

Abstract
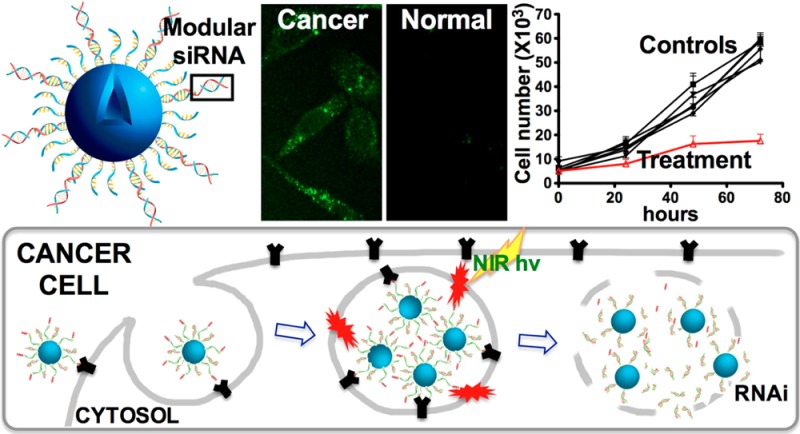
We have combined a versatile and powerful route to deliver nucleic acids with peptide-based cell-specific targeting. siRNA targeting the polo-like kinase gene is in clinical trials for cancer treatment, and here we deliver this RNA selectively to cancer cells displaying the neuropilin-1 epitope using gold nanoshells. Release of the siRNA from the nanoparticles results from irradiation with a pulsed near-infrared laser, which also provides efficient endosomal escape within the cell. As a result, our approach requires 10-fold less material than standard nucleic acid transduction materials and is significantly more efficient than other particle-based methods. We also describe a particle–nucleic acid design that does not rely on modified RNA, thereby making the preparation of these materials more efficient and much less expensive. These improvements, when combined with control over when and where the siRNA is released, could provide the basis for diverse cell biological studies.
Keywords: Hollow gold nanoshells, siRNA delivery, near-infrared laser, targeted, modular design, endosome escape
Since its discovery in 1998,1 RNA interference (RNAi) has been recognized for its potential to control the flow of genetic information. Much effort has been invested in the promise of therapeutic applications of gene silencing, yet the routine use of RNAi for disease treatment or prevention still calls for novel methods of delivery with spatial and temporal control.2,3 Current techniques in nucleic acid delivery include viral vectors and lipid vesicles, which have enhanced both biostability and bioavailability.3−11 One of the most difficult hurdles to overcome for RNAi using nonviral vectors is endosomal escape into the cytosol, which is necessary for gene silencing to occur.12−16 siRNA is prevented from reaching the site of action due to encapsulation in endosomes, and eventually the siRNA is vulnerable to degradation enzymes in late endosomes or expelled from the cell by exocytosis.12−14,17−20 The efficacy of siRNA delivery is largely governed by the ease and rapidity of the escape from endolysosomes.21 We recently developed a highly efficient approach to release siRNA from both the carrier and endosome by using hollow gold nanoshells (HGNs) as carriers and near-infrared (NIR) light as the release activator.11,42 The great advantage of using NIR light to induce release is that cells in culture, as well as tissue, blood, and so forth are relatively transparent to 650–900 nm wavelength light, allowing NIR transmission in soft tissues at depths up to 10 cm.22,23 Laser illumination at 800 nm resonant with the nanoparticle plasmon triggers the disassembly of the engineered surface-coat, releasing the drug molecules by thermalizing the gold–thiol bond. Escape of the drug from the endosomes occurs by localized cavitation that physically breaches the vesicles’ membrane11,24 but does not affect the integrity of the cell plasma membrane. Here, we delivered therapeutic siRNA specifically to cancer cells through the use of a cancer-cell specific targeting peptide. Peptides provide advantages over other targeting approaches (e.g., antibodies) due to their small size, synthetic versatility, and cell and tissue specificity.25−27 We targeted the polo-like kinase (PLK1) expressed by the plk1 gene in prostate cancer cells, critical to centrosome maturation during mitosis, to induce a pro-apoptotic pathway.28 RNAi-based plk1 gene down-regulation forms the basis of phase I/II clinical trials for the treatment of solid tumors.29 We also improve on HGN delivery of siRNA by developing a more versatile and modular nucleic acid architecture that does not require thiolated RNA, increasing the sequence space that may be explored. These novel architectural features are likely to have significant impact on the biomedical use of nanoparticles and RNAi for spatially patterned or cell selective gene expression.
Assembly and Characterization of the Delivery Vehicle
We designed a scheme for attaching peptides to the outside of a thiolated nucleic acid monolayer that can be expanded to include a variety of RNA cargo (Figure 1B). Multiple copies of siRNA against plk1 were conjugated to the surface of the ∼50 nm HGN either directly or supported by a single strand DNA linker through a quasi-covalent (Au–S) bond, simultaneously maximizing drug content, stability, and linkers for multivalent presentation of peptides (Figure 1B). We employed a peptide following the C-end rule (CendR), RPARPAR (RP),30 that binds specifically to the neuropilin-1 (NRP-1) receptor overexpressed by certain types of cancer cells. The siRNA and RP coated HGNs internalize into endosomes (Figure 1C). Upon pulsed NIR laser irradiation, the Au–S bond linking the siRNA to the HGN surface is ablated while the conversion of light energy to heat produces a transient vapor bubble that ruptures the endosome without damaging the siRNA or the cell, releasing the cargo into the cytosol (Figure 1C). This combination of steps results in highly efficient transfer of siRNA with specificity from the both the targeting peptide and the laser irradiation to ensure RNAi function occurs only in the doubly targeted cells (see below).
Figure 1.
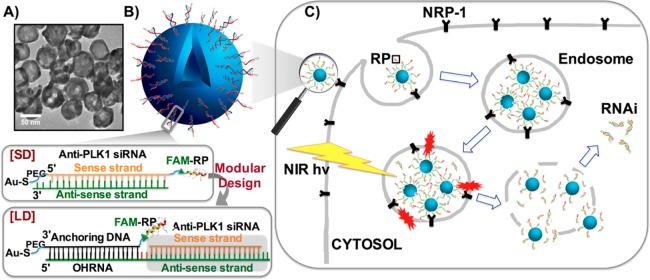
HGN-siRNA-RP. Hollow gold nanoshell (HGN)-siRNA-targeting peptide (RP) architecture with original or modular design and schematic of nanoparticle uptake, laser-activated siRNA delivery pathway in PPC-1 cells. (A) Transmission electron microscopy image of HGN showing the hollow center surrounded by a higher contrast rim. (B) Schematic of the original HGN-SD-RP architecture and second-generation architecture for modular HGN-LD-RP to achieve versatile siRNA assembly. A fluorescein fluorescent label is added to both SD and LD to help quantify delivery. (C) Schematic of nanoparticle uptake and laser-activated siRNA delivery pathway. Endocytosis is promoted by the RP, RPARPAR. Femtosecond pulses of near-IR light separate the nucleic acids (either “short” duplex (SD) or “long” duplex (LD) in B) from the HGN at the Au–S bond, followed by vapor bubble formation as the light energy is converted to heat. The vapor bubbles burst the endosome, releasing the siRNA to the cytosol, allowing for gene silencing.
To prepare the siRNA functionalized nanoparticles (HGN-SD-RP, Figure 1B) for gene knockdown, siRNA sense strands containing 5′ thiol modifications were assembled onto the citrate-passivated HGN surface using a low pH-induced self-assembly method.34 RNA strands were added to HGNs in low pH (3.0) sodium citrate buffer to neutralize the negatively charged phosphate backbone and maximize the assembly rate. Antisense RNA strands were then hybridized to the sense RNAs, resulting in ∼2300 ± 600 siRNA duplexes per particle (Supporting Information Figures S2A and S3). The residual HGN surface was then passivated with thiol-polyethylene glycol (PEG)-amine (3 kDa) and 6-mercapto-1-hexanol (MCH). The RP peptide, carrying a fluorescein dye (FAM) for tracking and characterization as well as a cysteine for conjugation, was incorporated to the HGN-siRNA via short PEG linkers to bridge the amine on the 3′ sense strand and the thiol (cysteine) on the peptide to enable cancer cell-specific targeting (Supporting Information Figure S2A). Initially the citrate HGN showed a resonance at ∼710 nm, which red-shifted to ∼810 nm as the layers were assembled (Supporting Information Figure S2C).
Irradiation of the final product HGN-SD-RP with pulsed laser light (1 kHz repetition rate, ∼120 fs pulse length) at 800 nm at a laser power density of 2.4 W/cm2 for 10 s released ∼85% of total siRNA (Supporting Information Figure S5A, 100% release achieved by HGN dissolution using KCN). The absorbed energy caused the Au–S bonds to be cleaved by hot electrons and the subsequent heating of the Au lattice.11,35 Importantly, the siRNA remained hybridized and biochemically active after the laser release (Supporting Information Figure S5B).
Endosomal Escape of siRNA
The down-regulation effect of plk1-siRNA released from HGNs was tested using the epithelial prostate cancer cell line PPC-1, which overexpresses the NRP-1 receptor targeted by the RP peptide. Western blot and cell viability assays were used to assess and quantify down-regulation of plk1 (Figure 2). Irradiation with the femtosecond-pulsed NIR laser (2.4 W/cm2 for 10 s) of cells exposed to HGN-SD-RP led to 70% loss of cell viability at 72 h (Figure 2A, sample H), similar to the effect of unconjugated siRNA transfected using commercial Lipofectamine RNAiMAX (Lipo). The Lipo treatment required ∼13 fold more siRNA (0.37 nM siRNA compared with 5 nM siRNA for 5000 cells) (Figure 2A, sample G). Like other nanoparticle approaches that also require considerably more siRNA, Lipo lacks an efficient escape mechanism from endosomes. Cells exposed to the same laser conditions in the absence of HGN-SD-RP showed no loss of viability (Figure 2A, sample B). HGN-siRNA-Q-RP (where Q indicates Quasar570 dye, placed here on 5′ antisense RNA, which blocked siRNA activity against plk1, Supporting Information Figure S6) and HGN-dsDNA25bp-RP (loaded nonfunctional dsDNA in place of the siRNA) were employed as negative controls and were also used to test whether the local heating caused by NIR laser irradiation in the presence of HGNs, induced cellular damage (Figure 2A, samples E,F). The targeting peptide was necessary for the internalization of nanoparticles into PPC-1 cells, as laser treatment of cells exposed to HGN-siRNA without RP did not show any knockdown or cell death (Figure 2A, sample C). Figure 2A shows that the HGN-SD-RP was the only construct to cause significant loss of cell viability upon laser exposure. Cells exposed to HGN-SD-RP but not to the NIR laser showed no effect, demonstrating that laser irradiation was required to activate the biological response in the cells, providing for both spatial and temporal control of knockdown.11 The minimum laser exposure power and time for effective siRNA release in cells was 2.4 W/cm2 for 10 s (Supporting Information Figure S7). Western blot analysis confirmed the down-regulated plk1 expression level in PPC-1 cells treated with HGN-SD-RP and NIR laser after 48 and 72 h. We observed a ∼60–70% decrease of the PLK1/β-actin band intensity in Figure 2B for lanes G and H compared to untreated cells (lane A) and to cells carrying HGN-SD-RP but not irradiated by the laser (lane D). This result likely underestimated the knockdown, as only live cells were used for the Western analysis.
Figure 2.
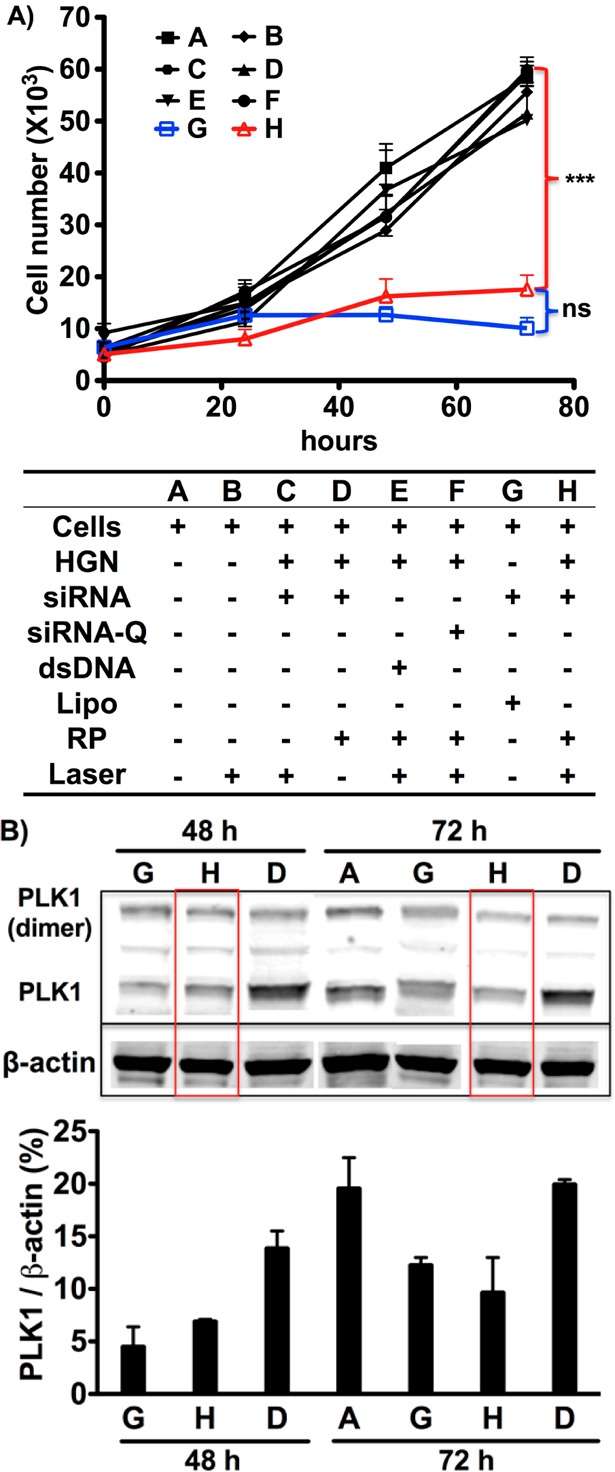
Functional plk1-siRNA released from HGN-SD-RP by NIR-laser (2.4 W/cm2 for 10 s) leads to loss of PPC-1 cell viability and down-regulation of PLK1 protein levels. (A) NIR-laser treatment of PPC-1 cells having internalized HGN-SD-RP (H) causes a significant decrease of cell viability similar to the effect of lipofectamine (G) but at much lower RNAi concentration. A series of controls (defined in the text) are shown in the table underneath the growth curve. ***, p < 0.001; ns, not significant. (B) Western blot analysis showing knockdown of plk1 gene expression in PPC-1 cells. Red boxes highlight the down-regulated expression of plk1 in cells with laser-released siRNA from HGN-SD-RP. The column graph underneath shows the band intensity ratio of PLK1 to β-actin in Western blot image. The HGN-SD-RP provided the similar level of plk1 knockdown as lipofectamine.
FAM-labeled HGN-SD-RP was readily internalized into PPC-1 cells (Figure 3A). The siRNA release was assessed using both fluorescence confocal microscopy and flow cytometry. The FAM label on the HGN-SD-RP is ∼50% quenched at distances of ∼10 nm or less from the gold; when the SD-RP is released by the laser, the fluorescence roughly doubles in intensity.36 Femtosecond pulsed laser irradiation caused both a significant increase and expansion of FAM fluorescence (from individual puncta to more uniform, diffuse) in the cytosol of each cell, indicative of the release of siRNA-RP in individual cells as shown by confocal microscopy (Figure 3A–C). Regions of interest were selected, enclosing either single cells (Regions 2, 3, and 4 in Figure 3A,B) or the glass slide background (Region Blank 1) to conduct pixel intensity analysis (Figure 3D). Forty to sixty percent of the pixels in cells showed greater than 1-fold increase in intensity, whereas almost 100% of the pixels in the Blank 1 region showed no fluorescence increase (ratio ∼ 1) following laser treatment. Flow cytometry showed an increase in average intensity (Figure 3E). We noted that ∼30% of the PPC-1 cell population after HGN-SD-RP internalization showed a significant fluorescence signal, defined by being above the brightest 1% of the unlabeled control cells. The percentage increased to ∼70% after laser irradiation (Figure 3E) due to a 2.5-fold increase in mean cell intensity that was consistent with maximal release of the SD-RP from the HGN. This also suggests that at least 70% of the cells efficiently internalized HGN-SD-RP.
Figure 3.

NIR laser-activated release of siRNA from HGN-SD-RP in PPC-1 cells demonstrated by confocal fluorescence microscopy and flow cytometry. (A) Fluorescence microscopy (FAM channel) of HGN-SD-RP taken up by PPC-1 cells. HGNs are recognizable as bright dots associated with each cell mostly collecting around the perinuclear area. (B) The same area of the cell monolayer shown in (A) after NIR pulsed laser irradiation efficiently releases HGN cargo. (C) Difference map of (A,B). Prelaser and postlaser images are overlaid and color scale chosen such that purple indicates an increase, green a decrease, and gray scale no change in pixel intensity. Scale bar is 50 μm. (D) Fluorescence intensity ratios of (B) to (A) for pixels in the selected four red regions. Blank 1 is the glass slide background outside the cells. Region 2, 3, and 4 are drawn along the edge of the single cell outline, respectively. Forty to sixty percent of pixels in regions 2, 3, and 4 have greater fold-increase in intensity than Blank 1 (ratio ∼ 1), while the remaining area where pixels show no increase are predominately nuclear regions. The expansion of bright pixels in perinuclear area postlaser suggests endosome release caused by laser irradiation. (E) PPC-1 cells were incubated with HGN-SD-RP then cell fluorescence intensity was assessed by flow cytometry before (red) and after (blue) pulsed NIR-laser treatment compared to fully untreated (no HGN-SD-RP, no laser) PPC-1 (black). Bars indicate the percentage of cells within the range of fluorescence intensity. Approximately seventy percent of the cell population showed a significantly enhanced fluorescence signal after particle internalization and laser release, defined by being above the brightest 1% of the unlabeled control cells.
The minimum HGN-SD-RP dosage used for efficient siRNA knockdown and cell death (Supporting Information Figure S8) was 6.5 pM nanoparticles carrying 15 nM siRNA for 2 × 105 cells (∼4000 nanoparticles per cell). The reported concentration represented the amount of HGN-SD-RP available per cell; the amount internalized was likely lower. Even so, the concentration was orders of magnitude less than other nanoparticle approaches that required 106–107 nanoparticles or more per cell to get effective knockdown of the respective genes.31−33 Meanwhile, the plk1-siRNA dosage needed for maximum cell viability loss by this method was ∼10 times less than other reported plk1-siRNA delivery methods.28,37−41 Lower particle and siRNA concentrations likely resulted from the combination of efficient particle internalization due to peptide targeting and endocytosis and efficient siRNA escape from the endosomes via vapor bubble formation and endosome rupture.
Prostate Cancer Cell-Specific Targeting
Normal human prostate epithelial RWPE-1 cells lack the NRP-1 receptor on the cell surface, resulting in negligible HGN-SD-RP internalization (Figure 4A). By flow cytometry only ∼1% RWPE-1 cells were above threshold fluorescence intensity, compared to 30% of PPC-1, which express NRP-1 (Figure 4B). As expected, there was no down-regulation of plk1 or loss of cell viability on laser treatment of RWPE-1 cells (Figure 4C). However, RWPE-1 cells were sensitive to plk1-siRNA, as shown using the nonselective Lipo transfection of the siRNA construct.
Figure 4.
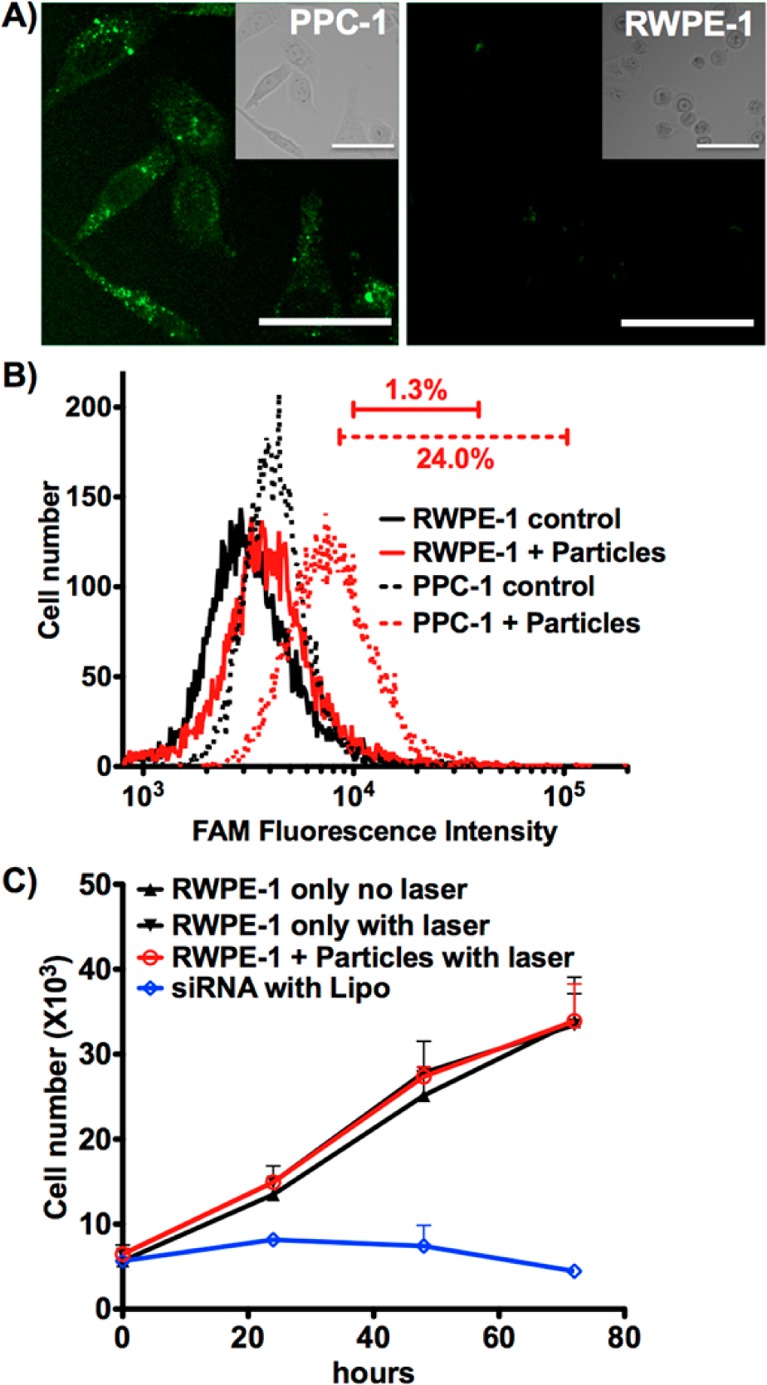
Targeted delivery of HGN-SD-RP. (A) HGN-SD-RP are internalized into PPC-1 cells but not in RWPE-1. PPC-1 cells show punctate fluorescence from FAM on HGN-SD-RP. RWPE-1 cells exposed to the same concentration of nanoparticles show none. Upper right inset, bright field; scale bars, 50 μm. (B) Flow cytometry assessment of PPC-1 and RWPE-1 cells fluorescence. Control indicates cells only. Cells + Particles indicates cells that were incubated with HGN-SD-RP. Only PPC-1 showed uptake with bars indicating the percent of cells within the range of fluorescence. (C) siRNA delivered to RWPE-1 cells by Lipo induces decreased viability, indicating susceptibility to plk1 knockdown. RWPE-1 cells incubated with HGN-SD-RP following laser irradiation do not show any decrease in viability compared to untreated controls, likely due to lack of HGN-SD-RP internalization. Cells were plated into 96-well plates for cell viability assay at 24, 48, and 72 h post laser release.
Modular RNA Assembly
The HGN-SD-RP (Figure 1B) relies on a thiolated RNA to attach to the SD-RP to the HGN, similar to other nanoparticle-based approaches.11,31−33 After validating the release efficiency, the targeting peptide specificity, and the siRNA function in knocking down plk1 expression, we created a new tethering molecule that could integrate all the above functionalities into a versatile modular architecture with greater flexibility toward delivering an generic siRNA cargo (HGN-LD-RP, Figure 1B) with the possibility of other peptide targeting sequences. Our goal was to develop a universal HGN-DNA assembly requiring only the addition of nonmodified (less expensive and more readily available) RNA. An anchoring thiol-DNA-amine strand was assembled on the HGN and later conjugated to the targeting peptide, as a replacement for the anchoring by thiol-RNA-amine. This core module was hybridized to a siRNA precursor designed with an overhang on its antisense strand complementary to the anchor sequence (OHRNA) (Figure 1B). We tested a variety of siRNA orientations and dye labels and positions keeping a constant OHRNA sequence (Supporting Information Figure S6A, structures E–G).
We compared the gene knockdown activity of the various combinations to conventional lipofectamine transfections. The 50 bp DNA-RNA “long” duplex (LD) was nearly as effective as the short siRNA (Supporting Information Figure S6B). Fluorescence-based quantification after KCN release of HGN-LD-RP showed that the number of larger LD strands per particle was ∼60% of the number of the smaller HGN-SD-RP strands (Supporting Information Figure S4). The down-regulation activity of the HGN-LD-RP was assessed on PPC-1 cells as with the HGN-SD-RP siRNA system. Cell exposure to the laser at 2.4 W/cm2 for 10 s resulted in 46% reduction of cell viability at 72 h in comparison with 70% from the HGN-SD-RP and from the same LD construct transfected by Lipo (Figure 5B). Cell uptake efficiency and cargo release were also evaluated by flow cytometry. Approximately 19% of the PPC-1 cells incubated with HGN-LD-RP show fluorescence intensity above threshold before laser irradiation. The percentage increased to ∼34% after laser treatment (Figure 5A). This suggests that although the LD construct retains the cell targeting ability, improvements may be necessary to increase the efficiency of cell uptake by modifying the peptide conjugation strategy, optimizing the overhang sequence, or tuning the hybridization conditions to increase the cargo density.
Figure 5.
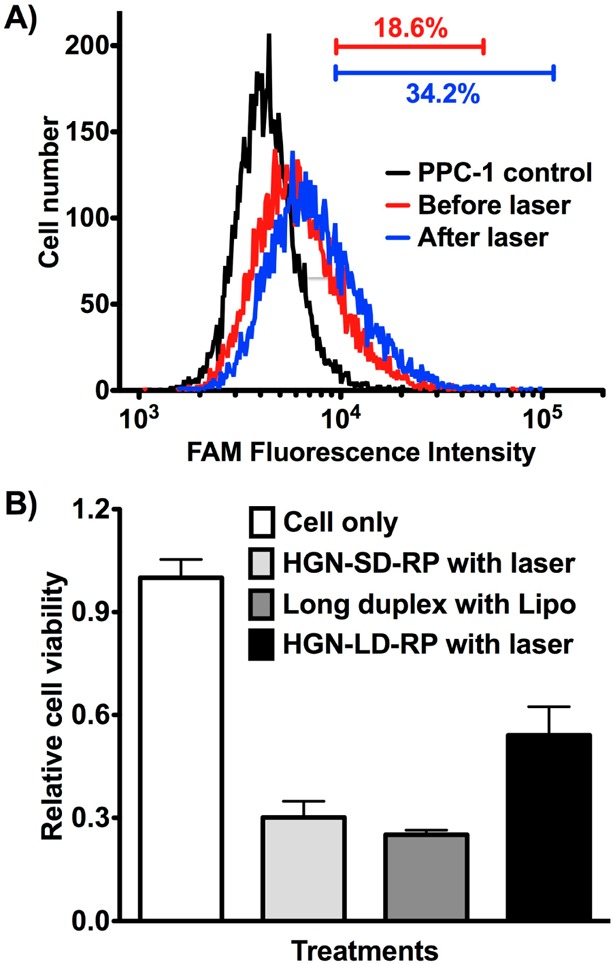
Cell uptake and knockdown efficiency assessment of the nanocarrier with new modular design (HGN-LD-RP). (A) HGN-LD-RP internalization detected by flow cytometry analysis of PPC-1 cells after incubation with HGN-LD-RP and laser treatment, as shown by the shift of the FAM intensity peak from the cell only control. Release is indicated by the intensity shift (compare blue and red). Bars indicate the percent of cells within the range of fluorescence intensity. (B) PPC-1 cell viability assay 72 h after laser treatment (2.4 W/cm2 for 10 s) indicates siRNA release and RNAi from modular HGN-LD-RP. Cell viability is expressed relative to untreated cells (cell only).
In summary, we have successfully developed two modular plasmonic siRNA nanocarriers coupled to hollow gold nanoshells for RNAi-mediated gene knockdown. The construct is engineered to specifically target cancer cells using the RPARPAR ligand against overexpressed NRP-1 receptor on the PPC-1 prostate cancer cell surface to promote cell internalization via endocytosis. The siRNA is released from the HGN and the endosome by femtosecond pulses of near-infrared light at 800 nm; the light energy is converted into heat, which thermalizes the thiol bonds holding the siRNA to the HGN, followed by vapor bubble formation that ruptures the endosomes without damaging the siRNA or the cell. This combination can provide cargo delivery with cellular level resolution at the desired time11,42 with ten times less siRNA than techniques with comparable transfection efficiency. Our results show minimum off-target toxicity as evidenced by the lack of cell viability changes with RWPE-1, which does not express NRP-1. Control of laser irradiation provides an additional targeting effect, as PPC-1 cells were also unaffected in the absence of laser treatment. Our approach shows high selectivity and RNAi delivery with high efficiency, versatility, and reduced cost. In particular, the modular design we introduce here provides a basis for future applications requiring only the annealing of unmodified siRNA precursor to previously prepared and generic HGN-DNA carriers. The combination of NIR laser-based release and endosomal escape, targeting peptide induced cell-specific internalization, and a versatile siRNA loading strategy substantially improves on our ability to use nanoparticles to target RNAi to specific cell types and even individual cells and furthers the possibility of using modular constructs for RNAi screening assays and for in vivo cancer therapy.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant R01 EB012637. The authors thank Erkki Ruoslahti, Renwei Chen, and Martin Moskovits for cell lines and helpful discussion and also thank Mary Raven’s help with two-photon microscopy, supported by NIH Grant S10RR022585. X.H. acknowledges support from California Institute for Regenerative Medicine (CIRM) Grant TG2-01151 and Chinese Scholarship Council (CSC) file number 2011674001. G.B.B. acknowledges support from the NIH (R01 CA 152327, T32 CA 121949).
Supporting Information Available
Materials and methods, particle characterization, and laser power and particle dosage optimization. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fire A.; Xu S. Q.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. Nature 1998, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Gartel A. L.; Kandel E. S. Biomol. Eng. 2006, 23, 17–34. [DOI] [PubMed] [Google Scholar]
- Stegh A. H. Integr. Biol. 2013, 5, 48–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi N. L.; Giljohann D. A.; Thaxton C. S.; Lytton-Jean A. K. R.; Han M. S.; Mirkin C. A. Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- Giljohann D. A., Seferos D. S., Prigodich A. E., Patel P. C., Mirkin C. A.. J. Am. Chem. Soc. 2009, 131, 2072-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvizo R.; Bhattacharya R.; Mukherjee P. Expert Opin. Drug Delivery 2010, 7, 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huschka R.; Barhoumi A.; Liu Q.; Roth J. A.; Ji L.; Halas N. J. ACS Nano 2012, 6, 7681–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon M. P.; Zhou M.; Li C. Acc. Chem. Res. 2011, 44, 947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z. Y.; Ji C. W.; Shi J. J.; Pridgen E. M.; Frieder J.; Wu J.; Farokhzad O. C. Angew. Chem., Int. Ed. 2012, 51, 11853–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. E.; Zuckerman J. E.; Choi C. H. J.; Seligson D.; Tolcher A.; Alabi C. A.; Yen Y.; Heidel J. D.; Ribas A. Nature 2010, 464, 1067–U1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun G. B.; Pallaoro A.; Wu G. H.; Missirlis D.; Zasadzinski J. A.; Tirrell M.; Reich N. O. ACS Nano 2009, 3, 2007–2015. [DOI] [PubMed] [Google Scholar]
- de Bruin K. G.; Fella C.; Ogris M.; Wagner E.; Ruthardt N.; Brauchle C. J. Controlled Release 2008, 130, 175–182. [DOI] [PubMed] [Google Scholar]
- Mintzer M. A.; Simanek E. E. Chem. Rev. 2009, 109, 259–302. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Choi S. H.; Kim S. H.; Park T. G. J. Controlled Release 2008, 125, 25–32. [DOI] [PubMed] [Google Scholar]
- Gilleron J.; Querbes W.; Zeigerer A.; Borodovsky A.; Marsico G.; Schubert U.; Manygoats K.; Seifert S.; Andree C.; Stoter M.; Epstein-Barash H.; Zhang L.; Koteliansky V.; Fitzgerald K.; Fava E.; Bickle M.; Kalaidzidis Y.; Akinc A.; Maier M.; Zerial M. Nat. Biotechnol. 2013, 31, 638–646. [DOI] [PubMed] [Google Scholar]
- Sahay G.; Querbes W.; Alabi C.; Eltoukhy A.; Sarkar S.; Zurenko C.; Karagiannis E.; Love K.; Chen D. L.; Zoncu R.; Buganim Y.; Schroeder A.; Langer R.; Anderson D. G. Nat. Biotechnol. 2013, 31, 653–U119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blidner R. A.; Svoboda K. R.; Hammer R. P.; Monroe W. T. Mol. BioSyst. 2008, 4, 431–440. [DOI] [PubMed] [Google Scholar]
- Oishi M.; Kataoka K.; Nagasaki Y. Bioconjugate Chem. 2006, 17, 677–688. [DOI] [PubMed] [Google Scholar]
- Zuber G.; Dauty E.; Nothisen M.; Belguise P.; Behr J. P. Adv. Drug Delivery Rev. 2001, 52, 245–253. [DOI] [PubMed] [Google Scholar]
- Panyam J.; Labhasetwar V. Pharm. Res. 2003, 20, 212–220. [DOI] [PubMed] [Google Scholar]
- Vasir J. K.; Labhasetwar V. Adv. Drug Delivery Rev. 2007, 59, 718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A.; Mackey M. A.; El-Sayed M. A.; Bellamkonda R. V. ACS Nano 2011, 5, 4919–4926. [DOI] [PubMed] [Google Scholar]
- Weissleder R. Nat. Biotechnol. 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- Lukianova-Hleb E. Y.; Belyanin A.; Kashinath S.; Wu X. W.; Lapotko D. O. Biomaterials 2012, 33, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Adv. Mater. 2012, 24, 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Greenwald D. R.; Ruoslahti E. Science 2010, 328, 1031–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugahara K. N.; Teesalu T.; Karmali P. P.; Kotamraju V. R.; Agemy L.; Girard O. M.; Hanahan D.; Mattrey R. F.; Ruoslahti E. Cancer Cell 2009, 16, 510–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassie J. P.; Liu X. Y.; Thomas G. S.; Whitaker R. M.; Thiel K. W.; Stockdale K. R.; Meyerholz D. K.; McCaffrey A. P.; McNamara J. O. 2nd; Giangrande P. H. Nat. Biotechnol. 2009, 27, 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Rao D. D.; Senzer N.; Nemunaitis J. Pharm. Res. 2011, 28, 2983–2995. [DOI] [PubMed] [Google Scholar]
- Teesalu T.; Sugahara K. N.; Kotamraju V. R.; Ruoslahti E. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16157–16162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D.; Giljohann D. A.; Chen D. L.; Massich M. D.; Wang X. Q.; Iordanov H.; Mirkin C. A.; Paller A. S. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Hao L. L.; Hurst S. J.; Mirkin C. A. J. Am. Chem. Soc. 2012, 134, 16488–16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.; Zhang G.; Zhang R.; Flores L. G. 2nd; Huang Q.; Gelovani J. G.; Li C. Cancer Res. 2010, 70, 3177–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Servos M. R.; Liu J. W. J. Am. Chem. Soc. 2012, 134, 7266–7269. [DOI] [PubMed] [Google Scholar]
- Huang X. H.; El-Sayed I. H.; Qian W.; El-Sayed M. A. J. Am. Chem. Soc. 2006, 128, 2115–2120. [DOI] [PubMed] [Google Scholar]
- Acuna G. P.; Bucher M.; Stein I. H.; Steinhauer C.; Kuzyk A.; Holzmeister P.; Schreiber R.; Moroz A.; Stefani F. D.; Liedl T.; Simmel F. C.; Tinnefeld P. ACS Nano 2012, 6, 3189–3195. [DOI] [PubMed] [Google Scholar]
- Yang X. Z.; Dou S.; Wang Y. C.; Long H. Y.; Xiong M. H.; Mao C. Q.; Yao Y. D.; Wang J. ACS Nano 2012, 6, 4955–4965. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Patel T. R.; Fu M.; Bertram J. P.; Saltzman W. M. Biomaterials 2012, 33, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W. Y.; Jia Z. F.; Truong N. P.; Prasadam I.; Xiao Y.; Monteiro M. J. Biomacromolecules 2013, 14, 3386–3389. [DOI] [PubMed] [Google Scholar]
- Yang X. Z.; Dou S.; Sun T. M.; Mao C. Q.; Wang H. X.; Wang J. J. Controlled Release 2011, 156, 203–211. [DOI] [PubMed] [Google Scholar]
- Xiang B.; Dong D. W.; Shi N. Q.; Gao W.; Yang Z. Z.; Cui Y.; Cao D. Y.; Qi X. R. Biomaterials 2013, 34286976–6991. [DOI] [PubMed] [Google Scholar]
- Morales D. P.; Braun G. B.; Pallaoro A.; Chen R. W.; Huang X.; Zasadzinski J.; Reich N. O.. Targeted intracellular delivery of proteins with spatial and temporal control. Submitted for publication. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.