

Abstract
Selective inhibitors of myosin or actin function and confocal microscopy were used to test the role of an actomyosin complex in controlling morphology, trafficking, and fusion of tubulovesicles (TV) containing H-K-ATPase with the apical secretory canaliculus (ASC) of primary-cultured rabbit gastric parietal cells. In resting cells, myosin IIB and IIC, ezrin, and F-actin were associated with ASC, whereas H-K-ATPase localized to intracellular TV. Histamine caused fusion of TV with ASC and subsequent expansion resulting from HCl and water secretion; F-actin and ezrin remained associated with ASC whereas myosin IIB and IIC appeared to dissociate from ASC and relocalize to the cytoplasm. ML-7 (inhibits myosin light chain kinase) caused ASC of resting cells to collapse and most myosin IIB, F-actin, and ezrin to dissociate from ASC. TV were unaffected by ML-7. Jasplakinolide (stabilizes F-actin) caused ASC to develop large blebs to which actin, myosin II, and ezrin, as well as tubulin, were prominently localized. When added prior to stimulation, ML-7 and jasplakinolide prevented normal histamine-stimulated transformations of ASC/TV and the cytoskeleton, but they did not affect cells that had been previously stimulated with histamine. These results indicate that dynamic pools of actomyosin are required for maintenance of ASC structure in resting cells and for trafficking of TV to ASC during histamine stimulation. However, the dynamic pools of actomyosin are not required once the histamine-stimulated transformation of TV/ASC and cytoskeleton has occurred. These results also show that vesicle trafficking in parietal cells shares mechanisms with similar processes in renal collecting duct cells, neuronal synapses, and skeletal muscle.
Keywords: F-actin, membrane dynamics, nonmuscle myosin II, vesicle trafficking
when gastric parietal cells from rabbits are isolated and cultured, their highly invaginated apical canalicular membranes are endocytosed and converted into several separate apical secretory canaliculi (ASC). These ASC remain separated from the basolateral membrane and also from the H-K-ATPase-containing tubulovesicles (TV) during extended culture. During secretagogue-induced stimulation of acid secretion by these parietal cells, H-K-ATPase-containing TV located in the cytosol fuse with the ASC membranes, leading to a large increase in area of these ASC membranes and activation of both K and Cl channels and the H-K-ATPase (27, 30, 38, 50, 77). These stimulation-associated morphological changes involve changes in both the structure and dynamics of the actin cytoskeleton located directly underneath the apical microvilli (27–29). Profound ultrastructural changes in the F-actin-containing microfilaments occur during the secretory cycle of parietal cells, i.e., resting → stimulated → return to resting (6). Such changes in the actin cytoskeleton may be controlled through interactions with myosin II (nonmuscle myosin II) exerting mechanical and tensile forces (40), but this has not been tested.
Myosin II consists of two heavy chains, two essential light chains (ELCs), and two regulatory light chains (RLCs). Myosin II motor activity and parallel filament assembly are regulated by the phosphorylation of residues localized on the RLCs and heavy chains (21, 55, 70, 72). Phosphorylation of RLC via myosin light chain kinase (MLCK) (7) initiates the binding of myosin II to filamentous actin. Myosin II can self-assemble into minifilaments and interact with actin to generate tensile force and respond to biochemical stimuli (41, 70). Myosin II and actin are enriched in and near the apical membrane of many epithelial cells where myosin II regulates actin turnover and generates contractile/tensile forces to maintain tissue shape and morphology (41). A dynamic actomyosin network is also required for cytokinesis, cell migration, polarity, epithelial morphogenesis, organelle positioning, and membrane vesicle trafficking (5, 35, 36, 41, 44, 46, 49, 51, 70, 74).
Previous studies in parietal cells have suggested that F-actin (4, 28, 76) and myosin II (75, 79) are both present in the microvilli of the ASC membrane of parietal cells, but their roles and interactions remain incompletely described. Use of actin inhibitors, cytochalasins B and D (bind barbed end of F-actin and depolymerize F-actin) and latrunculin B (sequesters G-actin and depolymerizes F-actin), showed that a dynamic pool of F-actin played an important role in translocation and fusion of H-K-ATPase-containing TV with apical microvilli (4, 6, 28). However, the role of F-actin in morphology of ASC in resting cells and interactions between actin and myosin II in ASC-TV fusion in stimulated cells were not determined. Some contradictory data indicated that myosin II was not involved in TV-ASC interactions during histamine stimulation: 1) histamine caused changes in myosin IIA distribution near the basolateral, not the apical, membranes (79) and 2) neither ML-7 (MLCK inhibitor) nor 2,3-butanedione monoxime (myosin II ATPase inhibitor) had any effect on TV translocation and fusion with ASC during histamine stimulation (79).
The goal of the present study was to test the roles of myosin II and F-actin in parietal cell function by confocal imaging of both fixed, immunostained and live parietal cells. Localization of F-actin; myosin IIA, IIB, and IIC; H-K-ATPase; and ezrin were analyzed in fixed cells. Dynamic changes in ASC were imaged in live cells by using either a novel probe of ASC membranes, adenoviral-expressed green fluorescent protein (GFP)-PLC1δ-PH, which binds to phosphatidylinositol (4,5) bisphosphate [PI(4,5)P2], or the previously characterized adenoviral-expressed yellow fluorescent protein (YFP)-ezrin (11, 18, 22, 81). Inhibitors of myosin II and F-actin were used to determine the role of the actomyosin complex on the structure, distribution, and function of ASC and H-K-ATPase-containing TV in resting and histamine-stimulated cells.
MATERIALS AND METHODS
Primary culture of rabbit gastric parietal cells.
All procedures and treatments of animals were approved by the University of California Berkeley Institutional Animal Care and Use Committee. New Zealand White rabbits were sedated with a subcutaneous cocktail containing ketamine (100 mg/ml) and xylazine (20 mg/ml). Pentobarbital (Nembutal) was administered intravenously to achieve anesthesia, and a midline abdominal incision was made. The gastric vasculature was perfused with PBS via retrograde cannulation of the abdominal aorta. The stomach was removed, the mucosa was scraped to remove cells, and the cells were minced and digested with collagenase (Worthington type 4, 1 mg/ml) in MEM supplemented with 1 mg/ml BSA for 30 min. The digestion was filtered through cheesecloth to remove undigested tissue. Gastric glands and large clumps of cells settled out in 10–15 min by gravity, leaving a dense suspension of individual cells that were subjected to a 40-μm cell strainer. Intact cells were recovered by centrifuging the suspension (3×, 100 g, 5 min) followed by resuspension in fresh medium A, which contained DMEM-F-12 (Life Technologies, Grand Island, NY), supplemented with 20 mM HEPES, 0.2% BSA, 10 mM glucose, 1× SITE (selenium, insulin, transferrin) medium (Sigma, St. Louis, MO), 1 mM glutamine, 100 U/ml penicillin-streptomycin (from Penicillium/Streptomyces griseus), 400 μg/ml gentamycin (Micromonospora purpurea). These procedures resulted in a cell suspension that was 70–75% parietal cells. Cells were plated and cultured on coverslips coated with Matrigel (BD Bio Sciences, San Jose, CA) as described (15, 16) in 12-well plates and incubated at 37°C in culture medium A.
Recombinant adenovirus constructs and infection of parietal cells.
A recombinant adenovirus (rAD) containing tagged with yellow fluorescent protein (YFP-ezrin) was used as previously described (81). GFP-PLC1δ-PH (kind gift from Prof. Tobias Meyer, Stanford University), widely used for labeling PI(4,5)P2 (20, 69, 78), was used to label ASC membranes. To make a rAD/green fluorescent protein (GFP)-tagged phospholipase C1δ pleckstrin homology domain (GFP-PLC1δ-PH) expression system, human PLC1δ-PH with GFP gene was amplified by PCR from pEGFP-N1/PLC1δ-PH and inserted into AdMax shuttle vector pDC311. The resulting plasmid pDC311/PLC1δ-PH and viral vector pBHGloxDE1,3Cre were cotransfected into HEK293 cells to obtain rAD/GFP-PLC1δ-PH.
Immunofluorescence and rAD infection.
Cells used for immunolocalization were fixed (4% formaldehyde in PBS 10 min) and washed three times with PBS. They were then permeabilized (0.25% Triton X-100 in PBS, 10 min), blocked (2% bovine serum albumin in PBS), and incubated with antibodies against mouse anti-ezrin (dilution 1:100) or rabbit anti-ezrin (1:100) (4A5, Covance, Berkeley, CA and Cell Signaling Technology, Danvers, MA), mouse anti-H-K-ATPase (1:1,000) (2G11, Affinity Bioreagents, Boulder, CO), rabbit anti-myosin heavy chain isoforms [myosin IIA (1:50), myosin IIB (1:200) and myosin IIC (1:50) (Cell Signaling Technology, Danvers, MA) or myosin regulatory light chain (MRLC; 1:100)(Thermo Scientific, Rockford, IL)], or mouse anti-tubulin (1:200) (Developmental Studies Hybridoma Bank, Iowa City, IA). Primary antibodies were visualized by Alexa Fluor-labeled goat secondary antibodies (1:500, Alexa Fluor 488 for ezrin and tubulin, Alexa Fluor 555 for H-K-ATPase, MRLC and myosin IIA, B, C) (Life Technologies). Actin was visualized using Alexa Fluor 647-labeled phalloidin (Life Technologies) in most experiments. Because phalloidin and jasplakinolide compete for similar binding sites in F-actin (8), mouse anti-actin antibody (1:200) (Neomarkers, Fremont, CA) were used to localize actin in experiments using jasplakinolide. After application of secondary antibodies, cells on the coverslips were washed with PBS and drained of excessive PBS by holding each coverslip on its edge with forceps and draining it onto a Kimwipe. Coverslips were air dried before placing them on the glass slides. This final step caused cells to become flat and thin. Finally, coverslips were mounted on the glass slides with mounting medium (Fluoromount-G, Southern Biotech, Birmingham, AL) for immunofluorescence.
For experiments to visualize ezrin or PLC1δ-PH in living cells, cultured parietal cells were infected with rAD to express YFP-ezrin or GFP-PLC1δ-PH 2 h postplating, followed by incubation (37°C, 12 h) and washing with fresh medium without viruses; >80% of parietal cells expressed the constructs.
Confocal microscopy.
Fluorescence and bright-field images of fixed/stained and live parietal cells were collected on a Zeiss 710 confocal microscope using Zeiss software. Parietal cells were left untreated or were treated with the secretory stimulant histamine (100 μM) or with inhibitors of cytoskeletal function, ML-7 (10 μM), blebbistatin (50 μM), Y-27632 (10 μM), jasplakinolide (3 μM), or nocodazole (20 μM). The molecular targets and mechanisms of actions of these drugs/inhibitors that affect myosin II, F-actin, and microtubule activity are summarized in Table 1. Images for movies were collected every 60 s. Figures and movies were constructed by use of Adobe Photoshop and Microsoft Movie Maker. All images shown represent typical observations made in 20–50 cells isolated from 3–10 animals.
Table 1.
Drugs used in this study, and their targets and actions
| Drugs | Targets | Actions |
|---|---|---|
| ML-7 | Myosin light chain kinase (MLCK) | Inhibition |
| Blebbistatin | Nonmuscle myosin II ATPase | Inhibition |
| Y-27632 | Rho-associated coiled-coil kinase (ROCK) | Inhibition |
| Jasplakinolide | F-actin | Stabilization |
| Nocodazole | Microtubule | Depolymerizaton |
As observed in the confocal microscope, fixed and stained parietal cells were routinely only 2–3 μm thick but 21–36 μm in diameter. These flattened cells also had flattened ASC and associated proteins, resulting in generating the equivalent optical effect of sandwiching individual x–y planes into a single z plane. Thus ASC appeared to be “filled” rather than as hollow structures surrounded by the proteins of interest. We therefore quantitated the effects of histamine and the drugs on ASC structure by measuring two diameters of ASCs in the different conditions, one measurement through the longest axis and a second measurement 90° to this first measurement. This approach allowed us to quantitate both the size and shape of the ASCs.
Western blotting.
Western blot experiments were conducted as described previously (48). Cell scrapings were used for Western blots to assay the expression of myosin heavy chain. Equal amounts of protein were loaded onto 4–12% SDS-PAGE gels. Proteins were transferred onto membranes and probed with antibodies to myosin II heavy chain isoforms A, B, or C. Results were visualized with chemiluminescence reagent (Western Lightning Plus ECL chemiluminescence reagent, PerkinElmer, Waltham, MA).
RESULTS
Localization of myosin II in resting and stimulated parietal cells.
Western blots showed that myosin II isoforms A, B, and C were present in lysates from control and histamine-stimulated parietal cell-enriched fractions from gastric mucosa (Fig. 1). Immunofluorescence microscopy was used to localize myosin II isoforms along with actin, ezrin, and H-K-ATPase in parietal cells. Myosin IIA localized to the cytoplasm near the basolateral membrane (Fig. 2A), similar to previous observations (79). In contrast, myosin IIB and IIC and MRLC predominantly colocalized with F-actin and ezrin in/near ASC in resting, control parietal cells (Fig. 2, B–D). These results indicated that myosin IIB and IIC were more likely than myosin IIA to be involved in morphological transformations at ASC and TV during histamine stimulation. Myosin IIB and IIC had nearly identical staining patterns under all conditions examined, but, given the more distinct localization of myosin IIB with ASC and the documented role of myosin IIB in polarized cell function and secretion (33, 52, 67), it was chosen as the focus of this work.
Fig. 1.
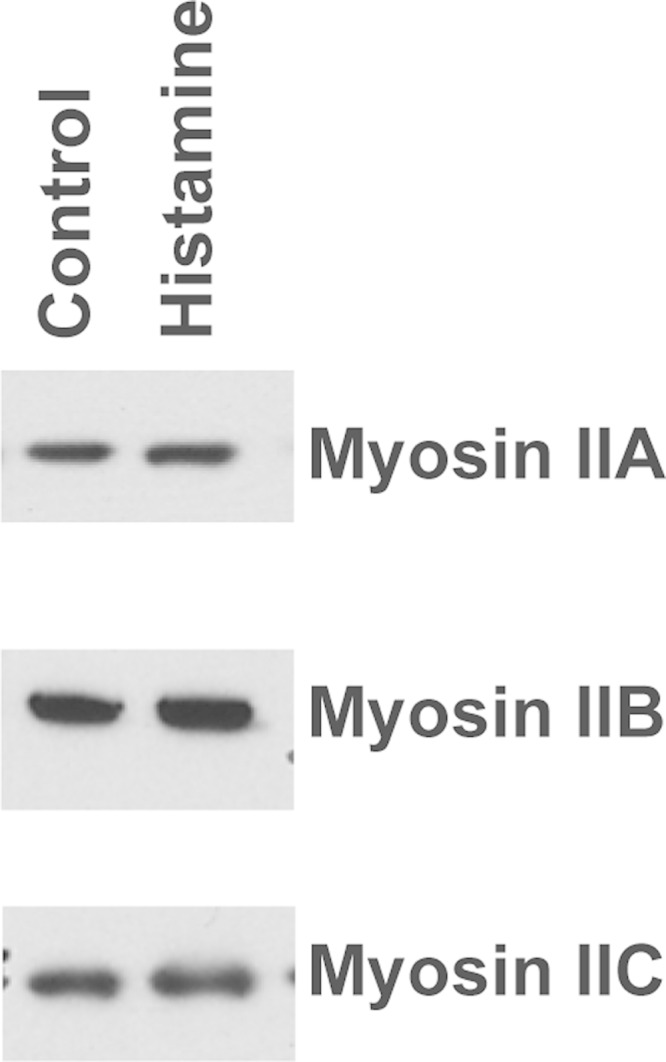
Expression of myosin II isoforms. Western blot of myosin IIA, IIB, and IIC in parietal cells. Isolated cells from rabbit stomachs were treated with either 100 μM cimetidine (Control) or 100 μM histamine (Histamine) for 30 min, then processed for Western blots to assay myosin heavy chain. Equal amounts of protein were loaded onto 4–12% SDS-PAGE gels. Proteins were transferred onto membranes and probed with antibodies to myosin heavy chain A, B, or C. Results were visualized with chemiluminescence reagent. There were no differences in myosin band intensities between control and histamine-stimulated cells.
Fig. 2.

Localization of myosin II, MRLC, actin, and ezrin in resting parietal cells. F-actin was stained by phalloidin conjugated with Alexa Fluor 647. Immunostaining was performed on ezrin with mouse anti-ezrin and myosin II by use of either rabbit anti-myosin IIA (A), rabbit anti-myosin IIB (B), rabbit anti-myosin IIC (C), or rabbit anti-MRLC (D) followed by Alexa Fluor-labeled goat secondary antibodies in resting parietal cells. Myosin IIB and IIC and MRLC colocalized with actin and ezrin in apical secretory canaliculus (ASC), but myosin IIA was localized near the basolateral membrane in resting parietal cells.
As summarized in Fig. 3, A and B, actin, ezrin, and myosin IIB were prominently associated with ASC in resting cells, whereas H-K-ATPase was distributed mostly in cytoplasmic TV. In histamine-stimulated cells, ASC stained with H-K-ATPase and became markedly expanded (Table 2) (by fusion of HK-ATPase-rich TV and HCl secretion); actin and ezrin remained associated with this expanded ASC, similar to previous observations (1, 16, 27, 43, 60). Myosin IIB changed its localization following histamine stimulation from being associated with ASC (where ezrin and actin are localized) to instead becoming dispersed to the cytoplasm. There was also some punctate myosin IIB staining near the basal membrane.
Fig. 3.

Effects of histamine and histamine + NaSCN on localization of myosin IIB, actin, ezrin, and H-K-ATPase in parietal cells. Parietal cells were treated with cimetidine (100 μM) (Control), histamine (100 μM), or histamine + NaSCN (3 mM NaSCN) together for 30 min, followed by fixation and staining for F-actin and ezrin as described in Fig. 2 and H-K-ATPase with mouse anti-H-K-ATPase (2G11) followed by Alexa Fluor-labeled goat secondary antibody (A) or F-actin, ezrin, and myosin IIB (B). F-actin and ezrin by were localized to ASCs in both resting and histamine-stimulated cells. Myosin IIB was localized to ASC in resting parietal cells and to the cytosol in histamine-stimulated cells. H-K-ATPase localized to tubulovesicles (TV) in cytosol in resting cells and to expanded ASC in histamine-stimulated cells. Cells treated with histamine + NaSCN together showed collapsed ASC, and ezrin, actin, and H-K-ATPase remained associated with the shrunken ASC, and myosin IIB remained dispersed to the cytosol. The less distinct staining of ezrin in images also stained for H-K-ATPase resulted from nonspecific binding of polyclonal ezrin antibody (i.e., compared with monoclonal antibody used in other images).
Table 2.
Quantitation of ASC morphology from confocal images
| Apical Vacuole Diameter L, μm | Apical Vacuole Diameter W, μm | Diameter L/Diameter W | |
|---|---|---|---|
| Control | 4.8 ± 0.69 (110;3) | 4.6 ± 0.51 (110;3) | 1.04 |
| Histamine | 20.8 ± 0.73 (102;3)* | 21.1 ± 1 (102;3)* | 0.99 |
| ML-7 | 2.7 ± 0.29 (108;3)* | 2.4 ± 0.25 (108;3)* | 1.10 |
| ML-7 + Histamine | 2.6 ± 0.3 (100;3)† | 2.8 ± 0.33 (100;3)† | 0.93 |
| Histamine + ML-7 | 21.8 ± 1.2 (96;3)* | 20.1 ± 0.95 (96;3)* | 1.08 |
| Histamine + NaSCN− | 6.9 ± 1.2 (74;3)† | 7.2 ± 0.95 (74;3)† | 0.95 |
| Y-27632 | 4.0 ± 0.51 (112;3) | 3.8 ± 0.23 (112;3) | 1.05 |
| Y-27632 + Histamine | 21.2 ± 0.73 (105;3)* | 19.3 ± 0.93 (105;3)* | 1.10 |
| Blebbistatin | 3.9 ± 0.33 (110;3) | 4.3 ± 0.72 (110;3) | 0.91 |
| Blebbistatin + Histamine | 22.0 ± 0.9 (113;3)* | 21.3 ± 0.81 (113;3)* | 1.03 |
| Jasplakinolide | 12.7 ± 1.44 (101;3)* | 3.4 ± 0.45 (101;3)* | 3.70* |
| Jasplakinolide +Histamine | 13.2 ± 1.3 (104;3)† | 3.7 ± 0.29(104;3)† | 3.60† |
| Histamine + Jasplakinolide | 19.5 ± 2.0 (90;3)* | 20.5 ± 2.2 (90;3)* | 0.95 |
| ML-7 + Jasplakinolide | 2.1 ± 0.19 (97;3)*‡ | 2.3 ± 0.31 (97;3)*‡ | 0.91 |
| Blebbistatin + Jasplakinolide | 4.3 ± 0.39 (78;3)‡ | 3.9 ± 0.45 (78;3)‡ | 1.10 |
| Nocodazole + Jasplakinolide | 3.6 ± 0.31 (75;3)*‡ | 1.5 ± 0.21 (75;3)*‡ | 2.40*‡ |
Cells were treated as follows: Control (100 μM cimetidine), Histamine (100 μM histamine for 30 min), ML-7 (10 μM ML-7 for 30 min), ML-7 + Histamine (10 μM ML-7 for 30 min followed by 100 μM histamine for 30 min), Histamine + ML-7 (100 μM histamine for 30 min followed by 10 μM ML-7 for 30 min), Histamine + NaSCN (100 μM histamine +3 mM NaSCN together for 30 min), Y-27632 (10 μM Y-27632 for 30 min), Y-27632 + Histamine (10 μM Y-27632 for 30 min followed by 100 μM histamine for 30 min), Blebbistatin (50 μM blebbistatin for 30 min), Blebbistatin + Histamine (50 μM blebbistatin for 30 min followed by 100 μM histamine for 30 min), Jasplakinolide (3 μM jasplakinolide for 60 min), Jasplakinolide + Histamine (3 μM jasplakinolide for 30 min followed by 100 μM histamine for 30 min), Histamine + Jasplakinolide (100 μM histamine for 30 min followed by 3 μM jasplakinolide for 30 min), ML-7 + Jasplakinolide (10 μM ML-7 for 30 min followed by 3 μM jasplakinolide for 30 min), Blebbistatin + Jasplakinolide (50 μM blebbistatin for 30 min followed by 3 μM jasplakinolide for 30 min), Nocodazole + Jasplakinolide (20 μM nocodazole for 30 min followed by 3 μM jasplakinolide for 30 min). Following these treatments, cells were fixed and stained (see materials and methods). Diameters of apical secretory canaliculus (ASC) were measured (from ezrin or myosin IIB staining) along the long axis and then at 90° to this in confocal micrographs of ASCs, and the ratio of the 2 measurements was calculated. Averages ± SD are reported for 75–113 ASCs measured in parietal cells from 3 different experiments each. Statistical comparisons:
P < 0.05 for comparison to control;
P < 0.05 for comparsion to histamine;
P < 0.05 for comparison to jasplakinolide.
We tested whether the apparent redistribution of myosin IIB from ASC to cytosol resulted simply from the swelling associated with histamine stimulated HCl plus H2O secretion into ASC. Studies in histamine-stimulated cells that had been treated with the protonophore SCN− (32) to eliminate HCl accumulation showed that ASC were less expanded than in histamine-treated cells but, like histamine-treated cells, ASC contained H-K-ATPase and were decorated with F-actin but not with myosin IIB (Fig. 3, A and B, bottom rows). This result confirmed that histamine caused dispersal of myosin IIB from ASC to the cytoplasm even in the absence of swelling of the ASC.
Pharmacological inhibition of myosin II by ML-7 alters ASC morphology in resting cells and trafficking of TV upon histamine stimulation.
Histamine-stimulated redistribution of myosin IIB and IIC from ASC to the cytosol suggested that these myosin II isoforms might play important roles in mediating movement and/or fusion of H-K-ATPase-containing TV to ASC membranes. This hypothesis was tested by using three inhibitors: ML-7, Y-27632, and blebbistatin (Table 1). ML-7 inhibits MLCK, which inhibits phosphorylation of MRLC and prevents myosin minifilament assembly and, therefore, actomyosin interactions. Y-27632 is a Rho kinase inhibitor that also inhibits phosphorylation of MRLC (66, 70). Blebbistatin blocks the ATPase activity of myosin II (39) and would alter actomyosin interactions if myosin II head groups and ATP turnover were involved in regulating myosin-actin interactions in parietal cells. Resting parietal cells were treated with 10 μM ML-7, then fixed and stained for actin, ezrin, myosin IIB, and H-K-ATPase (Fig. 4, A and B, top rows). Comparisons of images of ML-7-treated resting cells in Fig. 4, A and B, with images of control resting cells in Fig. 3, A and B, suggest that the main effect of ML-7 was to cause ASC containing actin, ezrin, and myosin IIB to shrink (Table 2). In addition, some of the myosin IIB appeared to relocalize to the basolateral membrane. ML-7 had no apparent effect on the localization of H-K-ATPase in TV.
Fig. 4.

Effects of ML-7 on actin, ezrin, myosin IIB, and ASC morphology in resting and histamine-stimulated parietal cells. Cells were treated with cimetidine (100 μM) followed by ML-7 (10 μM) for 30 min or ML-7 for 30 min followed by histamine (100 μM) for 30 min (ML-7 + Histamine) or with histamine for 30 min followed by ML-7 for 30 min (Histamine + ML-7). Cells were then fixed and stained for F-actin, ezrin, and myosin IIB (A) or F-actin, ezrin, and H-K-ATPase (B) as described in legends to Figs. 2 and 3.
The shrinkage of ASC upon the addition of ML-7 shown in Figs. 4, A and B, and 6, A and B, and summarized in Table 2 was unexpected. To validate and characterize this process further, two fluorescent fusion proteins, YFP-ezrin (81) and GFP-PLC1δ-PH [used for labeling PI(4,5)P2 (20, 69, 78) in membranes of live cells], were used to label ASC membranes (Fig. 6, A and B).
Fig. 6.

ML-7 causes ASC to shrink in resting parietal cells. Live cell fluorescence and DIC imaging of parietal cells expressing GFP-PLC1δ-PH (A) and YFP-ezrin (B) during treatment with 0.1% DMSO (Control) or ML-7 (10 μM) for 30 min. GFP-PLC1δ-PH and ezrin both localized to ASCs (arrows), which became shrunken/shriveled in the ML-7-treated compared with control cells.
Figure 5 shows typical fluorescence images of GFP-PLC1δ-PH (Fig. 5A) and YFP-ezrin (Fig. 5B) and the corresponding interference (DIC) images of live, cimetidine-treated (100 μM) rabbit parietal cells 40 h after infection and then in the same cells following removal of cimetidine and treatment for 30 min with histamine (100 μM). GFP-PLC1δ-PH was present most prominently in ASC but also in basolateral membranes. There was similarly intense YFP-ezrin labeling in the ASC membrane and weak labeling in the basolateral membrane, consistent with previous data (80, 82). The brighter fluorescence of both GFP-PLC1δ-PH and YFP-ezrin in ASC compared with basolateral membranes may have resulted from the greater surface area from the infoldings of microvilli in ASC compared with the smoother basolateral membranes, or a higher content of PI(4,5)P2 and ezrin in ASC. Time-lapse microscopy of parietal cells during histamine stimulation showed gradual expansion of ASC membranes as monitored with either GFP-PLC1δ-PH (Supplemental Movie S1) or YFP-ezrin (Supplemental Movie S2). The expansion of ASC likely resulted from fusion of TV with ASC and activation of H-K-ATPase and associated K and Cl channels in TV, resulting in accumulation of HCl and swelling of the ASC. Persistent labeling of ASC with GFP-PLC1δ-PH suggested that the specific content of PI(4,5)P2 in these membranes did not change appreciably during histamine stimulation, although the total amount of PI(4,5)P2 in these membranes may have increased owing to addition of ASC membrane and production of PI(4,5)P2. The data also indicated that GFP-PLC1δ-PH and YFP-ezrin were reliable markers of ASC in living parietal cells.
Fig. 5.

Localization of green fluorescent protein (GFP)-PLC1δ-PH and ezrin tagged with yellow fluorescent protein (YFP-ezrin) in live parietal cells: control and histamine stimulation. Primary cultures of parietal cells were infected with adenovirus (rAD) expressing GFP-PLC1δ-PH (A) and YFP-ezrin (B) 2 h after plating. Cells were maintained for 40 h at 37°C and treated for 30 min with either cimetidine (100 μM) to assure the resting state (Control) or with histamine (100 μM) to induce HCl secretion (Histamine). Fluorescence and differential interference (DIC) images are shown for the same cells in both control and histamine-treated states. Apical membrane vacuoles (arrows) became enlarged and occupied most of the cytoplasm in the stimulated compared with the resting state. GFP-PLC1δ-PH was localized to both ASC and basolateral membrane, whereas YFP-ezrin localized primarily to ASC membrane in resting and stimulated cells.
As shown in Fig. 6, A and B, and Supplementary Movies 3 and 4, time-lapse imaging of live, resting parietal cells showed that ML-7 caused ASC to collapse within 20 min as visualized by using GFP-PLC1δ-PH and YFP-ezrin. Similar experiments were performed on parietal cells that were fixed and stained for actin, ezrin, myosin IIB, and H-K-ATPase (Fig. 4, A and B, top row).
Interestingly, when added prior to histamine, ML-7 blocked the normal stimulatory effects of histamine (Fig. 4, A and B, middle row). Thus ML-7 blocked the recruitment of H-K-ATPase to ASC (H-K-ATPase remained localized to TV in the cytosol) and prevented the expansion of ASC (Table 2). In contrast, results published previously showed that ML-7 did not block the recruitment of H-K-ATPase to ASC in histamine-stimulated cells (79). This apparent discrepancy may have resulted from different experimental procedures. Zhou et al. (79) added histamine and ML-7 together and then monitored morphology, whereas we pretreated cells with ML-7 for 30 min and monitored morphology of both resting cells and also cells that were treated subsequently with histamine. We conclude that the effect of ML-7 on parietal cell function during the secretory cycle may depend critically on the timing of the addition of the inhibitor. Supporting these potential time-dependent effects are additional results in which treatment with histamine followed by ML-7 showed normal, stimulated-like appearance of actin, ezrin, myosin II, and H-K-ATPase (Fig. 4, A and B, bottom row; Table 2). These last data indicated that, in contrast to the dramatic effects of ML-7 on resting parietal cells and blocking stimulation-associated changes, once histamine had induced the stimulated morphology, ML-7 had little effect.
Previous studies showed that ML-7 had protonophore-like activity in isolated gastric tubulovesicles (2, 61, 79), which might have contributed to our results by preventing expansion of ASC in cells treated first with ML-7 and then histamine. However, results in Figs. 3, 4, and 6, Supplementary Movies S3 and S4, and Table 2 showed that effects of the known protonophore NaSCN and ML-7 were quite different: 1) Adding ML-7 to resting cells caused ASCs to collapse (Figs. 4A and 6 and Supplementary Movies S3 and S4), whereas NaSCN had no effect (Ref. 1; P. Natarajan, unpublished observations). 2) In the presence of SCN−, histamine caused fusion of H-K-ATPase containing TV with ASC (Fig. 3A, Ref. 1), whereas, in the presence of ML-7, histamine-induced fusion of TV with ASC was prevented (Fig. 4A). 3) Adding ML-7 to cells stimulated with histamine did not cause ASCs to collapse (Fig. 4B), inconsistent with ML-7 eliciting a protonophore action. On the basis of these observations we concluded that NaSCN and ML-7 had distinct effects on ASC and that the effects of ML-7 to shrink ASC in resting cells were mediated at least in part through blocking MLCK. However, further work will be required to determine unambiguously that ML-7-induced shrinkage of ASC in resting cells results solely from ML-7 block of MLCK activity.
In contrast to the effects of ML-7, neither blebbistatin nor Y-23632 had any effect on morphology of ASC or distribution of ezrin, actin, and myosin IIB in resting parietal cells (Fig. 7A and Table 2) and also did not alter the normal stimulatory effect of histamine (Fig. 7B and Table 2). These studies emphasized the selectivity of the inhibitory effects of ML-7 and indicated that the MRLC in parietal cells is phosphorylated by MLCK but not by Rho kinase.
Fig. 7.

Blebbistatin and Y-27632 do not affect ASC morphology of resting parietal cells or responses of parietal cells to histamine. A: resting parietal cells were treated with blebbistatin (50 μM) or Y-27632 (10 μM) for 30 min followed by immunostaining for F-actin, ezrin, and myosin IIB as described in Fig. 2. B: parietal cells were treated with blebbistatin or Y-27632 for 30 min, then stimulated with histamine (100 μM) for 30 min followed by fixation and immunostaining for F-actin, ezrin, and myosin IIB as described in Fig. 2.
Effects of jasplakinolide: a dynamic pool of F-actin is also required for ASC morphology in resting cells and for trafficking of TV during histamine stimulation.
Effects of ML-7 to cause collapse/shrinkage of ASC in resting parietal cells (Fig. 4, A and B, Table 2) were consistent with the idea that a dynamic pool of myosin IIB was involved in maintaining ASC structure in resting cells. Previous studies using the F-actin disruptors cytochalasin and latrunculin showed that actin filaments were required for the morphological transformation of ASC during histamine stimulation of parietal cells (4, 6, 56). As a complement to the effects of the F-actin disrupters, we tested the actin-stabilizing drug jasplakinolide (34) on morphology of ASC and TV in both resting cells and cells treated with histamine. Effects of jasplakinolide were tested both in live cell imaging to identify time course of changes in ASC in resting cells and in fixed/stained cells to identify changes in cytoskeletal network associated with ASC and H-K-ATPase in TV of both resting and histamine-stimulated cells.
In live cells expressing GFP-PLC1δ-PH (Fig. 8A; Supplemental Movie S5) or YFP-ezrin (Fig. 8B; Supplemental Movie S6), jasplakinolide caused blebbing of ASC toward and even extending to the basolateral membranes of parietal cells. Blebs started with small, smooth, spherical shapes that gradually increased in size during the next 15 min. Jasplakinolide has been reported to induce blebbing of plasma membranes toward the extracellular space (“outward directed”) in macrophages and cancer cells (26, 58). In parietal cells, jasplakinolide caused blebbing of ASCs toward the cytosol (“inward directed”) to such an extent that the blebs appeared to push on and cause eversion of the basolateral membranes of parietal cells (outward directed).
Fig. 8.

Jasplakinolide causes blebbing and shrinkage of ASC membranes: localization of GFP-PLC1δ-PH and YFP-ezrin in control and jasplakinolide-treated live parietal cells. Parietal cells expressing GFP-PLC1δ-PH (A) and YFP-ezrin (B) were treated with 0.3% DMSO (Control) or jasplakinolide (3 μM) for 30 min. Fluorescence and corresponding DIC images are shown. Jasplakinolide caused membrane blebbing (arrowhead) from 1 part of ASC, which appeared to shrink (arrow) compared with the resting state. C: parietal cells were incubated with 3 μM jasplakinolide for 30 min followed by fixation and staining for actin using mouse anti-actin antibody and Alexa Fluor-labeled goat secondary antibody; ezrin and myosin IIB were labeled as described in Fig. 3. Actin colocalized with ezrin (top) and myosin IIB (bottom).
Fixed/stained cells were used to test for distribution of cytoskeletal components in ASC-associated blebs and H-K-ATPase in TV during treatment with jasplakinolide. Comparison of images of jasplakinolide-treated cells in Figs. 8C and 9, A and B, with control cells in Figs. 2 and 3, A and B, showed that in resting parietal cells jasplakinolide caused relocalization of actin, ezrin, and myosin IIB from ASC to blebs (also see Table 2). These blebs assumed a hornlike appearance in fixed cells, likely resulting from fixation-induced shrinkage (13, 19). Because there is no distinct ASC in jasplakinolide-treated cells, we confirmed that jasplakinolide-treated cells were parietal cells by using H-K-ATPase staining (P. Natarajan, unpublished observations). As shown in Fig. 9B, these jasplakinolide-induced projections also contained microtubules/tubulin. Cells that were treated first with jasplakinolide and then histamine (Fig. 9C) had similar appearance as cells treated with jasplakinolide alone (Fig. 9A; also Table 2). In contrast, cells treated first with histamine and then jasplakinolide (Fig. 9D) looked nearly identical to cells that had been stimulated only with histamine (Fig. 3, A and B, middle rows; also Table 2). These results showed that jasplakinolide blocked histamine-stimulated transformation, but jasplakinolide did not affect morphology of parietal cells once they had been stimulated. The effects of jasplakinolide suggest that the normal localization and dynamic behavior of F-actin, myosin IIB, ezrin, and microtubules are required to maintain normal vacuolar morphology of resting parietal cells and prime the cells for histamine-stimulated transformation.
Fig. 9.

Jasplakinolide alters the localization of ezrin and myosin and morphology of ASC and inhibits histamine-stimulated ASC membrane expansion. Parietal cells were treated with jasplakinolide (3 μM) for 60 min (A and B) or jasplakinolide for 30 min followed by histamine (100 μM) for 30 min (Jasplakinolide + Histamine) (C) or with histamine for 30 min followed by jasplakinolide for 30 min (Histamine + Jasplakinolide) (D); cells were then fixed and stained for ezrin and myosin IIB as described in Fig. 2 (A, C, and D) or tubulin by using mouse anti-tubulin followed by Alexa Fluor-labeled goat secondary antibody (B). Arrows indicate membrane protrusion containing ezrin, myosin IIB, and tubulin.
We also used jasplakinolide in combination with inhibitors of myosin II (blebbistatin and ML-7) and microtubules (nocodazole) to test the roles of these cytoskeletal elements in forming the blebs. Comparison of Fig. 10, A and B, with Fig. 9A showed that pretreatment of resting parietal cells with either blebbistatin or ML-7 reduced blebbing caused by jasplakinolide (Table 2). Nocodazole also reduced the blebbing caused by jasplakinolide (Fig. 10C vs. Fig. 9A). Overall, these results indicated that myosin II, F-actin, ezrin, and microtubules may all be involved in generating the blebs or relocalization of these proteins during jasplakinolide treatment. However, similar to lack of effect of ML-7 in stimulated cells, jasplakinolide had no effect on appearance of cells that had been pretreated with histamine.
Fig. 10.

Effects of combinations of jasplakinolide, blebbistatin, ML-7, and nocodazole on parietal cell ezrin, myosin IIB, and tubulin. Parietal cells were treated with blebbistatin (50 μM) for 30 min followed by jasplakinolide (3 μM) for another 30 min (Blebbistatin + Jasplakinolide) (A) or with ML-7 (10 μM) for 30 min followed by jasplakinolide for 30 min (ML-7 + Jasplakinolide) (B) or with nocodazole (20 μM) for 4 h followed by jasplakinolide for 30 min (Nocodazole + Jasplakinolide) (C); cells were then fixed and stained for ezrin, myosin IIB, and tubulin as described in Fig. 9.
DISCUSSION
The major conclusions of this work are that dynamic pools of myosin IIB and F-actin (e.g., treadmilling) are important for maintaining ASC structure in resting parietal cells and for providing trafficking of TV to and fusion with ASC during histamine stimulation. A schematic model (Fig. 11) summarizes our proposal for the roles of the dynamic pools of myosin and actin in parietal cells.
Fig. 11.

Model for role of actomyosin complex in ASC morphology and TV trafficking in parietal cells. i: F-actin and myosin IIB assembled into filaments form an actomyosin complex that surrounds ASC in resting parietal cells; F-actin is attached to the ASC (green membrane) by ezrin. H-K-ATPase-containing TV (red membranes) are localized to the cytoplasm. ii: Histamine stimulation causes myosin IIB to dissociate from the ASC, whereas TV translocate from the cytosol to and fuse with ASC (which become yellow to denote fusion and mixing of TV with ASC). This fusion event is associated with activation of H-K-ATPase (and associated ion channels, not shown), leading to secretion of HCl and enlargement of ASC. F-actin and ezrin remain associated with ASC whereas myosin IIB dissociates from ASC in histamine-stimulated cells. iii: ML-7 causes ASC to shrink/collapse owing to F-actin depolymerization and dissociation of ezrin and myosin from ASC; these effects also prevent normal activation by histamine. iv: Jasplakinolide leads to stabilization of F-actin, which lengthens and, in association with myosin and microtubules, helps to form bleblike membrane protrusions of ASC that then prevent normal trafficking and fusion of TV with ASC. This effect of jasplakinolide is blocked when cells are pretreated with ML-7 or blebbistatin or nocodazole, indicating a role for myosin IIB and microtubules in the formation of the blebs.
In resting parietal cells (Fig. 11i), myosin IIB assembles into filaments that interact with F-actin to provide a dynamic actomyosin network that surrounds and connects to ASC (green line/membrane), with actin undergoing myosin IIB-regulated treadmilling. Myosin II-regulated actin treadmilling/turnover occurs during many membrane dynamic events (31, 37, 42, 47, 52, 59, 62, 71, 74), and resting parietal cells may require the dynamic actomyosin complex surrounding ASC to provide the force/tension required to resist an unknown collapsing force on the ASC, thereby helping to keep them patent in the resting cells. Ezrin connects these dynamic actin filaments with ASC membranes (3). H-K-ATPase-containing TV (red membrane) are localized to the cytosol, separated from the ASC, and these actomyosin filaments may also provide tracks along which TV move up to and fuse with ASC.
Histamine treatment (Fig. 11ii) results in remodeling of the actomyosin cytoskeleton surrounding ASC and translocation of TV to and their subsequent fusion with ASC (fusion and mixing of red TV membranes with green ASC membranes is shown by yellow membrane), thereby delivering the H-K-ATPase to ASC. Once TV fusion has occurred, myosin II is released from ASC. Further expansion of ASC after TV fusion results from HCl and water accumulation.
Blocking MLCK with ML-7 in resting cells (Fig. 11iii) likely allowed an unopposed phosphatase to dephosphorylate MRLC, leading to inhibition of myosin filament assembly, depolymerization of F-actin, and concomitant loss of ezrin from ASC to the cytosol, and thus disassembly of the actomyosin network that is required for maintenance of normal morphology of ASC in resting cells and for normal trafficking and fusion of TV to ASC during histamine stimulation. Because ML-7 has protonophore activity (61), it is possible that some of ML-7's effects on ASC morphology resulted from its combined action as a protonophore and inhibitor of MLCK. Further investigations are warranted to address this issue. Myosin II assembly and localization at the cleavage furrow during cytokinesis is regulated by myosin II heavy chain phosphorylation (54), and threonine phosphorylation of myosin heavy chain disassembles myosin filaments and alters the localization of myosin II (21, 23, 68). However, tests of histamine-stimulated changes in heavy chain phosphorylation, function, and/or localization of myosin IIB in parietal cells will require specific anti-phospho-heavy chain antibodies, which are currently not available. The lack of an effect of Y-23632 (blocks Rho kinase) suggests that Rho kinase is not involved in regulating myosin II phosphorylation in parietal cells. Jasplakinolide (F-actin stabilizing agent) caused actomyosin filaments to lengthen (Fig. 11iv), pulling ASC into inward-directed blebs of ASC that also contained ezrin and microtubules. A coordinated interaction of F-actin, myosin IIB, ezrin, and microtubules may be required to pull on and generate the inward-directed blebs of ASC membranes. The formation of these jasplakinolide-dependent blebs from ASC is also consistent with the conclusion that a dynamic F-actin network is important for maintaining normal ASC morphology in resting parietal cells.
Interestingly, jasplakinolide-induced inward bleb formation was prevented in cells that had been treated with ML-7 or blebbistatin, indicating that formation of these inward membrane blebs required myosin II activity. Myosin II activity is involved in outward bleb formation in other cells (12, 13, 19, 25, 26, 53, 58, 73). In addition, formation of blebs in glioblastoma cells (45) required growing microtubules in addition to actin and myosin II, consistent with observations here showing that nocodazole inhibits bleb formation. Such coordination between an actomyosin complex and the microtubular system has been proposed to control membrane dynamics during cell migration, cytokinesis, and polarity (9, 10, 14, 24). These results also suggest that the localization and/or activity of myosin IIB in parietal cells may regulate the localization of other cytoskeletal components such as ezrin or microtubules (or vice versa).
Jasplakinolide, when added prior to histamine, also blocked histamine-stimulated transformations in parietal cells, consistent with previous experiments showing the importance of the actin network for histamine-induced trafficking and fusion of TV with apical microvilli in parietal cells (4, 6, 28). The present data therefore indicate that dynamic pools of both F-actin and myosin II were important in normal stimulation-associated membrane transformations in parietal cells (4, 28, 56). Rapid hormone- or Ca2+-regulated trafficking of membrane vesicles in renal cortical collecting duct cells, presynaptic neurons, and muscle cells (17, 57, 63–65) are also blocked by either ML-7 or jasplakinolide, indicating that the actomyosin network is important for proper trafficking of membrane vesicles in many cell types in addition to parietal cells.
An important negative result is that once parietal cells had become fully activated by histamine, cells became resistant to ML-7 and jasplakinolide, suggesting that the inhibitory effect of these drugs is selective and relative to the physiological state of parietal cells. The lack of effect of the cytoskeletal inhibitors on structure of histamine-stimulated cells may result from histamine-induced dissociation of myosin from the ASC. In any case, our data indicate that the ASC and associated cytoskeleton behaves quite differently in resting cells (which have a dynamic, rapidly turning over actomyosin network) vs. histamine-stimulated cells (which appear to have a more stable actomyosin network). Overall, our data indicate that in normal parietal cells the coordination of the activities of the dynamic pools of myosin IIB and F-actin, perhaps as a complex, are required to maintain normal ASC morphology and a physiological state primed for histamine-stimulated translocation of TV and their fusion to ASC. Once parietal cells have achieved their stimulated morphology, the dynamic pools of actin and myosin are dispensable.
GRANTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases Grant AM-10141 to J. G. Forte.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
P.N., J.G.F., and T.E.M. conception and design of research; P.N., J.M.C.J., J.E.R., S.L.N., and M.R. performed experiments; P.N. analyzed data; P.N., J.M.C.J., and T.E.M. interpreted results of experiments; P.N. prepared figures; P.N. drafted manuscript; P.N., C.T.O., and T.E.M. edited and revised manuscript; P.N., C.T.O., and T.E.M. approved final version of manuscript.
Supplementary Material
ACKNOWLEDGMENTS
This study and paper were completed following Professor Forte's death on November 19, 2012. We believe the paper accurately presents his views and ideas.
REFERENCES
- 1.Agnew BJ, Duman JG, Watson CL, Coling DE, Forte JG. Cytological transformations associated with parietal cell stimulation: critical steps in the activation cascade. J Cell Sci 112: 2639–2646, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Akagi K, Nagao T, Urushidani T. Responsiveness of beta-escin-permeabilized rabbit gastric gland model: effects of functional peptide fragments. Am J Physiol Gastrointest Liver Physiol 277: G736–G744, 1999. [DOI] [PubMed] [Google Scholar]
- 3.Algrain M, Turunen O, Vaheri A, Louvard D, Arpin M. Ezrin contains cytoskeleton and membrane binding domains accounting for its proposed role as a membrane-cytoskeletal linker. J Cell Biol 120: 129–139, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ammar DA, Nguyen PN, Forte JG. Functionally distinct pools of actin in secretory cells. Am J Physiol Cell Physiol 281: C407–C417, 2001. [DOI] [PubMed] [Google Scholar]
- 5.Andzelm MM, Chen X, Krzewski K, Orange JS, Strominger JL. Myosin IIA is required for cytolytic granule exocytosis in human NK cells. J Exp Med 204: 2285–2291, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black JA, Forte TM, Forte JG. The effects of microfilament disrupting agents on HCl secretion and ultrastructure of piglet gastric oxyntic cells. Gastroenterology 83: 595–604, 1982. [PubMed] [Google Scholar]
- 7.Bresnick AR. Molecular mechanisms of nonmuscle myosin-II regulation. Curr Opin Cell Biol 11: 26–33, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem 269: 14869–14871, 1994. [PubMed] [Google Scholar]
- 9.Burnette DT, Ji L, Schaefer AW, Medeiros NA, Danuser G, Forscher P. Myosin II activity facilitates microtubule bundling in the neuronal growth cone neck. Dev Cell 15: 163–169, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol 11: 237–251, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavey M, Lecuit T. Imaging cellular and molecular dynamics in live embryos using fluorescent proteins. Methods Mol Biol 420: 219–238, 2008. [DOI] [PubMed] [Google Scholar]
- 12.Charras G, Paluch E. Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9: 730–736, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. Life and times of a cellular bleb. Biophys J 94: 1836–1853, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol 11: 62–74, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Chew CS. Parietal cell culture: new models and directions. Annu Rev Physiol 56: 445–461, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Chew CS, Ljungstrom M, Smolka A, Brown MR. Primary culture of secretagogue-responsive parietal cells from rabbit gastric mucosa. Am J Physiol Gastrointest Liver Physiol 256: G254–G263, 1989. [DOI] [PubMed] [Google Scholar]
- 17.Chou CL, Christensen BM, Frische S, Vorum H, Desai RA, Hoffert JD, de Lanerolle P, Nielsen S, Knepper MA. Non-muscle myosin II and myosin light chain kinase are downstream targets for vasopressin signaling in the renal collecting duct. J Biol Chem 279: 49026–49035, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Crivat G, Taraska JW. Imaging proteins inside cells with fluorescent tags. Trends Biotechnol 30: 8–16, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham CC. Actin polymerization and intracellular solvent flow in cell surface blebbing. J Cell Biol 129: 1589–1599, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature 443: 651–657, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Egelhoff TT, Lee RJ, Spudich JA. Dictyostelium myosin heavy chain phosphorylation sites regulate myosin filament assembly and localization in vivo. Cell 75: 363–371, 1993. [DOI] [PubMed] [Google Scholar]
- 22.Ellenberg J, Lippincott-Schwartz J, Presley JF. Dual-colour imaging with GFP variants. Trends Cell Biol 9: 52–56, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Even-Faitelson L, Ravid S. PAK1 and aPKCzeta regulate myosin II-B phosphorylation: a novel signaling pathway regulating filament assembly. Mol Biol Cell 17: 2869–2881, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol 9: 299–309, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol 181: 879–884, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flannagan RS, Harrison RE, Yip CM, Jaqaman K, Grinstein S. Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J Cell Biol 191: 1205–1218, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forte JG, Black JA, Forte TM, Machen TE, Wolosin JM. Ultrastructural changes related to functional activity in gastric oxyntic cells. Am J Physiol Gastrointest Liver Physiol 241: G349–G358, 1981. [DOI] [PubMed] [Google Scholar]
- 28.Forte JG, Ly B, Rong Q, Ogihara S, Ramilo M, Agnew B, Yao X. State of actin in gastric parietal cells. Am J Physiol Cell Physiol 274: C97–C104, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Forte JG, Zhu L. Apical recycling of the gastric parietal cell H,K-ATPase. Annu Rev Physiol 72: 273–296, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Forte TM, Machen TE, Forte JG. Ultrastructural changes in oxyntic cells associated with secretory function: a membrane-recycling hypothesis. Gastroenterology 73: 941–955, 1977. [PubMed] [Google Scholar]
- 31.Guha M, Zhou M, Wang YL. Cortical actin turnover during cytokinesis requires myosin II. Curr Biol 15: 732–736, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Gutknecht J, Walter A. SCN− and HSCN transport through lipid bilayer membranes. A model for SCN− inhibition of gastric acid secretion. Biochim Biophys Acta 685: 233–240, 1982. [DOI] [PubMed] [Google Scholar]
- 33.He Y, Beatty A, Han X, Ji Y, Ma X, Adelstein RS, Yates JR, 3rd, Kemphues K, Qi L. Nonmuscle myosin IIB links cytoskeleton to IRE1alpha signaling during ER stress. Dev Cell 23: 1141–1152, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holzinger A, Meindl U. Jasplakinolide, a novel actin targeting peptide, inhibits cell growth and induces actin filament polymerization in the green alga Micrasterias. Cell Motil Cytoskeleton 38: 365–372, 1997. [DOI] [PubMed] [Google Scholar]
- 35.Jerdeva GV, Wu K, Yarber FA, Rhodes CJ, Kalman D, Schechter JE, Hamm-Alvarez SF. Actin and non-muscle myosin II facilitate apical exocytosis of tear proteins in rabbit lacrimal acinar epithelial cells. J Cell Sci 118: 4797–4812, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaksonen M, Toret CP, Drubin DG. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 7: 404–414, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Konstantinidis DG, Pushkaran S, Johnson JF, Cancelas JA, Manganaris S, Harris CE, Williams DA, Zheng Y, Kalfa TA. Signaling and cytoskeletal requirements in erythroblast enucleation. Blood 119: 6118–6127, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kopic S, Geibel JP. Update on the mechanisms of gastric acid secretion. Curr Gastroenterol Rep 12: 458–464, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Kovacs M, Toth J, Hetenyi C, Malnasi-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem 279: 35557–35563, 2004. [DOI] [PubMed] [Google Scholar]
- 40.Lecuit T, Lenne PF, Munro E. Force generation, transmission, and integration during cell and tissue morphogenesis. Annu Rev Cell Dev Biol 27: 157–184, 2011. [DOI] [PubMed] [Google Scholar]
- 41.Levayer R, Lecuit T. Biomechanical regulation of contractility: spatial control and dynamics. Trends Cell Biol 22: 61–81, 2012. [DOI] [PubMed] [Google Scholar]
- 42.Ma X, Kovacs M, Conti MA, Wang A, Zhang Y, Sellers JR, Adelstein RS. Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc Natl Acad Sci USA 109: 4509–4514, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangeat P, Gusdinar T, Sahuquet A, Hanzel DK, Forte JG, Magous R. Acid secretion and membrane reorganization in single gastric parietal cell in primary culture. Biol Cell 69: 223–231, 1990. [DOI] [PubMed] [Google Scholar]
- 44.Matsumura F. Regulation of myosin II during cytokinesis in higher eukaryotes. Trends Cell Biol 15: 371–377, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Meshki J, Douglas SD, Hu M, Leeman SE, Tuluc F. Substance P induces rapid and transient membrane blebbing in U373MG cells in a p21-activated kinase-dependent manner. PloS One 6: e25332, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miserey-Lenkei S, Chalancon G, Bardin S, Formstecher E, Goud B, Echard A. Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat Cell Biol 12: 645–654, 2010. [DOI] [PubMed] [Google Scholar]
- 47.Murthy K, Wadsworth P. Myosin-II-dependent localization and dynamics of F-actin during cytokinesis. Curr Biol 15: 724–731, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Nakada SL, Crothers JM, Jr, Machen TE, Forte JG. Apical vacuole formation by gastric parietal cells in primary culture: effect of low extracellular Ca2+. Am J Physiol Cell Physiol 303: C1301–C1311, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nightingale TD, Cutler DF, Cramer LP. Actin coats and rings promote regulated exocytosis. Trends Cell Biol 22: 329–337, 2012. [DOI] [PubMed] [Google Scholar]
- 50.Okamoto CT, Forte JG. Vesicular trafficking machinery, the actin cytoskeleton, and H+-K+-ATPase recycling in the gastric parietal cell. J Physiol 532: 287–296, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pollard TD, Cooper JA. Actin, a central player in cell shape and movement. Science 326: 1208–1212, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, Rumbaugh G. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67: 603–617, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ridley AJ. Life at the leading edge. Cell 145: 1012–1022, 2011. [DOI] [PubMed] [Google Scholar]
- 54.Robinson DN, Spudich JA. Towards a molecular understanding of cytokinesis. Trends Cell Biol 10: 228–237, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Ronen D, Ravid S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J Biol Chem 284: 24948–24957, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenfeld GC, McAllister E, Thompson WJ. Cytochalasin inhibition of isolated rat gastric parietal cell function. J Cell Physiol 109: 53–57, 1981. [DOI] [PubMed] [Google Scholar]
- 57.Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci 19: 1317–1323, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Senderowicz AM, Kaur G, Sainz E, Laing C, Inman WD, Rodriguez J, Crews P, Malspeis L, Grever MR, Sausville EA, Duncan KL. Jasplakinolide's inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J Natl Cancer Inst 87: 46–51, 1995. [DOI] [PubMed] [Google Scholar]
- 59.Solecki DJ, Trivedi N, Govek EE, Kerekes RA, Gleason SS, Hatten ME. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron 63: 63–80, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soroka CJ, Chew CS, Hanzel DK, Smolka A, Modlin IM, Goldenring JR. Characterization of membrane and cytoskeletal compartments in cultured parietal cells: immunofluorescence and confocal microscopic examination. Eur J Cell Biol 60: 76–87, 1993. [PubMed] [Google Scholar]
- 61.Sugita Y, Nagao T, Urushidani T. Nonspecific effects of the pharmacological probes commonly used to analyze signal transduction in rabbit parietal cells. Eur J Pharmacol 365: 77–89, 1999. [DOI] [PubMed] [Google Scholar]
- 62.Sun SX, Walcott S, Wolgemuth CW. Cytoskeletal cross-linking and bundling in motor-independent contraction. Curr Biol 20: R649–R654, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terada Y, Simerly C, Schatten G. Microfilament stabilization by jasplakinolide arrests oocyte maturation, cortical granule exocytosis, sperm incorporation cone resorption, and cell-cycle progression, but not DNA replication, during fertilization in mice. Mol Reprod Devel 56: 89–98, 2000. [DOI] [PubMed] [Google Scholar]
- 64.Tong P, Khayat ZA, Huang C, Patel N, Ueyama A, Klip A. Insulin-induced cortical actin remodeling promotes GLUT4 insertion at muscle cell membrane ruffles. J Clin Invest 108: 371–381, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Torok D, Patel N, Jebailey L, Thong FS, Randhawa VK, Klip A, Rudich A. Insulin but not PDGF relies on actin remodeling and on VAMP2 for GLUT4 translocation in myoblasts. J Cell Sci 117: 5447–5455, 2004. [DOI] [PubMed] [Google Scholar]
- 66.Totsukawa G, Wu Y, Sasaki Y, Hartshorne DJ, Yamakita Y, Yamashiro S, Matsumura F. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J Cell Biol 164: 427–439, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ubukawa K, Guo YM, Takahashi M, Hirokawa M, Michishita Y, Nara M, Tagawa H, Takahashi N, Komatsuda A, Nunomura W, Takakuwa Y, Sawada K. Enucleation of human erythroblasts involves non-muscle myosin IIB. Blood 119: 1036–1044, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Leeuwen FN, van Delft S, Kain HE, van der Kammen RA, Collard JG. Rac regulates phosphorylation of the myosin-II heavy chain, actinomyosin disassembly and cell spreading. Nat Cell Biol 1: 242–248, 1999. [DOI] [PubMed] [Google Scholar]
- 69.Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta 1761: 957–967, 2006. [DOI] [PubMed] [Google Scholar]
- 70.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10: 778–790, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walcott S, Sun SX. Active force generation in cross-linked filament bundles without motor proteins. Phys Rev E Stat Nonlin Soft Matter Phys 82: 050901, 2010. [DOI] [PubMed] [Google Scholar]
- 72.Wang A, Ma X, Conti MA, Adelstein RS. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem Soc Trans 39: 1131–1135, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wiggan O, Shaw AE, DeLuca JG, Bamburg JR. ADF/cofilin regulates actomyosin assembly through competitive inhibition of myosin II binding to F-actin. Dev Cell 22: 530–543, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilson CA, Tsuchida MA, Allen GM, Barnhart EL, Applegate KT, Yam PT, Ji L, Keren K, Danuser G, Theriot JA. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature 465: 373–377, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolosin JM, Okamoto C, Forte TM, Forte JG. Actin and associated proteins in gastric epithelial cells. Biochim Biophys Acta 761: 171–182, 1983. [DOI] [PubMed] [Google Scholar]
- 76.Yao X, Chaponnier C, Gabbiani G, Forte JG. Polarized distribution of actin isoforms in gastric parietal cells. Mol Biol Cell 6: 541–557, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yao X, Forte JG. Cell biology of acid secretion by the parietal cell. Annu Rev Physiol 65: 103–131, 2003. [DOI] [PubMed] [Google Scholar]
- 78.Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol 65: 761–789, 2003. [DOI] [PubMed] [Google Scholar]
- 79.Zhou R, Watson C, Fu C, Yao X, Forte JG. Myosin II is present in gastric parietal cells and required for lamellipodial dynamics associated with cell activation. Am J Physiol Cell Physiol 285: C662–C673, 2003. [DOI] [PubMed] [Google Scholar]
- 80.Zhu L, Hatakeyama J, Chen C, Shastri A, Poon K, Forte JG. Comparative study of ezrin phosphorylation among different tissues: more is good; too much is bad. Am J Physiol Cell Physiol 295: C192–C202, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu L, Liu Y, Forte JG. Ezrin oligomers are the membrane-bound dormant form in gastric parietal cells. Am J Physiol Cell Physiol 288: C1242–C1254, 2005. [DOI] [PubMed] [Google Scholar]
- 82.Zhu L, Zhou R, Mettler S, Wu T, Abbas A, Delaney J, Forte JG. High turnover of ezrin T567 phosphorylation: conformation, activity, and cellular function. Am J Physiol Cell Physiol 293: C874–C884, 2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.