

Abstract
Background
Atopic dermatitis (AD) is classified as extrinsic (ADe) and intrinsic (ADi), representing approximately 80% and 20% of patients, respectively. While sharing a similar clinical phenotype, only ADe is characterized by high serum IgE. Since most AD patients exhibit high IgE, an “allergic”/IgE-mediated disease pathogenesis was hypothesized. However, current models associate AD with T-cell activation, particularly Th2/Th22 polarization, and epidermal barrier defects.
Objective
To define if both variants share a common pathogenesis.
Methods
We stratified 51 severe AD patients as ADe (42) and ADi (9) (with similar mean disease activity/SCORAD), and analyzed the molecular and cellular skin pathology of lesional and non-lesional ADi and ADe using gene-expression (RT-PCR) and immunohistochemistry.
Results
A significant correlation between IgE levels and SCORAD (r=0.76, p<10−5) was found only in ADe. Marked infiltrates of T-cells and dendritic cells and corresponding epidermal alterations (K16, Mki67, S100A7/A8/A9) defined lesional skin of both variants. However, higher activation of all inflammatory axes (including Th2) was detected in ADi, particularly Th17 and Th22-cytokines. Positive correlations between Th17-related molecules and SCORAD were only found in ADi, while only ADe showed positive correlations between SCORAD and Th2-cytokines (IL-4, IL-5), and negative correlations with differentiation products (loricrin, periplakin).
Conclusions
Although differences in Th17 and Th22 activation exist between ADi and ADe, we identified common disease-defining features of T-cell activation, production of polarized cytokines, and keratinocyte responses to immune products. Our data indicate that a Th2 bias is not the sole cause of high IgE in ADe, with important implications for similar therapeutic interventions.
Clinical Implications
Both extrinsic and intrinsic AD variants might be treated with T-cell targeted therapeutics or agents that modify keratinocyte responses.
Keywords: atopic dermatitis, eczema, extrinsic, intrinsic, IgE, T-cell, human skin, keratinocytes, S100 proteins
Introduction
Atopic Dermatitis (AD) is a common inflammatory skin disease that together with asthma and allergic rhinitis forms the atopic triad.1 Both AD and asthma are sub-typed as extrinsic and intrinsic, representing approximately 80% and 20% of adult atopic patients, respectively. Extrinsic AD is characterized primarily by high serum IgE, as well as a personal and familial history of atopy and specific IgEs to food or aeroallergens. Intrinsic AD shares a similar clinical phenotype but exhibits normal serum IgE, absence of other atopic diseases, and lack of allergen-specific IgEs.2,3 Although peripheral eosinophilia can be seen in both, higher recruitment of eosinophils to inflamed tissues4 and prolonged eosinophil life span have been observed in extrinsic AD.5,6
Since the majority of AD patients exhibit high IgE levels, an “allergic”, IgE-mediated disease pathogenesis has been historically hypothesized. However, current models also associate AD with T-cell activation, particularly Th2/Th22 polarization, with a Th1 component in chronic AD, and a possible contribution of Th17.7 Thus, high levels of the Th2 cytokines (IL-4, IL-13) in AD skin lesions could influence immunoglobulin class switching, promoting excessive IgE production.8,9 These cytokines have also been identified as inhibitors of epidermal differentiation and production of antimicrobial peptides (AMPs).10–12
Inconsistent differences between intrinsic and extrinsic AD have been reported on a limited array of Th1- and Th2-related cytokines in PBMCs, lesional skin, and atopic patch test sites, as well as the expression of FCεRI on antigen presenting cells.13–18 Furthermore, prior comparisons did not measure the more recently discovered Th17 and Th22 T-cells.
Thus, the T-cell activation pathways and cytokine circuits in skin lesions of intrinsic versus extrinsic AD patients have not been well defined and a global analysis of the molecular and cellular skin pathology of intrinsic and extrinsic AD is unavailable. In this study, we compared the skin structure, cellular infiltrates, and molecular markers between intrinsic and extrinsic AD. Our data identified common disease-defining features of intrinsic and extrinsic AD, including marked T-cell and dendritic cell (DC) skin infiltration and activation, that are associated with epidermal alterations (i.e hyperplasia, and barrier abnormalities). Overall, we have found a higher immune activation in intrinsic as compared to extrinsic AD, particularly of the Th17 and Th22 immune axes. Additionally, both intrinsic and extrinsic AD lesions showed marked Th2 activation (high levels of IL-4/IL-13 expression), suggesting that a Th2 bias is not the sole cause of high IgE levels in extrinsic disease.
METHODS
Patient Population
For this study, we grouped several cohorts of AD patients previously published by our group7,19–23 as well as three new patients (n total=51). Chronic lesional (>72 hours duration with lichenification) and non-lesional (≥ 10 cm from active lesions) AD skin biopsies and serum samples were collected for these studies under Institutional Review Board-approved protocols. Patients were stratified into extrinsic (42 total, 23 males, 19 females, age=15–81 years; mean age=36.3 years) and intrinsic (9 total, 8 males, 1 female, age=20–67 years; mean age=40.3 years) AD categories. To define extrinsic and intrinsic status, we primarily used IgE level, with values >200 kU/L defining extrinsic AD and <200 kU/L defining intrinsic AD. A summary of the demographics and clinical characteristics is presented in Table 1 and Table E1 (See Supplementary Table E1 in the Online Repository). Serum IgE levels ranged between 26–93 kU/L, mean=52.1 kU/L in the intrinsic group and between 200-70364 kU/L; mean=3577 kU/L in the extrinsic group (Reference range=0–200 kU/L; p=1.14×10−15). Eosinophil counts were significantly increased in the extrinsic group as compared with the intrinsic group (Extrinsic range=0.30%–13.60%, mean=6%; Intrinsic range=1.20%–6.70%, mean=2.9%; reference range=0% to 7%; p=0.006). Scoring of Atopic Dermatitis (SCORAD) index used to evaluate disease severity was similar between the intrinsic and extrinsic patients (Intrinsic range=42–76, mean=54; Extrinsic range=28–97.5, mean=53; p=0.46). 2 patients with extrinsic AD were found to have filaggrin mutations (1 heterozygous for the R501X allele; 1 heterozygous for the 2282del4 allele), while 19 extrinsic patients did not. However, 21 extrinsic patients predated our FLG genotyping studies and mutation information was unavailable.19,21,24 No filaggrin gene mutations were found in the 9 intrinsic AD patients. No associations between IgE status and age (p=0.49, t-test), sex, or family history were found (p=0.07, p=1 respectively, Fisher’s Exact test).
Table 1. Patient Characteristics and Demographics.
Summary of clinical characteristics and demographics of atopic dermatitis (AD) patients, categorized as intrinsic AD by IgE levels <200kU/L and extrinsic AD with IgE levels 200kU/L. IgE levels and eosinophil counts differed significantly between the groups (p=1.4×10−15 and 0.006, respectively), while mean Scoring of atopic dermatitis (SCORAD) was very similar among the groups (p=0.46).
| Intrinsic | Extrinsic | p-value | |
|---|---|---|---|
| IgE (kU/L)* | 52.1 ± 22.7 | 3,576.2 ± 11,429 | 1.4×10−15 |
| SCORAD* | 54 ± 10.6 | 53 ± 17.3 | 0.46 |
| Eosinophils (%)* | 2.9 ± 0.02 | 6 ± 0.03 | 0.006 |
| Total Number of Patients (n) | 9 | 42 | - |
| Gender (Males/Females) (n) | 8 / 1 | 23 / 19 | 0.07 |
| Family or Personal History of Atopy (Positive/Negative/Unreported) (n) | 3 / 6 / 0 | 8 / 14 / 20 | 1 |
Mean±SD.
Skin biopsies were not taken from skin that was clinically judged as infected. No systemic or topical treatments were allowed for ≥4 weeks prior to biopsies (see Table E1 in the Online Repository). Tissue samples were frozen in OCT medium for immunohistochemistry (IHC) and liquid nitrogen for RNA extraction. Histologic and real-time PCR (RT-PCR) analyses were previously performed on available non-lesional and lesional skin samples 7,19–23 with some additional analyses performed for the present study. Due to limited tissue availability some analyses were only performed on a limited number of patients (See Methods section in the Online Repository).
Immunohistochemistry and RT-PCR
IHC was performed on cryostat tissue sections using purified mouse anti-human monoclonal antibodies (see Table E2 and Methods in the Online Repository). Epidermal thickness was quantified and positive cells per millimeter were counted manually using computer-assisted image-analysis software (ImageJ 1.42; NIH, Bethesda, MD).7,19–23
Sample preparation for RT-PCR
RNA was extracted for RT-PCR, which was performed with EZ-PCR Core Reagents (Life Technologies, Grand Island, NY), and custom primers were generated for analysis (See primer list in Methods in the Online Repository).
Statistical analyses
The RT-PCR values normalized to hARP were transformed to the log2 scale (Values of 0 were substituted with 20% of the minimum value observed for that gene within each study). Since data were gathered from multiple studies, the existence of a batch, or study effect must be addressed and data adjusted. As some studies did not enroll intrinsic patients, all available extrinsic patients (defined by high IgE status) have been used to estimate the batch/study effect; and these estimators have then been employed to adjust the batch effect on all patients. Subsequently, only data from patients with chronic AD and known IgE status were retained for further analyses.7,19–23
As in previous publications7,19–23, both expression values (in log2) and cell counts were modeled by a linear mixed-effects model, with Tissue (Lesional/Non-Lesional) and IgE Category as the fixed effect and a random intercept for each patient. The comparisons of interest were tested using contrasts. Some markers were excluded from the analyses due to sample size limitations (minimum sample size criteria for inclusion in analysis was: n=4 per group for statistical comparisons between groups, and n=6 for the correlation analyses). All analyses were carried out using statistical language R (www.R-project.org). Word clouds were created using the homonym package R with word sizes scaled by log2-fold change in the different comparisons.
RESULTS
We studied a group of 51 patients, of which 42 were classified as extrinsic and 9 as intrinsic AD. The disease severity (mean SCORAD) was very similar among these groups (53 and 54 for extrinsic and intrinsic AD, respectively; p=0.46) (see patient characteristics in Tables 1 and E1). In the extrinsic group, a significant correlation was found between increased IgE levels and higher disease activity as detected by the SCORAD (r=0.76, p=3.7×10−7); such a correlation was not found in the intrinsic group (Figure 1).
Figure 1.

Scatter plot of the correlation between IgE and Scoring of atopic dermatitis (SCORAD) in extrinsic (red) and intrinsic (blue) atopic dermatitis (AD) categories; lines represent linear regression within each group. Only extrinsic AD exhibits a significant Pearson correlation between SCORAD and IgE (R=0.76, p=3.7×10−7), which is lacking in intrinsic AD (R=0.11, p=0.77).
The cellular, molecular and gene-expression differences between the intrinsic and extrinsic AD transcriptomes were analyzed, defining the gene expression differences in lesional and non-lesional disease phenotypes of extrinsic and intrinsic patients as the respective AD transcriptomes as previously described (Figures 2–5).7,22,23
Figure 2.

Representative H&Es and immunohistochemistry of lesional and non-lesional skin of extrinsic and intrinsic atopic dermatitis (AD) patients. A; H&E demonstrates similar hyperplasia in extrinsic and intrinsic lesions. B–F; Large T-cell (CD3+), myeloid dendritic cell (DC) (CD11c+), mature DC (CD83+), inflammatory DC (FCεRI+), and atopic DC (OX40L+) infiltrates are evident in lesional skin of both groups. (10x). ANL, non-lesional; AL, lesional.
Figure 5.

Word-clouds representing the atopic dermatitis (AD) phenotypes, as determined by lesional and non-lesional differences (by IHC, RT-PCR) for (A, D) intrinsic and (B, E) extrinsic AD. Word sizes are proportional to fold-change (FCH) differences, which are scaled relative to the numbers in black. Significance, up/down-regulation indicated by color (see key). Scatter-plots compare extrinsic with intrinsic phenotypes, in C) IHC and F) RT-PCR. Circle diameter represents relative differences; darker colors represent increased significance.
Marked Epidermal Hyperplasia and Increased cellular infiltrates in lesional skin characterize both atopic dermatitis (AD) subtypes
Both intrinsic and extrinsic AD lesions are characterized by marked increases in T-cells (CD3+, CD8+; p<0.005 for both) and myeloid DCs (CD11c+; p=0.003 for intrinsic and 0.02 for extrinsic disease) compared with non-lesional skin (Figures 2 and 3). Epidermal hyperplasia was detected to a similar extent in both variants based on measures of epidermal thickness (Figure 3A; p=0.86), MKi67 staining (Figure 3B; p=0.16), and mRNA expression of K16 (Figure 4A; p=0.65). mRNA expression of terminal differentiation products were similar between both variants (Supplementary Figure E2 in the Online Repository).
Figure 3.

Bar-plots of cell-counts (immunohistochemistry) in lesional and non-lesional intrinsic and extrinsic atopic dermatitis (AD). A–B, Similar epidermal hyperplasia (Thickness, Ki67+). C–L, Both variants showed significant increases (greatest in intrinsic patients) in T-cells (CD3+, CD8+), various dendritic cells (DCs), and Langerhans cells (CD11c+, CD1a+, CD83+, CD1c+, FcεRI+, TRAIL+, OX40L+) in lesional versus non-lesional skin. M–O, Neutrophils (NE) were increased in intrinsic lesions; extrinsic lesions exhibited increased eosinophils (MBP+) and plasmacytoid DCs (BDCA2+). Degree of significance between any comparisons indicated if p<0.1. Asterisks in parenthesis (top-right corners) represent significance of the interaction term (Anova model); if present, the AD phenotypes greatly differ between the variants. Mean±SEM. *p<0.10, **p<0.05, ***p<0.01, ****p<0.001. ANL, non-lesional; AL, lesional.
Figure 4.

Bar-plots of mRNA expression in lesional and non-lesional skin from intrinsic and extrinsic patients. A-DD, Similar hyperplasia and activation of all inflammatory axes characterize lesional versus non-lesional skin in both variants. Intrinsic lesions exhibited more significant increases in markers of inflammation, Th1, Th22, Th17, and Tregs. Degree of significance between any comparisons indicated if p<0.1. Asterisks in parenthesis (top-right corners) represent significance of the interaction term (Anova model); if present, the AD phenotypes greatly differ between the variants. Mean±SEM. *p<0.10, **p<0.05, ***p<0.01, ****p<0.001. ANL, non-lesional; AL, lesional.
Additionally, both AD variants also show similar increases in DC subsets, though some differences were detected in the magnitude of T-cells and DC infiltrates (Figures 2–3). Higher infiltrates of T-cells (CD3+, CD8+), myeloid DCs (CD11c+), Langerhans cells (LCs; CD1a+), and mature DCs (CD83+) were detected in intrinsic AD (p<0.05 for all). Less significant differences were observed for inflammatory dendritic epidermal cells/IDECs (FcεRI+, CD206+), resident DCs (CD1c+), atopic DCs (OX40L+, TRAIL). Neutrophils (Neutrophil Elastase+) were more abundant in intrinsic AD (Figure 3M; Supplementary Figure E1B in the Online Repository; p=0.009). Counts of eosinophils as detected by major basic protein (MBP+; Figure 3N and Supplementary Figure E1C in the Online Repository) and plasmacytoid DCs marked by blood dendritic cell antigen 2 (BDCA2+; Figure 3O) were greatly increased in lesional extrinsic AD (p=0.08 and 0.008, respectively) compared with intrinsic AD. Overall, larger cellular infiltrates were detected in intrinsic AD (Figures 2–3, E Figure 1).
More robust immune activation in intrinsic atopic dermatitis
Intrinsic and extrinsic AD were both associated with significant increases between lesional and non-lesional AD in gene expression levels of Th2-(IL-10, IL-13), Th22-(IL-22), and Th1-(IFN) defining cytokines. The IL-17/Th17-(IL-17, IL23p40, and CCL20; p=0.002, 0.05, and 0.004 respectively) and Th9-(IL-9) related products were significantly increased only in lesional intrinsic AD skin. Moreover, when comparing lesional skin in intrinsic and extrinsic AD, significant increases were detected in expression levels of genes related to inflammation (MMP12; p=0.006), as well as Th2-(CCL18, CCL22, TSLPR, OX40L), Th22-(IL-22), IFN/Th1-(MX-1), Th17-(IL-17, CCL20), and Treg-(FOXP3, p=0.06) related molecules. IL-19 (p=0.12) was the only cytokine that was increased in extrinsic versus intrinsic lesional AD skin (Figures 4).
Overall, lesional skin in intrinsic AD shows similar or higher Th2 and Th1 activity but significantly more robust Th22 and Th17 immune responses compared with extrinsic AD. Intrinsic AD showed a strong Th17 molecular fingerprint in lesional skin (Figures 2–4, Supplementary Figure E3A–B in the Online Repository). Non-lesional skin appeared, overall, to be very similar between the two groups, and only a small number of genes (OX40L, FOXP3, IL-33; p=0.09, 0.08, and 0.08 respectively) showed any appreciable differences between intrinsic and extrinsic AD (Figures 4L, 4DD, Supplementary Figure E3CD in the Online Repository).
Epidermal and T-cell pathway activation defines the atopic dermatitis transcriptome regardless of IgE status
To best visualize similarities and differences between extrinsic and intrinsic AD, we have created word clouds with the respective protein (by immunohistochemistry) and mRNA gene expression (by RT-PCR) AD phenotypes and scatter plots of the differences between these phenotypes (see Methods and Online Repository for details). The word clouds are scaled proportionately, with numbers added (in black) to illustrate relative fold change (FCH) differences. Significance is conveyed by color (Figure 5A–F). The diameter of the circles in the scatter plots is proportionate to the relative FCHs, and greater significance of the phenotype differences are emphasized by darker colors.
Overall, as illustrated in Figure 5A–F, the intrinsic AD phenotype is characterized by both higher leukocyte cell counts and gene expression for a subset of inflammatory genes. However, only selected cellular infiltrates and genes showed significant differences in the intrinsic versus extrinsic AD phenotypes (Figure 5C, F). Of particular interest are significant increases in expression of IL-17A (p=0.002), found only in the intrinsic AD transcriptome (Figure 5F).
Both intrinsic and extrinsic AD phenotypes showed significant activation of mRNA gene expression of the S100 epidermal responses and the Th1, Th2, and Th22 inflammatory pathways by RT-PCR. Contrary to our expectation, not only there was no Th2 bias in the extrinsic group, but we actually found higher immune activation of all immune axes, including Th2, in the intrinsic group (Figure 5D–F).
Associations with disease activity in intrinsic and extrinsic atopic dermatitis
To correlate disease activity (determined by the SCORAD) with variables measured by IHC and RT-PCR in lesional skin, we used the Pearson correlational coefficient. In the intrinsic AD group, mRNA and protein expression of several inflammatory mediators in lesional tissues was correlated with the SCORAD (Figure 6A, Supplementary Tables E3–4 in the Online Repository). These included inflammatory mediators, such as interferon-α (r=0.81, p=0.03, n=7), S100A12 (S100 Calcium-Binding Protein A12/Enrage; r=0.75, p=0.05, n=7), Matrix metalloproteinase 12 (MMP12; r=0.66, p=0.07, n=8), Tumor Necrosis Factor Ligand Superfamily 10 (TRAIL; r=0.75, p=0.05, n=7), and the Th17-associated chemokine CCL20 (r=0.68, p=0.09, n=7) (Figure 6A, Supplementary Tables E3–4 in the Online Repository).
Figure 6.
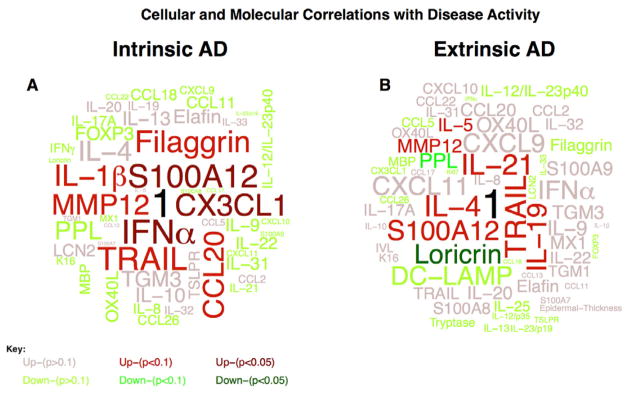
Word-clouds representing correlations between cellular and molecular markers (IHC, RT-PCR) and disease severity (SCORAD) in lesional A) intrinsic and B) extrinsic atopic dermatitis. Words in the clouds are proportional to the correlation coefficient between markers and SCORAD and are color-coded for significance and up/down-regulation (see key). A perfect correlation is indicated by 1 (black).
In the extrinsic group, high correlation coefficients between SCORAD and lesional skin expression were noted for inflammatory mediators (i.e. S100A12), Th2-related cytokines such as IL-4 (r=0.67, p=0.07, n=8), and the Th2-promoting cytokines IL-21 (r=0.67, p=0.07, n=8)25 and IL-19 (r=0.62, p=0.09, n=8).26,27 Furthermore, a strong negative correlation was detected only in the extrinsic group between the SCORAD and mRNA expression of terminal differentiation genes such as LOR (r=−0.59, p=0.02, n=16) and PPL (r=−0.43, p=0.09, n=16) (Figure 6B, Tables E3–4 in Online Repository).
Discussion
Although intrinsic AD represents a minority of AD patients, it is important to clarify whether it represents a single disease spectrum along with extrinsic AD or whether different immune mechanisms underlie the two variants.
Numerous studies have tried to define differences and similarities in pathogenic mechanisms of extrinsic and intrinsic AD.2,4,14,16,28–30 Such studies have suggested common disease features of epidermal hyperplasia and increased infiltration of T-cells and DC subsets, but indicated a Th2 bias and increased eosinophils in extrinsic AD.2,14,16,31 While previous studies of T-cell polarization in skin lesions of extrinsic and intrinsic AD have focused on Th1 and Th2 subsets/cytokines, activation of novel T-cell subsets, Th22 and Th17 T-cells might also be important for disease pathogenesis. While AD becomes increasingly recognized as associated with Th22, the role of Th17 T-cell in AD is still controversial.32–34 A few studies showed an increased Th17 axis in AD35,36, but overall the Th17 pathway is much less activated in AD compared to psoriasis.20 IL-17 and IL-22 cytokines derived from the newly recognized T-cell subsets have major effects on epidermal keratinocytes (i.e. regulating the transcription of S100A7, A8, and A9 mRNAs in human keratinocytes).37,38 The onset of acute AD shows significantly higher synthesis of these cytokines, coupled with marked epidermal activation and increased detection of the S100s gene expression, in comparison with uninvolved AD skin.7 Additionally, the Th2 (IL-4, IL-13, IL-31) and Th22 (IL-22) cytokines were shown to suppress epidermal differentiation.10,37,39 Thus, polar cytokines might interact to produce the pathologic epidermal phenotype in AD.7,37,38,40 However, the relative balance of T-cell activation and cytokine circuits of the novel T-cell subsets (Th17, Th22) in skin lesions of extrinsic and intrinsic AD have not been characterized.16,41,42
The present study provides novel insights into T-cell subsets that are activated in extrinsic versus intrinsic AD. IL-22 mRNA expression is significantly elevated in lesional versus non-lesional skin of both AD variants, although expression level is significantly higher in lesional intrinsic compared to extrinsic skin. Production of IL-17 cytokine and its related products IL-12/IL-34p40, Elafin and CCL20 are also highly up-regulated in intrinsic AD compared to extrinsic lesional skin. S100A7, A8, A9, and A12 mRNAs are highly up-regulated in lesional skin versus non-lesional skin of both intrinsic and extrinsic AD. S100A9 and S100A12 mRNA levels are higher in intrinsic versus extrinsic lesions, which might be explained by the higher expression of both IL-17 and IL-22 in intrinsic lesional skin and their potential synergistic effects on production of these S100s. Production of cathelicidin/LL-37 has also been reported to be lower in extrinsic compared to intrinsic skin lesions.13,43 Since LL-37 is strongly induced by IL-17 in human keratinocytes, the previously noted difference in LL-37 expression in intrinsic AD might be explained by higher levels of IL-17. Additional evidence for higher IL-17 activation in intrinsic lesions is suggested by the mRNA expression of the IL-17-regulated CCL20 and Elafin, as well as greater numbers of neutrophils in intrinsic AD, likely reflecting an increased Th17/IL-17 axis in intrinsic disease. Epidermal hyperplasia, which was comparably elevated in intrinsic and extrinsic skin lesions (Figures 3A, 4A), could be directly stimulated by IL-22 over-production in skin lesions; additionally, however, it could also be influenced by other cytokines such as IL-19, IL-20, and IL-24.44,45 From the study of IL-17 blockade in psoriasis46, it also appears that this cytokine can regulate keratinocyte hyperplasia, but most likely through secondary effects on induction of other cytokines, e.g, IL-19, IL-36, and IL-23, that have direct effects on keratinocyte growth.47,48
The cytokine environment within skin lesions might also influence immunoglobulin class switching. Several studies support the idea that high levels of IgE, produced by activated B-cells in extrinsic AD, are regulated by increased production of IL-13 or IL-4 cytokines by skin-homing T-cells.2,14,29,30 Conversely, higher production of interferon-γ in T-cells from intrinsic AD could suppress IgE production while stimulating IgG4 levels.2,14,16,29,30 Hence, from past work, one could hypothesize that differential activation of Th2 T-cells in skin lesions of intrinsic versus extrinsic disease might be the cellular basis for increased IgE production in extrinsic patients. Instead of the expected Th2 bias in extrinsic compared with intrinsic AD skin lesions, our study showed that the mRNA levels for the Th2 cytokines IL-4, IL-5, IL-13, and IL-31 were similarly elevated in skin lesions from both AD forms. Thus, the regulation of IgE levels is more complex than Th2 activation in the skin. Consistent with the concept of interferon-γ as a suppressive cytokine for Th2 activation48 and IgE production14,29, we measured higher mRNA levels for this cytokine and its induced molecules (CXCL9, CXCL10, and MX-1) in lesional intrinsic versus extrinsic AD skin. We also showed increased levels of the regulatory T-cell marker FOXP3 in lesional intrinsic skin, which has recently been shown to suppress allergic inflammation through induction of IgG4 and inhibition of IgE49, reflecting an alternative mechanism for the attenuated IgE production in intrinsic AD. Another potential consideration is that IL-17A cytokine blocks the Th2 cytokine effects through negative regulation of the TSLP immune pathway50, ultimately suppressing IgE production in intrinsic AD. Additionally, regulation of IgE production might also take place outside the skin, e.g. cytokine levels in the blood affecting circulating B-cells, which are more activated in AD compared to normal controls, or that levels of cytokines in lymph nodes affect B-cell class switching and IgE production.8,51–53 Since we observed a correlation between the SCORAD and IgE levels in extrinsic AD, we should also consider mechanisms by which IgE might increase cellular immune activation in the skin. Although we did not detect increased protein expression of the high affinity IgE receptor (FcεRI) in lesional extrinsic AD skin, its increased expression, previously noted on IDECs by flow cytometry,3,54,55 might provide a molecular target for IgE binding that potentially influences more complex immunological circuits in the skin. Although a few reports showed inconclusive effects of anti IgE targeting,56–58 the benefit from such an intervention still needs to be determined, particularly for extrinsic AD.
The hypothesized model of barrier inhibition via activation of Th2 cytokines10,11,59,60 could be operative in both extrinsic and intrinsic AD. However, our results noted strong correlations between disease activity/SCORAD and Th2 cytokines (IL-4, and IL-5), paralleled by negative correlations with barrier products (i.e LOR, PPL, and FLG) only in extrinsic AD (Figure 6B). In contrast, intrinsic AD demonstrated strong correlations between the SCORAD and Th1/IFN related genes (i.e. IL-1β, and IFNα) and the IL-17-related CCL20 chemokine (Figure 6A). Thus, in intrinsic AD, there may be competing effects of Th1, Th17 and Th2 cytokines on epidermal differentiation, so that cytokine-effect models may differ in this disease variant. Emerging studies of IL-4R (REGN668/clinical.trials.gov) and TSLPR blockade (MK8226-003/clinicaltrials.gov), which are being conducted in patients with both AD forms, may help clarify the physiological effects of Th2 cytokines on skin disease and associated barrier pathology.
We acknowledge a few inherent limitations of this study, including a retrospective design, several patient cohorts stratified for the extrinsic versus intrinsic analysis, and an unbalanced sample size. Nevertheless, our study defines molecular characteristics of both disease variants with significant differences between intrinsic and extrinsic AD. Our data, supported by the similarly effective therapeutic responses of extrinsic and intrinsic AD patients to Cyclosporine A, a broad T-cell suppressant61, suggest that both AD forms are primarily T-cell driven. Beyond the Th2 antagonism that could be used regardless of disease variant, the high expression of IL-17, IL-22, and IL-23/IL-12p40 suggest that use of other selective antagonists against these cytokines might have potential therapeutic benefits in AD, as directed by similarities and differences between its extrinsic and intrinsic forms.
Supplementary Material
Representative lesional and non-lesional samples from intrinsic and extrinsic patients showing neutrophil and eosinophil immunostaining (by Neutrophil Elastase and Major Basic Protein, respectively). A) H&Es of depicted samples. B) Significance of neutrophil infiltrates was greater in intrinsic versus extrinsic lesional (p=0.008 and 0.08, respectively) C) increased eosinophils detected in extrinsic versus intrinsic lesional skin (p=0.08 and 0.30, respectively).
Bar-plots illustrating lesional and non-lesional gene-expression (RT-PCR) of terminal differentiation products (A)Filaggrin, (B)Loricrin, (C)Transglutaminase-3 (TGM3), and (D)Periplakin. Overall, similar expression of terminal differentiation markers was found between lesional and non-lesional skin, as well as between intrinsic and extrinsic disease. Bars=SEM. *p<0.10, **p<0.05, ***p<0.01, ****p<0.001.
Word-clouds and scatter-plots representing mRNA expression differences (RT-PCR) for intrinsic versus extrinsic atopic dermatitis (AD) in A–B) lesional and C–D) non-lesional skin. Numbers (black) serve as fold-change scales for word sizes, which are sized according to expression differences between variants. Significance and up/down-regulation indicated by color (see key). In scatter-plots, circle diameter represents relative differences; darker colors represent increased significance.
Acknowledgments
Funding: JGK, MSF, and ND were supported by grant number 5UL1RR024143-02 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. EGY was supported by the Dermatology Foundation Physician Scientist Career Development Award.
Abbreviations
- AD
Atopic Dermatitis
- Th1
Type 1 Helper T Cell
- Th2
Type 2 Helper T Cell
- Th17
Type 17 Helper T Cell
- Th22
Type 22 Helper T cell
- Treg
Regulatory T-cell
- SCORAD
Scoring of Atopic
- IL-x
Interleukin (x identifies the type)
- CCL-x
CC Chemokine Ligand-x (x identifies the type)
- CXCL-x
CXC Chemokine-x (x identifies the type)
- IL4RA
Interleukin 4 Receptor α
- IL23p40
Interleukin 23 Subunit p40
- IgE
Immunoglobulin E
- IgG
Immunoglobulin G
- TNF
Tumor Necrosis Factor
- AMPs
antimicrobial peptides
- FceRI
Fc Epsilon Receptor 1
- DCs
Dendritic Cells
- LC
Langerhans cells
- IDEC
Inflammatory dendritic epidermal cells
- PBMC
Peripheral blood mononuclear cell
- MBP
Major Basic Protein
- BDCA2
Blood dendritic cell antigen 2
- FOXP3
Forkhead box P3
- FCH
Fold change
- hARP
Human acidic ribosomal protein
- IHC
Immunohistochemistry
- RT-PCR, Real
time PCR
- IFN
Interferon
- LOR
Loricrin
- PPL
Periplakin
- FLG
Filaggrin
- TSLP
Thymic Stromal Lymphopoietin
- TSLPR
Thymic Stromal Lymphopoietin Receptor
- IL-12/23p40
Common p40 subunit of Interleukins-12 and -23
- IL-4R
IL-4 Receptor
- K16
Keratin 16
- S100A-(7,8, 9, 12)
S100-Calcium binding protein A(7, 8, 9, 12)
- MMP12
Matrix metalloproteinase 12
- TRAIL
Tumor Necrosis Factor Ligand Superfamily 10
Footnotes
Disclosures: The authors have declared that they have no conflict of interest related to this study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guttman-Yassky E, Nograles K, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis—Part I: Clinical and pathologic concepts. J Allergy Clin Immunol. 2011;127:1110–8. doi: 10.1016/j.jaci.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 2.Akdis CA, Akdis M. Immunological differences between intrinsic and extrinsic types of atopic dermatitis. Clin Exp Allergy. 2003 Dec;33(12):1618–21. doi: 10.1111/j.1365-2222.2003.01803.x. [DOI] [PubMed] [Google Scholar]
- 3.Schmid-Grendelmeier P, Simon D, Simon HU, Akdis CA, Wüthrich B. Epidemiology, clinical features, and immunology of the “intrinsic” (non-IgEmediated) type of atopic dermatitis (constitutional dermatitis) Allergy. 2001 Sep;56(9):841–9. doi: 10.1034/j.1398-9995.2001.00144.x. [DOI] [PubMed] [Google Scholar]
- 4.Jeong CW, Ahn KS, Rho N-K, Park YD, Lee D-Y, Lee J-H, et al. Differential in vivo cytokine mRNA expression in lesional skin of intrinsic vs. extrinsic atopic dermatitis patients using semiquantitative RT-PCR. Clin Exp Allergy. 2003 Dec;33(12):1717–24. doi: 10.1111/j.1365-2222.2003.01782.x. [DOI] [PubMed] [Google Scholar]
- 5.Simon HU, Blaser K. Inhibition of programmed eosinophil death: a key pathogenic event for eosinophilia? Immunol Today. 1995 Feb;16(2):53–5. doi: 10.1016/0167-5699(95)80086-7. [DOI] [PubMed] [Google Scholar]
- 6.Wedi B, Raap U, Lewrick H, Kapp A. Delayed eosinophil programmed cell death in vitro: A common feature of inhalant allergy and extrinsic and intrinsic atopic dermatitis. J Allergy Clin Immunol. 1997 Oct;100(4):536–43. doi: 10.1016/s0091-6749(97)70147-7. [DOI] [PubMed] [Google Scholar]
- 7.Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J Allergy Clin Immunol. 2012 Aug; doi: 10.1016/j.jaci.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulsen LK, Hummelshoj L. Triggers of IgE class switching and allergy development. Ann Med. 2007;39(6):440–56. doi: 10.1080/07853890701449354. [DOI] [PubMed] [Google Scholar]
- 9.Oettgen HC, Geha RS. IgE in asthma and atopy: cellular and molecular connections. J Clin Investi. 1999 Oct 1;104(7):829–35. doi: 10.1172/JCI8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howell MD, Fairchild HR, Kim BE, Bin L, Boguniewicz M, Redzic JS, et al. Th2 Cytokines Act on S100/A11 to Downregulate Keratinocyte Differentiation. J Invest Dermatol. 2008 Apr 3;128(9):2248–58. doi: 10.1038/jid.2008.74. [DOI] [PubMed] [Google Scholar]
- 11.Kim BE, Leung DYM, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008 Mar;126(3):332–7. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin Immunol. 2006 Dec;121(3):332–8. doi: 10.1016/j.clim.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Leung D, Novak N, Boguniewicz M, Bieber T. Elevated levels of IL-10 in intrinsic (IAD) and extrinsic atopic dermatitis (EAD) is associated with deficiency in anti-microbial peptide (AMP) production. J Allergy Clin Immunol. 2004 Feb;113(2):S96. [Google Scholar]
- 14.Kabashima-Kubo R, Nakamura M, Sakabe J-I, Sugita K, Hino R, Mori T, et al. A group of atopic dermatitis without IgE elevation or barrier impairment shows a high Th1 frequency: possible immunological state of the intrinsic type. J Dermatol Sci. 2012 Jul;67(1):37–43. doi: 10.1016/j.jdermsci.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Park CO, Lee HJ, Lee JH, Wu WH, Chang NS, Hua L, et al. Increased expression of CC chemokine ligand 18 in extrinsic atopic dermatitis patients. Exp Dermatol. 2008 Jan;17(1):24–9. doi: 10.1111/j.1600-0625.2007.00634.x. [DOI] [PubMed] [Google Scholar]
- 16.Tokura Y. Extrinsic and intrinsic types of atopic dermatitis. J Dermatol Sci. 2010 Apr;58(1):1–7. doi: 10.1016/j.jdermsci.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 17.Kerschenlohr K, Decard S, Przybilla B, Wollenberg A. Atopy patch test reactions show a rapid influx of inflammatory dendritic epidermal cells in patients with extrinsic atopic dermatitis and patients with intrinsic atopic dermatitis. J Allergy Clin Immunol. 2003 Apr;111(4):869–74. doi: 10.1067/mai.2003.1347. [DOI] [PubMed] [Google Scholar]
- 18.Akdis CA, Akdis M, Simon D, Dibbert B, Weber M, Gratzl S, et al. T cells and T cell-derived cytokines as pathogenic factors in the nonallergic form of atopic dermatitis. J Invest Dermatol. 1999 Oct;113(4):628–34. doi: 10.1046/j.1523-1747.1999.00720.x. [DOI] [PubMed] [Google Scholar]
- 19.Guttman-Yassky EE, Lowes MA, Fuentes-Duculan JJ, Whynot JJ, Novitskaya II, Cardinale II, et al. Major differences in inflammatory dendritic cells and their products distinguish atopic dermatitis from psoriasis. J Allergy Clin Immunol. 2007 May 1;119(5):1210–7. doi: 10.1016/j.jaci.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, et al. Low Expression of the IL-23/Th17 Pathway in Atopic Dermatitis Compared to Psoriasis. J Immunol. 2008;181:7420–7. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guttman-Yassky E, Suárez-Fariñas M, Chiricozzi A, Nograles KE, Shemer A, Fuentes-Duculan J, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009 Dec;124(6):1235–58. doi: 10.1016/j.jaci.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 22.Tintle S, Shemer A, Suárez-Fariñas M, Fujita H, Gilleaudeau P, Sullivan-Whalen M, et al. Reversal of atopic dermatitis with narrow-band UVB phototherapy and biomarkers for therapeutic response. J Allergy Clin Immunol. 2011 Sep;129(1):76–85. doi: 10.1016/j.jaci.2011.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suárez-Fariñas M, Tintle SJ, Shemer A, Chiricozzi A, Nograles KE, Cardinale I, et al. Non-lesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127:954–64. doi: 10.1016/j.jaci.2010.12.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guttman-Yassky E, Vugmeyster Y, Lowes MA, Chamian F, Kikuchi T, Kagen M, et al. Blockade of CD11a by Efalizumab in Psoriasis Patients Induces a Unique State of T-Cell Hyporesponsiveness. J Invest Dermatol. 2008 Jan 31;128(5):1182–91. doi: 10.1038/jid.2008.4. [DOI] [PubMed] [Google Scholar]
- 25.Wurster AL, Rodgers VL, Satoskar AR, Whitters MJ, Young DA, Collins M, et al. Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J Exp Med. 2002 Oct 7;196(7):969–77. doi: 10.1084/jem.20020620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, et al. Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. Int Immunopharmacol. 2004 May;4(5):615–26. doi: 10.1016/j.intimp.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Gallagher G. Interleukin-19: multiple roles in immune regulation and disease. Cytokine Growth Factor Rev. 2010 Oct;21(5):345–52. doi: 10.1016/j.cytogfr.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Rho N-K, Kim W-S, Lee D-Y, Lee J-H, Lee E-S, Yang J-M. Immunophenotyping of inflammatory cells in lesional skin of the extrinsic and intrinsic types of atopic dermatitis. Br J Dermatol. 2004 Jul;151(1):119–25. doi: 10.1111/j.1365-2133.2004.06027.x. [DOI] [PubMed] [Google Scholar]
- 29.Akdis M, Akdis CA, Weigl L, Disch R, Blaser K. Skin-homing, CLA+ memory T cells are activated in atopic dermatitis and regulate IgE by an IL-13-dominated cytokine pattern: IgG4 counter-regulation by CLA-memory T cells. J Immunol. 1997 Nov 1;159(9):4611–9. [PubMed] [Google Scholar]
- 30.Akdis M, Simon HU, Weigl L, Kreyden O, Blaser K, Akdis CA. Skin homing (cutaneous lymphocyte-associated antigen-positive) CD8+ T cells respond to superantigen and contribute to eosinophilia and IgE production in atopic dermatitis. J Immunol. 1999 Jul 1;163(1):466–75. [PubMed] [Google Scholar]
- 31.Park J-H, Choi Y-L, Namkung J-H, Kim W-S, Lee J-H, Park H-J, et al. Characteristics of extrinsic vs. intrinsic atopic dermatitis in infancy: correlations with laboratory variables. Br J Dermatol. 2006 Oct;155(4):778–83. doi: 10.1111/j.1365-2133.2006.07394.x. [DOI] [PubMed] [Google Scholar]
- 32.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009 Jun;123(6):1244–52. e2. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis—Part II: Immune cell subsets and therapeutic concepts. J Allergy Clin Immunol. 2011 Jun;127(6):1420–32. doi: 10.1016/j.jaci.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, Noh G, Lee S, Youn Y, Rhim J. Atopic dermatitis and cytokines: recent patents in immunoregulatory and therapeutic implications of cytokines in atopic dermatitis--part I: cytokines in atopic dermatitis. Recent Pat Inflamm Allergy Drug Discov. 2012 Sep;6(3):222–47. doi: 10.2174/187221312802652820. [DOI] [PubMed] [Google Scholar]
- 35.Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008 Nov;128(11):2625–30. doi: 10.1038/jid.2008.111. [DOI] [PubMed] [Google Scholar]
- 36.Toda M, Leung DYM, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, et al. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol. 2003 Apr;111(4):875–81. doi: 10.1067/mai.2003.1414. [DOI] [PubMed] [Google Scholar]
- 37.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suárez-Fariñas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008 Nov;159(5):1092–102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boniface K, Bernard F-X, Garcia M, Gurney AL, Lecron J-C, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005 Mar 15;174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 39.Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Lüscher-Firzlaff J, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol. 2012 Feb;129(2):426–33. 433.e1–8. doi: 10.1016/j.jaci.2011.10.042. [DOI] [PubMed] [Google Scholar]
- 40.Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006 Oct 2;203(10):2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novak N, Bieber T. Allergic and nonallergic forms of atopic diseases. J Allergy Clin Immunol. 2003 Aug;112(2):252–62. doi: 10.1067/mai.2003.1595. [DOI] [PubMed] [Google Scholar]
- 42.Werfel T. The Role of Leukocytes, Keratinocytes, and Allergen-Specific IgE in the Development of Atopic Dermatitis. J Invest Dermatol. 2009 Apr 9;129(8):1878–91. doi: 10.1038/jid.2009.71. [DOI] [PubMed] [Google Scholar]
- 43.Howell MD, Novak N, Bieber T, Pastore S, Girolomoni G, Boguniewicz M, et al. Interleukin-10 Downregulates Anti-Microbial Peptide Expression in Atopic Dermatitis. J Invest Dermatol. 2005 Oct;125(4):738–45. doi: 10.1111/j.0022-202X.2005.23776.x. [DOI] [PubMed] [Google Scholar]
- 44.Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007 Feb 15;178(4):2229–40. doi: 10.4049/jimmunol.178.4.2229. [DOI] [PubMed] [Google Scholar]
- 45.He M, Liang P. IL-24 transgenic mice: in vivo evidence of overlapping functions for IL-20, IL-22, and IL-24 in the epidermis. J Immunol. 2010 Feb 15;184(4):1793–8. doi: 10.4049/jimmunol.0901829. [DOI] [PubMed] [Google Scholar]
- 46.Krueger JG, Fretzin S, Suárez-Fariñas M, Haslett PA, Phipps KM, Cameron GS, et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J Allergy Clin Immunol. 2012 Jul;130(1):145–9. doi: 10.1016/j.jaci.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindroos J, Svensson L, Norsgaard H, Lovato P, Moller K, Hagedorn PH, et al. IL-23-mediated epidermal hyperplasia is dependent on IL-6. J Invest Dermatol. 2011 May;131(5):1110–8. doi: 10.1038/jid.2010.432. [DOI] [PubMed] [Google Scholar]
- 48.Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. 2011 Feb 1;186(3):1495–502. doi: 10.4049/jimmunol.1001001. [DOI] [PubMed] [Google Scholar]
- 49.Meiler F, Klunker S, Zimmermann M, Akdis CA, Akdis M. Distinct regulation of IgE, IgG4 and IgA by T regulatory cells and toll-like receptors. Allergy. 2008 Nov;63(11):1455–63. doi: 10.1111/j.1398-9995.2008.01774.x. [DOI] [PubMed] [Google Scholar]
- 50.Bogiatzi SI, Guillot-Delost M, Cappuccio A, Bichet J-C, Chouchane-Mlik O, Donnadieu M-H, et al. Multiple-checkpoint inhibition of thymic stromal lymphopoietin–induced TH2 response by TH17-related cytokines. J Allergy Clin Immunol. 2012 Jul;130(1):233–5. doi: 10.1016/j.jaci.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 51.Obayashi K, Doi T, Koyasu S. Dendritic cells suppress IgE production in B cells. Int Immunol. 2007 Feb;19(2):217–26. doi: 10.1093/intimm/dxl138. [DOI] [PubMed] [Google Scholar]
- 52.Corry DB, Kheradmand F. Induction and regulation of the IgE response. Nature. 1999 doi: 10.1038/35037014. [DOI] [PubMed] [Google Scholar]
- 53.Akdis M, Akdis CA. IgE class switching and cellular memory. Nat Immunol. 2012 Apr;13(4):312–4. doi: 10.1038/ni.2266. [DOI] [PubMed] [Google Scholar]
- 54.Oppel T, Schuller E, Günther S, Moderer M, Haberstok J, Bieber T, et al. Phenotyping of epidermal dendritic cells allows the differentiation between extrinsic and intrinsic forms of atopic dermatitis. Br J Dermatol. 2000 Dec;143(6):1193–8. doi: 10.1046/j.1365-2133.2000.03887.x. [DOI] [PubMed] [Google Scholar]
- 55.Novak N, Kruse S, Kraft S, Geiger E, Kluken H, Fimmers R, et al. Dichotomic Nature of Atopic Dermatitis Reflected by Combined Analysis of Monocyte Immunophenotyping and Single Nucleotide Polymorphisms of the Interleukin-4/Interleukin-13 Receptor Gene: The Dichotomy of Extrinsic and Intrinsic Atopic Dermatitis. J Invest Dermatol. 2002 Oct;119(4):870–5. doi: 10.1046/j.1523-1747.2002.00191.x. [DOI] [PubMed] [Google Scholar]
- 56.Kim DH, Park KY, Kim BJ, Kim MN, Mun SK. Anti-immunoglobulin E in the treatment of refractory atopic dermatitis. Clin Exp Dermatol. 2012 Oct 22; doi: 10.1111/j.1365-2230.2012.04438.x. [DOI] [PubMed] [Google Scholar]
- 57.Park S-Y, Choi M-R, Na J-I, Youn S-W, Park K-C, Huh C-H. Recalcitrant atopic dermatitis treated with omalizumab. Ann Dermatol. 2010 Aug;22(3):349–52. doi: 10.5021/ad.2010.22.3.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heil PM, Maurer D, Klein B, Hultsch T, Stingl G. Omalizumab therapy in atopic dermatitis: depletion of IgE does not improve the clinical course - a randomized, placebo-controlled and double blind pilot study. J Dtsch Dermatol Ges. 2010 Dec;8(12):990–8. doi: 10.1111/j.1610-0387.2010.07497.x. [DOI] [PubMed] [Google Scholar]
- 59.Howell MD, Kim BE, Boguniewicz M, Leung DYM. J Allergy Clin Immunol. 1. Vol. 119. Elsevier; 2007. Jan, Modulation of Filaggrin by Th2 Cytokines in the Skin of Atopic Dermatitis (AD) pp. S283–3. [Google Scholar]
- 60.Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007 Jul;120(1):150–5. doi: 10.1016/j.jaci.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haw S, Shin M-K, Haw C-R. The Efficacy and Safety of Long-term Oral Cyclosporine Treatment for Patients with Atopic Dermatitis. Ann Dermatol. 2010 Feb;22(1):9–15. doi: 10.5021/ad.2010.22.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative lesional and non-lesional samples from intrinsic and extrinsic patients showing neutrophil and eosinophil immunostaining (by Neutrophil Elastase and Major Basic Protein, respectively). A) H&Es of depicted samples. B) Significance of neutrophil infiltrates was greater in intrinsic versus extrinsic lesional (p=0.008 and 0.08, respectively) C) increased eosinophils detected in extrinsic versus intrinsic lesional skin (p=0.08 and 0.30, respectively).
Bar-plots illustrating lesional and non-lesional gene-expression (RT-PCR) of terminal differentiation products (A)Filaggrin, (B)Loricrin, (C)Transglutaminase-3 (TGM3), and (D)Periplakin. Overall, similar expression of terminal differentiation markers was found between lesional and non-lesional skin, as well as between intrinsic and extrinsic disease. Bars=SEM. *p<0.10, **p<0.05, ***p<0.01, ****p<0.001.
Word-clouds and scatter-plots representing mRNA expression differences (RT-PCR) for intrinsic versus extrinsic atopic dermatitis (AD) in A–B) lesional and C–D) non-lesional skin. Numbers (black) serve as fold-change scales for word sizes, which are sized according to expression differences between variants. Significance and up/down-regulation indicated by color (see key). In scatter-plots, circle diameter represents relative differences; darker colors represent increased significance.