

Abstract
Many of the factors that contribute to CD8+ T cell immunodominance hierarchies during viral infection are known. However, the functional differences that exist between dominant and subdominant epitope-specific CD8+ T cells remain poorly understood. In this study, we characterized the phenotypic and functional differences between dominant and subdominant simian immunodeficiency virus (SIV) epitope-specific CD8+ T cells restricted by the major histocompatibility complex (MHC) class I allele Mamu-A*01 during acute and chronic SIV infection. Whole genome expression analyses during acute infection revealed that dominant SIV epitope-specific CD8+ T cells had a gene expression profile consistent with greater maturity and higher cytotoxic potential than subdominant epitope-specific CD8+ T cells. Flow-cytometric measurements of protein expression and anti-viral functionality during chronic infection confirmed these phenotypic and functional differences. Expression analyses of exhaustion-associated genes indicated that LAG-3 and CTLA-4 were more highly expressed in the dominant epitope-specific cells during acute SIV infection. Interestingly, only LAG-3 expression remained high during chronic infection in dominant epitope-specific cells. We also explored the binding interaction between peptide:MHC (pMHC) complexes and their cognate TCRs to determine their role in the establishment of immunodominance hierarchies. We found that epitope dominance was associated with higher TCR:pMHC affinity. These studies demonstrate that significant functional differences exist between dominant and subdominant epitope-specific CD8+ T cells within MHC-restricted immunodominance hierarchies and suggest that TCR:pMHC affinity may play an important role in determining the frequency and functionality of these cell populations. These findings advance our understanding of the regulation of T cell immunodominance and will aid HIV vaccine design.
Author Summary
MHC-restricted CD8+ T cell populations that bind viral proteins are often present at different frequencies. It is thought that those virus-specific CD8+ T cells that are present at the highest frequency are predominantly responsible for eliciting control of viral infections. While the number of virus-specific CD8+ T cells is undoubtedly important, the functionality of these cells may also play an anti-viral role. It is not known if high-frequency virus-specific CD8+ T cells are more functionally effective against viral infection than those present at low frequencies. In this study, we characterized the functional differences between the SIV-specific cells present at high versus low frequencies in rhesus monkeys infected with simian immunodeficiency virus (SIV). We found that the high- and low-frequency SIV-specific cells had different functional capacities during acute and chronic SIV infection. We also found that the affinity with which a cell interacts with viral proteins may contribute to these functional differences. These findings further our understanding of anti-viral immune responses and may help to inform HIV vaccine development.
Introduction
Virus-specific CD8+ T cells contribute to the control of HIV and SIV replication [1], [2] and are therefore an important component of a protective immunity. In infected or vaccinated individuals, the frequencies of different viral epitope-specific CD8+ T cells vary considerably [3], [4]. Epitopes restricted by the same MHC Class I allele can be ranked in an immunodominance hierarchy based on the relative frequencies of their respective epitope-specific CD8+ T cells [5]. The determinants of immunodominance hierarchies have been explored in the past in efforts to enhance the magnitude of particular epitope-specific CD8+ T cell responses through vaccination [6], [7]. However, such investigations have primarily focused on determining the mechanisms underlying the establishment of immunodominance [8]–[12]; less is known about the resulting functional differences between dominant and subdominant epitope-specific CD8+ T cells.
CD8+ T cell polyfunctionality is often associated with superior viral control [13]. Numerous studies have reported that individuals with superior control of HIV replication and delayed disease progression have higher frequencies of polyfunctional CD8+ T cells and therefore such cells are thought to be an important component of a protective immune response during HIV infection [14]–[17]. Therefore, HIV vaccine development is particularly focused on the identification of vaccine strategies that will generate highly polyfunctional, and therefore protective, responses [18]. In order to do so, a better understanding of the factors that contribute to the polyfunctionality of a CD8+ T cell response is needed.
Few studies have evaluated the functional differences between dominant and subdominant epitope-specific CD8+ T cells. Studies in DNA-immunized mice showed that subdominant epitope-specific CD8+ T cells were less cytotoxic, but produced more cytokines, than dominant epitope-specific CD8+ T cells during acute infection [19]. Moreover, a cryptic epitope-specific CD8+ T cell population exhibited an altered maturation phenotype when compared to cells comprising the dominant response [20]. A study of the functionality of CD8+ T cells within the HLA-B27-restricted immunodominance hierarchy in HIV-infected individuals found higher-frequency epitope-specific CD8+ T cells were associated with superior in vitro viral suppression; although, subsequent functional studies could not identify a particular function associated with this suppressive capacity [21]. Additional knowledge of functional differences between dominant and subdominant epitope-specific CD8+ T cell responses is needed for enhancing CD8+ T cell epitope-specific responses by vaccination.
In the present studies, we performed longitudinal gene expression analysis in SIV-infected Mamu-A*01 + rhesus monkeys to assess potential functional differences between dominant and subdominant epitope-specific CD8+ T cells during acute infection. We demonstrate that dominant epitope-specific CD8+ T cells display gene expression patterns consistent with a more mature phenotype and harbor greater cytotoxic potential than subdominant epitope-specific CD8+ T cells during acute infection. Protein expression analyses of CD8+ T cells sampled during chronic infection confirmed the presence of these functional differences between dominant and subdominant epitope-specific CD8+ T cells. Finally, dominant and subdominant epitope TCR:pMHC affinities were correlated with relative immunodominance. These findings advance our understanding of the basis underlying immunodominance and may better inform T cell-based HIV vaccine design.
Results
p11C- and p54AS-specific CD8+ T cell frequencies and viral loads during acute SIVmac251 infection
We infected six Mamu-A*01 + rhesus monkeys with SIVmac251. The Mamu-A*01-restricted dominant SIV Gag p11C- and subdominant SIV Env p54AS-specific CD8+ T cells were quantified and sorted by flow cytometry on days 14, 21, 28, 35, 42, 56, and 70 post-inoculation. The Mamu-A*01-restricted CD8+ T cell response is one of the best characterized CD8+ T cell responses in SIV-infected rhesus monkeys [22], [23]. The p11C epitope (Gag CM9) has been used widely as a model dominant epitope in studies of SIV-specific immunity [24]–[26]. p54AS (Env TL9) is a well-described subdominant epitope that, from our experience, was the most likely to be elicited in all Mamu-A*01 + rhesus monkeys at a frequency that was high enough such that we could collect sufficient numbers of cells to facilitate our gene expression measurements.
The differences in frequency between the p11C- and p54AS-specific CD8+ T cells were apparent upon detection in the peripheral blood (day 14) and were maintained throughout the duration of infection (Figure 1A and Figure S1). The plasma viral loads peaked at approximately 7 logs of viral RNA copies/mL by day 14 and reached setpoint of approximately 5.5 logs of viral RNA/mL by day 28 (Figure 1B).
Figure 1. Frequencies of p11C- and p54AS-specific CD8+ T cells and plasma viral loads during primary infection.
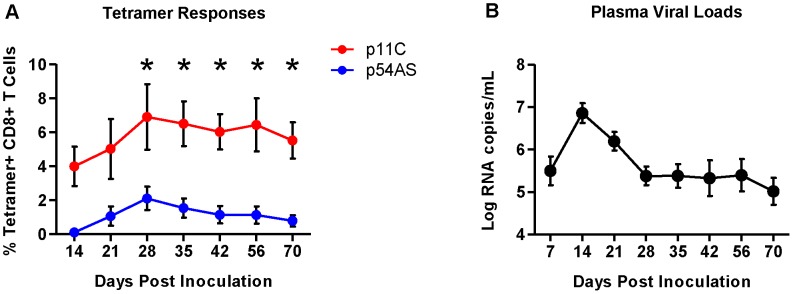
A) Mean frequencies of the p11C- and p54AS-specific CD8+ T cells in peripheral blood. * significant at p≤0.05. B) Plasma SIV RNA levels in the peripheral blood.
We measured RNA expression on the sorted epitope-specific cell populations at various timepoints post infection. We also sorted total naïve CD8+ T cells (CD95−CD28+) before challenge (day 0) and measured RNA expression in these cells to establish a baseline expression level for each transcript. We generated expression data from both epitope-specific cell populations from all six monkeys only on days 14, 21, 56 and 70. We obtained expression data for both epitope-specific populations in only some of the monkeys on the three other time points: days 35 and 42 (4 of 6 monkeys) and day 28 (5 of 6 monkeys). Therefore, sometimes we observed trends suggesting differential expression (Figure S2) that did not achieve statistical significance possibly due to reduced power.
To characterize the functional differences between the dominant p11C- and subdominant p54AS-specific cells based on their RNA expression profiles, we focused expression analysis on genes known to be associated with specific CD8+ T cell functions. These genes were grouped into the following categories: maturation, cytotoxicity, cell cycle and apoptosis, and cytokines and chemokines (Table 1). Genes were considered differentially expressed if they satisfied both of the following criteria: 1) expression was significantly different between the two epitope-specific cells on at least one timepoint and 2) the median fold difference of expression at this timepoint (p11C/p54AS) was at least ±1.5. Technical replicates were not conducted; gene expression was measured once per monkey per timepoint.
Table 1. Genes compared between dominant and subdominant epitope-specific CD8+ T cells.
| Maturation | Cytotoxicity | Cytokines and Chemokines | Cell Cycle and Apoptosis | |||||
| B3GAT1 | AP3B1 | CCL3 | AIFM1 | BRCA2 | CDKN1A | HDAC2 | NAIP | SMAD2 |
| BCL6 | C1ORF9 | TNFSF11 | AIFM2 | BTRC | CDKN1B | HDAC3 | NCAPD2 | SMAD3 |
| BMI1 | CSPG5 | CCL7 | AKT1 | CABIN1 | CDKN1C | HRAS | NCAPG | SMAD4 |
| CCR7 | CTSC | CCL4 | ANAPC1 | CASP1 | CDKN2A | HRK | NCAPH | SMAD5 |
| CD27 | EBAG9 | CCL5 | ANAPC10 | CASP10 | CDKN2B | HTRA2 | NFKB1 | SMAD6 |
| CD28 | GNLY | TGFB1 | ANAPC11 | CASP12 | CDKN2C | HUS1 | NFKB2 | SMAD7 |
| CXCR3 | GZMA | IL12A | ANAPC13 | CASP2 | CDKN2D | ID1 | NOL3 | SMAD9 |
| EOMES | GZMB | MIF | ANAPC16 | CASP3 | CEBPA | ID2 | NRAS | SMC2 |
| GFI1 | GZMH | IL22 | ANAPC2 | CASP6 | CEBPB | ID3 | NUSAP1 | SMC4 |
| ID2 | GZMK | IL16 | ANAPC4 | CASP7 | CENPF | ID4 | PCNA | SNRPE |
| IL2RA | IQGAP1 | IL26 | ANAPC5 | CASP8 | CFLAR | IGF1 | PEA-15 | SOD2 |
| IL7R | JAKMIP1 | IL29 | ANAPC7 | CASP9 | CHEK1 | IGFBP3 | PKMYT1 | SP1 |
| ITGAL | LAMP2 | IL1A | APAF1 | CCNA1 | CHEK2 | IRF1 | PLK1 | STK11 |
| KLF2 | LYST | IFNW1 | APC | CCNA2 | CKS1B | IRF2 | PMAIP1 | SUV39H1 |
| KLRG1 | M6PR | IL10 | ARAF | CCNB1 | CKS2 | IRF3 | PPARA | TACC3 |
| MBD2 | PRF1 | SPP1 | ARHGAP19 | CCNB2 | CPEB1 | IRF4 | PPKACA | TCL1A |
| PRDM1 | RAB27A | TNFSF14 | ATM | CCNC | CUL1 | IRF8 | PPKACB | TERT |
| SELL | SMPD1 | IFNB1 | ATR | CCND1 | DAP | IRF9 | PRKAR1A | TFDP1 |
| SPN | SNAP23 | XCL2 | AURKB | CCND2 | DAXX | JUN | PRKAR1B | TFDP2 |
| TBX21 | SNAP25 | XCL1 | BAD | CCND3 | DIABLO | JUNB | PRKAR2A | THAP5 |
| XBP1 | SRGN | OSM | BAK1 | CCNDBP1 | DLG1 | KAT2B | PRKAR2B | TNFR1 |
| STOML2 | CX3CL1 | BARD1 | CCNE1 | DLG3 | KIT | PRKDC | TNFRSF10A | |
| STX11 | CCL20 | BAX | CCNE2 | DLG4 | KLF10 | PRPS1 | TNFRSF10B | |
| STXBP2 | IL17D | BBC3 | CCNF | E2F1 | KLF4 | RAC1 | TNFRSF21 | |
| SYTL1 | IL17F | BCL2 | CCNG2 | E2F2 | KLF5 | RAD1 | TNFRSF25 | |
| SYTL2 | IL17A | BCL2A1 | CCNH | E2F3 | KLF6 | RAF1 | TNFSF10 | |
| TFF1 | IFNG | BCL2L1 | CDC16 | E2F4 | KRAS | RALA | TP53 | |
| TRIP10 | LTB | BCL2L10 | CDC20 | E2F5 | MAP2K1 | RALB | TP53AIP1 | |
| UNC13D | LTA | BCL2L11 | CDC23 | E2F6 | MAP2K2 | RALGDS | TP73 | |
| VAMP7 | TNF | BCL2L14 | CDC25A | E2F7 | MAP3K5 | RB1 | TRADD | |
| VAMP8 | FASLG | BCL2L2 | CDC25B | ECT2 | MAP3K7 | RBL1 | TRAF2 | |
| CXCL11 | BCL3 | CDC25C | EGR1 | MAPK1 | RBL2 | TRIM25 | ||
| CXCL10 | BCL6 | CDC27 | EIF4EBP1 | MAPK14 | REL | TSC1 | ||
| IL18 | BID | CDC2L2 | ENDOG | MAPK7 | RELA | TSC2 | ||
| CXCL5 | BIK | CDC42 | EP300 | MAX | RELB | TSC22D1 | ||
| IRF4 | BIN1 | CDC42EP4 | FADD | MCL1 | RGL1 | TTK | ||
| CCL18 | BIRC2 | CDCA8 | FAS | MDM2 | RGL2 | UBE2C | ||
| IFNA1 | BIRC3 | CDK1 | FASLG | MDM4 | RHEB | UHRF1 | ||
| IL13 | BIRC5 | CDK10 | FBX05 | MEF2A | RHEBL1 | WEE1 | ||
| CSF2 | BIRC6 | CDK11A | FLT3 | MEF2B | RHOA | XIAP | ||
| IL6 | BIRC7 | CDK11B | FOSL1 | MEF2C | RING1 | YWHAB | ||
| CXCL9 | BIRC8 | CDK13 | FOSL2 | MEF2D | RPS6KB1 | YWHAE | ||
| IL4 | BLK | CDK16 | FOXO1 | MGA | RPS6KB2 | YWHAG | ||
| IL5 | BMF | CDK2 | FOXO3 | MKI67 | RRAS | YWHAH | ||
| IL2 | BNIP3 | CDK3 | FOXO4 | MNAT1 | RRAS2 | YWHAQ | ||
| IL3 | BNIP3L | CDK4 | FZR1 | MXD1 | SCRIB | YWHAZ | ||
| IL8 | BOK | CDK5 | GABPA | MXD3 | SFN | YY1 | ||
| BRAF | CDK6 | GADD45A | MYB | SKP2 | ZMIZ1 | |||
| BRCA1 | CDK7 | HDAC1 | MYC | SMAD1 | ||||
Maturation-associated gene expression
Of the genes involved in CD8+ T cell maturation, IL7R (CD127/IL-7 receptor, refseq accession # XM_937367.1), SELL (CD62L/L-selectin, NM_000655.3), and CCR7 (NM_001838.2) met our criteria for differential expression following SIV infection (Figure 2A). While the trends of expression of these three genes between p11C- and the p54AS-specific cells were similar over time, all of these genes were found to be expressed at relatively higher levels in the subdominant p54AS-specific cells compared to the dominant p11C-specific cells. IL7R expression was higher in the p54AS-specific cells with significant fold differences of 1.8 on day 14, 1.5 on day 56, and 1.9 on day 70. SELL expression was also higher in the p54AS-specific cells with significant fold differences of 1.8 on day 14, 2.0 on day 21, and 1.7 on day 56. CCR7 was more highly expressed in the p54AS-specific cells with a significant fold difference of 2.4 on day 14. While the expression of IL7R and SELL tended to be higher in the p54AS-specific cells for all timepoints examined, the expression for CCR7 appeared only to be different on day 14 (Figure 2A and Figure S2A).
Figure 2. Genes differentially expressed between dominant p11C- and subdominant p54AS-specific CD8+ T cells.

The median of the normalized expression values, measured in fluorescence units, of A) maturation, B) cytotoxicity, and C) proliferation and apoptosis genes that were found to be differentially expressed between the p11C- and p54AS-specific cells on at least one time point are shown for each timepoint evaluated during the first 70 days following SIVmac251 infection. The expression of these genes by total naïve CD8+ T cells, measured on day 0, is also shown. For those genes with more than one probe on the BeadChip, the numerical probe IDs are included in the gene name. * indicates that expression met the criteria for differential expression on that timepoint.
Cytotoxicity-associated gene expression
Of the genes involved in cytotoxicity, GZMB (granzyme B, NM_004131.3), GZMK (granzyme K, NM_002104.2), and LAMP2 (CD107b, NM_013995.1) met our criteria for differential expression following SIV infection (Figure 2B). LAMP2 was measured on the BeadChip by three different probes, two of which measured differential expression (probes 1659753 and 1752351). Expression levels of each of the three cytotoxicity-associated genes were found to be increased in the dominant p11C-specific cells compared to the subdominant p54AS-specific cells. On day 14, the p11C-specific cells exhibited higher expression of both GZMB (significant fold difference of 1.5) and GZMK (significant fold difference of 1.9). Although the trends in differential expression of these two granzyme genes were still apparent on day 21 and 28, differences were subsequently lost (Fig. 2B and Figure S2B). The two LAMP2 probes detected similar trends of expression of this gene. However, probe 1659753 detected significant differential expression on day 14 (1.5-fold) while probe 1752351 detected significant differential expression on day 70 (1.8-fold).
Cell cycle and apoptosis-associated gene expression
Of the cell cycle and apoptosis-associated genes, nine met our criteria for differential expression following SIV infection (Figure 2C and Figure S2C). Two of these genes, BIRC3 (AIP1/API2/c-IAP2, NM_001165.3) and CASP2 (caspase-2, NM_032983.2), are involved in apoptosis. BIRC3, which encodes a protein with multiple anti-apoptotic functions [27], was differentially expressed on day 14, showing 1.8-fold higher expression in the p54AS-specific cells. CASP2, which encodes a pro-apoptotic protease [28], was differentially expressed on day 14, showing 1.7-fold higher expression in the p11C-specific cells.
The remaining seven differentially expressed genes are involved in proliferation: UBE2C (ubiquitin-conjugating enzyme E2C, NM_181800.1), AURKB (aurora B kinase, NM_004217.2), CCND2 (cyclin D2, NM_001759.2), CDC42 (cell division cycle 42, NM_001039802.1), NUSAP1 (nucleolar and spindle associated protein 1, NM_018454.5), UHRF1 (ubiquitin-like with PHD and ring finger domains 1, NM_001048201.1), and ATM (ataxia telangiectasia, NM_000051.3). UBE2C and AURKB encode pro-proliferative genes involved in the regulation of the anaphase-promoting complex [29], [30]; both were more highly expressed in the p11C-specific cells on day 56 with 1.6- and 1.5- fold differences, respectively. CCND2, CDC42, NUSAP1, and UHRF1 also encode pro-proliferative genes [31]–[35]. All of these genes showed differential expression on day 70 and were more highly expressed in the p11C-specific cells: CCND2 by 1.6-fold, CDC42 by 1.9-fold, NUSAP1 by 2.7-fold, and UHRF1 by 2.5-fold. Finally, ATM, an anti-proliferative serine/threonine protein kinase [36], was differentially expressed on day 21 and was more highly expressed in the p54AS-specific cells with by 1.5-fold.
In summary, we detected differential expression of genes involved in CD8+ T cell maturation, cytotoxicity, and cell cycle and apoptosis suggesting that functional differences may exist between the dominant p11C-specific and the subdominant p54AS-specific cells. Decreased expression of the genes encoding CD127, CD62L, and CCR7 and the increased expression of genes encoding granzyme B, granzyme K, and CD107b (LAMP2) in the dominant p11C-specific cells compared to the subdominant p54AS-specific cells suggested that the p11C-specific cells were more mature and possessed greater cytotoxic potential than the p54AS-specific cells. Moreover, we observed that the p11C-specific cells had higher expression of pro-proliferative genes and an increased expression of a pro-apoptotic molecule.
The dominant epitope-specific CD8+ T cell population was enriched with cells that exhibit a more mature phenotype
Functional assays were performed on PBMCs isolated from chronically infected monkeys to confirm and extend the observations of differential gene expression patterns. In addition to the dominant p11C- and subdominant p54AS-specific cells, we studied an additional CD8+ T cell population of even lower frequency that recognizes the Mamu-A*01-restricted SIV Pol p68A (STPPLVRLV, Pol SV9621) subdominant epitope [23]. Additionally, we analyzed cells from monkeys infected with a different SIV isolate, SIVsmE660 [37]. The p54 epitope from the SIVsmE660 (TVPWPNETL) virus differs by two amino acids from the p54 epitope from SIVmac251 (TVPWPNASL) virus [22] and we refer to this epitope as p54E660. The immunodominance hierarchy of the p11C-, p54AS/E660-, and p68A-specific cells during chronic SIVsmE660 infection was similar to SIVmac251-infected monkeys (Figure 3A and Figure S3).
Figure 3. Phenotype and cytotoxic potential of dominant and subdominant epitope-specific cells during chronic SIVmac251 and SIVsmE660 infection.

The frequency, cell surface phenotype, and ex vivo perforin and granzyme B content were evaluated for p11C-, p54AS/E660-, and p68A-specific cells, identified with Mamu-A*01 tetramers, from peripheral blood of chronically infected SIVmac251- and SIVsmE660- infected rhesus monkeys. A) Frequencies of the p11C-, p54AS/E660-, and p68A-specific cells from (top) SIVmac251- and (bottom) SIVsmE660- infected rhesus monkeys. Error bars indicate median ± interquartile range. SIVmac251 frequencies measured between weeks 37–50, except for 133-06 which died early and data presented are from week 18. SIVsmE660 frequencies are measured between weeks 19–22. B) Phenotyping of cells from SIVmac251- (top, n = 4) and SIVsmE660-infected monkeys (bottom, n = 8) based on cell surface expression of CCR7 and CD28. Positivity for CCR7 and CD28 is indicated by the + and − signs below the bar graph. Cells were categorized as central memory (CCR7+CD28+), transitional memory (CCR7−CD28+), or effector memory (CCR7−CD28−). All tetramer-positive cells were CD95+. Phenotyping was conducted between weeks 44–78 for SIVmac251 and 33–46 for SIVsmE660. C) Measurement of percent of ex vivo perforin+granzyme B+ cells from SIVmac251- (Left, n = 5) or SIVsmE660-infected monkeys (right, n = 8). D) Per-cell expression of perforin and granzyme B, measured by geometric mean of fluorescence (GMF) of perforin and granzyme B staining on cells from SIVmac251- (left, n = 5) and SIVsmE660-infected monkeys (right, n = 8). Perforin and granzyme B measurements were conducted between weeks 63–83 for SIVmac251 and 41–49 for SIVsmE660. In parts B–D, error bars represent mean ± SEM. * and **, significant at p≤0.05 and 0.01, respectively, after Bonferroni correction. NS, not significant.
The maturation of tetramer-positive cell populations from SIVmac251- and SIVsmE660-infected monkeys was determined by measuring the surface expression of CCR7 and CD28. Cells were categorized as central memory (CCR7+CD28+), transitional memory (CCR7−CD28+), or fully differentiated effector memory (CCR7−CD28−) as previously described for rhesus monkeys [38]. All tetramer-positive cells were CD95+ (not shown). The dominant p11C-specific cells displayed a greater proportion of the more mature effector memory phenotype (CCR7−CD28−) compared to the subdominant p54AS-specific cells (Figure 3B and Figure S4). This is consistent with gene expression studies in SIVmac251-infected monkeys that expressed CCR7, CD62L, and SELL at lower levels in the p11C-specific cells, suggesting they were more mature than the p54AS-specific cells.
Additionally, we found that the more subdominant p68A-specific cells were phenotypically less mature compared to the subdominant p54AS/E660-specific cells (Figure 3B). Of the three epitope-specific cell populations, the p68A-specific population was the most enriched with cells of the less mature phenotypes and were the least enriched with cells of the mature effector memory phenotype. This trend was observed in both SIVmac251- and SIVsmE660-infected monkeys. While the distribution of phenotypes was similar between cells from SIVmac251- and SIVsmE660-infected monkeys, the sample size for SIVmac251 (n = 4) was much smaller than that of SIVsmE660 (n = 8) and was not sufficiently powered to detect significance.
To confirm that differences in gene expression of cytolytic molecules were similarly reflected in protein production, we measured the intracellular content of perforin and granzyme B within each tetramer-positive population. The dominant p11C-specific populations had a greater frequency of cells containing perforin and granzyme B than the subdominant p54AS/E660-specific population, which in turn had a greater frequency of perforin- and granzyme B-containing cells than the subdominant p68A-specific population (Figure 3C and Figure S5). The p11C-specific cells also contained more perforin and granzyme B per-cell (reflected by higher geometric mean fluorescence (GMF)) than the p54AS/E660-specific cells, which in turn contained more of these proteins than the p68A-specific cells (Figure 3D and Figure S5). These observations were consistent with previous findings that demonstrated not only that more mature cells are more likely to express perforin and granzymes [39], but also that more mature cells have more perforin and granzyme per cell than less mature cells [40].
The dominant p11C-specific CD8+ T cells exhibited decreased antigen-specific expansion compared to subdominant epitope-specific CD8+ T cells
Although differences in expression of genes involved in cell cycle and apoptosis were observed, it was unclear what impact these genes had on cell expansion upon antigen stimulation during chronic infection. To explore this, PBMCs from monkeys chronically infected with SIVmac251 or SIVsmE660 were peptide-stimulated and expansion of the epitope-specific cells was assessed over 14 days. We found that the dominant p11C-specific cells had reduced capacity to expand when compared to both of the subdominant epitope-specific cells (Figure 4 and Figure S6). This trend was consistent between cells from SIVsmE660- and SIVmac251-infected monkeys and is in agreement with prior reports that effector memory CD8+ T cells have limited proliferative capacity compared to less mature memory CD8+ T cells [41].
Figure 4. The dominant p11C-specific cells exhibited decreased antigen-specific expansion compared to subdominant epitope-specific cells.

PBMCs from monkeys chronically-infected with either SIVmac251 (A) or SIVsmE660 (B) were stimulated in vitro with either p11C (red), p54E660/AS (blue), or p68A (green) peptide, harvested on days 3, 4, 5, 6, 8, 10, 12, and 14 following stimulation, and measured by flow cytometry to calculate the percent of tetramer-positive CD8+ T cells. Expansion was calculated as the fold change of the percent of each tetramer-positive population on each day, relative to day 0. Data from three SIVmac251- and three SIVsmE660-infected monkeys are shown. Measurements were conducted between weeks 40–52 for SIVmac251 and 31–44 for SIVsmE660.
The dominant p11C-specific CD8+ T cell population contained a lower frequency of cytokine- and chemokine-producing cells than the subdominant epitope-specific CD8+ T cell populations
Our gene expression data did not demonstrate differences in cytokine or chemokine expression between the p11C- and p54AS-specific cells. However, cytokine genes often require re-stimulation immediately prior to measurement [42]. Therefore, we considered that differences in cytokine expression between dominant and subdominant epitope-specific cells might be observed following antigen stimulation. To investigate, we stimulated PBMCs with epitope peptides and measured IL-2, TNFα, IFNγ, and MIP1-β production in standard intracellular cytokine staining (ICS) assays (Figure S7). Lower frequencies of cells expressing each of these cytokines and chemokines were present in the dominant p11C-specific cell population compared to the subdominant epitope-specific cell populations (Figure 5A). Furthermore, the subdominant epitope-specific populations were more polyfunctional, exhibiting larger proportions of cells expressing three or four functions, compared to the dominant p11C-specific population (Figure 5B and 5C).
Figure 5. Dominant p11C-specific population contained lower frequency of cytokine- and chemokine-producing cells than subdominant epitope-specific populations.

PBMCs from monkeys chronically-infected with either SIVmac251 (n = 4) or SIVsmE660 (n = 5) were stimulated with either p11C, p54AS/E660, or p68A peptides and intracellular staining was used to assess production of the chemokine MIP-1β and cytokines IFNγ, TNFα, and IL-2. Bars represent mean ± SEM. A) Individual cytokine and chemokine production. Top, SIVmac251. Bottom, SIVsmE660. B) Polyfunctional analysis. Positivity for each cytokine/chemokine is indicated by the dots below the bar graph. Vertical bars are grouped into 4, 3, 2 or 1 function (indicated by the pink, light blue, purple, and orange horizontal bars, respectively). C) Polyfunctionality charts. Left, SIVmac251. Right, SIVsmE660. Each slice represents the percentage of tetramer-positive cells expressing between 1 and 4 functions. Data were collected between weeks 36–42 for SIVmac251 and 14–25 for SIVsmE660.
Expression of exhaustion-associated genes in dominant and subdominant CD8+ T cells
To assess differences in functionality that may be due to different extents of exhaustion, we compared the expression of genes encoding exhaustion-associated molecules between the p11C- and p54AS-specific cells during acute infection (day 14), at the onset of chronic infection (day 70), and during late chronic infection (week 31 or 32). The genes LAG3 (NM_002286.4) and CTLA4 (NM_005214.3) were significantly more highly expressed in the dominant p11C-specific cells during acute infection by 3.2- and 1.5-fold, respectively (Table 2). However, no genes were found differentially expressed on day 70. During late chronic infection, LAG3 was expressed at increased levels in the dominant p11C-specific cells (1.7-fold). Therefore, the dominant p11C-specific cells showed higher expression of exhaustion-associated genes than the subdominant p54AS-specific cells.
Table 2. Expression of exhaustion-associated genes.
| Median Fold Change (p11C/p54AS) | |||
| Gene (Protein) | Day 14 | Day 70 | Week 31/32 |
| cd244 (2B4) | −1.1 | −1.1 | 1.0 |
| havcr2 (TIM-3) | −1.0 | −1.1 | 1.1 |
| lag3 (LAG-3) | 3.2* | 1.0 | 1.7* |
| pdcd1 (PD-1) | −1.1 | −1.0 | −1.1 |
| ctla4 (CTLA-4) | 1.0 | 1.0 | −1.0 |
| ctla4 | 1.5* | 1.0 | −1.0 |
| ctla4 | −1.1 | −1.1 | 1.0 |
| prdm1 (BLIMP-1) | 1.1 | 1.1 | 1.1 |
| prdm1 | −1.0 | −1.2 | −1.0 |
| prdm1 | 1.0 | −1.1 | −1.0 |
| prdm1 | −1.2 | −1.1 | −1.0 |
| cd160 (CD160) | −1.1 | −1.2 | −1.2 |
| tbx21 (T-bet) | 1.1 | −1.1 | −1.1 |
| eomes (Eomesodermin) | −1.0 | −1.7 | −1.1 |
| klrg1 (KLRG1) | −1.1 | 1.1 | 1.2 |
| klrg1 | 1.1 | −1.1 | −1.0 |
* p≤0.05.
High TCR:pMHC affinity correlates with the epitope dominance
We were interested in identifying mechanisms underlying the differences in the frequency and function of epitope-specific cells within an immunodominance hierarchy. Since TCR:pMHC interactions can influence T cell activation, proliferation, and function [43]–[49], we considered that TCR:pMHC interactions might play a role in the establishment of the magnitude or of the functionality of CD8+ T cells within an immunodominance hierarchy. We employed a surface plasmon resonance (SPR)-based technique developed in our laboratory that assesses the interaction of TCRs from a polyclonal epitope-specific CD8+ T cell population with monomeric pMHC complexes [50] to evaluate the role of TCR:pMHC binding in determining the Mamu-A*01-restricted epitope immunodominance hierarchy.
For initial measurements of the binding of the p11C-, p54E660-, and p68A-specific TCRs to their respective monomeric pMHC complexes, we sorted total CD8+ T cells from seven Mamu-A*01+ SIVsmE660-infected monkeys. Detergent-resistant micro-domain (DRM) preparations from sorted cells, which were enriched in TCR complexes, were immobilized onto a Biacore L1 chip. Initial assays demonstrated that specific binding to DRM preparations could be detected using monomers constructed with the dominant p11C and subdominant p54E660 epitope peptides (Figure 6A and Figure S8). The binding signal from the p54E660 monomers was weaker than that of the p11C monomers at the same concentration. We were unable to detect specific binding of the monomers constructed with the more subdominant p68A epitope peptide at any of the concentrations of monomers (25–150 μg/mL) that were initially evaluated (Figure 6A).
Figure 6. TCR:pMHC measurements.

DRMs were purified from total CD8+ T cells sorted from seven chronically-infected SIVsmE660-infected monkeys. The DRMs were evaluated for specific binding, measured in resonance units (RU), to pMHC monomers constructed with p11C, p54E660, and p68A epitope peptides and Mamu-A*01. Data are representative of the binding observed from all seven monkeys evaluated. A) Initial experiments to detect p11C, p54E660, and p68A monomer binding to DRMs. 100 μg/mL p11C (red), 100 μg/mL p54E660 (blue), and 150 μg/mL p68A (green) pMHC monomer binding are overlaid from experiments conducted on separate Biacore Chips. Binding of the p68A:Mamu-A*01 monomer to the DRMs was not observed at any concentration of monomer tested (25–200 μg/mL). B) Titrations of p11C (top) and p54E660 (bottom) peptide:Mamu-A*01 monomers for calculation of binding kinetics and affinity. Overlaid sensograms of the binding of p11C and p54E660 pMHC monomers to DRMs purified from total CD8+ T cells are shown. Monomer binding was evaluated using pMHC concentrations ranging from 25 to 200 μg/mL. The black curve shows the Langmuir fitted curve that was used to calculate kinetics. C) Detection of p68A peptide:Mamu-A*01 monomer binding to DRMs. p68A-specific CD8+ T cells were collected from multiple tetramer-specific flow cytometric cell sorts and pooled for DRM purification. Titrations of p68A pMHC monomers were performed at concentrations ranging from 150 to 1000 μg/mL. Binding to DRMs from monkey ZD57 at 1000 μg/mL is shown and is representative of the four monkeys that were evaluated. All readings have been normalized by subtracting the binding of the control monomer TL8 run at the same concentrations as the experimental monomers.
We performed titrations of the p11C and p54E660 pMHC monomers on DRM preparations from all seven monkeys (Figure 6B and Figure S9) and used a 1∶1 Langmuir curve fitting analysis to calculate the association rate (ka) and the dissociation rate (kd) of the TCR:pMHC binding interaction (Table 3). The p11C:Mamu-A*01 monomers bound to the DRM samples with a median kon of 7.38×103/Ms (range 3.56–28.6×103/Ms), which was faster than the p54E660:Mamu-A*01 monomers that had a median kon of 0.96×103/Ms (range 0.66–1.87×103/Ms). The dissociation rate of p11C:Mamu-A*01 monomers from DRMs (median koff 0.024/s, range 0.015–0.030/s) was not substantially different from that of p54E660:Mamu-A*01 monomers (median koff of 0.033/s, range 0.021–0.061/s). The apparent dissociation constants (Kd) were derived from the kinetic rate constant values and showed that the p11C:Mamu-A*01 monomers had a lower median Kd (2.0 μM, range 1.0–6.8 μM) than the p54E660:Mamu-A*01 monomers (32 μM, range 22–43 μM). Thus, monomers constructed with the dominant p11C peptide epitope exhibited faster association rates to chip-immobilized DRMs, resulting in higher affinities (lower Kd) to TCRs than monomers constructed with the subdominant p54E660 epitope peptide.
Table 3. TCR:pMHC binding values.
| kon (×103M−1s−1) | koff (s−1) | Kd (μM) | |
| p11C | 7.38 (3.56–28.6) | 0.024 (0.015–0.030) | 2.0 (1.0–6.8) |
| p54E660 | 0.96 (0.66–1.87) | 0.033 (0.021–0.061) | 32 (22–43) |
| p68A | ≥10* | ≥1‡ | ≥100† |
Values are the median (range) from seven monkeys.
kon, association rate.
koff, dissociation rate.
Kd, equilibrium dissociation constant. Calculated using the equation koff/kon.
* kon of the p68A monomers was estimated as being at least as fast as the fastest measured monomer, p11C.
koff of p68A monomers was estimated as being at least as fast as the limit of detection of the Biacore instrument, approx.1/s.
Kd of p68A monomer binding calculated using estimated values of the kon and koff using the equation koff/kon.
We speculated that the inability to detect the binding of p68A:Mamu-A*01 monomers to the DRM preparations may be due to a substantially lower affinity of the p68A:Mamu-A*01 complex for its cognate TCRs. The difficulty in detecting binding was also likely exacerbated by the low frequency of the p68A-specific cells, which would have resulted in underrepresentation of the p68A-specific TCRs in the DRMs. Therefore, we pooled DRMs from p68A-specific cells sorted from multiple time points. These samples contained two- to six-fold more p68A-specific cells than samples from the initial experiments.
Using these DRM preparations containing more p68A-specific cells and using higher concentrations of p68A:Mamu-A*01 monomers (>100 μg/mL), we detected specific DRM binding (Figure 6C and Figure S10). However, specific monomer binding exhibited extremely fast association and dissociation rates and was detected only at very high concentrations. As a result, reliable Kd values could not be measured using global curve fitting analyses. Our previous measurement of p11C and p54E660 pMHC monomer binding to DRMs indicated that p11C had the fastest association rate. The association rate of the p68A monomer to its respective TCRs was at least as fast as the association of the quantifiable p11C monomer, although likely to be even faster. Therefore, we estimated that the kon for p68A was at least 10×103/Ms. Knowing that the Biacore instrument's lower limit of detection of dissociation is about 1/s, we estimated that the koff of the p68A:Mamu-A*01 monomer from its respective TCRs was at least as fast as 1/s. Using these estimated kon and koff, the apparent Kd of the p68A monomer was estimated be at least 100 μM (Table 3).
Therefore, the calculated Kd values were lowest for p11C monomers, intermediate for p54E660 monomers, and estimated to be highest for p68A monomers. In summary, epitope dominance was associated with the hierarchy of TCR affinities. Affinity differences were driven by the fast association rate of the dominant p11C epitope:Mamu-A*01 complex to its respective TCRs and likely fast dissociation rate of the more subdominant p68A:Mamu-A*01 complex.
Discussion
The development of an effective vaccine against a pathogen such as HIV-1 will require a detailed understanding of the interplay between epitope immunodominance and the functional capacities of epitope-specific CD8+ T cells. The present study was undertaken to explore this relationship in SIV-infected rhesus monkeys.
It is important to note that our longitudinal gene expression analyses were conducted with a restricted sample size of six monkeys. In addition, because of the large numbers of cells required for RNA isolation, we were unable to perform phenotyping or functional assays during acute infection. Despite these clear limitations, we were able to detect a number of significant differences in gene expression between the dominant and subdominant epitope-specific populations during acute infection. Baron et al had similar findings, in a murine model, where they characterized a dominant (H7a) and a cryptic (HY) H2-Db–restricted epitope-specific CD8+ T cell population [20]. In the murine model, the cryptic epitope-specific cells had higher IL7R expression during primary expansion and higher L-selectin/SELL on all timepoints evaluated. Similarly, we found that both IL7R and SELL were higher in the subdominant p54AS-specific cells. Additionally, Baron et al found that GZMA was highly expressed in the dominant epitope-specific CD8+ T cells. We found a trend toward higher expression of GZMA and significantly higher expression of GZMB and GZMK in the dominant epitope-specific populations. This suggests that common gene networks are involved in the regulation of dominant immunologic responses.
We also evaluated the expression of exhaustion-associated genes between the dominant and subdominant epitope-specific cells. We found that the dominant p11C-specific cells had increased expression of genes encoding LAG-3 and CTLA-4 during acute infection, both of which are known to be upregulated upon TCR stimulation [51], [52]. The role of these exhaustion-associated genes during acute infection is not well established and it is unclear to what extent these molecules impair CD8+ T cell function during acute HIV/SIV infection. Their increased expression on the dominant p11C-specific cells during acute infection suggests that these cells were experiencing a greater level of activation driven by greater stimulation through their higher-affinity TCRs. On day 70, no differences in gene expression were noted between the dominant and subdominant epitope-specific cells. However, by late chronic infection, the dominant p11C-specific cells showed increased levels of LAG-3 transcripts. LAG-3 is a well-described marker of exhausted CD8+ T cells during chronic viral infections [53], [54], including HIV [55]. However, while LAG-3 expression on CD8+ T cells has been shown to contribute to immune suppression in certain mouse models [56], its suppressive effect during HIV/SIV infection remains unclear [57], [58]. Additional studies are needed to confirm the cell surface expression of this protein and determine the extent to which increased expression contributes to functional differences between dominant and subdominant SIV/HIV epitope-specific cells.
In the present study, we found that dominant epitope-specific cells had greater cytotoxic potential, whereas the subdominant epitope-specific cells displayed enhanced proliferation capacity combined with increased cytokine and chemokine production. Importantly, these dominant epitope-specific cells had higher TCR affinity, a measure that typically correlates with functional avidity [59]–[61]. The ability of HIV/SIV epitopes with higher functional avidity to induce T cells that are less polyfunctional with lower proliferative capacity has been described by Harari et al [62] and Viganò et al [63]. These studies also corroborate our findings that cells of increased avidity also exhibited increased expression of exhaustion-associated molecules. Similarly, Conrad et al found that dominant TCR clonotypes exhibited reduced in vitro cytokine production and survival compared to the subdominant clonotypes that was associated with increased TCR avidity and increased expression of PD-1 [64]. These studies support the notion that reduced CD8+ T cell avidity promotes the development of polyfunctional T cells that under conditions of persistent high antigenemia may be able to partially control viral replication. This is also supported by the fact that increased avidity is significantly associated with viral escape [65]–[67] and increased induction of cell death and exhaustion [68], [69]. Collectively, these studies are consistent with our findings and suggest that to generate polyfunctional T cells after vaccination, the antigens used cannot confer extremely high TCR affinity; otherwise exhausted and monofunctional T cells will be generated.
The finding that the dominant p11C-specific cells were not highly polyfunctional is surprising considering that these cells are thought to be predominantly responsible for control of SIV viral replication [70], [71]. This reduced polyfunctionality may be compensated by a number of other cell characteristics. First, they may possess higher functional avidity, a characteristic that also correlates strongly with HIV control [15], [60], [72], [73] and higher TCR affinity [59]–[61]. Second, the dominant epitope-specific cells are present at substantially higher frequencies and contain greater amounts of perforin and granzyme than the subdominant epitope-specific cells. Thus on a percentage basis, the former population has an increased capacity to kill virus-infected cells. Finally, they may have greater TCR cross-reactivity to epitope variants, a characteristic that is thought to be an important component of durable HIV control by CD8+ T cells [73], [74]. In support of this possibility, Mothe et al found that HIV controllers had higher functional avidity that was associated with higher cross-reactivity of CD8+ T cells to HIV epitope variants compared to non-controllers [73]. Additionally, Bennett et al generated a panel of mutant TCRs specific for the HIV epitope SL9 and found that those TCR mutants with higher TCR avidities generally were more cross-reactive to SL9 variants than those with lower avidities [61]. In contrast, the previously-mentioned study by Conrad et al found that the high TCR avidity of the dominant clonotypes had reduced cross-reactivity than subdominant clonotypes [64]. Future studies should compare the TCR cross-reactivity to variant epitopes between the dominant p11C- and the subdominant epitope-specific cells to determine the extent that it contributes to the protective capacity of the p11C-specific cells.
We found that TCR affinity was predictive of frequency of the dominant and subdominant epitope-specific cells that are generated. The role of TCR:pMHC interactions in determining immunodominance hierarchies has been largely unexplored. A limited number of murine studies that have addressed this by measuring TCR dissociation rates using peptide:MHC class I tetramers have either found a lack of association between TCR affinity and epitope dominance [75] or found an association in the opposite direction [76]. The discordance between these studies and ours may be that for the measurement of the TCR dissociation rate the mouse studies used pMHC tetramers as opposed to monomers used in the present study. It is known that the accuracy of pMHC tetramers to predict physiologic TCR:pMHC binding affinities is limited [77], [78]; The multi-valency of tetramers combined with their propensity to aggregate can complicate accurate measurement of these interactions [79]. Moreover, the present study found that overall binding affinity (which incorporates both association and dissociation rate), rather than dissociation rate alone, was predictive of epitope dominance. In the above-mentioned murine studies, only the dissociation rate was assessed, leaving the association rate and affinity unknown. Slower association rates have been previously attributed to induced-fit or thermodynamic entropic penalty [80]–[82] in TCR binding and as such, pMHC epitopes that bind with faster association rates may outcompete those that bind with slower kinetic rates and gain dominance in response. Therefore, it is possible that association rate also played a role in the immunodominance hierarchies in these mouse studies and its measurement may have been informative about the particular parameter of the TCR:pMHC interactions that was important for immunodominance.
The present study found that high-frequency dominant epitope-specific CD8+ T cells had reduced proliferation and cytokine production but increased cytotoxicity potential compared to low-frequency subdominant epitope-specific CD8+ T cells. This was associated with higher TCR affinity and increased expression of exhaustion-associated genes in the dominant epitope-specific cells. The engineering of higher-affinity TCRs has been explored for enhancement of HIV immunity [61], [83]. However, our findings suggest that high TCR affinity may increase susceptibility to exhaustion and reduced functionality, although it may also promote higher cytotoxic potential. Since both proliferation and cytokine production as well as cytotoxicity are associated with superior control of HIV replication, antigen targets of engineered TCRs or antigens that are included in an HIV vaccine should be of both high and low affinity. The high-affinity antigens will promote epitope-specific CD8+ T cells of high frequency with high cytotoxic potential while the low-affinity antigens will promote CD8+ T cells that are more polyfunctional and proliferative and that have decreased potential for exhaustion.
Materials and Methods
Ethics statement
Indian-origin rhesus monkeys used in this study were maintained according to the guidelines of the National Institutes of Health (NIH) Guide to the Care and Use of Laboratory Animals and the approval of the Institutional Animal Care and Use Committee (IACUC) of Harvard Medical School (protocol # 03503) and the NIH. The institution also accepts as mandatory the Public Health Service Policy on Humane care and use of Laboratory Animals by Awardee Institutions and the NIH Principles for the Utilization and Care of Vertebrate Animals used in Testing, Research and Training. The New England Primate Research Center (NEPRC) is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). The NEPRC has developed a comprehensive environmental enrichment, psychological well-being, and minimization of distress plans for primates that is available for inspection by the United States Animal and Plant Health Inspection Service (APHIS) and to officials of any pertinent funding agency. The Harvard Medical School IACUC documents NEPRC compliance with the plan during semiannual facility inspections.
Animals
Mamu-A*01-positive, Mamu-B*17-negative, Mamu-B*08-negative, Indian-origin rhesus monkeys (Macaca mulatta) were selected for these studies by PCR-based MHC typing as previously described [84]. All monkeys were infected intrarectally with either SIVmac251 or SIVsmE660. SIVmac251 infection was administered by a single high dose challenge. SIVsmE660 was administered by a repeated low-dose challenge.
Antibodies and flow cytometry reagents
Conjugated antibodies and staining reagents included MIP-1β-PE, CD3-PB, CD3-PE-Cy7, CD3-PerCP-Cy5.5, CD3-APC-Cy7, CD3-Horizon V450, CD4-PerCP-Cy5.5, CD4-AmCyan, CD4-FITC, CD8α-APC, CD8α-APC-Cy7, CD8α-AlexaFluor700, CD8α-FITC, CD8α-APC-H7, CD69- ECD (Beckman Coulter), CD20-Horizon V450, CCR7-FITC (R&D Systems), CD95-APC, CCR7-PerCP-Cy5.5, CD28-PE-Cy7 (eBioscience), granzyme B-AlexaFluor700, perforin-FITC (MabTech), IFNγ-PE-Cy7, TNFα- AlexaFluor700, IL-2-APC, CD95-PE, CD95-APC, and Aqua LIVE/DEAD Fixable Dead Cell Stain (Invitrogen). All reagents are from BD Biosciences unless indicated otherwise. For construction of monomers and tetramers, the following peptides were synthesized and purified to >95% by HPLC by New England Peptide LLC: p11C (CTPYDINQM), p54AS (TVPWPNASL), p54E660 (TVPWPNETL), p68A (STPPLVRLV), and TL8 (TTPESANL). The monomers and tetramers were prepared as previously described [85], [86]. Tetramers were prepared using either streptavidin-PE (Prozyme), -APC (Prozyme), -AlexaFluor488 (Invitrogen), or -Qdot655 (Invitrogen). Monomers used in surface plasmon resonance studies were further quantified using an RC DC protein kit (Bio-Rad).
RNA extraction and microarray analysis
Peripheral blood was collected on days 14, 21, 28, 35, 42, 56, 70, and at week 31 or 32 post-inoculation with SIVmac251. Plasma viral RNA from these samples was measured using an ultra-sensitive branched DNA amplification assay (Siemens Diagnostics, Berkeley, CA). PBMC were stained with p11C and p54AS tetramers and CD3 and CD8 antibodies at 4°C. Tetramer-positive single CD3+CD8+ lymphocytes were sorted to ≥95% purity into RNAprotect (Qiagen) at 4°C. RNA was isolated using a Trizol (Invitrogen) extraction protocol. Briefly, 0.8 ml of Trizol were added to the cell pellet and incubated for 5 minutes at room temperature and 0.16 ml of chloroform were added, shaken vigorously by hand for 15 seconds, incubated at room temperature for 2–3 minutes and centrifuged at 13,000 rpm for 15 minutes at 4°C. The colorless upper aqueous phase was carefully collected and transferred to a new tube containing 2 μl of linear acrylamide for mixing. An equal volume of isopropyl alcohol was then added and mixed. The mixture was incubated at room temperature for 10 minutes and centrifuged at 13,000 rpm for 10 minutes at 4°C. The supernatant was collected and the RNA was washed with 1 ml of 70% ethanol and centrifuged at 10,500 rpm for 5 minutes at 4°C. The supernatant was completely removed and the RNA pellet was allowed to air-dry. The RNA was then resuspended in RNase-free water and stored at −80°C. RNA integrity was tested using an Agilent Bioanalyzer. RNA was then amplified using the TargetAmp 2-Round Biotin-aRNA Amplification Kit 3.0 (Epicentre Biotechnologies) according the manufacturer's instructions. Amplified biotinylated antisense-RNA (aRNA) was resuspended in RNase-free water and stored at −80°C. Nanodrop ND-1000 was used to determine the biotinylated aRNA concentration and an Agilent Bioanalyzer was used to determine its integrity. Amplified aRNA was hybridized to Illumina Human HT-12 Expression BeadChips according to the manufacturer's instructions and was stained with Streptavidin Cy3 for detection (Illumina, San Diego, CA, USA). The Human HT-12 BeadChip assays 48,000 transcripts. The BeadChips were built with sequences derived from the National Center for Biotechnology Information Reference Sequence (NCBI RefSeq) database (Build 36.2, Release 22). Arrays were scanned according to the manufacturer's instructions. Processing of the raw array data was performed using Illumina BeadStudio software. Matlab (Mathworks, Natick, MA, USA) was used to perform statistical analysis of gene expression data. The entire expression dataset was first quantile-normalized. Fold-differences of individual transcripts were computed as the ratios of the median expression levels of the p11C- to those of the p54AS-specific CD8+ T cells (p11C/p54AS). When p54AS expression values were larger than p11C values, the negative reciprocal was calculated. A Wilcoxon signed-rank test was used to evaluate the significance of the differences in gene expression between p11C- and p54AS-specific CD8+ T cells. Genes whose expression was significantly different between the p11C- and p54AS-specific cell samples and whose median fold change difference was at least ±1.5 were considered differentially expressed. The Minimum Information about a Microarray Experiment (MIAME) criteria have been met by these experiments. The complete dataset is available in the Gene Expression Omnibus, accession number GSE54001.
Flow cytometric analysis
In order to collect and analyze comparable numbers of each CD8+ T cell specificity, different numbers of PBMCs were used for each p11C, p54, and p68A sample. Input numbers of PBMCs were dependent on the expected relative frequency of each epitope-specific population. For example, if the expected frequencies of the p11C-, p54-, and p68A-specific CD8+ T cells were 10%, 1%, and 0.1%, respectively, then the ratio of the numbers of cells used for each sample was 1∶10∶100. Staining volumes, washing volumes, and amounts of staining reagents used were adjusted for each sample to ensure the same cell-to-reagent concentration ratios were used for all samples. For quantification of epitope-specific CD8+ T cells, PBMCs were stained with tetramer and CD3, CD4, and CD8 antibodies. For phenotyping, PBMCs were additionally stained with CD20, CD95, CD28, and CCR7 antibodies. For ex vivo measurement of perforin and granzyme B, PBMCs were stained with tetramer, CD3, CD4, and CD8 antibodies and then stained intracellularly with perforin and granzyme B antibodies as previously described [87]. FMO samples were also prepared to set the gates for perforin and granzyme B gating. All flow cytometric data were collected on an LSRII (BD) and analyzed using FlowJo (Tree star). Statistical analysis of flow cytometric data was conducted in GraphPad Prism 5. Wilcoxon signed rank test was used to compare groups. P-values ≤0.05 were considered significant. Bonferroni correction was used for multiple comparisons with significant p-values ≤0.05/(number of comparisons) considered significant.
Cytokine and chemokine production
Measurements of IL-2, IFNγ, TNFα, and MIP-1β production by epitope-specific CD8+ T cells following stimulation of 1 nM peptide was performed in 5 mL FACS tubes as previously described [50]. Flow cytometric data were gated on single, live, CD3+CD4−CD8+CD69+ lymphocytes. Boolean gating was used to calculate percent of polyfunctional cells.
Expansion of epitope-specific CD8+ T cells
PBMCs, at approximately 3×106 lymphocytes/mL (measured using Guava EasyCyte automatic cytometer (Millipore)), were plated in flat-bottomed culture plates and stimulated for expansion with 1 nM epitope peptide. Approximately 1×106 PBMCs were also stained with tetramers and CD3, CD4, and CD8 antibodies, to measure the frequency each CD8+ T cell specificity before peptide-stimulated expansion (day 0). 20 U/mL IL-2 (Hoffmann-LaRoche) was added to stimulated cells on day 3, and media supplemented with IL-2 was changed thereafter as needed. Samples were harvested on days 3, 4, 5, 6, 8, 10, 12, and 14 post-stimulation for staining with aqua LIVE/DEAD, tetramer and CD3, CD4, and CD8 antibodies for flow cytometric analysis. Flow cytometric data were gated on single, live, CD3+CD4−CD8+ tet+ lymphocytes.
Surface plasmon resonance (SPR) measurements
Freshly isolated PBMCs were enriched for CD8+ T lymphocytes using Miltenyi's magnetic-activated cell sorting (MACS) kit for isolation of untouched NHP CD8+ T cells according to the manufacturer's instructions. Isolated CD8+ T cells were then stained at 4°C with tetramers and CD3 and CD8 antibodies. Sorting was performed on an Aria flow cytometer/cell sorter (BD). For initial TCR binding analyses, cells were only stained with the TL8 tetramers and those cells that were positive for TL8 were excluded from the sorted CD3+CD8+ lymphocytes. For repeated TCR binding analysis for enrichment of p68A-specific TCRs, cells were stained with TL8, p54E660, and p68A tetramers, and multiple populations were simultaneously sorted: p54E660− p68A− TL8−, p54E660+ p68A− TL8−, and p54E660− p68A+ TL8−. These sorted populations were subsequently combined during purification of the DRMs. For samples with low total cell number, extra DRMs from cells not specific for any of the epitopes being evaluated were added to provide extra mass to the pellet during centrifugation. These extra DRMs were obtained from sorted single CD3+CD8+ lymphocytes that were negative for all evaluated epitope specificities (p11C−p54E660−p68A−TL8−). All sorted cells were resuspended in a 4°C solution of protease inhibitors (1 μg/mL each leupeptin, pepstatin, and aprotinin) and stored at −80°C until analysis. DRM purification was carried out as described previously [50] with the modification that the repeat samples for measurements of p68A were conducted at concentrations up to 1,000 μg/mL of monomer. The specific binding signal was obtained by subtracting the non-specific signal from TL8 pMHC control monomer binding to the TL8-depleted DRM preparation when injected at the same concentration from the p11C, p54E660, or p68A binding signal. The global curve fitting to the Langmuir equation was used to derive kinetic rate constants (kon and koff) for calculation of equilibrium dissociation constant Kd as previously described [88], [89]. All SPR measurements were carried out on a Biacore 3000 instrument, and data analyses were performed using BIAevaluation 4.1 software (GE Healthcare).
Supporting Information
Gating of p11C- and p54AS-specific CD8+ T cells sorted for microarray analyses. Gates for p11C (y-axis) and p54AS (x-axis) tetramers on samples sorted from each of the six Mamu-A*01 + rhesus monkeys (107-06, 112-06, 125-06, 133-06, 142-06, and 202-06) used in the gene expression analysis. Plots are gated on single CD3+CD8+ lymphocytes and percent of tetramer-positive cells are shown on each plot for the p11C- (upper left) and p54AS- (middle right) positive cells. 50,000 CD8+ T cells events are displayed for all samples except where indicated in upper right corner of plots. The data file for 107-06 on day 56 was corrupt and not available for re-analysis.
(TIF)
Individual values of genes differentially expressed between dominant p11C- and subdominant p54AS-specific CD8+ T cells. Shown are the individual normalized expression, measured in fluorescence units, for each A) maturation, B) cytotoxicity, and C) proliferation and apoptosis gene that was determined to be differentially expressed between p11C- and p54AS-specific cells. Plots include values for which its matching pair is missing and therefore were not used in determination of differential expression. Fold change values are indicated in upper left corner. An asterisk under fold change values is present if that gene met the criteria for differential expression on that timepoint. For those genes with more than one probe on the BeadChip, the numerical probe IDs are included in the gene name. Red, p11C. Blue, p54AS.
(PDF)
Staining and gating of tetramers for quantification of epitope-specific cells in Mamu-A*01 immunodominance hierarchies. Gating of p11C-, p54AS/E660-, and p68A-specific CD8+ T cells from A) SIVmac251- and B) SIVsmE660-infected animals. 50,000 CD8+ T cell events are displayed on each flow plot. SIVmac251 frequencies were measured between weeks 37–50, except for 133-06 which died early and data used are from week 18. SIVsmE660 frequencies were measured between weeks 19–22.
(TIF)
Representative flow staining and gating for phenotyping. Representative staining and gating for phenotyping of tetramer-positive cells from (A) SIVmac251- and (B) SIVsmE660-infected monkeys. Left, Staining and gating of tetramer-positive cells among CD8+ T cells (40,000 CD8+ T cell events are shown). Right, staining and gating of CD28 (y-axis) and CCR7 (x-axis) cell surface proteins among the tetramer-positive cells (1,000 tetramer-positive events are shown). Phenotyping was conducted between weeks 44–78 for SIVmac251 and 33–46 for SIVsmE660.
(TIF)
Representative staining and gating of perforin and granzyme B. Mamu-A*01 tetramers were used to identify p11C-, p54AS/E660-, and p68A-specific cells among PBMCs from monkeys chronically-infected with either SIVmac251 or SIVsmE660. Perforin and granzyme B content within the tetramer-positive cells were measured by flow cytometry. A) FMOs used to set gates for perforin (left) and granzyme B (right). For SIVmac251 (B) and SIVsmE660 (D), representative staining and gating of tetramer-positive cells (left) and perforin+granzyme B+ cells among the tetramer-positive cells (right). 250,000 CD8+ T cells are shown on flow plots of tetramer staining. 1,000 tetramer-positive events are shown on flow plots of perforin and granzyme B staining. For SIVmac251 (C) and SIVsmE660 (E), histograms illustrating fluorescence intensity of perforin (top) and granzyme B (bottom) within the p11C (red), p54AS/E660 (blue), and p68A (green) tetramer-positive cells. FMOs shown as filled gray histograms. Measurements were conducted between weeks 63–83 for SIVmac251 and 41–49 for SIVsmE660.
(TIF)
Representative staining and gating of tetramers during in vitro expansion. PBMCs from monkeys chronically-infected with either SIVmac251 (A) or SIVsmE660 (B) were stimulated in vitro with either p11C, 54E660/AS, or p68A peptide, harvested on days 3, 4, 5, 6, 8, 10, 12, and 14 following stimulation, and measured by flow cytometry to calculate the percent (upper right on each flow plot) of tetramer-positive CD8+ T cells. Expansion was calculated as the fold change (middle left on each flow plot) of the percent of each tetramer-positive population on each day, relative to day 0. Number of CD8+ T cell events displayed are the same for pllC, p54AS/E660, and p68A plots within a given day. Measurements were conducted between weeks 40–52 for SIVmac251 and 31–44 for SIVsmE660.
(TIF)
Representative staining and gating for cytokine and chemokine production. PBMCs from monkeys chronically-infected with either SIVmac251 (A) or SIVsmE660 (B) were stimulated with either p11C, p54AS/E660, or p68A peptides and intracellular staining was used to assess production of the chemokine MIP-1β and the cytokines IFNγ, TNFα, and IL-2. Left, representative staining and gating of tetramer-positive cells (25,000 CD8+ T cell events are shown). Right, corresponding staining and gating of MIP-1β, IFNγ, TNFα, and IL-2 (250 tetramer-positive events are shown). Individual gates were then used in a Boolean analysis for assessment of polyfunctionality. Data was collected between weeks 36–42 for SIVmac251 and 14–25 for SIVsmE660.
(PDF)
Detection of specific binding of p11C and p54E660 peptide:Mamu-A*01 monomers to DRMs. DRMs were purified from total CD8+ T cells sorted from seven chronically-infected SIVsmE660-infected monkeys. The DRMs were evaluated for specific binding, measured in resonance units (RU), to pMHC monomers constructed with p11C, p54E660, and p68A epitope peptides and Mamu-A*01. Shown are overlaid readings of the binding of p11C (red) and p54E660 (blue) pMHC monomers at 100 μg/mL. p68A:Mamu-A*01 monomer binding above background was not detected at any concentration and is not shown. Readings have been normalized by subtracting the binding of the control monomer TL8 run at the same concentrations as the experimental monomers.
(TIF)
Titrations of p11C and p54E660 peptide:Mamu-A*01 monomers for calculation of binding kinetics and affinity. Shown are sensograms indicating the binding of p11C (left) and p54E660 (right) pMHC monomers to DRMs purified from total CD8+ T cells sorted from seven SIVsmE660-infected monkeys. p11C monomers were run at 25 (green), 50 (pink), 100 (blue), and 200 (red) μg/mL. The ARB0 plot for p11C shows a 150 μg/mL (light blue) run in place of the 100 μg/mL. The AP34, ZD57, and A6V031 plots for p11C do not show the 200 μg/mL run. p54E660 monomers were run at 25, 50, 150, and 200 μg/mL for AP54, ARB0, 8B1, and AS47 and at 25, 100, and 200 μg/mL for AP34, ZD57, and A6V031. The ZD57 plot includes an additional 50 μg/mL run. A Langmuir curve was fit to each binding curve at each concentration and was used to calculate binding kinetics. Readings have been normalized by subtracting the binding of the control monomer TL8 run at the same concentrations as the experimental monomers.
(PDF)
Detection of p68A monomer binding. p68A-specific CD8+ T cells were collected from multiple tetramer-specific flow cytometric cell sorts and pooled for DRM purification. Titrations of p68A pMHC monomers were performed at concentrations ranging from 150 to 1000 μg/mL. The highest concentration evaluated is shown. Binding of the control monomer TL8 at the same concentration has been subtracted from all readings.
(TIF)
Acknowledgments
We thank our collaborators at the Duke Center for Human Genome Variation: Dr. Kevin Shianna for Illumina BeadChip hybridizations and Rachel Lovingood for measuring viral loads. We also thank Michelle Lifton, Katie L. Furr, Sarah Schaake, Kevin Carlson, and Leila Eslamizar for flow cytometric assistance. We thank Dr. Evan M. Cale, Dr. James B. Whitney, and Dr. Keith A. Reimann for their critical advice.
Funding Statement
This work was supported by the NIH NIAID CHAVI #U19 AI067854. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Walker CM, Moody DJ, Stites DP, Levy JA (1986) CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science 234: 1563–1566. [DOI] [PubMed] [Google Scholar]
- 2. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, et al. (1999) Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283: 857–860. [DOI] [PubMed] [Google Scholar]
- 3. Loffredo JT, Bean AT, Beal DR, Leon EJ, May GE, et al. (2008) Patterns of CD8+ immunodominance may influence the ability of Mamu-B*08-positive macaques to naturally control simian immunodeficiency virus SIVmac239 replication. J Virol 82: 1723–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu XG, Addo MM, Rosenberg ES, Rodriguez WR, Lee PK, et al. (2002) Consistent patterns in the development and immunodominance of human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T-cell responses following acute HIV-1 infection. J Virol 76: 8690–8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yewdell JW, Bennink JR (1999) Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17: 51–88. [DOI] [PubMed] [Google Scholar]
- 6. Akram A, Inman RD (2012) Immunodominance: a pivotal principle in host response to viral infections. Clin Immunol 143: 99–115. [DOI] [PubMed] [Google Scholar]
- 7. Chen W, McCluskey J (2006) Immunodominance and immunodomination: critical factors in developing effective CD8+ T-cell-based cancer vaccines. Adv Cancer Res 95: 203–247. [DOI] [PubMed] [Google Scholar]
- 8. Cao W, Myers-Powell BA, Braciale TJ (1996) The weak CD8+ CTL response to an influenza hemagglutinin epitope reflects limited T cell availability. J Immunol 157: 505–511. [PubMed] [Google Scholar]
- 9. Haeryfar SM, Hickman HD, Irvine KR, Tscharke DC, Bennink JR, et al. (2008) Terminal deoxynucleotidyl transferase establishes and broadens antiviral CD8+ T cell immunodominance hierarchies. J Immunol 181: 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Messaoudi I, Guevara Patino JA, Dyall R, LeMaoult J, Nikolich-Zugich J (2002) Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science 298: 1797–1800. [DOI] [PubMed] [Google Scholar]
- 11. Belz GT, Stevenson PG, Doherty PC (2000) Contemporary analysis of MHC-related immunodominance hierarchies in the CD8+ T cell response to influenza A viruses. J Immunol 165: 2404–2409. [DOI] [PubMed] [Google Scholar]
- 12. Deng Y, Yewdell JW, Eisenlohr LC, Bennink JR (1997) MHC affinity, peptide liberation, T cell repertoire, and immunodominance all contribute to the paucity of MHC class I-restricted peptides recognized by antiviral CTL. J Immunol 158: 1507–1515. [PubMed] [Google Scholar]
- 13. Harari A, Dutoit V, Cellerai C, Bart PA, Du Pasquier RA, et al. (2006) Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol Rev 211: 236–254. [DOI] [PubMed] [Google Scholar]
- 14. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, et al. (2006) HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107: 4781–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, et al. (2007) Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med 204: 2473–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Critchfield JW, Lemongello D, Walker DH, Garcia JC, Asmuth DM, et al. (2007) Multifunctional human immunodeficiency virus (HIV) gag-specific CD8+ T-cell responses in rectal mucosa and peripheral blood mononuclear cells during chronic HIV type 1 infection. J Virol 81: 5460–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferre AL, Hunt PW, Critchfield JW, Young DH, Morris MM, et al. (2009) Mucosal immune responses to HIV-1 in elite controllers: a potential correlate of immune control. Blood 113: 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perreau M, Levy Y, Pantaleo G (2013) Immune response to HIV. Curr Opin HIV AIDS 8: 333–340. [DOI] [PubMed] [Google Scholar]
- 19. Rodriguez F, Slifka MK, Harkins S, Whitton JL (2001) Two overlapping subdominant epitopes identified by DNA immunization induce protective CD8(+) T-cell populations with differing cytolytic activities. J Virol 75: 7399–7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baron C, Meunier MC, Caron E, Cote C, Cameron MJ, et al. (2006) Asynchronous differentiation of CD8 T cells that recognize dominant and cryptic antigens. J Immunol 177: 8466–8475. [DOI] [PubMed] [Google Scholar]
- 21. Payne RP, Kloverpris H, Sacha JB, Brumme Z, Brumme C, et al. (2010) Efficacious early antiviral activity of HIV Gag- and Pol-specific HLA-B 2705-restricted CD8+ T cells. J Virol 84: 10543–10557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen TM, Mothe BR, Sidney J, Jing P, Dzuris JL, et al. (2001) CD8(+) lymphocytes from simian immunodeficiency virus-infected rhesus macaques recognize 14 different epitopes bound by the major histocompatibility complex class I molecule mamu-A*01: implications for vaccine design and testing. J Virol 75: 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Newberg MH, McEvers KJ, Gorgone DA, Lifton MA, Baumeister SH, et al. (2006) Immunodomination in the evolution of dominant epitope-specific CD8+ T lymphocyte responses in simian immunodeficiency virus-infected rhesus monkeys. J Immunol 176: 319–328. [DOI] [PubMed] [Google Scholar]
- 24. Seth A, Ourmanov I, Schmitz JE, Kuroda MJ, Lifton MA, et al. (2000) Immunization with a modified vaccinia virus expressing simian immunodeficiency virus (SIV) Gag-Pol primes for an anamnestic Gag-specific cytotoxic T-lymphocyte response and is associated with reduction of viremia after SIV challenge. J Virol 74: 2502–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price DA, Asher TE, Wilson NA, Nason MC, Brenchley JM, et al. (2009) Public clonotype usage identifies protective Gag-specific CD8+ T cell responses in SIV infection. J Exp Med 206: 923–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto T, Johnson MJ, Price DA, Wolinsky DI, Almeida JR, et al. (2012) Virus inhibition activity of effector memory CD8(+) T cells determines simian immunodeficiency virus load in vaccinated monkeys after vaccine breakthrough infection. J Virol 86: 5877–5884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wrzesien-Kus A, Smolewski P, Sobczak-Pluta A, Wierzbowska A, Robak T (2004) The inhibitor of apoptosis protein family and its antagonists in acute leukemias. Apoptosis 9: 705–715. [DOI] [PubMed] [Google Scholar]
- 28. Kumar S (2009) Caspase 2 in apoptosis, the DNA damage response and tumour suppression: enigma no more? Nat Rev Cancer 9: 897–903. [DOI] [PubMed] [Google Scholar]
- 29. Nguyen HG, Chinnappan D, Urano T, Ravid K (2005) Mechanism of Aurora-B degradation and its dependency on intact KEN and A-boxes: identification of an aneuploidy-promoting property. Mol Cell Biol 25: 4977–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stewart S, Fang G (2005) Destruction box-dependent degradation of aurora B is mediated by the anaphase-promoting complex/cyclosome and Cdh1. Cancer Res 65: 8730–8735. [DOI] [PubMed] [Google Scholar]
- 31. Satyanarayana A, Kaldis P (2009) Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 28: 2925–2939. [DOI] [PubMed] [Google Scholar]
- 32. Olson MF, Ashworth A, Hall A (1995) An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269: 1270–1272. [DOI] [PubMed] [Google Scholar]
- 33. Iyer J, Moghe S, Furukawa M, Tsai MY (2011) What's Nu(SAP) in mitosis and cancer? Cell Signal 23: 991–998. [DOI] [PubMed] [Google Scholar]
- 34. Fang Z, Xing F, Bronner C, Teng Z, Guo Z (2009) ICBP90 mediates the ERK1/2 signaling to regulate the proliferation of Jurkat T cells. Cell Immunol 257: 80–87. [DOI] [PubMed] [Google Scholar]
- 35. Tien AL, Senbanerjee S, Kulkarni A, Mudbhary R, Goudreau B, et al. (2011) UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem J 435: 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, et al. (1992) A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 71: 587–597. [DOI] [PubMed] [Google Scholar]
- 37. Letvin NL, Rao SS, Montefiori DC, Seaman MS, Sun Y, et al. (2011) Immune and Genetic Correlates of Vaccine Protection Against Mucosal Infection by SIV in Monkeys. Sci Transl Med 3: 81ra36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, et al. (2002) Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168: 29–43. [DOI] [PubMed] [Google Scholar]
- 39. Takata H, Takiguchi M (2006) Three memory subsets of human CD8+ T cells differently expressing three cytolytic effector molecules. J Immunol 177: 4330–4340. [DOI] [PubMed] [Google Scholar]
- 40. Tomiyama H, Matsuda T, Takiguchi M (2002) Differentiation of human CD8(+) T cells from a memory to memory/effector phenotype. J Immunol 168: 5538–5550. [DOI] [PubMed] [Google Scholar]
- 41. Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, et al. (2001) Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410: 106–111. [DOI] [PubMed] [Google Scholar]
- 42. Slifka MK, Rodriguez F, Whitton JL (1999) Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 401: 76–79. [DOI] [PubMed] [Google Scholar]
- 43. Valitutti S, Muller S, Dessing M, Lanzavecchia A (1996) Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J Exp Med 183: 1917–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kersh GJ, Kersh EN, Fremont DH, Allen PM (1998) High- and low-potency ligands with similar affinities for the TCR: the importance of kinetics in TCR signaling. Immunity 9: 817–826. [DOI] [PubMed] [Google Scholar]
- 45. Holler PD, Kranz DM (2003) Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity 18: 255–264. [DOI] [PubMed] [Google Scholar]
- 46. Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM (1994) Kinetics of T-cell receptor binding to peptide/I-Ek complexes: correlation of the dissociation rate with T-cell responsiveness. Proc Natl Acad Sci U S A 91: 12862–12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tian S, Maile R, Collins EJ, Frelinger JA (2007) CD8+ T cell activation is governed by TCR-peptide/MHC affinity, not dissociation rate. J Immunol 179: 2952–2960. [DOI] [PubMed] [Google Scholar]
- 48. Lyons DS, Lieberman SA, Hampl J, Boniface JJ, Chien Y, et al. (1996) A TCR binds to antagonist ligands with lower affinities and faster dissociation rates than to agonists. Immunity 5: 53–61. [DOI] [PubMed] [Google Scholar]
- 49. Ueno T, Tomiyama H, Fujiwara M, Oka S, Takiguchi M (2004) Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. J Immunol 173: 5451–5457. [DOI] [PubMed] [Google Scholar]
- 50. Cale EM, Bazick HS, Rianprakaisang TA, Alam SM, Letvin NL (2011) Mutations in a dominant Nef epitope of simian immunodeficiency virus diminish TCR:epitope peptide affinity but not epitope peptide:MHC class I binding. J Immunol 187: 3300–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, et al. (1990) LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med 171: 1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, et al. (1992) Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med 176: 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, et al. (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27: 670–684. [DOI] [PubMed] [Google Scholar]
- 54. Takamura S, Tsuji-Kawahara S, Yagita H, Akiba H, Sakamoto M, et al. (2010) Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J Immunol 184: 4696–4707. [DOI] [PubMed] [Google Scholar]
- 55. Price P, Keane N, Gray L, Lee S, Gorry PR, et al. (2006) CXCR4 or CCR5 tropism of human immunodeficiency virus type 1 isolates does not determine the immunological milieu in patients responding to antiretroviral therapy. Viral Immunol 19: 734–740. [DOI] [PubMed] [Google Scholar]
- 56. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, et al. (2009) Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lim AY, Price P, Beilharz MW, French MA (2006) Cell surface markers of regulatory T cells are not associated with increased forkhead box p3 expression in blood CD4+ T cells from HIV-infected patients responding to antiretroviral therapy. Immunol Cell Biol 84: 530–536. [DOI] [PubMed] [Google Scholar]
- 58. Pena J, Jones NG, Bousheri S, Bangsberg DR, Cao H (2013) Lymphocyte Activation Gene-3 Expression Defines a Discrete Subset of HIV-Specific CD8 T Cells That Is Associated with Lower Viral Load. AIDS Res Hum Retroviruses [epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD (1999) Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol 162: 2227–2234. [PubMed] [Google Scholar]
- 60. Iglesias MC, Almeida JR, Fastenackels S, van Bockel DJ, Hashimoto M, et al. (2011) Escape from highly effective public CD8+ T-cell clonotypes by HIV. Blood 118: 2138–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bennett MS, Joseph A, Ng HL, Goldstein H, Yang OO (2010) Fine-tuning of T-cell receptor avidity to increase HIV epitope variant recognition by cytotoxic T lymphocytes. AIDS 24: 2619–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harari A, Cellerai C, Enders FB, Kostler J, Codarri L, et al. (2007) Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc Natl Acad Sci U S A 104: 16233–16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vigano S, Enders FB, Miconnet I, Cellerai C, Savoye AL, et al. (2013) Rapid perturbation in viremia levels drives increases in functional avidity of HIV-specific CD8 T cells. PLoS Pathog 9: e1003423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Conrad JA, Ramalingam RK, Smith RM, Barnett L, Lorey SL, et al. (2011) Dominant clonotypes within HIV-specific T cell responses are programmed death-1high and CD127low and display reduced variant cross-reactivity. J Immunol 186: 6871–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, et al. (2004) HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10: 282–289. [DOI] [PubMed] [Google Scholar]
- 66. O'Connor DH, Allen TM, Vogel TU, Jing P, DeSouza IP, et al. (2002) Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med 8: 493–499. [DOI] [PubMed] [Google Scholar]
- 67. Vogel TU, Friedrich TC, O'Connor DH, Rehrauer W, Dodds EJ, et al. (2002) Escape in one of two cytotoxic T-lymphocyte epitopes bound by a high-frequency major histocompatibility complex class I molecule, Mamu-A*02: a paradigm for virus evolution and persistence? J Virol 76: 11623–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Molldrem JJ, Lee PP, Kant S, Wieder E, Jiang W, et al. (2003) Chronic myelogenous leukemia shapes host immunity by selective deletion of high-avidity leukemia-specific T cells. J Clin Invest 111: 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Van Parijs L, Abbas AK (1998) Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science 280: 243–248. [DOI] [PubMed] [Google Scholar]
- 70. Barouch DH, Kunstman J, Glowczwskie J, Kunstman KJ, Egan MA, et al. (2003) Viral escape from dominant simian immunodeficiency virus epitope-specific cytotoxic T lymphocytes in DNA-vaccinated rhesus monkeys. J Virol 77: 7367–7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Barouch DH, Kunstman J, Kuroda MJ, Schmitz JE, Santra S, et al. (2002) Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415: 335–339. [DOI] [PubMed] [Google Scholar]
- 72. Berger CT, Frahm N, Price DA, Mothe B, Ghebremichael M, et al. (2011) High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J Virol 85: 9334–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mothe B, Llano A, Ibarrondo J, Zamarreno J, Schiaulini M, et al. (2012) CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7: e29717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Turnbull EL, Lopes AR, Jones NA, Cornforth D, Newton P, et al. (2006) HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J Immunol 176: 6130–6146. [DOI] [PubMed] [Google Scholar]
- 75. St Leger AJ, Peters B, Sidney J, Sette A, Hendricks RL (2011) Defining the herpes simplex virus-specific CD8+ T cell repertoire in C57BL/6 mice. J Immunol 186: 3927–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. La Gruta NL, Doherty PC, Turner SJ (2006) A correlation between function and selected measures of T cell avidity in influenza virus-specific CD8+ T cell responses. Eur J Immunol 36: 2951–2959. [DOI] [PubMed] [Google Scholar]
- 77. Fahmy TM, Bieler JG, Edidin M, Schneck JP (2001) Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity 14: 135–143. [PubMed] [Google Scholar]
- 78. Xiao Z, Mescher MF, Jameson SC (2007) Detuning CD8 T cells: down-regulation of CD8 expression, tetramer binding, and response during CTL activation. J Exp Med 204: 2667–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang XL, Altman JD (2003) Caveats in the design of MHC class I tetramer/antigen-specific T lymphocytes dissociation assays. J Immunol Methods 280: 25–35. [DOI] [PubMed] [Google Scholar]
- 80. Boniface JJ, Reich Z, Lyons DS, Davis MM (1999) Thermodynamics of T cell receptor binding to peptide-MHC: evidence for a general mechanism of molecular scanning. Proc Natl Acad Sci U S A 96: 11446–11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Willcox BE, Gao GF, Wyer JR, Ladbury JE, Bell JI, et al. (1999) TCR binding to peptide-MHC stabilizes a flexible recognition interface. Immunity 10: 357–365. [DOI] [PubMed] [Google Scholar]
- 82. Garcia KC, Radu CG, Ho J, Ober RJ, Ward ES (2001) Kinetics and thermodynamics of T cell receptor- autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 98: 6818–6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, et al. (2008) Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med 14: 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Knapp LA, Lehmann E, Piekarczyk MS, Urvater JA, Watkins DI (1997) A high frequency of Mamu-A*01 in the rhesus macaque detected by polymerase chain reaction with sequence-specific primers and direct sequencing. Tissue Antigens 50: 657–661. [DOI] [PubMed] [Google Scholar]
- 85. Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, et al. (1996) Phenotypic analysis of antigen-specific T lymphocytes. Science 274: 94–96. [DOI] [PubMed] [Google Scholar]
- 86. Kuroda MJ, Schmitz JE, Barouch DH, Craiu A, Allen TM, et al. (1998) Analysis of Gag-specific cytotoxic T lymphocytes in simian immunodeficiency virus-infected rhesus monkeys by cell staining with a tetrameric major histocompatibility complex class I-peptide complex. J Exp Med 187: 1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Makedonas G, Banerjee PP, Pandey R, Hersperger AR, Sanborn KB, et al. (2009) Rapid up-regulation and granule-independent transport of perforin to the immunological synapse define a novel mechanism of antigen-specific CD8+ T cell cytotoxic activity. J Immunol 182: 5560–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Alam SM, Davies GM, Lin CM, Zal T, Nasholds W, et al. (1999) Qualitative and quantitative differences in T cell receptor binding of agonist and antagonist ligands. Immunity 10: 227–237. [DOI] [PubMed] [Google Scholar]
- 89. Alam SM, Travers PJ, Wung JL, Nasholds W, Redpath S, et al. (1996) T-cell-receptor affinity and thymocyte positive selection. Nature 381: 616–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gating of p11C- and p54AS-specific CD8+ T cells sorted for microarray analyses. Gates for p11C (y-axis) and p54AS (x-axis) tetramers on samples sorted from each of the six Mamu-A*01 + rhesus monkeys (107-06, 112-06, 125-06, 133-06, 142-06, and 202-06) used in the gene expression analysis. Plots are gated on single CD3+CD8+ lymphocytes and percent of tetramer-positive cells are shown on each plot for the p11C- (upper left) and p54AS- (middle right) positive cells. 50,000 CD8+ T cells events are displayed for all samples except where indicated in upper right corner of plots. The data file for 107-06 on day 56 was corrupt and not available for re-analysis.
(TIF)
Individual values of genes differentially expressed between dominant p11C- and subdominant p54AS-specific CD8+ T cells. Shown are the individual normalized expression, measured in fluorescence units, for each A) maturation, B) cytotoxicity, and C) proliferation and apoptosis gene that was determined to be differentially expressed between p11C- and p54AS-specific cells. Plots include values for which its matching pair is missing and therefore were not used in determination of differential expression. Fold change values are indicated in upper left corner. An asterisk under fold change values is present if that gene met the criteria for differential expression on that timepoint. For those genes with more than one probe on the BeadChip, the numerical probe IDs are included in the gene name. Red, p11C. Blue, p54AS.
(PDF)
Staining and gating of tetramers for quantification of epitope-specific cells in Mamu-A*01 immunodominance hierarchies. Gating of p11C-, p54AS/E660-, and p68A-specific CD8+ T cells from A) SIVmac251- and B) SIVsmE660-infected animals. 50,000 CD8+ T cell events are displayed on each flow plot. SIVmac251 frequencies were measured between weeks 37–50, except for 133-06 which died early and data used are from week 18. SIVsmE660 frequencies were measured between weeks 19–22.
(TIF)
Representative flow staining and gating for phenotyping. Representative staining and gating for phenotyping of tetramer-positive cells from (A) SIVmac251- and (B) SIVsmE660-infected monkeys. Left, Staining and gating of tetramer-positive cells among CD8+ T cells (40,000 CD8+ T cell events are shown). Right, staining and gating of CD28 (y-axis) and CCR7 (x-axis) cell surface proteins among the tetramer-positive cells (1,000 tetramer-positive events are shown). Phenotyping was conducted between weeks 44–78 for SIVmac251 and 33–46 for SIVsmE660.
(TIF)
Representative staining and gating of perforin and granzyme B. Mamu-A*01 tetramers were used to identify p11C-, p54AS/E660-, and p68A-specific cells among PBMCs from monkeys chronically-infected with either SIVmac251 or SIVsmE660. Perforin and granzyme B content within the tetramer-positive cells were measured by flow cytometry. A) FMOs used to set gates for perforin (left) and granzyme B (right). For SIVmac251 (B) and SIVsmE660 (D), representative staining and gating of tetramer-positive cells (left) and perforin+granzyme B+ cells among the tetramer-positive cells (right). 250,000 CD8+ T cells are shown on flow plots of tetramer staining. 1,000 tetramer-positive events are shown on flow plots of perforin and granzyme B staining. For SIVmac251 (C) and SIVsmE660 (E), histograms illustrating fluorescence intensity of perforin (top) and granzyme B (bottom) within the p11C (red), p54AS/E660 (blue), and p68A (green) tetramer-positive cells. FMOs shown as filled gray histograms. Measurements were conducted between weeks 63–83 for SIVmac251 and 41–49 for SIVsmE660.
(TIF)
Representative staining and gating of tetramers during in vitro expansion. PBMCs from monkeys chronically-infected with either SIVmac251 (A) or SIVsmE660 (B) were stimulated in vitro with either p11C, 54E660/AS, or p68A peptide, harvested on days 3, 4, 5, 6, 8, 10, 12, and 14 following stimulation, and measured by flow cytometry to calculate the percent (upper right on each flow plot) of tetramer-positive CD8+ T cells. Expansion was calculated as the fold change (middle left on each flow plot) of the percent of each tetramer-positive population on each day, relative to day 0. Number of CD8+ T cell events displayed are the same for pllC, p54AS/E660, and p68A plots within a given day. Measurements were conducted between weeks 40–52 for SIVmac251 and 31–44 for SIVsmE660.
(TIF)
Representative staining and gating for cytokine and chemokine production. PBMCs from monkeys chronically-infected with either SIVmac251 (A) or SIVsmE660 (B) were stimulated with either p11C, p54AS/E660, or p68A peptides and intracellular staining was used to assess production of the chemokine MIP-1β and the cytokines IFNγ, TNFα, and IL-2. Left, representative staining and gating of tetramer-positive cells (25,000 CD8+ T cell events are shown). Right, corresponding staining and gating of MIP-1β, IFNγ, TNFα, and IL-2 (250 tetramer-positive events are shown). Individual gates were then used in a Boolean analysis for assessment of polyfunctionality. Data was collected between weeks 36–42 for SIVmac251 and 14–25 for SIVsmE660.
(PDF)
Detection of specific binding of p11C and p54E660 peptide:Mamu-A*01 monomers to DRMs. DRMs were purified from total CD8+ T cells sorted from seven chronically-infected SIVsmE660-infected monkeys. The DRMs were evaluated for specific binding, measured in resonance units (RU), to pMHC monomers constructed with p11C, p54E660, and p68A epitope peptides and Mamu-A*01. Shown are overlaid readings of the binding of p11C (red) and p54E660 (blue) pMHC monomers at 100 μg/mL. p68A:Mamu-A*01 monomer binding above background was not detected at any concentration and is not shown. Readings have been normalized by subtracting the binding of the control monomer TL8 run at the same concentrations as the experimental monomers.
(TIF)
Titrations of p11C and p54E660 peptide:Mamu-A*01 monomers for calculation of binding kinetics and affinity. Shown are sensograms indicating the binding of p11C (left) and p54E660 (right) pMHC monomers to DRMs purified from total CD8+ T cells sorted from seven SIVsmE660-infected monkeys. p11C monomers were run at 25 (green), 50 (pink), 100 (blue), and 200 (red) μg/mL. The ARB0 plot for p11C shows a 150 μg/mL (light blue) run in place of the 100 μg/mL. The AP34, ZD57, and A6V031 plots for p11C do not show the 200 μg/mL run. p54E660 monomers were run at 25, 50, 150, and 200 μg/mL for AP54, ARB0, 8B1, and AS47 and at 25, 100, and 200 μg/mL for AP34, ZD57, and A6V031. The ZD57 plot includes an additional 50 μg/mL run. A Langmuir curve was fit to each binding curve at each concentration and was used to calculate binding kinetics. Readings have been normalized by subtracting the binding of the control monomer TL8 run at the same concentrations as the experimental monomers.
(PDF)
Detection of p68A monomer binding. p68A-specific CD8+ T cells were collected from multiple tetramer-specific flow cytometric cell sorts and pooled for DRM purification. Titrations of p68A pMHC monomers were performed at concentrations ranging from 150 to 1000 μg/mL. The highest concentration evaluated is shown. Binding of the control monomer TL8 at the same concentration has been subtracted from all readings.
(TIF)