl terminus, and the fragment after incubation with TMP comigrated with RNAs containing a 5′-triphosphate. This confirmed that the products were triphosphorylated at their 5′-terminus. Quantitation of the signals after different triphosphorylation reaction times resulted in single-exponential time dependence, revealing pseudo-first order rate constants between 0.01 and 0.03 min−1 for each of the eight analyzed ribozymes. Although most ribozymes reacted to a final extent of 30–50%, one ribozyme (clone R5_3C21) reacted to >80%. This clone was chosen for further analysis.
Figure 3.

Kinetic analysis of the eight most promising ribozyme clones. (A) Schematic of the assay using the 8–17 DNAzyme. After reaction with TMP, internally [32P]-radiolabeled ribozymes were cleaved by the DNAzyme. This freed the eight 5′-terminal nucleotides, facilitating gel separation of triphosphorylated and unreacted RNAs. (B) Autoradiogram of products after the DNAzyme reaction and separation by denaturing 22% PAGE. An RNA that was not exposed to TMP (5′-OH) and an RNA that was transcribed with a 5′-terminal triphosphate (5′-PPP) were used as negative and positive controls, respectively. The incubation times with TMP are indicated on the top. The long fragment of the cleaved ribozymes (174 nt) and possible remaining uncleaved ribozymes (182 nt) migrated much slower than the 8-nt fragments. The 8-nt fragments were separated based on their phosphorylation status. The particular autoradiogram was from analysis of clone R8_35C18A. (C) Determination of triphosphorylation kinetics from signals as shown in (B). The percent of triphosphorylation of the 8-mer was plotted as function of the incubation time with TMP. Single-exponential fits are shown as black or gray lines. Error bars are standard deviations from three experiments. Symbols are explained in (D). Single-exponential curve fits are shown in black lines for filled symbols and gray lines for empty symbols. (D) Symbols and clone names used in (C), together with the parameters obtained by curve fits, the maximal percentage of reacted ribozyme (Max.) and the observed pseudo-first order rate constant (kobs).
If the ribozymes were able to triphosphorylate single free nucleosides, they could directly generate nucleoside triphosphates. To test whether the eight most active ribozymes could triphosphorylate free nucleosides, they were truncated at their 5′-terminus by one nucleotide (to accommodate the free nucleoside) and incubated with C14-radiolabeled guanosine. The products were separated by Polyethylenimine (PEI) cellulose thin layer chromatography (TLC) analysis and analyzed by autoradiography (Supplementary Figure S5). None of the tested ribozymes generated a detectable signal for guanosine triphosphate (GTP). The result was the same when the ribozymes were not truncated, or when they were incubated at pH 9.5 or at different temperatures (40°C and −20°C) (data not shown).
Truncation of ribozyme R5_3C21
We removed fragments from different portions of the 182-nt long R5_3C21 ribozyme to test whether a shorter fully active ribozyme could be obtained (Figure 4). The activity of the resulting truncated ribozymes was measured with the gel shift assay that directly monitored the 5′-triphosphorylation with the help of the 8–17 DNAzyme. The choice of deleted ribozyme fragments was based on the predicted secondary structures of the full-length ribozyme (58). When portions of the 5′-domain of the ribozyme were deleted, triphosphorylation activity was abolished (truncation 1 and 2). When portions of the 3′-domain were deleted, full triphosphorylation activity was retained (truncations 3 and 4). This allowed truncating the ribozyme to 96 nt, whereas further truncation to 92 nt caused a slight reduction in activity (truncation 5). Therefore, the further experiments used truncation 4 of ribozyme R5_3C21, with a length of 96 nt. This ribozyme was termed TPR1.
Figure 4.
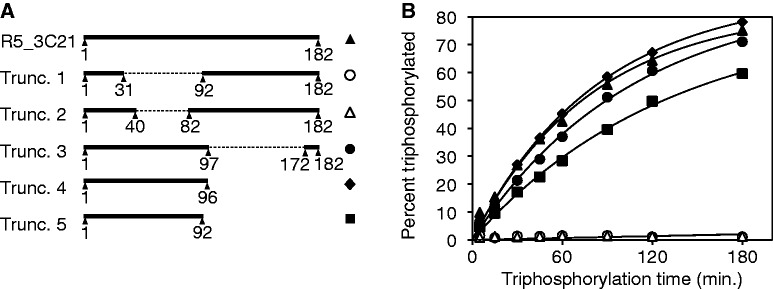
Truncation analysis of the ribozyme clone R5_3C21. (A) Schematic of the tested truncations. The numbering is relative to the complete sequence of the 182-nt long initial isolate (Supplementary Figure S3). Dotted lines indicate internal segments that were removed. Symbols to the right of each construct are consistent with the symbols in (B). (B) Triphosphorylation kinetics for the initial isolate and five truncated sequences. The kinetics was determined with the DNAzyme assay (Figure 3). Black lines represent single-exponential curve fits to the data. The symbols of truncations 1 and 2 are overlapping near the x-axis.
Triphosphorylation of short RNAs in trans
We were interested to see whether the TPR1 ribozyme would be able to triphosphorylate a short substrate RNA in trans (Figure 5). For this purpose, a short hairpin structure near the ribozyme 5′-terminus was separated at its loop, and the duplex was elongated to different lengths (6–9 bp). This was done using substrates with a length of 12, 13, 14 and 15 nt that showed complementarity to the corresponding ribozyme 5′-termini. After incubating the radiolabeled substrates with the corresponding TPR1 ribozymes, the products were separated on denaturing polyacrylamide gels and the signals quantitated. The results showed that the TPR1 ribozyme was triphosphorylating the substrates in trans. The shortest substrate, which formed a duplex of 6 bp with the ribozyme, resulted in slow reaction kinetics. In contrast, longer substrates that formed duplexes of 7–9 bp showed the same kinetics and reacted to the same extent of reaction as the cis-acting TPR1 ribozyme. The duplex length of 8 bp was chosen for further experiments, corresponding to a 14-nt substrate RNA. When 6 of the 8 bp in this duplex were modified (see ‘Materials and Methods’ section for the sequences), the reaction kinetics did not change significantly (0.023 min−1 in the mutant compared with 0.020 min−1 in the wild-type). This suggests that the existence of the duplex but not the identity of its paired bases was important to mediate the trans-reaction.
Figure 5.

Trans-triphosphorylation of substrate RNAs, by truncated versions of the R5_3C21 ribozyme. (A) Secondary structure schematic of a 14-nt substrate recognized by the ribozyme with an 8 bp substrate recognition duplex. The TMP is shown to highlight the reacting 5′-hydroxyl group. The body of the ribozyme is not shown for clarity (see Figure 6). (B) Autoradiogram of reaction products between substoichiometric concentrations of [32P]-radiolabeled substrate and TMP, catalyzed by 5.5 μM of the ribozyme, after separation on denaturing 22.5% PAGE. The particular image is from the reaction with a substrate recognition duplex of 8 bp. (C) Reaction kinetics of the trans-reaction with different lengths of the substrate recognition duplex. Symbols in the graph correspond to a duplex length of 6 bp (empty circles), 7 bp (empty diamonds), 8 bp (filled triangles) and 9 bp (empty squares). Curved lines are single-exponential fits to the data. (D) Mass spectral analysis of the substrate and the product of the trans-triphosphorylation reaction. The 14-nt substrate shown in (A) was analyzed before (gray lines) and after (black lines) incubation with the trans-acting ribozyme under triphosphorylation reaction conditions. The expected mass increase by 5′-triphosphate was 239.94 Da. Substrate and product carried a 2′-3′ cyclic phosphate due to their method used to synthesize them.
Several lines of evidence suggested that the TPR1 ribozyme was generating 5′-triphosphate groups: the reaction between nucleoside 5′-hydroxyl groups and TMP was established previously (31), the reacted ribozymes could be ligated by the R3C ligase ribozyme (Supplementary Figure S4) and the ribozyme reaction products showed the same gel shift as a 5′-triphosphate (Figure 3B). In addition, the trans-reaction presented a good opportunity for a more rigorous test. The mass of the short substrate RNA was analyzed before and after the reaction with ribozyme and TMP (Figure 5D). The expected mass difference was 239.94 Da. Satisfyingly, the observed mass difference (239.83 Da) was in excellent agreement with the prediction, thereby confirming that the in vitro selected ribozymes used TMP to triphosphorylate the RNA 5′-hydroxyl groups.
Secondary structure analysis
To identify single-stranded nucleotides in the ribozyme, we subjected it to SHAPE analysis with 1-methyl-7-nitroisatoic anhydride (1M7) (59). This reagent covalently reacts with 2′-hydroxyl groups that can adopt a specific geometry, and can therefore discriminate flexible nucleotides from double-stranded nucleotides, which is used to determine RNA secondary structures (60). The 1M7 reactivities with our ribozyme identified several flexible regions and several regions that were probably base paired (Figure 6A). These constraints were used to build a secondary structure model that contained a central four-helix junction (Figure 6B). Four-helix junctions were identified previously for RNA molecules such as between the spliceosomal RNAs U2 and U6 (61) and in the internal ribosomal entry site of HCV (62).
Figure 6.

Secondary structure analysis for the trans-triphosphorylating TPR1 ribozyme. (A) Autoradiograms of SHAPE analysis products, after reacting the ribozymes with 1M7 and subjecting them to reverse transcription with a 5′-[32P]-radiolabeled primer. The left image shows a separation by 20% PAGE to separate short products, whereas the right image shows a separation by 10% PAGE to show the longer products. Each image shows three lanes, where (−) denotes a negative control with DMSO, (+) denotes the SHAPE reaction with 1M7 dissolved in DMSO and M denotes a marker lane with three 5′-radiolabeled DNAs that have the identical sequence as the expected reverse transcription products. The primer has a length of 12 nt, base paired to 12 nt that were appended to the ribozyme 3′-terminus. The SHAPE signal is shifted by one nucleotide relative to the length of the reverse transcription product because the reverse transcriptase stops at the nucleotide before the SHAPE modification. (B) The secondary structure was based on two types of analysis. Filled triangles indicate a strong signal from SHAPE analysis, whereas empty triangles indicate weaker signals. (C) Two base pairs suggested by the SHAPE data were tested by single mutations of each base partner, and double mutation that should have restored activity for correct base pairs. The observed reaction rate was determined as in Figure 5 and given for each mutant.
To test whether the two short helices formed, the predicted base pairs between C10 and G33 on the ribozyme and between C5 on the substrate and G69 on the ribozyme were chosen for analysis (Figure 6C). At each base pair, three mutants were constructed and tested for activity. The single mutations removed the expected base pair and were expected to decrease activity. The double mutants were expected to restore the base pair and catalytic activity. As predicted, there was no detectable activity for any of the single mutations ribozyme variants tested. The C5G/G69C double mutation restored ∼50% of the activity, but the C10G/G33C double mutation showed only slight activity. This confirmed the formation of the base pair between C5 and G69. It is possible that the C10/G33 base pair formed but that the identity of the bases was also important for activity.
The sequence 5′-GGG-3′ (G32–G34) could base pair to the sequence 5′-CCCCC-3′ (C8–C12) in three possible registers, not only in the register described above (C10/G33). To test whether the helix formed in any of the two alternative registers, base covariation experiments were performed at C9/G33 and C11/G33. No single mutant and no double mutant showed detectable activity (data not shown), in contrast to the register C10/G33. This suggested that the base pair between C10 and G33 was formed, and that the identity of C10 and/or G33 was also required for maximum activity.
Dependence of kobs on [TMP], [Mg2+] and pH
To identify the reaction conditions at which the TPR1 ribozyme worked most efficiently, the dependence of the observed reaction rate on the concentration of TMP, free magnesium ions and pH were investigated (Figure 7). TMP appeared to chelate an equimolar amount of Mg2+ (data not shown); therefore the excess of [Mg2+] over [TMP] in our experiments was called the free [Mg2+]. When the concentration of free magnesium ions was kept constant at 50 mM, an increase in the TMP concentration led to an increase in the observed rate to 0.03 min−1 at 100 mM [TMP], at pH 8.1. The [TMP] dependence was consistent with a 1:1 stoichiometry between ribozyme and TMP (curve fit in Figure 7A). Increases in the magnesium concentration at a constant TMP concentration of 100 mM led to a further increase in the reaction rate, with a kobs of 0.16 min−1 at 400 mM of free [Mg2+], at pH 8.1 (Figure 7B). The pH dependence of the reaction was tested under these conditions [100 mM [TMP] and 400 mM free [Mg2+]], as well as under the reaction conditions used during the selection [50 mM [TMP] and 50 mM free [Mg2+]] (Figure 7C). In these reactions three buffers were used: 2-(N-morpholino)ethanesulfonic acid (MES)/NaOH, HEPES/NaOH and Tris–HCl. For all reactions, the observed triphosphorylation rate increased linearly with the pH in the region between pH 5.5 and 8.5, with a slope of about one. This is consistent with a single deprotonation step being limiting for the reaction, perhaps the deprotonation of the RNA 5′-hydroxyl group in preparation for the nucleophilic attack on TMP.
Figure 7.
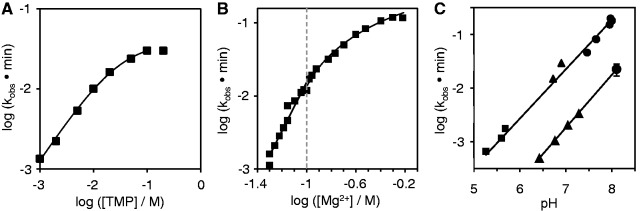
Dependence of reaction kinetics on reaction conditions, for the trans-splicing TPR1 ribozyme. (A) Dependence of the observed reaction rate on the concentration of TMP, with a free [Mg2+] of 50 mM at pH 8.1. The maximal rate is reached at 100 mM [TMP]. The black line shows the fit of the Michaelis–Menten equation kobs = (kcat * [TMP]/(KM + [TMP]) to data points from 1–100 mM [TMP], with kcat = 0.039 min−1 and KM = 30 mM. (B) Dependence of the observed reaction rate on the concentration of free magnesium ions. This is the excess of magnesium ions over the TMP concentration (here 100 mM), at pH 8.1. The dotted line separates reactions with [Mg2+]/[TMP] < 1 (left) from reactions with [Mg2+]/[TMP] > 1 (right). The fastest rate is obtained at 400 mM free [Mg2+]. The half-maximal rate of 0.068 min−1 was observed at 150 mM free [Mg2+]. (C) Dependence of the observed reaction rate on the pH, at 100 mM [TMP] and 400 mM free [Mg2+] (top line) and at 50 mM [TMP] and 50 mM free [Mg2+] (bottom line). The slopes of the two lines were 0.92 (top line) and 0.99 (bottom line), respectively. The symbols denote the buffer systems MES/NaOH (squares), HEPES/NaOH (triangles) and Tris–HCl (circles). The data point in the lower data set at pH 8.1 is the average of seven independent experiments, with error bars denoting their standard deviation.
DISCUSSION
We established an in vitro selection strategy for ribozymes with a novel activity, the triphosphorylation of RNA 5′-hydroxyl groups. The procedure identified more than a dozen independent ribozymes. One ribozyme was characterized in more detail. This ribozyme was able to work in trans, and its product was confirmed by mass spectroscopy. Its secondary structure appears to include a four-way helical junction. Its optimal reaction rate was at 100 mM TMP, 400 mM free [Mg2+] and increased pH.
The observed rate with the TPR1 ribozyme was 0.16 min−1 under optimal conditions (100 mM [TMP], 400 mM free [Mg2+], pH 8.1) (Figure 7). As comparison, the rate of the uncatalyzed reaction (kuncat) in the absence of Mg2+ was estimated to be 3.4 × 10−4 min−1, at 500 mM [TMP] and pH 12 [Figure 2 in (31)]. Because the pH is limiting for the uncatalyzed reaction, its rate at pH 8.1 was assumed to be 103.9-fold slower, at 4.3 × 10−8 min−1. In addition, higher concentrations of TMP lead to faster reactions for the uncatalyzed reaction (31). Because the influence of [TMP] on kuncat may be non-linear, we assumed a 3-fold lower kuncat at 100 mM [TMP] compared with 500 mM [TMP]. This rate of 1.4 × 10−8 min−1 is 1.1 × 107-fold below the rate of 0.16 min−1 for the ribozyme-catalyzed reaction. Mg2+ ions accelerate the uncatalyzed reaction (63) but no kinetic data are available for this Mg2+ acceleration. Therefore, the value for kcat/kuncat of ∼107 describes the rate enhancement of the ribozyme-catalyzed reaction in the presence of magnesium relative to the rate of the uncatalyzed reaction without magnesium.
None of the six tested ribozymes showed triphosphorylation activity of free nucleosides (Supplementary Figure S5). It may be possible to overcome this in future experiments because many more ribozyme clones from our selection can now be tested for this activity. The fact that most of the isolated 36 clones appeared as single clones (Supplementary Figure S3) suggests that many more active sequences can be isolated. However, RNA world organisms could have relied on the triphosphorylation of RNAs instead of free nucleosides to drive RNA polymerization: activation groups at the nucleoside 5′-phosphate are only necessary for RNA polymerization in the 5′–3′ direction. In contrast, RNA polymerization in the 3′–5′ direction does not require nucleotide activation because chain elongation proceeds via the reaction of the activated RNA primer 5′-terminus with the nucleoside 3′-hydroxyl group. This principle has been shown with an evolved variant of the class I ligase ribozyme that uses nucleoside triphosphates (NTPs) for the extension of RNA primers in both the 3′–5′ and 5′–3′ direction (64). This suggests that instead of NTP, a nucleoside could be used to extend the growing primer 5′-terminus. In the second step, the new RNA 5′-terminus could be triphosphorylated. Therefore, RNA polymerization in an RNA world organism could have proceeded via the alternating triphosphorylation of the RNA primer, and the extension of the primer 5′-terminus with a templated nucleoside. This polymerization in the 3′–5′ direction may have had an advantage for RNA world organisms: Nucleoside triphosphates are necessary for 5′–3′ elongation, but their negative charge destabilizes the binding of the monomer to the template strand due to charge repulsion with the phosphodiester backbone of the template strand. This difference in affinity can be inferred from measurements of stacking interactions in nucleosides and NTPs (65). In contrast, RNA polymerization in the 3′–5′ direction could use the uncharged nucleosides for the primer extension step, with a corresponding increase in monomer affinity to the template strand. Therefore, the alternating RNA 5′-triphosphorylation and extension with a nucleoside could have been an efficient RNA polymerization strategy for RNA world organisms.
In addition to promoting RNA polymerization, RNA world organisms could have used RNA 5′-triphosphates for several other reactions. This could have included RNA–RNA ligation (42,50,66,67), the formation of phosphoamidate bonds (45), RNA capping (46,68) and amino acid activation (69). Together with the results of this study, this suggests that RNA world organisms could have used TMP to thermodynamically drive a wide range of reactions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [molecular biophysics training grant T32 GM008326 to J.E.M.]; Hellman Family foundation [awards 2011/12 and 2012/13 to U.F.M.]; This material is based upon work supported by the National Aeronautics and Space Administration under Grant/cooperative agreement [No. NNX13AJ09G] issued through the Science Mission Directorate, Research Opportunities in Space and Earth Sciences -2011 (ROSES-2011), in Astrobiology/Exobiology (to U.F.M.). Funding for open access charge: NASA grant [NNX13AJ09G].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Chengguo Yao for pilot experiments with the hammerhead ribozyme, Manny Ares for the generous gift of 1M7 for SHAPE analysis and Ram Krishnamurthy for helpful discussions.
REFERENCES
- 1.Woese CR. The Genetic Code. The Molecular Basis for Genetic Expression. New York: Harper & Row; 1967. [Google Scholar]
- 2.Crick FHC. The origin of the genetic code. J. Mol. Biol. 1968;38:367–379. doi: 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- 3.Orgel LE. Evolution of the genetic apparatus. J. Mol. Biol. 1968;38:381–393. doi: 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- 4.Robertson MP, Joyce GF. The origins of the RNA world. Cold Spring Harb. Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a003608. pii: a003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szostak JW. The eightfold path to non-enzymatic RNA replication. J. Syst. Chem. 2012;3:2. [Google Scholar]
- 6.Wacey D, Kilburn MR, Saunders M, Cliff J, Brasier MD. Microfossils of sulphur-metabolizing cells in 3.4-billion-year-old rocks of Western Australia. Nat. Geosci. 2011;4:698–702. [Google Scholar]
- 7.Von Kiedrowski G. A self-replicating hexadeoxynucleotide. Angew. Chem. Int. Ed. Engl. 1986;25:932–935. [Google Scholar]
- 8.Orgel LE. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004;39:99–123. doi: 10.1080/10409230490460765. [DOI] [PubMed] [Google Scholar]
- 9.Hargreaves WR, Deamer DW. Liposomes from ionic, single-chain amphiphiles. Biochemistry. 1978;17:3759–3768. doi: 10.1021/bi00611a014. [DOI] [PubMed] [Google Scholar]
- 10.Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science. 2001;292:1319–1325. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- 11.Szostak JW. An optimal degree of physical and chemical heterogeneity for the origin of life? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:2894–2901. doi: 10.1098/rstb.2011.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sczepanski JT, Joyce GF. Synthetic evolving systems that implement a user-specified genetic code of arbitrary design. Chem. Biol. 2012;19:1324–1332. doi: 10.1016/j.chembiol.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez RA, Orgel LE. Studies in prebiotic synthesis. V. Synthesis and photoanomerization of pyrimidine nucleosides. J. Mol. Biol. 1970;47:531–543. doi: 10.1016/0022-2836(70)90320-7. [DOI] [PubMed] [Google Scholar]
- 14.Powner MW, Gerland B, Sutherland JD. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature. 2009;459:239–242. doi: 10.1038/nature08013. [DOI] [PubMed] [Google Scholar]
- 15.Powner MW, Sutherland JD, Szostak JW. Chemoselective multicomponent one-pot assembly of purine precursors in water. J. Am. Chem. Soc. 2010;132:16677–16688. doi: 10.1021/ja108197s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joyce GF, Schwartz AW, Miller SL, Orgel LE. The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl Acad. Sci. USA. 1987;84:4398–4402. doi: 10.1073/pnas.84.13.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnamurthy R. Role of pK(a) of nucleobases in the origins of chemical evolution. Acc. Chem. Res. 2012;45:2035–2044. doi: 10.1021/ar200262x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hud NV, Cafferty BJ, Krishnamurthy R, Williams LD. The origin of RNA and “my grandfather's axe". Chem. Biol. 2013;20:466–474. doi: 10.1016/j.chembiol.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 19.Chen MC, Cafferty BJ, Mamajanov I, Gallego I, Khanam J, Krishnamurthy R, Hud NV. Spontaneous prebiotic formation of a beta-ribofuranoside that self-assembles with a complementary heterocycle. J. Am. Chem. Soc. 2013 doi: 10.1021/ja410124v. (epub ahead of print, doi:10.1021/ja410124v) [DOI] [PubMed] [Google Scholar]
- 20.Verlander MS, Lohrmann R, Orgel LE. Catalysts for the self-polymerization of adenosine cyclic 2′, 3′-phosphate. J. Mol. Evol. 1973;2:303–316. doi: 10.1007/BF01654098. [DOI] [PubMed] [Google Scholar]
- 21.Rajamani S, Vlassov A, Benner S, Coombs A, Olasagasti F, Deamer D. Lipid-assisted synthesis of RNA-like polymers from mononucleotides. Orig. Life Evol. Biosph. 2008;38:57–74. doi: 10.1007/s11084-007-9113-2. [DOI] [PubMed] [Google Scholar]
- 22.Prabahar KJ, Cole TD, Ferris JP. Effect of phosphate activating group on oligonucleotide formation on montmorillonite: the regioselective formation of 3′,5′-linked oligoadenylates. J. Am. Chem. Soc. 1994;116:10914–10920. doi: 10.1021/ja00103a006. [DOI] [PubMed] [Google Scholar]
- 23.Prabahar KJ, Ferris JP. Adenine derivatives as phosphate-activating groups for the regioselective formation of 3′,5′-linked oligoadenylates on montmorillonite: possible phosphate-activating groups for the prebiotic synthesis of RNA. J. Am. Chem. Soc. 1997;119:4330–4337. doi: 10.1021/ja9700764. [DOI] [PubMed] [Google Scholar]
- 24.Lohrmann R, Orgel LE. Prebiotic synthesis: phosphorylation in aqueous solution. Science. 1968;161:64–66. doi: 10.1126/science.161.3836.64. [DOI] [PubMed] [Google Scholar]
- 25.Lohrmann R. Formation of nucleoside 5′-phosphoramidates under potentially prebiological conditions. J. Mol. Evol. 1977;10:137–154. doi: 10.1007/BF01751807. [DOI] [PubMed] [Google Scholar]
- 26.Sawai H, Orgel LE. Letter: Oligonucleotide synthesis catalyzed by the Zn-2+ ion. J. Am. Chem. Soc. 1975;97:3532–3533. doi: 10.1021/ja00845a050. [DOI] [PubMed] [Google Scholar]
- 27.Inoue T, Orgel LE. Oligomerization of (guanosine 5′-phosphor)-2-methylimidazolide on poly(C). An RNA polymerase model. J. Mol. Biol. 1982;162:201–217. doi: 10.1016/0022-2836(82)90169-3. [DOI] [PubMed] [Google Scholar]
- 28.Hanczyc MM, Fujikawa SM, Szostak JW. Experimental models of primitive cellular compartments: encapsulation, growth, and division. Science. 2003;302:618–622. doi: 10.1126/science.1089904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen IA, Salehi-Ashtiani K, Szostak JW. RNA catalysis in model protocell vesicles. J. Am. Chem. Soc. 2005;127:13213–13219. doi: 10.1021/ja051784p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budin I, Debnath A, Szostak JW. Concentration-driven growth of model protocell membranes. J. Am. Chem. Soc. 2012;134:20812–20819. doi: 10.1021/ja310382d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Etaix E, Orgel LE. Phosphorylation of nucleosides in aqueous solution using trimetaphosphate: Formation of nucleoside triphosphates. J. Carbohydr. Nucleosides Nucleotides. 1978;5:91–110. [Google Scholar]
- 32.Yamagata Y, Watanabe H, Saitoh M, Namba T. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature. 1991;352:516–519. doi: 10.1038/352516a0. [DOI] [PubMed] [Google Scholar]
- 33.Pasek MA, Kee TP, Bryant DE, Pavlov AA, Lunine JI. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew Chem. Int. Ed. Engl. 2008;47:7918–7920. doi: 10.1002/anie.200802145. [DOI] [PubMed] [Google Scholar]
- 34.Osterberg R, Orgel LE. Polyphosphate and trimetaphosphate formation under potentially prebiotic conditions. J. Mol. Evol. 1972;1:241–248. doi: 10.1007/BF01660243. [DOI] [PubMed] [Google Scholar]
- 35.Thilo E, Schulz G, Wichmann E-M. Die Konstitution des Grahamschen und Kurrolschen Salzes. Zeitschrift für Anorganische und Allgemeine Chemie. 1953;272:182–200. [Google Scholar]
- 36.Pasek MA, Harnmeijer JP, Buick R, Gull M, Atlas Z. Evidence for reactive reduced phosphorus species in the early Archean ocean. Proc. Natl Acad. Sci. USA. 2013;110:10089–10094. doi: 10.1073/pnas.1303904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feldmann W. Das Trimetaphosphat als Triphosphorylierungsmittel für Alkohole und Kohlenhydrate in wäßriger Lösung. Seine Sonderstellung unter den kondensierten Phosphaten. Chem. Ber. 1967;100:3850–3860. [Google Scholar]
- 38.Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 39.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 40.Hutchins CJ, Rathjen PD, Forster AC, Symons RH. Self-cleavage of plus and minus RNA transcripts of avocado sunblotch viroid. Nucleic Acids Res. 1986;14:3627–3640. doi: 10.1093/nar/14.9.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buzayan JM, Gerlach WL, Bruening G. Satellite tobacco ringspot virus RNA: A subset of the RNA sequence is sufficient for autolytic processing. Proc. Natl Acad. Sci. USA. 1986;83:8859–8862. doi: 10.1073/pnas.83.23.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartel DP, Szostak JW. Isolation of new ribozymes from a large pool of random sequences. Science. 1993;261:1411–1418. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 43.Lorsch JR, Szostak JW. In vitro evolution of new ribozymes with polynucleotide kinase activity. Nature. 1994;371:31–36. doi: 10.1038/371031a0. [DOI] [PubMed] [Google Scholar]
- 44.Wiegand TW, Janssen RC, Eaton BE. Selection of RNA amide synthases. Chem. Biol. 1997;4:675–683. doi: 10.1016/s1074-5521(97)90223-4. [DOI] [PubMed] [Google Scholar]
- 45.Baskerville S, Bartel DP. A ribozyme that ligates RNA to protein. Proc. Natl Acad. Sci. USA. 2002;99:9154–9159. doi: 10.1073/pnas.142153799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang F, Yarus M. 5′-RNA self-capping from guanosine diphosphate. Biochemistry. 1997;36:6557–6563. doi: 10.1021/bi970475b. [DOI] [PubMed] [Google Scholar]
- 47.Unrau PJ, Bartel DP. RNA-catalysed nucleotide synthesis. Nature. 1998;395:260–263. doi: 10.1038/26193. [DOI] [PubMed] [Google Scholar]
- 48.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 49.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 50.Rogers J, Joyce GF. The effect of cytidine on the structure and function of an RNA ligase ribozyme. RNA. 2001;7:395–404. doi: 10.1017/s135583820100228x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 52.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000;25:169–193. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 53.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price SR, Ito N, Oubridge C, Avis JM, Nagai K. Crystallization of RNA-protein complexes. I. Methods for the large-scale preparation of RNA suitable for crystallographic studies. J. Mol. Biol. 1995;249:398–408. doi: 10.1006/jmbi.1995.0305. [DOI] [PubMed] [Google Scholar]
- 55.Turner R, Shefer K, Ares M., Jr Safer one-pot synthesis of the ‘SHAPE' reagent 1-methyl-7-nitroisatoic anhydride (1m7) RNA. 2013;19:1857–1863. doi: 10.1261/rna.042374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uhlenbeck OC. A small catalytic oligoribonucleotide. Nature. 1987;328:596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 57.Santoro SW, Joyce GF. Mechanism and utility of an RNA-cleaving DNA enzyme. Biochemistry. 1998;37:13330–13342. doi: 10.1021/bi9812221. [DOI] [PubMed] [Google Scholar]
- 58.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mortimer SA, Weeks KM. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 2007;129:4144–4145. doi: 10.1021/ja0704028. [DOI] [PubMed] [Google Scholar]
- 60.Hajdin CE, Bellaousov S, Huggins W, Leonard CW, Mathews DH, Weeks KM. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl Acad. Sci. USA. 2013;110:5498–5503. doi: 10.1073/pnas.1219988110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sashital DG, Cornilescu G, McManus CJ, Brow DA, Butcher SE. U2-U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat. Struct. Mol. Biol. 2004;11:1237–1242. doi: 10.1038/nsmb863. [DOI] [PubMed] [Google Scholar]
- 62.Berry KE, Waghray S, Mortimer SA, Bai Y, Doudna JA. Crystal structure of the HCV IRES central domain reveals strategy for start-codon positioning. Structure. 2011;19:1456–1466. doi: 10.1016/j.str.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cheng C, Fan C, Wan R, Tong C, Miao Z, Chen J, Zhao Y. Phosphorylation of adenosine with trimetaphosphate under simulated prebiotic conditions. Orig. Life Evol. Biosph. 2002;32:219–224. doi: 10.1023/a:1016513114799. [DOI] [PubMed] [Google Scholar]
- 64.McGinness KE, Wright MC, Joyce GF. Continuous in vitro evolution of a ribozyme that catalyzes three successive nucleotidyl addition reactions. Chem. Biol. 2002;9:585–596. doi: 10.1016/s1074-5521(02)00136-9. [DOI] [PubMed] [Google Scholar]
- 65.Sigel H, Griesser R. Nucleoside 5′-triphosphates: self-association, acid-base, and metal ion-binding properties in solution. Chem. Soc. Rev. 2005;34:875–900. doi: 10.1039/b505986k. [DOI] [PubMed] [Google Scholar]
- 66.Ikawa Y, Tsuda K, Matsumura S, Inoue T. De novo synthesis and development of an RNA enzyme. Proc. Natl Acad. Sci. USA. 2004;101:13750–13755. doi: 10.1073/pnas.0405886101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robertson MP, Ellington AD. In vitro selection of an allosteric ribozyme that transduces analytes to amplicons. Nat. Biotechnol. 1999;17:62–66. doi: 10.1038/5236. [DOI] [PubMed] [Google Scholar]
- 68.Zaher HS, Watkins RA, Unrau PJ. Two independently selected capping ribozymes share similar substrate requirements. RNA. 2006;12:1949–1958. doi: 10.1261/rna.131306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kumar RK, Yarus M. RNA-catalyzed amino acid activation. Biochemistry. 2001;40:6998–7004. doi: 10.1021/bi010710x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.