


Abstract
Identifying differential features between conditions is a popular approach to understanding molecular features and their mechanisms underlying a biological process of particular interest. Although many tests for identifying differential expression of gene or gene sets have been proposed, there was limited success in developing methods for differential interactions of genes between conditions because of its computational complexity. We present a method for Evaluation of Dependency DifferentialitY (EDDY), which is a statistical test for differential dependencies of a set of genes between two conditions. Unlike previous methods focused on differential expression of individual genes or correlation changes of individual gene–gene interactions, EDDY compares two conditions by evaluating the probability distributions of dependency networks from genes. The method has been evaluated and compared with other methods through simulation studies, and application to glioblastoma multiforme data resulted in informative cancer and glioblastoma multiforme subtype-related findings. The comparison with Gene Set Enrichment Analysis, a differential expression-based method, revealed that EDDY identifies the gene sets that are complementary to those identified by Gene Set Enrichment Analysis. EDDY also showed much lower false positives than Gene Set Co-expression Analysis, a method based on correlation changes of individual gene–gene interactions, thus providing more informative results. The Java implementation of the algorithm is freely available to noncommercial users. Download from: http://biocomputing.tgen.org/software/EDDY.
INTRODUCTION
Since the emergence of high-throughput genomic profiling techniques, numerous statistical methods gained high popularity in biomedical studies to assess diverse features in biological samples. One of such statistical approaches is identifying variables with differential patterns between different conditions, where genomic entities (such as genes or proteins) are often modeled as target variables. Such methods can vary based on the definition of differentiality or what a target feature of comparison is, but the general idea is comparing probability distributions of a target feature across given conditions.
The simplest case of identifying differentiality is differential expression of a single gene, where each gene is independently tested for differential expression. There have been many studies with this approach of independent tests for individual genes. For comprehensive reviews of single gene test approaches see (1). The main drawback of single-gene test approaches is that they focus on individual genes instead of a set of genes, while a set of interacting genes constitutes a functional module in many biological systems. For this reason, a more beneficial approach is testing differentiality for a set of genes between conditions.
Considering that a joint probability distribution of a set of variables can provide more comprehensive view of underlying process, an ideal method to test differentiality of a set of genes between conditions is comparing the joint probability distributions of their activity levels. However, this ideal approach is not practical in many real situations owing to the complexity of the model to represent the joint probability distribution and the lack of available data to infer such complex models with sufficient reliability. For this reason, most of the methods to test the differentiality of a set of genes rely on heuristic approaches by focusing on specific features in the set of genes rather than considering the complete joint probability distributions.
Several methods have been proposed to test the differentiality of a gene set between conditions by considering differential expressions of genes in the gene set (2–4). Their methods take a common approach of computing differential expressions of genes in the target gene set and summarizing them into a single statistic that represents the differentiality of the gene set between conditions. Gene Set Enrichment Analysis (GSEA) (2) is a popular method of testing gene sets, where it computes the degree to which the expression of a gene set is specifically correlated to a target condition. GSEA has been successfully applied in recent studies, but it is designed to capture only the gene sets with consistent differential expressions (either over- or under-expression) under a target condition. Each gene in a biological pathway does not necessarily show differential expressions of one direction; therefore, there is a need for methods to evaluate relationships between genes in computing the statistics of differentiality.
The idea of network-driven activities of biological functions has gained more interests, as more evidence is found that biological systems can show highly diverse activity patterns because genes can interact differentially across specific molecular contexts (5). The simplest approach to evaluate such differential interactions is building separate networks for different conditions and comparing them (6–9). With the need for more statistical power to discriminate differential interactions, several studies proposed statistical methods to test the differentiality of individual interactions. Lai et al. (10) used an expected conditional F-statistic to test the differentiality of a gene–gene co-expression between conditions. The differential correlation approaches (11–13) used difference in correlation coefficients between a pair of variables across two conditions to identify differential interactions. Besides these methods for individual differential interactions, there have been recent studies to identify differential subnetworks across conditions. The general idea of such approach is using already known genetic interactions as a ground truth network and overlaying observed genomic data (e.g. messenger RNA expressions) of different conditions to statistically evaluate regions with differential genetic activities. Guo et al. (14) used an edge-based scoring measure to identify condition-responsive protein–protein interaction subnetwork. Hwang and Park (15) used a multivariate analysis of variance scoring method to find differentially expressed subnetworks. Kim et al. (16) represented networks with activity weight matrices, and nonnegative matrix factorization was used to find principal subnetworks. The COSINE method (17) computes a score from both of gene expressions and available gene interactions to find condition-specific subnetworks. Besides these methods using already known interactions, a few methods without using known interactions have been also proposed. The differential dependency network method (18,19) infers a local dependency model to represent the topology around each gene for each condition. A permutation test is used to compute the significance of local topology change between conditions. Ouyang et al. (20) modeled interactions coming into a gene with ordinary differential equations, and the difference in slopes of the models was compared across conditions to compute the difference in the magnitudes of local genetic relationships. These methods were designed to identify individual differential interactions or condition-specific subnetworks, but they were not designed to test gene sets for dependency variance across conditions. Choi and Kendziorski (21) proposed Gene Set Co-expression Analysis (GSCA), which computes a Euclidean distance between gene interaction correlation vectors from two different conditions as a discrepancy measure. GSCA was designed to test gene sets for interaction differentiality, but it can be too sensitive to minor correlation changes and can give biased results with respect to the size of gene sets.
In this article, we propose a method for Evaluation of Dependency DifferentialitY (EDDY), which is a statistical test for the differential dependency relationship of a set of genes between two given conditions. For each condition, possible dependency network structures are enumerated and their likelihoods are computed to represent a probability distribution of dependency networks. The difference between the probability distributions of dependency networks is computed between conditions, and its statistical significance is evaluated with random permutations of condition labels on the samples. The proposed method has been evaluated and compared with other methods through simulation studies and was applied to the gene expression data of glioblastoma multiforme (GBM) from The Cancer Genome Atlas (TCGA) to reveal the functional difference between the four subtypes of GBM. Simulation experiments show the validity of EDDY as well as its superior performance in identifying gene sets with differential interactions. From the application to the TCGA GBM data, the results show that the proposed method can identify novel gene sets that could not be found with GSEA, which is considered a representative method of considering only differential expressions, while providing many results specific to the subtypes of GBM. When compared with GSCA, which is an existing gene set test method that considers differential interactions, EDDY gives less-biased results that can be more informative.
MATERIALS AND METHODS
Outline of approach
The proposed method computes the discrepancy between probability distributions of dependency network structures for a given set of genes, across given samples of two different conditions, and evaluates its statistical significance. We assume that a set of genes is given as the target of a test, and the activity levels of the genes are represented with a set of variables V (each variable corresponds to each gene). For V, there are N (which is a finite number) possible dependency network structures g1, g2, … , gN for the variables. If we consider a discrete random variable G that can have g1, g2, … , gN as its discrete values, the posterior probability distribution 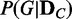 for data
for data 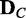 of a given condition C can represent the probability distribution of dependency network structures for V in the condition C. When two data sets,
of a given condition C can represent the probability distribution of dependency network structures for V in the condition C. When two data sets, 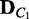 and
and 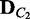 , are given for two different conditions C1 and C2, the divergence between the two corresponding probability distributions
, are given for two different conditions C1 and C2, the divergence between the two corresponding probability distributions 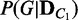 and
and 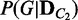 are computed as a measure of difference between the conditions. This approach is a generalization of comparing the best networks from different conditions by considering many possible dependency networks and their likelihoods. The benefit of this generalization is more reliable measure of discrepancy, especially when data are limited; thus, there is a high chance of finding many local optima for the best network. By considering many probable dependency networks instead of one local optimal network, our approach can represent the truer picture of dependencies at the cost of additional computation. The statistical significance of the divergence is computed using a permutation approach, by repeating the random shuffling of condition labels C1 and C2 and computing the divergence to evaluate the probability of obtaining the original or larger divergence by random chance. This outline of the method is illustrated in Figure 1.
are computed as a measure of difference between the conditions. This approach is a generalization of comparing the best networks from different conditions by considering many possible dependency networks and their likelihoods. The benefit of this generalization is more reliable measure of discrepancy, especially when data are limited; thus, there is a high chance of finding many local optima for the best network. By considering many probable dependency networks instead of one local optimal network, our approach can represent the truer picture of dependencies at the cost of additional computation. The statistical significance of the divergence is computed using a permutation approach, by repeating the random shuffling of condition labels C1 and C2 and computing the divergence to evaluate the probability of obtaining the original or larger divergence by random chance. This outline of the method is illustrated in Figure 1.
Figure 1.

The conceptual outline of EDDY. A target gene set and gene expression data of two conditions C1 and C2 are given as input. (STEP I-A) The probability distribution of dependency network likelihood is computed for each condition. (STEP I-B) The divergence between C1 and C2 is computed from the two probability distributions of dependency network likelihoods. (STEP II-A) Random data sets are built by shuffling sample condition labels. (STEP II-B) For each random data set, a random divergence is computed. The collection of all random divergences constitutes the null distribution of divergence. (STEP II-C) The P-value of the original divergence is evaluated in comparison with the computed null distribution of divergence.
Computing the posterior probability of each dependency network structure
Evaluating the probability distribution of 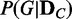 of a discrete random variable G requires computing the posterior probability
of a discrete random variable G requires computing the posterior probability 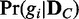 of each dependency network structure gi (
of each dependency network structure gi (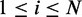 ) for the condition C. The exact computation of the posterior probability of a model (gi) given observation (
) for the condition C. The exact computation of the posterior probability of a model (gi) given observation (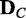 ) is not straightforward. Thus, we use an approach that uses only the likelihoods to compute the posterior probability. With the Bayes’ theorem and assumption of a uniform prior, the posterior probability
) is not straightforward. Thus, we use an approach that uses only the likelihoods to compute the posterior probability. With the Bayes’ theorem and assumption of a uniform prior, the posterior probability 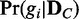 can be computed as follows with only likelihoods
can be computed as follows with only likelihoods 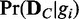 (see Supplementary Method S1 for details):
(see Supplementary Method S1 for details):
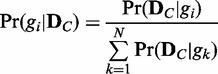 |
(1) |
In this work, we use the Bayesian network model of discrete random variables to compute network likelihoods, which is widely used in the field of computational biology due to its strong statistical foundation. The Bayesian network model assumes directed acyclic graphs (DAGs) for network structures, whereas real biological networks can have cycles such as feedback loops. However, it is not a limitation in this work, as we use the Bayesian network model to represent dependency relationships between genes rather than physical interactions. With this consideration, the computation of likelihood 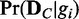 is done using the Bayesian Dirichlet equivalence uniform scoring method (22).
is done using the Bayesian Dirichlet equivalence uniform scoring method (22).
Even though we decided to use the Bayesian network model assuming discrete random variables, the rest of our formulations and algorithms are independent of model choices. Thus, other network and random variable models can be also used as long as the likelihood of a network structure can be computed based on the model of preference.
Approximate computation of probability distribution for dependency network structures
The exact computation of the probability distribution 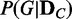 requires the enumeration of all possible N dependency network structures, g1, … , gN, and subsequent computation of their posterior probabilities,
requires the enumeration of all possible N dependency network structures, g1, … , gN, and subsequent computation of their posterior probabilities, 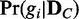 (
(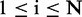 ). Such exact computation is possible for the case of small number of variables (genes), but it becomes computationally intractable as the number of variables increases. For example, the possible number of DAGs for five variables is ∼29 000, but it becomes ∼
). Such exact computation is possible for the case of small number of variables (genes), but it becomes computationally intractable as the number of variables increases. For example, the possible number of DAGs for five variables is ∼29 000, but it becomes ∼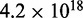 for 10 variables. For this reason, we take a heuristic approach to approximate the probability distribution of
for 10 variables. For this reason, we take a heuristic approach to approximate the probability distribution of 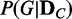 . In this approach, we assume that the probabilities of
. In this approach, we assume that the probabilities of 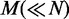 dependency structures are significantly high in either C1 or C2, and the rest of the dependency structures have similar low probabilities in both of the conditions and, thus, can be ignored, as they make little difference between the conditions. To ensure fairness for both conditions,
dependency structures are significantly high in either C1 or C2, and the rest of the dependency structures have similar low probabilities in both of the conditions and, thus, can be ignored, as they make little difference between the conditions. To ensure fairness for both conditions, 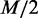 dependency structures are chosen from the condition C1 and the other
dependency structures are chosen from the condition C1 and the other 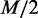 are chosen from the condition C2.
are chosen from the condition C2.
Selecting the top 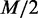 dependency network structures with the highest probabilities from a condition also requires computing the probabilities of all dependency network structures, which makes our approximate approach ineffective. To reduce such computational complexity, we use a heuristic method that proposes probable dependency structures by independently evaluating each dependency between two variables. In this method,
dependency network structures with the highest probabilities from a condition also requires computing the probabilities of all dependency network structures, which makes our approximate approach ineffective. To reduce such computational complexity, we use a heuristic method that proposes probable dependency structures by independently evaluating each dependency between two variables. In this method, 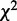 -test is applied to test the independence between every pair of two variables Vi and Vj (
-test is applied to test the independence between every pair of two variables Vi and Vj ( ), obtaining the resultant P-value pij (
), obtaining the resultant P-value pij (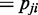 ). In case of assuming continuous valued random variable models, other proper statistical tests of independence for continuous variables can be used instead. When a probable dependency structure gk is proposed for
). In case of assuming continuous valued random variable models, other proper statistical tests of independence for continuous variables can be used instead. When a probable dependency structure gk is proposed for 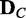 , an edge e between Vi and Vj is included with the following probability
, an edge e between Vi and Vj is included with the following probability 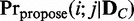 :
:
| (2) |
where 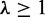 . With this definition of edge inclusion probability, an edge between two variables will be included in the proposed structure with higher probability when the dependency test between the two variables yields a lower P-value. Either direction of the edge eij or eji is randomly chosen with the same probability of 0.5 as long as it does not violate the acyclic property of DAG in gk. To reduce computational complexity in evaluating DAG structures, the maximum number of incoming edges is limited to a predetermined K. A formal description of this process is given in Supplementary Method S2 as an algorithm StructurePropose. This pairwise dependency-based method of structure proposal has a limitation in identifying full multivariate conditional dependency. However, the actual computation of network structure likelihoods is done in consideration of such combinatorial dependencies (with the Bayesian Dirichlet equivalence uniform scoring method), and sampling many network structures will further diminish such limitation.
. With this definition of edge inclusion probability, an edge between two variables will be included in the proposed structure with higher probability when the dependency test between the two variables yields a lower P-value. Either direction of the edge eij or eji is randomly chosen with the same probability of 0.5 as long as it does not violate the acyclic property of DAG in gk. To reduce computational complexity in evaluating DAG structures, the maximum number of incoming edges is limited to a predetermined K. A formal description of this process is given in Supplementary Method S2 as an algorithm StructurePropose. This pairwise dependency-based method of structure proposal has a limitation in identifying full multivariate conditional dependency. However, the actual computation of network structure likelihoods is done in consideration of such combinatorial dependencies (with the Bayesian Dirichlet equivalence uniform scoring method), and sampling many network structures will further diminish such limitation.
After using this method to collect up to M network structures for the cases of large number of variables, the probability distributions  and
and 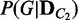 are computed by evaluating the likelihoods of network structures (a formal description of this process is given in Supplementary Method S3 as an algorithm ComputeDistribution).
are computed by evaluating the likelihoods of network structures (a formal description of this process is given in Supplementary Method S3 as an algorithm ComputeDistribution).
Computing the divergence between conditions and its statistical significance
Once the probability distributions of dependency network structures 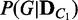 and
and  are computed, the divergence between the conditions C1 and C2 is measured using the Jensen–Shannon (JS) divergence (23), which is a popular method of measuring the divergence between two discrete probability distributions. Once the JS divergence value, JS, is obtained, its statistical significance is computed with a permutation approach. Condition labels of C1 and C2 are randomly reassigned to the samples of
are computed, the divergence between the conditions C1 and C2 is measured using the Jensen–Shannon (JS) divergence (23), which is a popular method of measuring the divergence between two discrete probability distributions. Once the JS divergence value, JS, is obtained, its statistical significance is computed with a permutation approach. Condition labels of C1 and C2 are randomly reassigned to the samples of 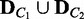 to build permuted sample sets
to build permuted sample sets 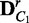 and
and 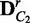 , and the same process is applied to compute a new divergence JSr. If JSr is larger than or equal to JS for t times out of T random permutations, the statistical significance P-value of JS is defined as t/T. This whole process is specifically defined in Algorithm 1: EDDY.
, and the same process is applied to compute a new divergence JSr. If JSr is larger than or equal to JS for t times out of T random permutations, the statistical significance P-value of JS is defined as t/T. This whole process is specifically defined in Algorithm 1: EDDY.
Algorithm 1 EDDY.
Require: V, 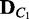 ,
, 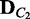 ,
, 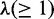 ,
, 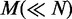 , T, K
Ensure:
JS, p
, T, K
Ensure:
JS, p
1: {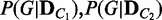 }
}  ComputeDistribution(V,
ComputeDistribution(V, 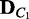 ,
, 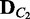 , λ, M, K)
, λ, M, K)
2: 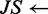 JensenShannon(
JensenShannon(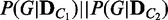 )
)
3: 
4:
5: for
 to T
do
to T
do
6: Build 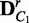 and
and 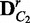 by randomly shuffling the condition labels
by randomly shuffling the condition labels
7: { }
}  ComputeDistribution(V,
ComputeDistribution(V, 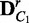 ,
, 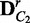 , λ, M, K)
, λ, M, K)
8: 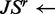 JensenShannon(
JensenShannon(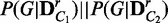 )
)
9:
10: if
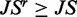 then
then
11: 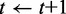
12: end if
13: end for
14:
15: 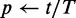
16: return JS and p
Simulation experiments
In this study, we prepared two simulation experiments. The first simulation (Simulation I) is to characterize the performance of EDDY with varying parameters such as sample size, network size and network differentiality. The second simulation (Simulation II) is to compare the performance of EDDY in identifying differential gene sets with that of other methods (GSCA and GSEA), using an interaction-focused synthetic data generation model. The outline of these two simulations is illustrated in Figure 2.
Figure 2.
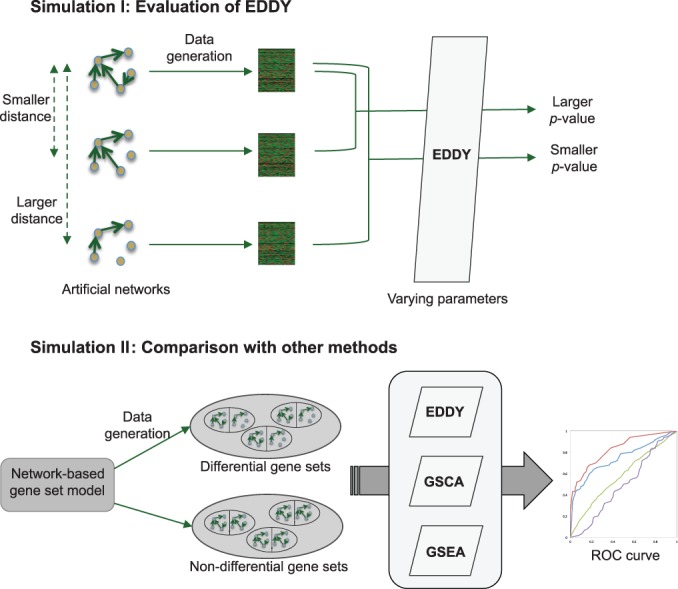
The outline of two simulation experiments. Simulation I is to evaluate the characteristics of EDDY in various configurations. Simulation II is to compare EDDY with other methods, based on an interaction-focused synthetic data generation model.
Simulation I: evaluating the characteristics of EDDY
We conducted simulation experiment to evaluate the ability of EDDY in discriminating two different conditions. In this simulation experiment, we consider 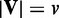 discrete random variables that can have three possible discrete values (−1, 0, 1). A Bayesian network B0 with
discrete random variables that can have three possible discrete values (−1, 0, 1). A Bayesian network B0 with 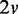 edges is randomly built with the v variables and randomly initialized conditional probability tables, and d samples are generated from B0 to constitute a data set
edges is randomly built with the v variables and randomly initialized conditional probability tables, and d samples are generated from B0 to constitute a data set 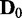 . To generate a data set of another condition for comparison, Bs is built by randomly removing
. To generate a data set of another condition for comparison, Bs is built by randomly removing 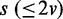 edges from B0, and d samples are generated from Bs for
edges from B0, and d samples are generated from Bs for 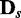 . In the process of edge removal, the conditional probability table of a variable that is affected by the edge removal is randomly reinitialized. The objective of this simulation experiment is to show that the divergence JS increases and its statistical significance P-value decreases as s, which represents the distance between two data sets in the sense of dependency relationship, increases.
. In the process of edge removal, the conditional probability table of a variable that is affected by the edge removal is randomly reinitialized. The objective of this simulation experiment is to show that the divergence JS increases and its statistical significance P-value decreases as s, which represents the distance between two data sets in the sense of dependency relationship, increases.
Different number of variables were tested with v = 5, 10, 20 and 50 as well as varying sample size with d = 50, 200 and 500. For the parameters of EDDY, M was chosen among 50, 200, 1000, 3000, 5000 and Nv (the number of all possible DAG structures for v nodes) according to v that can represent the size of a problem.  ,
, 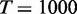 ,
, 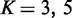 and
and  (which is the maximum value) were used throughout the simulation experiment.
(which is the maximum value) were used throughout the simulation experiment. 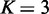 and 5 were used to evaluate the effect of limiting incoming edges on the performance (detailed results of these cases will be given in Supplementary Figures S1 and S2). For each case of testing
and 5 were used to evaluate the effect of limiting incoming edges on the performance (detailed results of these cases will be given in Supplementary Figures S1 and S2). For each case of testing 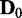 versus
versus 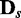 , the processes of building random Bayesian networks B0 and Bs, generation of data
, the processes of building random Bayesian networks B0 and Bs, generation of data 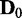 and
and 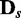 and applying EDDY was repeated 100 times to compute the average JS and P-values.
and applying EDDY was repeated 100 times to compute the average JS and P-values.
Another network comparison scenario has been also tested, where 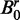 was built instead of Bs by randomly relocating the edges in B0, then used for comparison with B0. This scenario represents more general cases of comparison, where the networks generating given data sets may have more complex interaction discrepancies than simply missing interactions. For this simulation experiment, the number of edges in B0 was randomly determined between 0 and the maximum possible numbers. Brief summaries of the results from these additional simulation experiments will be given in the next section, and the detailed reports are given in Supplementary Figures S3–S9.
was built instead of Bs by randomly relocating the edges in B0, then used for comparison with B0. This scenario represents more general cases of comparison, where the networks generating given data sets may have more complex interaction discrepancies than simply missing interactions. For this simulation experiment, the number of edges in B0 was randomly determined between 0 and the maximum possible numbers. Brief summaries of the results from these additional simulation experiments will be given in the next section, and the detailed reports are given in Supplementary Figures S3–S9.
Simulation II: comparison of EDDY with other methods
To show the benefits and distinguished characteristics of EDDY, we compared the performance of EDDY with that of other methods in identifying differential gene sets using simulated data sets. There have been several studies of comparing multiple gene set test methods including (24–26) using simulated data sets. Their configurations vary, with the number of samples d in each condition from 20 to 500, the total number of genes from 100 to 1000 and the size of each gene set v from 10 to >40. However, they used similar methods to generate synthetic gene expression data assuming multivariate normal distributions and using covariance matrices. A differentially expressed gene (DEG) between two conditions is represented with two different mean values 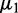 and
and 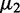 in two corresponding conditions, and differential gene sets between conditions are often defined by controlling the number of DEGs. Such a DEG-focused scenario can be appropriate in comparing methods focused on differential expressions. However, it has a limitation in comparing methods focused on gene interactions because differential interactions do not necessarily accompany differential expressions. For this reason, we used an interaction-focused synthetic data generation model for the simulation.
in two corresponding conditions, and differential gene sets between conditions are often defined by controlling the number of DEGs. Such a DEG-focused scenario can be appropriate in comparing methods focused on differential expressions. However, it has a limitation in comparing methods focused on gene interactions because differential interactions do not necessarily accompany differential expressions. For this reason, we used an interaction-focused synthetic data generation model for the simulation.
In this interaction-focused simulation, the expression levels of a gene set for a condition is generated from a Bayesian network model of continuous values. The expression levels of a gene set with v genes for the condition Ck are generated from a randomly built Bayesian network model Bk, where each node corresponds to a gene, 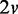 edges are randomly assigned and conditional probability tables are randomly initialized. For computational simplicity in data generation, each node has two possible discrete values (−1, 1), and they are later substituted with two different normal distributions during the data sampling process (e.g. when a value −1 is sampled for a gene, a value is randomly sampled from the corresponding normal distribution instead). The number of different edge connections between two Bayesian networks B1 and B2 of two conditions is randomly determined to a value higher than v (50%) for a differential gene set, and it is randomly determined to a value lower than
edges are randomly assigned and conditional probability tables are randomly initialized. For computational simplicity in data generation, each node has two possible discrete values (−1, 1), and they are later substituted with two different normal distributions during the data sampling process (e.g. when a value −1 is sampled for a gene, a value is randomly sampled from the corresponding normal distribution instead). The number of different edge connections between two Bayesian networks B1 and B2 of two conditions is randomly determined to a value higher than v (50%) for a differential gene set, and it is randomly determined to a value lower than 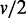 (25%) for a nondifferential gene set. As change in dependency (edge discrepancy) does not necessarily mean differential expressions, interaction-focused methods can be preferred in this scenario. For one synthetic data set, 50 differential gene sets and 50 nondifferential gene sets are prepared, where each gene set has v genes. Total 10 000 genes including the 100 gene sets are generated, and the genes that do not belong to the 100 gene sets are generated in the same way of generating nondifferential gene sets. Gene set sizes of
(25%) for a nondifferential gene set. As change in dependency (edge discrepancy) does not necessarily mean differential expressions, interaction-focused methods can be preferred in this scenario. For one synthetic data set, 50 differential gene sets and 50 nondifferential gene sets are prepared, where each gene set has v genes. Total 10 000 genes including the 100 gene sets are generated, and the genes that do not belong to the 100 gene sets are generated in the same way of generating nondifferential gene sets. Gene set sizes of 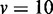 , 20 and 30 were considered, and two different normal distributions for gene expressions have the same variance of 1 but different mean values of 1 and 3.
, 20 and 30 were considered, and two different normal distributions for gene expressions have the same variance of 1 but different mean values of 1 and 3.
For each scenario, the simulation was repeated 100 times for each of GSEA, GSCA and EDDY, and their average false-/true-positive rates were evaluated by varying the P-value threshold then summarized as receiver operating characteristics (ROC) curves. For GSCA, Pearson correlation coefficient was used as a correlation measure. For EDDY,  ,
, 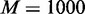 and 5000 dependency network structures of consideration and
and 5000 dependency network structures of consideration and  were used. As EDDY relies on the Bayesian network model with discrete random variables, the expression levels of each gene were standardized and quantized to three discrete values of (−1, 0, 1) using one standard deviation as a threshold. For all three methods, the same 1000 permutations were used to evaluate P-values.
were used. As EDDY relies on the Bayesian network model with discrete random variables, the expression levels of each gene were standardized and quantized to three discrete values of (−1, 0, 1) using one standard deviation as a threshold. For all three methods, the same 1000 permutations were used to evaluate P-values.
Identifying GBM subtype-specific gene sets with EDDY and comparison with other methods
EDDY and other two methods (GSEA and GSCA) were applied to the TCGA GBM gene expression data to identify subtype-specific gene sets. Gene expression data of GBM were obtained from TCGA for 202 samples with four previously reported GBM subtypes [54 classical, 58 mesenchymal, 33 neural and 57 proneural (27)], as well as 10 normal samples. The expression of 17 814 genes in the GBM samples were log-transformed, and standardized to z-scores using the 10 normal samples as a reference to convert the expression levels in GBM samples to the ratios to the mean expressions from normal samples. As we used the Bayesian network model assuming discrete random variables for EDDY, the standardized expression values were further quantized to three discrete values of ‘1’ (overexpression compared with normal), ‘0’ (no-change compared with normal) and ‘−1’ (underexpression compared with normal), by using one standard deviation as a threshold. Using higher thresholds for quantization rendered the gene expression values less informative (too consistent across all samples), thus higher thresholds were avoided in this experiment.
For all methods, the tests were done by comparing samples of subtype S versus the rest of the samples to identify gene sets that show distinct patterns in the subtype S. For gene sets of test targets, we collected 2101 canonical pathway gene sets and Gene Ontology (GO) gene sets of biological process and molecular function from MSigDB (2). In testing each gene set for a subtype versus the rest using EDDY,  ,
, 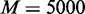 dependency network structures of consideration,
dependency network structures of consideration, 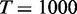 permutations and
permutations and 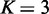 were used. To further reduce the computational cost, we filter out the genes with the changes in <10% of the samples after quantization, resulting only 13 884 genes for the analysis. Obtained P-values were false discovery rate (FDR)-corrected using the Benjamini and Hochberg’s method (28), and gene sets with FDR-corrected P < 0.05 were declared to be statistically significant.
were used. To further reduce the computational cost, we filter out the genes with the changes in <10% of the samples after quantization, resulting only 13 884 genes for the analysis. Obtained P-values were false discovery rate (FDR)-corrected using the Benjamini and Hochberg’s method (28), and gene sets with FDR-corrected P < 0.05 were declared to be statistically significant.
For comparison with conventional methods based on differential gene expression, we applied GSEA to identify gene sets for each subtype. Of the 2101 gene sets, 2067 gene sets (98.4%) with up to 500 genes were tested using GSEA. In running GSEA for each gene set, 1000 permutations were applied. From the result, P-values were FDR-corrected using the Benjamini and Hochberg’s method, and the same P-value threshold 0.05 was used for statistical significance. We also compared our result with that of GSCA, which is a method that evaluates the differentiality of interactions given a gene set, but based on simple pairwise correlations rather than assessing global topology of network structures. For GSCA, Pearson correlation coefficient was used as a correlation measure, and 1000 permutations were applied to compute statistical significance of measured discrepancy. The FDR-corrected P-value = 0.05 was used as a threshold for statistical significance. In applying GSEA and GSCA, the standardized gene expression data were used without quantization.
RESULTS
Simulation I: the characteristics of EDDY
Figure 3 shows JS and P-values by varying s, from applying EDDY with different parameters and data amounts, but with 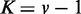 . In general, JS divergences increase and P-values decrease as the discrepancy increases between the Bayesian networks from which
. In general, JS divergences increase and P-values decrease as the discrepancy increases between the Bayesian networks from which 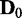 and
and 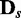 were generated, which meets our expectation.
were generated, which meets our expectation.
Figure 3.

JS divergence and P-values from applying EDDY to compare 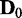 and
and 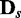 . (A)
. (A) 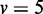 variables. (B)
variables. (B)  variables. (C)
variables. (C) 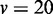 variables. (D)
variables. (D) 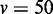 variables.
variables. 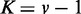 was used for all cases. Each point in a plot represents the average from 100 repetitions. Error bars indicate the 95% confidence interval of the average.
was used for all cases. Each point in a plot represents the average from 100 repetitions. Error bars indicate the 95% confidence interval of the average.
Regarding the number of dependency network structures M, using larger M gives higher JS divergence values and lower P-values as shown in Figure 3. This is because considering more dependency network structures improves the approximation of probability distributions 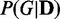 . Therefore, EDDY can distinguish two different data sets more correctly, and can recognize smaller discrepancies better. Compared with EDDY, the approach of considering only the best scoring networks suffers with low reliability. This is because many near-optimal networks can have similarly high scores in real applications, and it can lead to higher false positives.
. Therefore, EDDY can distinguish two different data sets more correctly, and can recognize smaller discrepancies better. Compared with EDDY, the approach of considering only the best scoring networks suffers with low reliability. This is because many near-optimal networks can have similarly high scores in real applications, and it can lead to higher false positives.
The effect of using more samples for the test is also evident from Figure 3. When smaller number of samples (e.g. 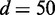 ) was used, the increase of JS and the decrease of P-values are less clear even if the discrepancy s in dependency is increased between the networks behind the data sets. However, as the available amount of samples increases (e.g. d = 200 and 500), the pattern of increasing JS and decreasing P-values becomes clearer. This indicates that the performance of EDDY in discriminating distinct data sets improves as we increase the number of available samples for a test.
) was used, the increase of JS and the decrease of P-values are less clear even if the discrepancy s in dependency is increased between the networks behind the data sets. However, as the available amount of samples increases (e.g. d = 200 and 500), the pattern of increasing JS and decreasing P-values becomes clearer. This indicates that the performance of EDDY in discriminating distinct data sets improves as we increase the number of available samples for a test.
Another observation is that more dependency network structures (larger M) may need to be considered as the problem size gets bigger (larger number of variables). From Figure 3A and B of 5 and 10 variable cases accordingly, considering. 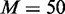 ∼1000 dependency network structures could make EDDY show properly increasing JS and decreasing P-values with increasing s. However, such patterns are not comparably clear for the case of 20 variables with the same amount of dependency network structures and sample sizes (the case of d = 200 in Figure 3C). Using more samples (the case of
∼1000 dependency network structures could make EDDY show properly increasing JS and decreasing P-values with increasing s. However, such patterns are not comparably clear for the case of 20 variables with the same amount of dependency network structures and sample sizes (the case of d = 200 in Figure 3C). Using more samples (the case of 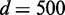 in Figure 3C) made JS and P-values more distinguishable, but increasing the amount of dependency network structures for consideration (M = 3000 and 5000) made EDDY provide much clearer pattern of varying JS and P-values. Using larger M values also produced reasonable performances for the case of 50 variables (Figure 3D). This is because the number of possible dependency network structures is significantly increased as the number of variables increases. Thus, it requires more dependency network structures to be considered for proper approximation of probability distributions
in Figure 3C) made JS and P-values more distinguishable, but increasing the amount of dependency network structures for consideration (M = 3000 and 5000) made EDDY provide much clearer pattern of varying JS and P-values. Using larger M values also produced reasonable performances for the case of 50 variables (Figure 3D). This is because the number of possible dependency network structures is significantly increased as the number of variables increases. Thus, it requires more dependency network structures to be considered for proper approximation of probability distributions 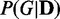 in the process of EDDY.
in the process of EDDY.
In addition to the aforementioned characteristics of JS and P-values, we also characterized how sensitive EDDY is in determining statistical significance with varying s. Of the 100 repetitions of EDDY for each comparison of 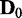 versus
versus 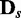 , the ratio of cases with P < 0.05 was computed. Figure 4 shows the ratio of such statistically significant cases by varying the discrepancy s. As expected from the P-value plots in Figure 3, the ratio of statistically significant cases increases with increasing network discrepancy s. Similarly to JS and P-values, increasing the number of samples or the number of dependency network structures of consideration clearly allows EDDY to identify more statistically significant cases. One observation is that EDDY identifies relatively small amount of statistically significant cases (true-positive cases) when the number of available samples or computational capacity is limited (small d or small M), or when the size of a problem is huge (large v). However, considering that statistically significant cases from the test of
, the ratio of cases with P < 0.05 was computed. Figure 4 shows the ratio of such statistically significant cases by varying the discrepancy s. As expected from the P-value plots in Figure 3, the ratio of statistically significant cases increases with increasing network discrepancy s. Similarly to JS and P-values, increasing the number of samples or the number of dependency network structures of consideration clearly allows EDDY to identify more statistically significant cases. One observation is that EDDY identifies relatively small amount of statistically significant cases (true-positive cases) when the number of available samples or computational capacity is limited (small d or small M), or when the size of a problem is huge (large v). However, considering that statistically significant cases from the test of 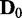 versus
versus 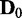 (the cases of
(the cases of 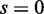 in Figure 4) correspond to false positives, it is important to note that EDDY provides low false positives regardless of conditions as nearly 0% cases are reported to be statistically significant for
in Figure 4) correspond to false positives, it is important to note that EDDY provides low false positives regardless of conditions as nearly 0% cases are reported to be statistically significant for 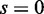 . The ratio of statistically significant cases from the tests of
. The ratio of statistically significant cases from the tests of 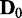 versus
versus 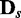 (
(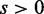 ) can be seen as sensitivity, although it needs to be considered differently with the case of conventional classification problems as the ratios in Figure 4 depends on the degree of discrepancy between conditions.
) can be seen as sensitivity, although it needs to be considered differently with the case of conventional classification problems as the ratios in Figure 4 depends on the degree of discrepancy between conditions.
Figure 4.

The ratio r that EDDY identifies 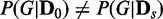 with P
with P
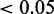 , of 100 repetitions. (A)
, of 100 repetitions. (A) 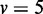 variables, (B)
variables, (B) 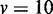 variables, (C)
variables, (C) 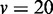 variables, and (D)
variables, and (D) 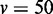 variables.
variables. 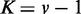 was used for all cases. Note that the cases of
was used for all cases. Note that the cases of 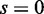 correspond to false positives as both
correspond to false positives as both 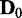 and
and 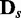 are derived from the same network, and those of
are derived from the same network, and those of 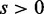 correspond to true positives.
correspond to true positives.
In summary, we have shown that the discrepancy detected by EDDY well correlates with the true dependency discrepancy behind the data sets. We were also able to observe that increasing the number of available samples or the number of dependency network structures of consideration improves performance, while requiring more computations. One important benefit of EDDY is its conservative behavior of giving low false positives; thus, we can get trustworthy results even for challenging problems.
We also evaluated the effect of using smaller K values for selected cases. Limiting the number of incoming edges to each variable restricts possible DAG structures, thus limits the ability of the algorithm to correctly approximate the probability distribution of dependency network structures. For this reason, limiting K to smaller values gives similar effects with considering less dependency network structures (smaller M) in approximating the network distribution, which results relatively lower discrepancy, higher P-values and lower calling rates of statistically significant cases (see Supplementary Figures S1 and S2). However, limiting K significantly improved the running speed of the algorithm (∼2-folds for 20 variables and 8-folds for 50 variables from our implementation), thus, helped to increase the scalability of the method. As the running time of the algorithm linearly increases with the considered amount of dependency network structures M, using practically small K with good amount of M can be a useful strategy for problems of large sizes.
Finally, we also evaluated the performance of EDDY when the topology of an original network was altered without reducing the number of edges, then used to generate a synthetic data set, as previously described. For the observed range of topology discrepancy, EDDY showed the similar behavior—increasing JS, decreasing P-values and increasing statistical significance calls, as the discrepancy in networks behind the data sets increases (see Supplementary Figures S3–S9 for more details).
Simulation II: comparison of EDDY with other methods
Figure 5 illustrates the ROC curves of GSCA, GSEA and EDDY from Simulation II, and Table 1 lists the area under curve values of the corresponding ROC curves in Figure 5.
Figure 5.

The comparison of ROC of GSCA, GSEA and EDDY in identifying differential gene sets from the interaction-focused simulation experiments. (A) 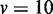 . (B)
. (B) 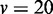 . (C)
. (C) 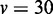 .
.
Table 1.
The area under curve values of GSCA, GSEA and EDDY in identifying differential gene sets from Simulation II
| Method | 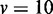 |
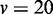 |
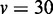 |
|---|---|---|---|
| GSCA | 0.5774 | 0.5822 | 0.5965 |
| GSEA | 0.4911 | 0.5574 | 0.6075 |
| EDDY (M = 1000) | 0.7440 | 0.6768 | 0.6704 |
| EDDY (M = 5000) | 0.8287 | 0.7580 | 0.7064 |
Bold face indicates top performance.
From the results of the interaction-focused simulation experiments (Figure 5), EDDY demonstrates superior performance than the other two methods. This is partly due to the fact that the data were generated from models assuming conditional dependencies in gene expressions rather than simple linear correlations, which is also assumed by the Bayesian network model that the current implementation of EDDY uses. The performance of EDDY declines with increasing the size of gene sets, but it improves with more computations (by using larger M as shown in Figure 5). Another observation is significantly lower false positive rates of EDDY than that of other methods (Supplementary Figure S10–S12).
This simulation study clearly indicates that EDDY significantly outperforms other methods when differential gene sets are defined in the sense of gene interactions, with significantly lower false-positive rates.
Besides the simulation scenario covered in this study, there can be various different simulation configurations depending on the methods to generate synthetic data sets and the definition of differential gene sets. However, it is not feasible to enumerate and cover all such different cases, and thus, they are left for future studies.
Comparison of EDDY with other methods in application to TCGA GBM gene expression data
Table 2 lists the number of statistically significant gene sets identified with the three different methods for each subtype. EDDY and GSEA produced different results, as EDDY identified 10 ∼ 22 gene sets for each subtype, whereas GSEA identified 245 gene sets for mesenchymal but just a few for other subtypes. Moreover, there is only one common gene set (for mesenchymal) between the results from the two methods. A possible hypothesis of GSEA identifying many gene sets only for mesenchymal is that mesenchymal is the most differentiated form of GBM (physiologically or genotypically) (27), and many genes are differentially expressed in mesenchymal compared with other subtypes. Compared with GSEA, the results of EDDY are relatively less biased to a specific subtype (for the lists of identified gene sets from EDDY and GSEA, see Supplementary Tables S1–S8).
Table 2.
The number of statistically significant gene sets for each subtype
| Method | Classical | Mesenchymal | Neural | Proneural |
|---|---|---|---|---|
| EDDY | 13 | 10 | 22 | 22 |
| GSEA | 1 (0) | 245 (1) | 6 (0) | 3 (0) |
| GSCA | 1590 (11) | 1432 (7) | 1681 (21) | 1563 (17) |
The number of common cases with EDDY is indicated in the parentheses.
Compared with the other two methods, GSCA identified much more gene sets as statistically significant, from 68 to 80% of the tested gene sets, making it almost noninformative (for the lists of identified gene sets from GSCA, see the supplementary file provided in http://biocomputing.tgen.org/software/EDDY). This becomes clearer from Figure 6, where the P-values from GSCA are much closer to 0 in general than that of EDDY and GSEA (for direct comparison of P-values from GSCA and EDDY, see Supplementary Figure S13). To better understand the result of GSCA showing too many cases of low P-values, we compared the P-values of EDDY and GSCA along with the sizes of gene sets, where both methods target differential genetic interactions. GSCA reported much lower P-values as the size of gene sets increases, while the result from EDDY did not show such bias related to the size of gene sets (Supplementary Figure S14). This characteristic of GSCA has been noted by the developers of GSCA (21), as GSCA could be sensitive to the sum of minor local correlation changes from many gene interactions of a large gene set. This can lead to GSCA reporting significantly lower P-values for larger gene sets, hence, biased results toward larger gene sets, as can be seen from the application to the TCGA GBM gene expression data (Supplementary Figure S14). We also show from the simulated comparisons in the previous subsection that GSCA reports higher false-positive rates than EDDY in general (Supplementary Figures S10–S12). Even though EDDY may be less sensitive (higher false negative) than GSCA, results from EDDY can be more informative once identified as statistically significant due to such low false positives. Moreover, the sensitivity of EDDY can be increased with more computational resources without increasing false positives, as shown from Simulation I that evaluated the characteristics of EDDY (Figure 4).
Figure 6.

Empirical cumulative distributions of P-values from applying three different gene set test methods to each subtype of TCGA GBM gene expression data. (A) From testing classical versus nonclassical. (B) From testing mesenchymal versus nonmesenchymal. (C) From testing neural versus nonneural. (D) From testing proneural versus nonproneural.
Gene sets identified with EDDY for each GBM subtype
Figure 7 illustrates which biological functions contribute to the gene sets identified with EDDY for the four subtypes of GBM and their proportions. All four GBM subtypes are associated with gene sets of biological functions that are popular in general cancer, such as cell cycle, immune system and signal transduction. Besides such cancer-generic functions, some functional groups are specifically associated with different subtypes. For the mesenchymal subtype (Figure 7B), gene sets that are related to cell migration/structure were identified. This fits well with the characteristics of the mesenchymal subtype, where cancer cells go through epithelial–mesenchymal transition and show migrative behavior. The neural subtype is exclusively associated with gene sets that are related to DNA damage repair (Figure 7C). The failure of DNA repair mechanism can lead to the formation of cancer through unregulated cell division, which can be related to cancer in general. Thus, it can be a viable next step to investigate the activities of DNA repair functions in GBM subtypes to find out the neural subtype-specific driving mechanism for the abnormality of this function. Another observation is that only the neural and proneural subtypes are associated with gene sets related to apoptosis, and the cause and effect of this abnormal activity of apoptotic gene sets in these two subtypes remain for future study.
Figure 7.
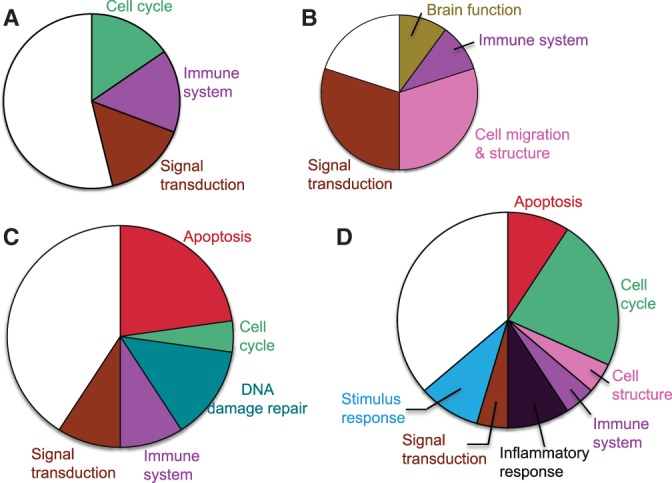
The relative amount of statistically significant gene sets identified with EDDY, based on their relevant functions. The size of each area is proportional to the number of gene sets that are related to the corresponding function. White blank area indicates the amount of gene sets with functions other than the specified functional groups. (A) Classical, (B) mesenchymal, (C) neural and (D) proneural.
CDKN2A and p53 roles in GBM revisited
We also investigated each gene set and found few cases that are consistent with the previous studies and were not identified with GSEA. From the result of applying EDDY, three selected pathway gene sets are illustrated in Figure 8, where their genetic relationships were distinct with statistical significance between a subtype sample and the rest of the samples. To present an intuitive visualization of how much difference exists regarding dependency relationships, we composed a representative dependency network of each pathway, which shows subtype-specific, nonsubtype specific (specific to the other samples than the subtype) and common dependencies. In the process of EDDY, 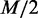 probable dependency network structures were proposed by Algorithm StructurePropose (see the ‘Materials and Methods’ section and Supplementary Method S2 for details) and collected for each case of subtype and nonsubtype samples. For each case, the frequency of edge connection from the
probable dependency network structures were proposed by Algorithm StructurePropose (see the ‘Materials and Methods’ section and Supplementary Method S2 for details) and collected for each case of subtype and nonsubtype samples. For each case, the frequency of edge connection from the 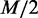 collected dependency network structures was evaluated for every gene–gene pair. If an edge existed in >95% dependency network structures, the edge was included for visualization in Figure 8. As Figure 8 shows, genes in the three pathways have many changes in dependency relationships between each corresponding subtype and nonsubtype cases. However, individual genes do not show clear differential expression between each subtype and nonsubtype cases as shown in heat maps of Figure 8. This implies that the dependency relationships between genes can be significantly different across conditions even when the overall expression of individual genes does not show clear differential patterns. Thus, using EDDY can be a better approach than using differential gene expression-based methods (e.g. GSEA) for such cases. Figure 8A shows the gene set for the ARF tumor suppressor pathway, identified from testing classical versus nonclassical. This is related to the findings of a previous study, where Verhaak et al. (27) reported that there is focal 9p21.3 homozygous deletion targeting CDKN2A in the classical subtype, and subsequently, RB pathway is almost exclusively affected through the CDKN2A deletion. This relationship between CDKN2A deletion and RB protein in the classical subtype can be seen in Figure 8A, where CDKN2A lost many dependency relationships with other genes in the classical subtype (dotted edges) possibly due to its deletion, while the only dependency relationship it acquired in the classical subtype is with RB1. Figure 8B illustrates the G2 pathway gene set and Figure 8C shows the p53 pathway gene set, where both were identified from testing proneural versus nonproneural. We found that these two cases can be related to the p53 mutation enrichment in the proneural subtype, which was also reported by Verhaak et al. (27). From these two pathway gene sets, p53 lost all of its dependencies with other genes in the proneural subtype, possibly due to its mutation. None of these three cases was identified using GSEA.
collected dependency network structures was evaluated for every gene–gene pair. If an edge existed in >95% dependency network structures, the edge was included for visualization in Figure 8. As Figure 8 shows, genes in the three pathways have many changes in dependency relationships between each corresponding subtype and nonsubtype cases. However, individual genes do not show clear differential expression between each subtype and nonsubtype cases as shown in heat maps of Figure 8. This implies that the dependency relationships between genes can be significantly different across conditions even when the overall expression of individual genes does not show clear differential patterns. Thus, using EDDY can be a better approach than using differential gene expression-based methods (e.g. GSEA) for such cases. Figure 8A shows the gene set for the ARF tumor suppressor pathway, identified from testing classical versus nonclassical. This is related to the findings of a previous study, where Verhaak et al. (27) reported that there is focal 9p21.3 homozygous deletion targeting CDKN2A in the classical subtype, and subsequently, RB pathway is almost exclusively affected through the CDKN2A deletion. This relationship between CDKN2A deletion and RB protein in the classical subtype can be seen in Figure 8A, where CDKN2A lost many dependency relationships with other genes in the classical subtype (dotted edges) possibly due to its deletion, while the only dependency relationship it acquired in the classical subtype is with RB1. Figure 8B illustrates the G2 pathway gene set and Figure 8C shows the p53 pathway gene set, where both were identified from testing proneural versus nonproneural. We found that these two cases can be related to the p53 mutation enrichment in the proneural subtype, which was also reported by Verhaak et al. (27). From these two pathway gene sets, p53 lost all of its dependencies with other genes in the proneural subtype, possibly due to its mutation. None of these three cases was identified using GSEA.
Figure 8.
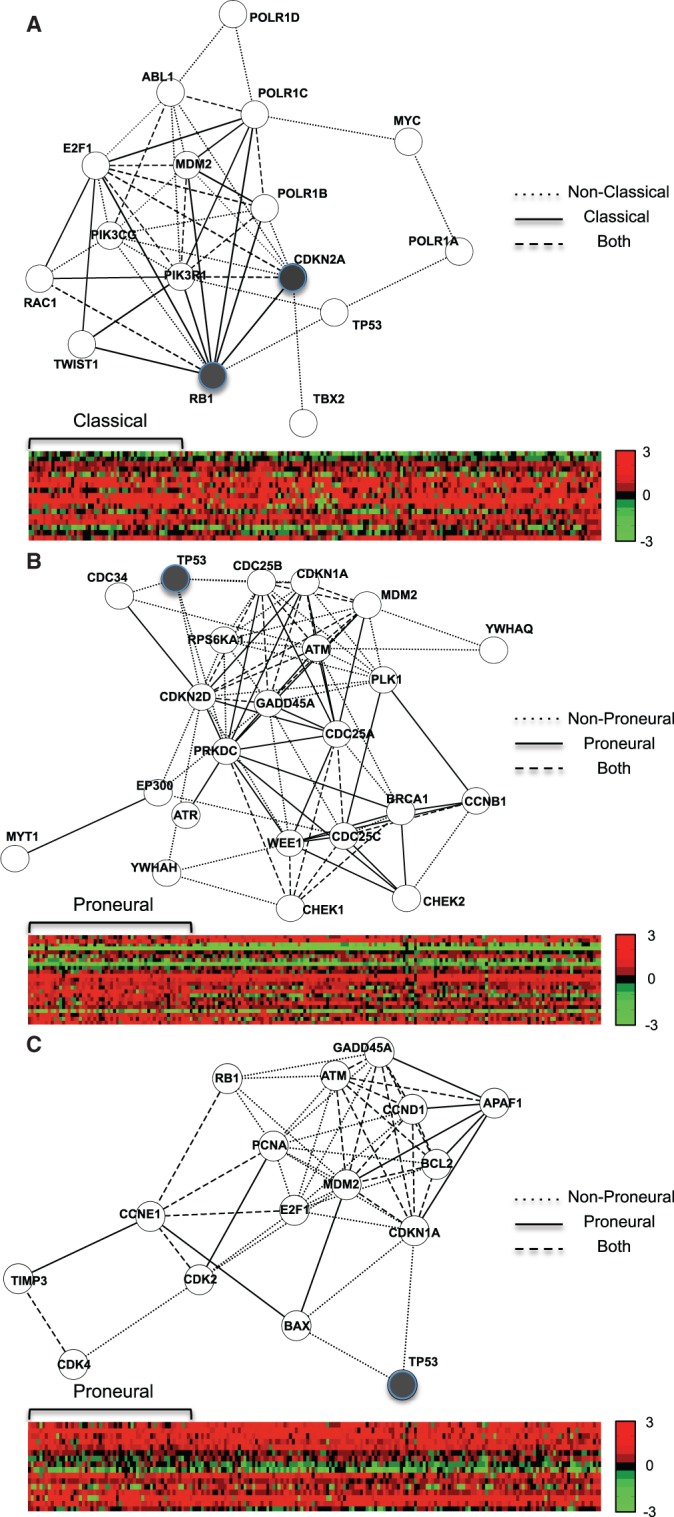
Selected pathway gene sets identified using EDDY. Each network is a visualization of representative dependency relationships. Dependency relationships only in nonsubtype are represented with dotted edges. Dependency relationships only in each subtype are represented with solid edges. Dependency relationships observed for both of subtype and nonsubtype were denoted with dashed edges. Each heat map shows the log-ratio of expressions compared with the average from normal samples. (red: higher-expression than normal, green: lower-expression than normal) (A) ARF pathway from classical versus nonclassical. (B) G2 pathway from proneural versus nonproneural. (C) p53 pathway from proneural versus nonproneural.
DISCUSSION
We proposed a method, EDDY, which is a statistical test method for a given gene set to evaluate the differentiality of dependencies between two conditions and its statistical significance. Unlike previous gene set test methods that evaluate only differential expressions, EDDY evaluates the discrepancy between conditions by considering the probability distribution of dependency networks. Compared with the previous methods to identify local differential interactions or condition-specific subnetworks, EDDY distinguishes itself with its functionality of testing gene sets for dependency differentiality, while those methods lack the functionality of testing gene sets. The proposed method has been evaluated through simulation experiments and it demonstrated that EDDY provides well-correlated results with the true discrepancy behind the synthetic data sets, while returning low false positives. When EDDY was compared with other methods through simulation studies, EDDY showed better performance than other methods when differential gene sets are defined in the sense of conditional dependency changes. We also applied EDDY to the TCGA GBM gene expression data to identify gene sets that show statistically distinct genetic relationships among the four subtypes of GBM, and its result was compared with the result of GSEA. We showed that EDDY can identify largely different gene sets with those from GSEA, while providing meaningful outcomes that are often consistent with previously reported findings in addition to potentially novel findings. EDDY has been also compared with GSCA, which is a gene set test method that evaluates differential interactions within a gene set using pairwise correlation measures. From the application to the real data set, GSCA showed biased results with the size of gene sets, while EDDY did not show such a behavior. From the extent of our knowledge, EDDY is the only method to identify gene sets with statistically significant changes in genetic interactions without the risk of getting high false positives.
A potential bottleneck of the proposed method is that it requires a significant amount of computational resources, especially for large gene sets. The necessity of such huge computation is mainly because large number of dependency network structures needs to be evaluated as the size of a gene set is increased. For this reason, we used heuristics to limit incoming edges to each gene and filtered genes based on expression variance to further reduce the computational cost in real applications. Furthermore, we are also planning our future work to develop a naive version of EDDY, which tests the relationship between each gene–gene pair independently across conditions and aggregate the statistics for a final result. Additionally, only two conditions were considered for the application of EDDY in this work. However, it is possible to extend the algorithm for more than two conditions, as the JS divergence can be extended for more than two discrete probability distributions, and such extension will be part of our future work.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute, National Institutes of Health (NIH) [29XS195, 1U01CA168397-01]. Funding for open access charge: NIH [1U01CA168397-01].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Michael Berens and Dr Jeff Kiefer for assistance in interpreting the result of biological applications. Access to high-performance computing facilities granted by Translational Genomics Research Institute is gratefully acknowledged. The content of this article does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
REFERENCES
- 1.Pan W. A comparative review of statistical methods for discovering differentially expressed genes in replicated microarray experiments. Bioinformatics. 2002;18:546–554. doi: 10.1093/bioinformatics/18.4.546. [DOI] [PubMed] [Google Scholar]
- 2.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma S, Kosorok MR. Identification of differential gene pathways with principal component analysis. Bioinformatics. 2009;25:882–889. doi: 10.1093/bioinformatics/btp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shojaie A, Michailidis G. Analysis of gene sets based on the underlying regulatory network. J. Comput. Biol. 2009;16:407–426. doi: 10.1089/cmb.2008.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Califano A. Rewiring makes the difference. Mol. Syst. Biol. 2011;7:463. doi: 10.1038/msb.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi JK, Yu U, Yoo OJ, Kim S. Differential coexpression analysis using microarray data and its application to human cancer. Bioinformatics. 2005;21:4348–4355. doi: 10.1093/bioinformatics/bti722. [DOI] [PubMed] [Google Scholar]
- 7.Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T. Conserved patterns of protein interaction in multiple species. Proc. Natl Acad. Sci. USA. 2005;102:1974–1979. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tischler J, Lehner B, Fraser AG. Evolutionary plasticity of genetic interaction networks. Nat. Genet. 2008;40:390–391. doi: 10.1038/ng.114. [DOI] [PubMed] [Google Scholar]
- 9.Gholami AM, Fellenberg K. Cross-species common regulatory network inference without requirement for prior gene affiliation. Bioinformatics. 2010;26:1082–1090. doi: 10.1093/bioinformatics/btq096. [DOI] [PubMed] [Google Scholar]
- 10.Lai Y, Wu B, Chen L, Zhao H. A statistical method for identifying differential gene–gene co-expression patterns. Bioinformatics. 2004;20:3146–3155. doi: 10.1093/bioinformatics/bth379. [DOI] [PubMed] [Google Scholar]
- 11.Hu R, Qiu X, Glazko G, Klebanov L, Yakovlev A. Detecting intergene correlation changes in microarray analysis: a new approach to gene selection. BMC Bioinformatics. 2009;10:20. doi: 10.1186/1471-2105-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mentzen W, Floris M, de la Fuente A. Dissecting the dynamics of dysregulation of cellular processes in mouse mammary gland tumor. BMC Genomics. 2009;10:601. doi: 10.1186/1471-2164-10-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonardson AS, Zhu J, Chen Y, Wang K, Lamb JR, Reitman M, Emilsson V, Schadt EE. The effect of food intake on gene expression in human peripheral blood. Hum. Mol. Genet. 2010;19:159–169. doi: 10.1093/hmg/ddp476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Z, Wang L, Li Y, Gong X, Yao C, Ma W, Wang D, Li Y, Zhu J, Zhang M, et al. Edge-based scoring and searching method for identifying condition-responsive protein–protein interaction sub-network. Bioinformatics. 2007;23:2121–2128. doi: 10.1093/bioinformatics/btm294. [DOI] [PubMed] [Google Scholar]
- 15.Hwang T, Park T. Identification of differentially expressed subnetworks based on multivariate anova. BMC Bioinformatics. 2009;10:128. doi: 10.1186/1471-2105-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim Y, Kim TK, Kim Y, Yoo J, You S, Lee I, Carlson G, Hood L, Choi S, Hwang D. Principal network analysis: identification of subnetworks representing major dynamics using gene expression data. Bioinformatics. 2011;27:391–398. doi: 10.1093/bioinformatics/btq670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma H, Schadt EE, Kaplan LM, Zhao H. Cosine: condition-specific sub-network identification using a global optimization method. Bioinformatics. 2011;27:1290–1298. doi: 10.1093/bioinformatics/btr136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Li H, Riggins RB, Zhan M, Xuan J, Zhang Z, Hoffman EP, Clarke R, Wang Y. Differential dependency network analysis to identify condition-specific topological changes in biological networks. Bioinformatics. 2009;25:526–532. doi: 10.1093/bioinformatics/btn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B, Tian Y, Jin L, Li H, Shih IeM, Madhavan S, Clarke R, Hoffman EP, Xuan J, Hilakivi-Clarke L, et al. Ddn: a cabig® analytical tool for differential network analysis. Bioinformatics. 2011;27:1036–1038. doi: 10.1093/bioinformatics/btr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ouyang Z, Song M, Güth R, Ha TJ, Larouche M, Goldowitz D. Conserved and differential gene interactions in dynamical biological systems. Bioinformatics. 2011;27:2851–2858. doi: 10.1093/bioinformatics/btr472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi YJ, Kendziorski C. Statistical methods for gene set co-expression analysis. Bioinformatics. 2009;25:2780–2786. doi: 10.1093/bioinformatics/btp502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buntine W. Proceedings of the Seventh Conference on Uncertainty in Artificial Intelligence. San Mateo, CA: Morgan Kaufmann Publishers; 1991. Theory refinement on bayesian networks; pp. 52–60. [Google Scholar]
- 23.Lin J. Divergence measures based on the shannon entropy. IEEE Trans. Inform. Theory. 1991;37:145–151. [Google Scholar]
- 24.Song S, Black M. Microarray-based gene set analysis: a comparison of current methods. BMC Bioinformatics. 2008;9:502. doi: 10.1186/1471-2105-9-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fridley BL, Jenkins GD, Biernacka JM. Self-contained gene-set analysis of expression data: an evaluation of existing and novel methods. PLoS One. 2010;5:e12693. doi: 10.1371/journal.pone.0012693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maciejewski H. Gene set analysis methods: statistical models and methodological differences. Brief. Bioinformatics. 2013 doi: 10.1093/bib/bbt002. (epub ahead of print, doi: 10.1093/bib/bbt002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in pdgfra, idh1, egfr, and nf1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.