

Abstract
Michaelis and Menten introduced to biochemistry the idea of time-scale separation, in which part of a system is assumed to be operating sufficiently fast compared to the rest that it may be assumed to have reached a steady state. This allows, in principle, the fast components to be eliminated, resulting in a simplified description of the system's behaviour. Similar ideas have been widely used in different areas of biology, including enzyme kinetics, protein allostery, receptor pharmacology, gene regulation and post-translational modification. However, the methods used have been independent and ad hoc. Here, we review the use of time-scale separation as a means to simplify the description of molecular complexity and discuss recent work which sets out a single framework which unifies these separate calculations. The framework offers new capabilities for mathematical analysis and helps to do justice to Michaelis and Menten's insights about individual enzymes in the context of multi-enzyme biological systems.
Time-scale separation in enzyme kinetics
2013 is the 100th anniversary of Leonor Michaelis and Maud Menten's paper which introduced their famous mathematical formula for the rate of an enzymatic reaction (40; 60). There are many instructive lessons in this paper (28) but I want to focus here on one particular aspect of what they did which has ramified through biochemistry, pharmacology, molecular biology and, now, systems biology. Michaelis and Menten considered the reaction scheme
| (1) |
in which free enzyme, E, binds reversibly to a substrate, S, to form an intermediate enzyme-substrate complex, ES, which then irreversible breaks down to free the enzyme and yield the product, P. The labels on the reactions are the rate constants, assuming mass-action kinetics. Michaelis and Menten derived from this scheme their rate formula
| (2) |
in which Vmax is the maximal rate of the reaction, Vmax = k3Etot, and KM = (k3 + k2)/k1 is the Michaelis-Menten constant.
There is something quite odd about the relationship between the reaction scheme in Eq. 1 and the rate formula in Eq. 2. The former involves the free enzyme E and the enzyme-substrate complex ES but these components have disappeared from the latter. The only vestige left of the enzyme is its total amount Etot in the expression for the maximal rate. The total amount does not change over the course of the reaction, so is a conserved quantity, not a dynamical variable. All other enzyme-related components have been eliminated.
To pull off this sleight-of-hand, Michaelis and Menten used a time-scale separation. They assumed that, under their in-vitro conditions, in which substrate was in considerable excess of enzyme, the enzyme-substrate complex would rapidly form and reach a quasi-steady state, in which d[ES]/dt = 0. We might say, informally, that the enzyme-related components are assumed to be fast variables, which rapidly reach steady state, while the substrate and product are slow variables, which adjust to this steady state. (Formally, in biochemical systems, it is the reactions which are fast or slow, relatively speaking, not the components, a point to which we will return below.) With a little algebra, which has struck terror into the hearts of generations of students, the enzyme-related components can be eliminated in favour of the total amount of enzyme Etot, from which Eq. 2 falls out.
I should make two historical points here. First, this is not quite what Michaelis and Menten did. They used a different time-scale separation—a rapid equilibrium assumption—and it was Briggs and Haldane who suggested the more appropriate steady-state assumption that is now standard (7). Second, they were not quite the first to use time-scale separation, as we will discuss below, but they were certainly the first in terms of influence.
Enzymologists rapidly took up the method of time-scale separation to analyse more complicated reaction schemes than that in Eq. 1 and there are now enzyme-rate formulas which cover a wide-range of enzymological contexts and include the impact of inhibitors and other kinds of effectors (79). An interesting feature of these formulas is that they are always rational functions in the slow variables. That is, the right hand side is a ratio in which the numerator and the denominator are both sums of products (polynomials) in the concentrations of the slow variables. This may not seem particularly remarkable for the original Michaelis-Menten formula in Eq. 2 but it is a striking and universal feature of more complex formulas. The necessary algebraic manipulations, which get very intricate very quickly, were eventually codified in the King-Altman method (44), to which we will return.
Eliminating variables like ES is, of course, a very good thing because, at the time of Michaelis and Menten, nobody knew anything about them. They were theoretical entities, suggested by the experimental data. It is often forgotten that Michaelis and Menten never characterised the enzyme-substrate complex (for the enzyme invertase which they studied) and they never measured its rates of assembly and disassembly (k1 and k2 in Eq. 1). The first person to do so, for the enzyme peroxidase, was Britton Chance, no less that thirty years after Michaelis and Menten (9). This did not stop enzymologists from enthusiastically using such theoretical entities in the intervening years. Biology is actually more theoretical than physics; biologists just like to pretend otherwise (29).
The Michaelis-Menten formula has been hugely important (40), as this Review Series for the FEBS Journal confirms. It is perhaps the one quantitative mathematical statement that any biologist working at the molecular level would be expected to know. Unfortunately, its very familiarity has bred, not respect, but, rather, ignorance. The elimination of the enzyme-substrate complex has meant that such complexes have been lost from view, so that enzyme sequestration is all too readily overlooked (6; 68). The formula is also widely used in the wrong contexts. In particular, Michaelis and Menten assumed that product formation was irreversible because they measured initial reaction rates when product was negligible. Yet, the formula is habitually used in contexts, such as phosphorylation and dephosphorylation cycles, in which the amount of product could be substantial (25). Michaelis and Menten would have been horrified. One of the goals of this review is to explain how we can start to do justice to what Michaelis and Menten taught us a century ago. Before setting out on that, let us examine some of the other contexts in which time-scale separation has been used.
Other applications of time-scale separation
Allosteric proteins
If we fast forward by fifty years from 1913 to 1963 we come to Jacques Monod, Jean-Pierre Changeux and François Jacob's famous paper (63) on what Monod would later call “microscopic cybernetics” (62). Monod, Changeux and Jacob pointed out that for a biosynthetic pathway to balance supply against demand through feedback inhibition, it was necessary for enzymes to be regulated by effectors that were chemically different from their normal substrates or products. They introduced the idea of allostery, in which an effector binds at a site distinct from the normal catalytic site and triggers or stabilises a conformational change in the enzyme which then alters its catalytic activity.
The general form of such an allosteric model is that an enzyme, or more generally a protein like hemoglobin that performs a transport function rather than catalysis, can exist in multiple conformations, T1, ··· , Tm, and a ligand (or several such) can bind to multiple sites on the protein. If, for instance, there is a single ligand with k binding sites, then there are 2k potential patterns of ligand binding, or “microstates”. Not all conformations need be equally accessible to the ligand but, in principle, there could be a total of m.2k relevant microstates. There is much internal complexity, with the microstates playing a similar role to the enzyme-substrate complexes in enzyme kinetics.
To analyse such a system, the time-scale separation is made in which it is assumed that conformational transitions and ligand binding have reached thermodynamic equilibrium. Some microstates may have high activity, others low activity and the overall activity of the protein is taken to be an average over the equilibrium distribution of microstates. For a transport protein like hemoglobin, an appropriate average is the fractional saturation: the proportion of sites that are bound by oxygen. Under the equilibrium assumption, the microstates can be eliminated, which is to say that, in a similar way to the intermediate complexes in enzyme kinetics, they can be calculated in terms of binding affinities and conserved quantities like the total amount of protein. This yields formulas for the average activity as a function of the ligand concentration. As with enzyme kinetics, the functions are rational.
Specific allosteric models make specific assumptions within this general setup. In Monod, Wyman and Changeux's “plausible model”, the protein is assumed to be a multimer that exists in two quaternary conformations, traditionally called “relaxed” and “tense”, in which the tertiary structure of the individual monomers does not change (64). The equilibrium is assumed to exist prior to ligand binding, so that ligand binding is a selective process that biases the equilibrium towards the relaxed or the tense conformation. In Koshland, Némethy and Filmer's more general model, tertiary changes to the monomers are permitted and, as might have been expected from Dan Koshland's previous introduction of the induced-fit mechanism (46), ligand binding is an instructive process which may induce conformational changes that did not previously exist (48). Although the MWC model is often treated as the standard description of allostery, presumably because the algebra is less terrifying than for the KNF model, the former cannot accommodate negative cooperativity, while the latter can, as Dan Koshland often pointed out (47).
A variety of other allosteric models have been put forward over the years which generalise and mix and match the MWC and KNF assumptions (31; 32; 65; 66). In each case, thermodynamic equilibrium is assumed as a time-scale separation between the fast microstates and the slow interaction of the protein with its environment but different methods are used to undertake the elimination of microstates and calculate the rational functions. In some cases, elementary algebra suffices, while in others, equilibrium statistical mechanics is called upon, as discussed below. Allostery remains a potent conceptual idea with wide application across biological disciplines, even if the meaning of the term has broadened from the rather precise ideas of those who pioneered it.
Gene regulation
Allostery was not, of course, the only breakthrough to which Monod and Jacob contributed. If Crick and Watson revealed the structure of DNA, with its profound implications for genetics and heredity, Monod, Jacob and Lwoff, through their studies of the lac operon and of λ phage, established that genes could be turned on or off in response to environmental signals (21). They were awarded the Nobel Prize in Physiology or Medicine in 1965 for this work. Although gene regulation was first revealed in unicellular microbes, it is fundamental to understanding multicellular development. A hepatocyte and a cardiomyocyte from the same organism have identical genotypes; their profound phenotypic difference arises from differential gene expression (16).
Let us fast forward again, by nearly twenty years, from allostery in 1963 to 1982, when Gary Ackers, Sandy Johnson and Madeleine Shea published their “quantitative model for gene regulation by λ phage repressor”. The λ repressor, or master regulator protein CI, binds to the right operator region of λ phage and thereby regulates the mutually exclusive expression of its own cI gene and the neighbouring cro gene. Expression of the former leads to the phage behaving as a lysogen, which replicates passively by hitching a ride on the bacterial DNA, while expression of the latter leads to the lytic state, in which the virus actively creates more copies of itself and eventually kills its host. The lysis-lysogeny decision is a classic cellular decision process (72) from which new insights continue to emerge (54).
Ackers, Johnson and Shea created the first mathematical model for how expression of cI depended on the concentration of λ repressor. It was known that λ repressor bound in dimerised form to three specific sites in the operator, giving 23 = 8 potential DNA microstates. The importance of cooperative interactions between repressor molecules bound at different sites had been uncovered in in-vitro experiments undertaken by Sandy Johnson in Mark Ptashne's lab at Harvard (38). The mathematical model was intended to reveal how that cooperativity contributed to the lysis-lysogeny decision. Ackers, Johnson and Shea made the very reasonable time-scale separation that repressor binding to DNA had reached thermodynamic equilibrium. They knew from the experimental studies that certain microstates repressed cI. They used equilibrium statistical mechanics to calculate the probability of each microstate in terms of a partition function, thereby eliminating the microstates and allowing calculation of the probability of repression of cI as a function of λ repressor concentration. The free energies of repressor binding to DNA and of cooperative interactions between repressor dimers at different sites, which are required to build up the partition function, were estimated from Johnson's previous experimental data. Here again, rational functions emerge from the calculation, albeit with numerical, as opposed to symbolic, coefficients.
The “thermodynamic formalism” developed in this work has been the starting point for much subsequent analysis of gene regulation. In later work, Shea and Ackers treated the RNA polymerase holoenzyme as another component that could bind DNA and thereby induce mRNA transcription (83). This allowed a more nuanced treatment of how transcription factor binding could influence gene expression through cooperative interactions with RNA polymerase. In effect, mRNA transcription is treated as a slow process which averages over the equilibrium distribution of microstates. The formalism can accommodate multiple transcription factors each binding at multiple sites, yielding insights into combinatorial regulation (8) and the evolution of regulatory circuits (81). Rob Phillips and colleagues elaborated the thermodynamic formalism for the bacterial context in (4; 5).
Gene regulation is quite different in eukaryotes. DNA is no longer naked but is wrapped around nucleosomes and compacted into chromatin, which may exhibit varying accessibility to transcription factors (24). Nucleosomes are dynamic entities that can be assembled, moved and disassembled and the tails of the histone proteins that make up the nucleosome octamers are festooned with a quite astonishing number of post-translational modifications (PTMs) (30). It has been suggested that these combinatorial PTMs influence gene expression through some kind of “code” (37; 67), or perhaps a “language” (3), whose biochemical basis remains obscure (71). Transcription factors both recruit and are recruited by accessory proteins and co-regulators, such as the massive Mediator complex and a variety of components which can reshape the local chromatin environment (39). This enormously complex machine is a testament to the creative powers of evolution and, presumably, a necessary adjunct to the huge expansion of gene regulatory complexity that is found in moving from bacteria to unicellular microbes like yeast to multicellular organisms such as animals (16).
Notwithstanding this increased complexity, the thermodynamic formalism has also been widely influential in studying eukaryotic gene regulation (78; 84). For instance, it has been used to analyse patterning in the early Drosophila embryo, where it seems that much can be explained by transcription factor binding (36; 70; 77; 90). It has even been extended to accommodate nucleosomes by treating them as components that bind to specific DNA sequences in competition with transcription factors (73).
Despite these successes, there is a fundamental problem with this approach. Eukaryotic mechanisms, such as nucleosome remodelling or histone PTM, are dissipative. They necessarily expend energy and may reach a steady state but never a thermodynamic equilibrium. The significance of dissipative mechanisms in the molecular realm was pointed out by John Hopfield in a seminal paper (35), in which he showed that certain information processing tasks (in his case, error reduction in transcription and translation) could not be undertaken at equilibrium but required dissipative expenditure of energy. It is very plausible that complex gene regulation in eukaryotes can undertake forms of information processing, such as precise embryonic patterning, which fundamentally depend on dissipative mechanisms. The thermodynamic formalism can shed no light on this because it is based on equilibrium assumptions. We will return to this issue below.
Pharmacology and receptor theory
Michaelis and Menten were not quite the first to use time-scale separation. They were anticipated in 1909 by an undergraduate student at Trinity College, Cambridge who was doing a research project with the physiologist John Newport Langley (33). The student was Archibald Vivian Hill and this was his first published paper, on which he was sole author. Hill would become one of the founders of biophysics and a winner of the Nobel Prize in Physiology and Medicine in 1922 for his work on muscle physiology. We also know him for the widely used and abused Hill function and attendant Hill coefficient that came out of his work on hemoglobin.
Langley had been one of the first to suggest that the action of drugs on tissues came about through an interaction of the drug with a specific molecular receptor in the tissue (49; 56). Hill considered the reversible binding of a drug L to a hypothetical receptor R,
This is just Michaelis and Menten's reaction scheme in Eq. 1 without the catalysis. Hill made the the time-scale separation that binding had reached equilibrium, while the downstream effect of ligand binding took place on a slower time scale through the equilibrium proportion of ligand-bound receptor. A similar but easier process of elimination to that of Michaelis and Menten allowed the latter proportion to be calculated and the familiar hyperbolic saturation of downstream effect with ligand concentration provided a good match to data that Hill acquired for the action of nicotine and of curare on muscle.
Here again, just as with Michaelis and Menten, the mathematical calculation provided evidence for the existence of a molecular receptor, although, here, it is R and not RL whose existence is being conjectured. Just as in enzyme kinetics the theoretical idea of a receptor proved irresistible to pharmacologists who quickly appreciated that quantitative measurements and equilibrium binding models were the key to unravelling how drugs acted (12). It would take nearly seventy years from Langley's suggestion before receptor molecules were actually shown to exist (29) during which time binding models were essential conceptual tools (53). Bob Lefkowitz, who was awarded the 2012 Nobel Prize in Chemistry for his isolation and characterisation of one of the first receptors, the beta-adrenergic G-protein coupled receptor, points out in his Nobel lecture1 the significance of the ternary-complex binding model (50) for understanding GPCRs. Similar equilibrium binding models are basic ingredients in modern quantitative pharmacology (42) and, as above, rational functions emerge from the elimination that describe receptor activation as a function of ligand concentration.
As David Colquhoun recounts (14), quantitative pharmacology seems to have occupied a parallel universe to very similar studies in enzyme kinetics and allosteric proteins, with surprisingly little cross-fertilisation despite similar underlying ideas. Models with distinct receptor conformations, strikingly close to the idea of protein allostery, emerged in classic studies by del Castillo and Katz in 1957 of the acetylcholine receptor (17), and played a key role in attempts to disentangle the notions of drug affinity and drug efficacy (13). Similar models, albeit more complicated, have been suggested to explain the “collateral efficacy” through which distinct ligands for GPCRs elicit distinct subsets of downstream effects (41). The study of ion channels, which provides some of the most exquisite quantitative data through patch clamping, has been one context in which the classical Monod-Wyman-Changeux models of protein allostery and the equilibrium binding models of pharmacology have coalesced, aided, no doubt, by Jean-Pierre Changeux whose work bridged the two fields (10; 20).
Post-translational modification
Histones are not the only proteins subject to PTM. Indeed, it is hard to find a cellular protein which is not post-translationally modified. Not only are there many types of modification, including phosphorylation, acetylation, ADP-ribosylation, GLcNAcylation, ubiquitination, sumoylation, etc (88), but there are often many sites on a protein subject to the same modification. PTM is fundamentally dissipative with forward modification and reverse de-modification being catalysed by separate enzymes. The free energy for driving such processes ultimately comes from background cellular processes which maintain the concentration of donor molecules (such as ATP in the case of phosphorylation) sufficiently high compared to the concentrations of their hydrolysis products (such as ADP and inorganic phosphate). PTM provides a way for the structure of a protein to be altered dynamically in response to changing cellular conditions. It is a mechanism of escape from the constraint of genetic coding which seems to have been as essential for evolutionary novelty as the gene regulation discussed above (71).
The potential combinatorial complexity in multisite PTM is staggering. Considering just phosphorylation, which is merely a binary on/off modification in contrast to the further complexity that comes from iterated polypeptide modifications like ubiquitination (71), a protein with n sites of modification has 2n potential modification states, or “mod-forms” (71). The serine/arginine repetitive matrix factor Srrm2 has 300 experimentally detected phosphorylation sites, as reported on Phospho.ELM (18). While this example is perhaps extreme, it is not unusual for mammalian signalling proteins to have tens of sites of modification. Clearly, not all mod-forms may be present in any context but this begs the question of which mod-forms are present. Evidence from a variety of contexts shows that distinct mod-forms can exert distinct downstream effects, for instance, by offering alternative recruitment patterns for modification-specific binding domains (71). It is, therefore, the distribution of mod-forms which determines the overall effect of a modified protein and this distribution is regulated by the collective actions of the network of forward- and reverse-modifying enzymes.
We have been developing mathematical approaches to analysing PTM. We followed the strategy that has been widely employed in the literature since “futile cycles” of phosphorylation and dephosphorylation were first mathematically analysed by Chock and Stadtman (82) and by Goldbeter and Koshland (23). We made the time-scale separation that modification and demodification are fast, while the downstream effects of modification are slow. In this way, modification and demodification can be assumed to have reached steady state. The time-scale separation is usually not mentioned explicitly in the literature but is always implicitly present whenever steady states are considered.
We found, to our surprise, that it was possible to eliminate much of the internal complexity at steady state (25; 58; 86). For instance, for a substrate S which is subject to modification by an enzyme E and demodification by an enzyme F on n sites, it was possible to write down a pair of equations for the free enzyme concentrations, [E] and [F],
| (3) |
which correspond to the conservation laws for the two enzymes (86). The salient point here is that these conservation laws can be expressed solely in terms of [E] and [F] (and the total amount of substrate, Stot, which is hidden away inside Φ1 and Φ2). This is true no matter what the value of n. The substrate mod-forms and the intermediate complexes arising from the enzyme mechanisms have all been eliminated. They can be calculated from [E] and [F] by rational functions, once again. This exponential reduction in complexity allows rigorous conclusions to be drawn, for systems with any number of sites, that were previously beyond reach (86).
These calculations were done by ad-hoc algebraic methods and it is fair to say, looking back, that we did not understand them. In particular, we were very puzzled as to why the rational functions that emerged had the property of “positivity”: they always gave a positive result for positive values of [E] and [F] (86, Supplementary Information). Of course, this is what they should do in order to be realistic. The rational functions in the previous examples also exhibit positivity. However, the algebraic methods that we used for PTM seemed to require miraculous cancellations for positivity to emerge. Miracles are a sign of some more fundamental principle at work. This first came to light for PTM (85) but its implications have turned out to be much broader (26), as we discuss below.
Time-scale separation in reality
Michaelis and Menten felt justified in making a time scale separation because under their experimental conditions, substrate was in excess over enzyme. The enzyme-substrate complex would be expected to form quickly and then remain fairly steady until much of the substrate had been consumed. The steady-state assumption seems very reasonable.
It is harder to justify the assumption in some of the other examples discussed above, especially as we move away from the in-vitro setting. For instance, in gene regulation, especially in eukaryotes, is it really the case that transcription factor binding to DNA has reached equilibrium before gene expression begins? Probably not. However, the time-scale separation assumption has still been extremely useful in offering a way to think about the system and providing quantitative formulas which can be experimentally tested. Gertz, Siggia and Cohen used the thermodynamic formalism to analyse libraries of synthetic promoters with up to five transcription factors in yeast, and found that it explained between 40-60% of the observed variation in gene expression (22). While this is a not a direct test of equilibrium, it suggests that the assumption is not awful.
As we move from individual enzymes to multi-enzyme systems, the value of time-scale separation lies more in providing a methodology for simplifying the description of molecular complexity, even when we might not know that the time scales are well separated. In other words, it shifts the burden of the argument from the assumptions we make to the conclusions we draw. In biological contexts where the experimental capabilities are well developed, such as gene regulation in model organisms like yeast and flies, this strategy has been very successful (43; 70; 90).
The mathematical justification for time-scale separation as a good approximation relies on the method of singular perturbation or the use of Tikhonov's Theorem, in which it is assumed that the separation between fast and slow components increases as a small parameter goes to zero (27). For simple biochemical systems like the Michaelis-Menten scheme in Eq. 1, appropriate parameters can be readily identified (75; 80). However, biochemical systems come to us with reactions which may be relatively fast or slow, not components; any given component may be influenced by a mix of fast and slow reactions. Reich and Sel'kov argued by example for the existence of a time-hierarchy of ultrafast, fast and slow components in core metabolism, to which Tikhonov's Theorem could be applied (74). More recently, Lee and Othmer, using the more general framework of Chemical Reaction Network Theory, showed how a time-scale separation on reactions could be reorganised into one on components, in a form suitable for Tikhonov's Theorem (51). It has also been a theoretical and experimental concern to know how long it takes for a steady state to be effectively reached. Easterby was the first to define a transition time to steady state (19) and later authors have extended this idea to wider classes of networks with more general boundary conditions, within a predominantly metabolic context; see, for instance, (55). While of interest in their own right, these issues lie outside the scope of the present paper, which is concerned with the process of elimination at steady state, to which we now turn.
The linear framework
Laplacian dynamics on graphs
The applications of time-scale separation discussed above cover a wide range of biological areas and differ considerably in their biochemical details. Some are assumed to be at thermodynamic equilibrium while others are assumed to be at steady state far from equilibrium. With the exception of the statistical mechanical procedures sometimes used at equilibrium, the calculations are undertaken by ad-hoc methods among which it is difficult to see any common ground. Nevertheless, there are tantalising similarities. In each example, time-scale separation enables some of the internal complexity to be eliminated, with these internal quantities being explicitly calculated in terms of rate constants and conserved quantities, and the resulting formulas are invariable rational functions which exhibit positivity.
These similarities are not an accident. All of the calculations above are instances of a single mathematical procedure, which we call “the linear framework” (26). This not only unifies many biological studies that were previously thought to be separate, it also provides a way to do new kinds of analysis.
The framework starts from a labelled, directed graph G, consisting of vertices 1, ··· , n and directed edges, , decorated with labels a (Figure 1). The graph should have no self loops, , and we will take for granted that it is connected, so that it does not consist of separate pieces between which there are no edges. For the moment, the labels are just symbols corresponding to positive numbers and having units of time–1.
Figure 1.
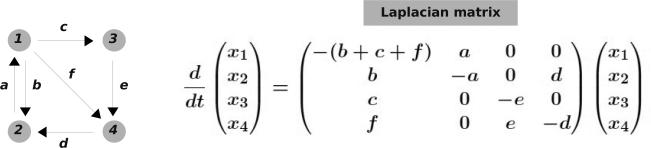
The linear framework. Left, a labelled directed graph, which is strongly connected. Right, Laplacian dynamics on this graph, in which the edges are treated as chemical reactions under mass-action kinetics. The resulting matrix is the Laplacian matrix of the graph.
We can define a dynamics on G as follows: place concentrations of material at the vertices and consider each edge to be a chemical reaction under mass-action kinetics with the label as the rate constant. The reaction will then convert i to j at a rate given by dxj/dt = axi, where xi is the concentration of material at vertex i. (Such concentrations are implicitly functions of time and we should write xi(t). We mostly avoid doing so as not to clutter up the notation. The meaning should be clear from the context.) Since each edge has only one source vertex, the dynamics is described by a linear equation
| (4) |
in which x is the column vector of concentrations and is a n × n matrix called the Laplacian matrix of G. (The notation A.B signifies the product of matrices A and B, with a column vector being treated as a n × 1 matrix.)
Concepts similar to the Laplacian were first introduced in Gustav Kirchhoff's studies of electrical circuits (45). Laplacian matrices have been widely studied in graph theory (59) but with differing conventions and normalisations. The “laplacian” appellation stems from the fact that, with suitable normalisation, they can be seen as discretisations of the Laplace operator (11).
It may seem implausible that such linear chemistry has anything to do with the nonlinear biochemical systems considered in the examples above. Nonlinearity is imported into the framework through the labels. These may be arbitrary rational expressions composed of actual biochemical rate constants and concentrations of actual biochemical species. The only constraint is the “uncoupling condition”, which states that any component whose concentration appears in a label cannot also be a component that is represented by a vertex in the graph. For instance, if the vertices represent fast components in a time-scale separation, then the labels could include the concentrations of slow components. Uncoupling is essential to preserve the linearity of the dynamics.
The key to applying the linear framework is to set up the labels in such a way that the uncoupling condition is satisfied and the linear Laplacian dynamics given by Eq. 4 reproduces the nonlinear dynamics of the actual biochemistry. Let us see how this works in enzyme kinetics for the reversible Michaelis-Menten scheme,
| (5) |
We construct a labelled, directed graph on the fast components, which, in this case, are the free enzyme E and the intermediate enzyme-substrate complex ES, with the labelled edges summarising the biochemistry in Eq. 5:
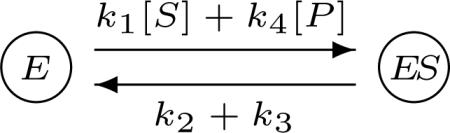 |
(6) |
This graph is strongly connected and the uncoupling condition is satisfied. The Laplacian dynamics is given by
| (7) |
and it is easy to see that these linear equations correspond precisely to the nonlinear equations coming from the reaction scheme in Eq. 5 under mass-action kinetics. You might say that nonlinearity with simple rate constants has been traded for linearity with complicated labels.
Laplacian matrices like that in Eq. 7 and Figure 1 have a characteristic structure with the sum of the entries in each column being zero. This is because of a conservation law. Since material is neither created nor destroyed but simply moved around the vertices, its total amount is conserved at all times,
| (8) |
It follows that 1.dx/dt = 0 where 1 is the all-ones row vector. Since x is arbitrary, it follows from Eq. 4 that .
Steady states and elimination
Time-scale separation requires calculation of a steady steady, corresponding to a vector x* such that dx*/dt = 0. Equivalently, from Eq. 4, . x* = 0, so that x* is in the kernel of the Laplacian: . The kernel depends in an interesting way on the structure of G. Recall that a graph is strongly connected if, given any two distinct vertices i and j, there is a sequence of consecutive edges from i to j all pointing in the same direction. Since i are j are arbitrary, there must be a corresponding directed sequence from j to i. The graph in Figure 1 is strongly connected, as are all the examples in Figure 2. It can be shown that, if G is strongly connected, then the kernel of the Laplacian is one dimensional (85, Lemma 1) or (61, Proposition 3):
| (9) |
Figure 2.

Labelled, directed graphs from different contexts. Labels are omitted for clarity. Black edges have labels which are biochemical rate constants, while blue edges have labels which are algebraic expressions that involve concentrations of other components. (a) enzyme kinetics, in which an enzyme E is irreversible and follows a random-order bi-bi mechanism, as studied in (89). (b) gene regulation, in which a single transcription factor binds to two sites. (c) protein allostery, in which a protein exists in two conformations, R and T with ligand binding at two sites. (d) post-translational modification, in which a substrate S is modified and unmodified in random order on two sites, as studied in (58). (e) receptor pharmacology, with the extended ternary-complex model, adapted from (76, Figure 10): R, receptor; R*, allosteric conformation; H, hormone; G, G-protein.
This result has remarkable implications. We can place any concentrations of matter at the vertices initially; there are as many degrees of freedom available as there are vertices in the graph. Once the dynamics gets under way, it eventually reaches a steady state. (This is true for any graph and any initial condition (61).) In a strongly-connected graph, once a steady-state has been reached, Eq. 9 tells us that only one generalised degree of freedom is left. Aside from this, the steady-state is completely determined by the structure of the graph and is independent of the initial conditions. Eq. 9 is the key to elimination.
To derive formulas for the eliminated quantities, it is necessary to calculate a standard basis element from the structure of G. We will see how to do this below and we will find that ρi is a rational function of the labels. If x* is any steady state of the dynamics then, because the kernel is one-dimensional, x* = λρ, where λ is some scalar, which represents the remaining generalised degree of freedom. It follows from the conservation law in Eq. 8 that λ = xtot/ρtot, where ρtot = ρ1 + ··· + ρn, so that
| (10) |
In other words, the steady-state value at any vertex can be eliminated in favour of the expression ρi/ρtot and the conserved quantity xtot. Since ρi is a rational function of the labels, so too is the fraction ρi/ρtot.
The examples discussed above can all be derived from Eq. 10. In other words, underlying each example is a labelled, directed graph of the form considered here (Figure 2). This graph may not be explicitly described in the corresponding papers but it can be constructed. In each case, the graph is strongly connected and satisfies the uncoupling condition. Eq 10 expresses the eliminated quantities as rational functions of the labels and the conserved total and this is where the rational functions in the examples all come from.
Eq. 9 and Eq. 10 are surprisingly powerful. A strongly connected graph can be arbitrarily complex and have an arbitrary number of vertices; nevertheless, any steady state has only one degree of freedom. The potential reduction in complexity is unbounded, making the elimination procedure in Eq. 10 of particular significance in tackling the enormous combinatorial complexity that we encounter at the molecular level. This is evident in post-translational modification, where the number of modification states may be astronomical. Nevertheless, at steady state, the effective algebraic complexity depends only on the number of enzymes, as in Eq. 3.
The two equations in Eq. 3 are derived from the conservation laws for the forward and the reverse enzyme in the post-translational modification system. These equations are highly nonlinear and are an aspect of the “linear” framework that we have glossed over up to now. The labels in the graph may contain concentrations of components which are not represented by vertices in the graph. How these labels are dealt with depends on the context. In enzyme kinetics, the labels contain the concentrations of slow components. Quantities of interest, like reaction rates, can be calculated from the eliminated components in terms of these slow components and nothing further is required (see below, for the calculation of a rate formula in Eq. 15). In ligand binding contexts, such as allosteric proteins or gene regulation, the ligand is itself a fast component, although not one that is represented by a vertex in the graph. The equilibrium value for the free ligand concentration can be calculated from the conservation law for the total amount of ligand. If ligand is in abundance, so that depletion can be ignored, then [L] ≈ Ltot; if not, the conservation equation must be solved to obtain the equilibrium value of [L]. In post-translational modification, the labels contain the concentrations of free enzymes which are also fast components. Here too, the steady-state values of these are specified by the conservation laws for the enzymes, as in Eq. 3. It is the nonlinearity of these equations which permits multiple solutions and the existence of multiple steady states (69; 86).
We see from this that the linear framework is not entirely linear. However, the linearity is perhaps its most surprising aspect. Linearity is largely invisible in the examples discussed above. One never thinks, for instance, of Michaelis and Menten's calculation as being linear. In retrospect, the linearity is crucial. It is what makes elimination of internal complexity feasible.
Calculating kernels
It remains to calculate a standard basis element . On the face of it, this should be an exercise in linear algebra. However, this relies on determinants, which are sums of terms with alternating signs. Miraculously, positive and negative terms somehow cancel out, leaving only a sum of positive terms. This is the origin of the positivity that was noted above. This miracle turns out to be a special property of the Laplacian matrix. It is easier to use two other procedures for calculating ρ, in which the positivity is immediately manifest without the need for any cancellations.
The simplest way to calculate ρ is when thermodynamic equilibrium can be assumed. In this case, the graph must have a particular structure which comes from the Principle of Detailed Balance (DB). This states that if a chemical reaction is at thermodynamic equilibrium then it must be reversible and the pair of reversible reactions must be at equilibrium independently of any other reactions in which the substrates and products are participating. DB was put forward in the chemical context by Gilbert Lewis to rule out thermodynamically paradoxical states in chemical reaction networks (52). He made the following charming argument based on chemical intuition and time-scale separation. He imagined that a reaction within a network had a catalyst which could increase its rate, and the rate of its reverse reaction, so much that this reaction was working much faster than others in the network. The reversible reaction would then be at equilibrium independently of the rest of the network. Not surprisingly, this argument did not convince the physicists! DB was subsequently shown to be a consequence of “microscopic reversibility”, or time-reversal symmetry, in the laws of physics (57) and must be considered as a fundamental physical principle. It imposes restrictions on the biochemical rate constants, as we will see below, and many remarkable conclusions have been drawn by failing to enforce it at equilibrium.
If a graph G represents a context in which thermodynamic equilibrium is reached, then each edge must be reversible. If we consider any pair of reversible edges
| (11) |
in an equilibrium state , then, according to DB, . The equilibrium state can then be calculated by choosing a reference vertex, say vertex 1, and choosing any path of reversible edges from 1 to i,
Applying DB repeatedly to each reversible pair, we find that
| (12) |
There may, of course, be more than one path of reversible edges from 1 to i. It is a consequence of DB that, no matter which path is used, we get the same answer for . It is necessary and sufficient for DB to hold for any equilibrium state that the graph G satisfies the “cycle condition”: given any cycle of reversible edges, the product of the labels going clockwise around the cycle must equal the product of the labels going counterclockwise (26). It is not difficult to see that this implies that the calculation in Eq. 12 is independent of the chosen path. The cycle condition is a fundamental constraint on the labels and, through them, on the underlying biochemical rate constants. Examples b, c and e in Figure 2, which are treated at equilibrium, must satisfy the cycle condition, while a and d, which are dissipative, would not have to.
If we let μi be the expression given by the product of the terms in brackets in Eq. 12 then . (Note that μ1 = 1.) Since is a common factor in each of these expressions, we can obtain a standard element in by setting ρi = μi. Here, the positivity is obvious.
The thermodynamic formalism is essentially an additive version of this calculation. The connection between the two methods comes through van't Hoff's law, which says that, for a reversible reaction like that in Eq. 11 which can reach equilibrium,
| (13) |
where ΔU is the free energy difference between the two vertices. The quantity μi, which is a product of terms of the form (a/b) in Eq. 12, corresponds through the exponential in Eq. 13 to a sum of free energies. At equilibrium, the thermodynamic formalism and the linear framework are equivalent and it is a matter of preference whether one calculates multiplicatively like a chemist using reactions or additively like a physicist using free energies (61).
It is another matter if the system does not reach thermodynamic equilibrium but instead reaches a steady state that is far from equilibrium. There is then no requirement for DB to hold and we can obtain graphs with irreversible edges. Equilibrium thermodynamics cannot help us.
When G is strongly connected, a standard basis element can be calculated by using the Matrix-Tree Theorem (MTT) (26; 61; 85): ρi is given by taking a spanning tree rooted at i, multiplying together the labels on its edges and then adding such expressions over all spanning trees rooted at i (26).
A spanning tree is a subgraph of G which contains every vertex in G (spanning) and which has no cycles when edge directions are ignored (tree). It is rooted at i if i is the only vertex with no outgoing edges. A graph is strongly connected if, and only if, every vertex has a rooted spanning tree (61, Lemma 1). If we let Θi(G) denote the set of spanning trees of G rooted at i, then in symbols the MTT implies that
| (14) |
Note that all the terms are positive. The MTT takes care of all the cancellations (61, Appendix).
For the reversible Michaelis-Menten scheme in Eq. 5, it is easy to see that there is only one spanning tree at each vertex and that, according to Eq. 14,
Applying the elimination formula in Eq. 10, we find that, for any steady state ,
Once a steady state has been reached, the rate of the reaction is given by
from which one obtains by substitution the reversible Michaelis-Menten rate formula
| (15) |
in which Vf = k3xtot, Vr = k2xtot, Kf = (k2 + k3)/k1 and Kr = (k2 + k3)/k4. The fast components, E and ES, have been eliminated, leaving only the slow components S and P and the rational function in Eq. 15.
The reversible Michaelis-Menten scheme is sufficiently simple that the steady state can be calculated without using the MTT but the MTT becomes invaluable as the graph becomes more complicated. It is, however, more demanding to apply than the equilibrium formula for ρi = μi given by Eq. 12. At equilibrium, only a single path to i is needed to calculate ρi; away from equilibrium all spanning trees rooted at i are needed to calculate ρi. The number of spanning trees increases rapidly as the graph becomes more complicated. Computational methods for enumerating them are sometimes helpful but the main importance of the MTT is that it gives a mathematical description of the steady state, which is then accessible to further analysis.
Theorems of Matrix-Tree type go back to Kirchhoff but the MTT which is relevant to us, for labelled, directed graphs, is due to Bill Tutte, one of the founders of modern graph theory (87). It has been independently re-discovered many times. Enzymologists will recognise the MTT as equivalent to King and Altman's “schematic procedure” (44), which remains the standard method for calculating enzyme rate formulas. Here, spanning trees are called “patterns”. Somewhat later, Terrell Hill re-discovered the MTT in his studies of non-equilibrium molecular systems (34). He called spanning trees “directional diagrams”. Amusingly, he thanked a correspondent for pointing that he was making a contribution to the “theory of graphs”. Neither he nor his correspondent seemed aware that the result had already been proved by one of the founders of that subject nearly twenty years earlier, let alone that it was also known to enzymologists! The MTT has also been independently re-discovered in economics, engineering, computer science and theoretical physics (for more background, see (61)), a testament to its fundamental importance and to the insidious compartmentalisation of modern science.
There is much to be gained by adopting the neutral language of mathematics, of graphs, Laplacians and spanning trees, in preference to the specialised language that might be used in a particular area of application. A common mathematical structure emerges which cuts across disciplines and it becomes much easier to transfer knowledge between them and to avoid re-inventing the wheel.
Looking forward
One of the advantages of doing chemistry, with reactions and graphs, in contrast to physics with free energies, is that one is not limited to equilibrium. The linear framework is applicable in contexts like enzyme kinetics or post-translational modification where the system is far from equilibrium. It can accommodate some of the dissipative mechanisms, such as nucleosome reorganisation and histone PTM, which arise in eukaryotic gene regulation, offering a new methodology that goes beyond the thermodynamic formalism. Sequences were sufficient to describe genomes and an elaborate mathematical and computational machinery has evolved to exploit them. Perhaps labelled, directed graphs will provide the appropriate mathematical object with which to describe genomic functionality. This is work in progress (1).
A more immediate problem leads us back to Michaelis and Menten. As pointed out above, the irreversible scheme in Eq. 1, is commonly used to study multi-enzyme systems in circumstances which would have horrified its authors. The dangers of doing this have been pointed out (6; 68). It is not only dangerous, it is also illegal. If P is appreciably present (which it was not for Michaelis and Menten), then it must be able to rebind to E, or detailed balance would be violated once the reaction reaches equilibrium. The usual reason given for ignoring thermodynamic reality is that the scheme is being used to represent a reaction which is irreversible under physiological circumstances, such as a kinase or phosphatase reaction. If so, a better solution would be to use the reaction scheme
| (16) |
which can be irreversible without being in a state of Original Thermodynamic Sin. There is, of course, a price to pay for such virtue, which is increased complexity and yet more rate constants whose values we do not know.
A further issue arises with Eq. 1 because it assumes single-substrate reactions. Yet, kinase reactions, which are frequently represented by Eq. 1, involve two substrates. It may be reasonable to ignore ATP as a dynamical variable, if, indeed, its concentration is kept constant by background cellular processes, but that does not address the order of substrate binding, which can give rise to additional intermediate complexes. It might be more appropriate to use a reaction scheme for which the corresponding graph looks something like that in Figure 2a.
The linear framework provides a systematic approach to this problem, at least for contexts like post-translational modification. We can consider a limited enzymological grammar, consisting of the reactions
| (17) |
which allows for multiple intermediates, Y1, ··· , YL. This accommodates Eq. 1, Eq. 16 and the reaction scheme behind Figure 2a and covers most of the enzymology that we would expect to find for post-translational modification and demodification involving metabolic modifications (89). The linear framework shows that, no matter how complex the reaction scheme that is derived from Eq. 17, the steady state behaviour can be summarised in four aggregated parameters, a “total generalised catalytic efficiency” (tgCE) and a “total generalised Michaelis-Menten constant” (tgMMC), one each for the reaction substrate S and one each for the reaction product P. In this way, we can distinguish between a reaction scheme that is irreversible, such as Eq. 16, in which the tgCE for P is zero (it cannot make S), but the tgMMC for P is not (it can still bind to E). In contrast, the Michaelis-Menten scheme in Eq. 1 is strongly irreversible, with both the tgCE and tgMMC for P being zero.
The distinction makes a difference. In one of the pioneering papers on “futile cycles” of phosphorylation and dephosphorylation, Albert Goldbeter and Dan Koshland showed that a single-site cycle is capable of unlimited ultrasensitivity as the enzymes become more saturated by the substrate (23). They assumed that the kinase and phosphatase followed the standard Michaelis-Menten scheme in Eq. 1. One can show, using the formulation just described, that, if the forward and reverse enzymes can be expressed in the grammar of Eq. 17 and are both strongly irreversible, then, no matter how complicated they are, unlimited ultrasensitivity continues to hold (89). However, if the enzymes are weakly irreversible—irreversible but not strongly so (ie: their tgMMCs for product are non-zero)—then the ultrasensitivity is always bounded and one can even calculate a bound for it (15).
Strong irreversibility is mathematically convenient because there are only two aggregated parameters to deal with per reaction, but it introduces an artifact, a singularity, which leads to infinite behaviour in the high-substrate limit (89). Weak irreversibility provides a more physiologically realistic and a more nuanced picture, in which the properties of the individual enzymes become significant. One can apply the mathematics with more confidence to actual enzymes with complicated enzymology (15).
This brings us to the main reason, I believe, why the original Michaelis-Menten scheme continues to exert such a hold, despite its limitations being known (6; 68). It allows us to pretend that all enzymes are the same, so that we can avoid paying attention to them in our rush to understand the behaviour of multi-enzyme systems (25). This does an injustice to Michaelis and Menten, which is compounded by our continuing to use their ideas in contexts which they would have known to be wrong. It is also a disservice to the enzymologists of the intervening century, who have done so much to disentangle the mechanisms of individual enzymes (2). And, not least, it is not a good way to educate the next generation. It may help to have a systematic method to deal with the additional complexity, which the linear framework now provides, but scientific cultures have a lot of inertia and it may be the next generation, indeed, who fully integrates enzymology and systems biology.
Time-scale separation may have been a convenient tool for Michaelis and Menten, who were dealing with a system of just four components, but it assumes much greater significance for us, as we contemplate molecular networks in which an individual protein may have thousands of different states of modification. Without some means to rise above this complexity, it is going to be extremely difficult to see the wood for the trees. Time-scale separation offers a way to do this, which may at least provide a starting point for thinking about a system and developing our intuitions, even if we do not know how well the time scales are separated in reality.
What the linear framework shows us is that, sometimes, and in the most signifi-cant examples of time-scale separation, we can undertake the elimination of internal complexity in a purely mathematical way, which depends only on the structure of the system, its graph. We can rise above some of the complexity. We can show, for instance, that futile cycles have certain behaviours, irrespective of the complexity of their enzymes (15; 89). That is a different kind of assertion to what we normally see in the literature. It is also considerably more powerful than what can be achieved by numerical simulation, in which all the details must be precisely specified. We can begin to discern in these developments, perhaps, a mathematical language through which biological principles can emerge from molecular complexity. I like to think that Michaelis and Menten would have approved.
Acknowledgements
I thank two anonymous reviewers for several helpful comments. The work outlined here was supported by NIH GM081578 and by NSF 0856285.
Footnotes
References
- 1.Ahsendorf T, Wong F, Eils R, Gunawardena J. A framework for modelling eukaryotic gene regulation that accommodates non-equilibrium mechanisms. 2012 doi: 10.1186/s12915-014-0102-4. In preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson KS. Detection and characterization of enzyme intermediates: utility of rapid chemical quench methodology and single enzyme turnover experiments. In: Johnson KA, editor. Kinetic Analysis of Macromolecules: A Practical Approach. Oxford University Press; Oxford, UK: 2003. pp. 19–47. [Google Scholar]
- 3.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–11. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 4.Bintu L, Buchler NE, Garcia GG, Gerland U, Hwa T, Kondev J, Kuhlman T, Phillips R. Transcriptional regulation by the numbers: applications. Curr. Opin. Gen. Dev. 2005;15:125–35. doi: 10.1016/j.gde.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bintu L, Buchler NE, Garcia GG, Gerland U, Hwa T, Kondev J, Phillips R. Transcriptional regulation by the numbers: models. Curr. Opin. Gen. Dev. 2005;15:116–24. doi: 10.1016/j.gde.2005.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blüthgen N, Bruggeman FJ, Legewie S, Herzel H, Westerhoff HV, Kholodenko BN. Effects of sequestration on signal transduction cascades. FEBS J. 2006;273:895–906. doi: 10.1111/j.1742-4658.2006.05105.x. [DOI] [PubMed] [Google Scholar]
- 7.Briggs GE, Haldane JBS. A note on the kinetics of enzyme action. Biochem. J. 1925;19:338–9. doi: 10.1042/bj0190338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchler N, Gerland U, Hwa T. On schemes of combinatorial transcription logic. Proc. Natl. Acad. Sci. USA. 2003;100:5136–41. doi: 10.1073/pnas.0930314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chance B. The kinetics of the enzyme-substrate compound of peroxidase. J. Biol. Chem. 1943;151:553–77. [PubMed] [Google Scholar]
- 10.Changeux JP, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–80. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 11.Chung FRK. Spectral Graph Theory. American Mathematical Society; 1997. Number 92 in Regional Conference Series in Mathematics. [Google Scholar]
- 12.Clark AJ. The Mode of Action of Drugs on Cells. Edward Arnold and Co.; 1933. [Google Scholar]
- 13.Colquhoun D. Binding, gating, affinity and efficacy. Br. J. Pharmacol. 1998;125:923–47. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colquhoun D. The quantitative analysis of drug-receptor interactions: a short history. Trends Pharmacol. Sci. 2006;27:149–57. doi: 10.1016/j.tips.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 15.Dasgupta T, Croll DH, Owen JA, Vander Heiden MG, Locasale JW, Alon U, Cantley LC, Gunawardena J. A fundamental trade off in covalent switching and its circumvention in glucose homeostasis. 2012 doi: 10.1074/jbc.M113.546515. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davidson EH. The Regulatory Genome: Gene Regulatory Networks in Development and Evolution. Academic Press; 2006. [Google Scholar]
- 17.del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc. R. Soc. Lond. B. 1957;146:369–81. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 18.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho. ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 2011;39:D261–7. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Easterby JS. Coupled enzyme assays: a general expression for the transient. Biochim. Biophys. Acta. 1973;293:552–8. doi: 10.1016/0005-2744(73)90362-8. [DOI] [PubMed] [Google Scholar]
- 20.Edelstein SJ, Schaad O, Henry E, Bertrand D, Changeux J-P. A kinetic mechanism for nicotinic acetylcholine receptors based on multiple allosteric transitions. Biol. Cybern. 1996;75:361–79. doi: 10.1007/s004220050302. [DOI] [PubMed] [Google Scholar]
- 21.Gann A. Jacob and Monod: from operons to evo-devo. Curr. Biol. 2010;20:R718–23. doi: 10.1016/j.cub.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 22.Gertz J, Siggia ED, Cohen BA. Analysis of combinatorial cis-regulation in synthetic and genomic promoters. Nature. 2009;457:215–8. doi: 10.1038/nature07521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldbeter A, Koshland DE. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA. 1981;78(11):6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grewal SIS, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 25.Gunawardena J. Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc. Natl. Acad. Sci. USA. 2005;102:14617–22. doi: 10.1073/pnas.0507322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunawardena J. A linear framework for time-scale separation in nonlinear biochemical systems. PLoS ONE. 2012;7:e36321. doi: 10.1371/journal.pone.0036321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunawardena J. Modelling of interaction networks in the cell: theory and mathematical methods. In: Egelman E, editor. Comprehensive Biophysics. Vol. 9. Elsevier; 2012. [Google Scholar]
- 28.Gunawardena J. Some lessons about models from Michaelis and Menten. Mol. Biol. Cell. 2012;23:517–9. doi: 10.1091/mbc.E11-07-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gunawardena J. Biology is more theoretical than physics. Mol. Biol. Cell. 2013;24:1827–9. doi: 10.1091/mbc.E12-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 31.Henry ER, Bettati S, Hofrichter J, Eaton WA. A tertiary two-state allosteric model for hemoglobin. Biophys. Chem. 2002;98:149–64. doi: 10.1016/s0301-4622(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 32.Herzfeld J, Stanley HE. A general approach to co-operativity and its application to the oxygen equilibrium of hemoglobin and its effectors. J. Mol. Biol. 1974;82:231–65. doi: 10.1016/0022-2836(74)90343-x. [DOI] [PubMed] [Google Scholar]
- 33.Hill AV. The mode of action of nicotine and curari determined by the form of the contraction curve and the method of temperature coefficients. J. Physiol. 1909;39:361–73. doi: 10.1113/jphysiol.1909.sp001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill TL. Studies in irreversible thermodynamics IV. Diagrammatic representation of steady state fluxes for unimolecular systems. J. Theoret. Biol. 1966;10:442–59. doi: 10.1016/0022-5193(66)90137-8. [DOI] [PubMed] [Google Scholar]
- 35.Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc. Natl. Acad. Sci. USA. 1974;71:4135–39. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janssens H, Hou S, Jaeger J, Kim A-R, Myasnikova E, Sharp D, Reinitz J. Quantitative and predictive model of transcriptional control of the drosophila melanogaster even skipped gene. Nat. Genet. 2006;38:1159–65. doi: 10.1038/ng1886. [DOI] [PubMed] [Google Scholar]
- 37.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 38.Johnson AD, Meyer BJ, Ptashne M. Interactions between dna-bound repressors govern regulation by λ phage repressor. Proc. Natl. Acad. Sci. USA. 1979;76:5061–5. doi: 10.1073/pnas.76.10.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson DG, Dent SYR. Chromatin: receiver and quarterback for cellular signals. Cell. 2013;152:685–9. doi: 10.1016/j.cell.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson KA, Goody RG. The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry. 2011;50:8264–9. doi: 10.1021/bi201284u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nature Rev. Drug Discov. 2005;4:919–27. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 42.Kenakin T. A Pharmacology Primer: Theory, Application and Methods. 3 edition Academic Press; London, UK: 2009. [Google Scholar]
- 43.Kim HD, O'Shea EK. A quantitative model of transcription factor-activated gene expression. Nat. Struct. Mol. Biol. 2008;15:1192–8. doi: 10.1038/nsmb.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King EL, Altman C. A schematic method of deriving the rate laws for enzyme-catalyzed reactions. J. Phys. Chem. 1956;60:1375–8. [Google Scholar]
- 45.Kirchhoff G. Über die Auflösung der Gleichungen, auf welche man bei der Untersuchung der linearen Verteilung galvanischer Ströme gefuührt wird. Ann. Phys. Chem. 1847;72:497–508. [Google Scholar]
- 46.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koshland DE, Hamadani K. Proteomics and models for enzyme cooperativity. J. Biol. Chem. 2002;277:46841–4. doi: 10.1074/jbc.R200014200. [DOI] [PubMed] [Google Scholar]
- 48.Koshland DE, Némethy G, Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–85. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 49.Langley JN. On the reactions of cells and of nerve-endings to certain poisons, chiefly as regards the reaction of striated muscle to nicotine and curari. J. Physiol. 1905;33:374–413. doi: 10.1113/jphysiol.1905.sp001128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J. Biol. Chem. 1980;255:7108–17. [PubMed] [Google Scholar]
- 51.Lee CH, Othmer HG. A multi-time scale analysis of chemical reaction networks: I. Deterministic systems. J. Math. Biol. 2010;60:387–450. doi: 10.1007/s00285-009-0269-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis GN. A new principle of equilibrium. Proc. Natl. Acad. Sci. USA. 1925;11:179–83. doi: 10.1073/pnas.11.3.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Limbird LE. The receptor concept: a continuing evolution. Mol. Interven. 2004;4:326–36. doi: 10.1124/mi.4.6.6. [DOI] [PubMed] [Google Scholar]
- 54.Little JW. Evolution of complex gene regulatory circuits by addition of refinements. Curr. Biol. 2010;20:R724–34. doi: 10.1016/j.cub.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 55.Lloréns M, Nuñno JC, Rodríguez Y, Meléndez-Hevia E, Montero F. Generalization of the theory of transition times in metabolic pathways: a geometrical approach. Biophys. J. 1999;77:23–36. doi: 10.1016/S0006-3495(99)76869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maehle A-H, Pruüll C-R, Halliwell RF. The emergence of the drug receptor theory. Nat. Rev. Drug. Discov. 2002;1:637–41. doi: 10.1038/nrd875. [DOI] [PubMed] [Google Scholar]
- 57.Mahan BH. Microscopic reversibility and detailed balance. J. Chem. Educ. 1975;52:299–302. [Google Scholar]
- 58.Manrai A, Gunawardena J. The geometry of multisite phosphorylation. Biophys. J. 2008;95:5533–43. doi: 10.1529/biophysj.108.140632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merris R. Laplacian matrices of graphs: a survey. Linear Algebra Appl. 1994;198:143–76. [Google Scholar]
- 60.Michaelis L, Menten M. Die kinetik der Invertinwirkung. Biochem. Z. 1913;49:333–69. [Google Scholar]
- 61.Mirzaev I, Gunawardena J. Laplacian dynamics on general graphs. Bull. Math. Biol. 2013 doi: 10.1007/s11538-013-9884-8. to appear. [DOI] [PubMed] [Google Scholar]
- 62.Monod J. Chance and Necessity: an Essay on the Natural Philosophy of Modern Biology. Alfred A. Knopf; New York: 1971. [Google Scholar]
- 63.Monod J, Changeux JP, Jacob F. Allosteric proteins and cellular control systems. J. Mol. Biol. 1963;6:306–29. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- 64.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 65.Motlagh HN, Li J, Thompson EB, Hilser VJ. Interplay between allostery and intrinsic disorder in an ensemble. Biochem. Soc. Trans. 2012;40:975–80. doi: 10.1042/BST20120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najdi TS, Yang CR, Shapiro BE, Hatfield GW, Mjolsness ED. Application of a generalised MWC model for the mathematical simulation of metabolic pathways regulated by allosteric enzymes. J. Bioinform. Comput. Biol. 2006;4:335–55. doi: 10.1142/s0219720006001862. [DOI] [PubMed] [Google Scholar]
- 67.Nightingale KP, O'Neill LP, Turner BM. Histone modifications: signalling receptors and potential elements of a heritable epigenetic code. Curr. Opin. Genet. Dev. 2006;16:125–36. doi: 10.1016/j.gde.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 68.Ortega F, Acerenza L, Westerhoff HV, Mas F, Cascante M. Product dependence and bifunctionality compromise the ultrasensitivity of signal transduction cascades. Proc. Natl. Acad. Sci. USA. 2002;99:1170–5. doi: 10.1073/pnas.022267399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ortega F, Garcés JL, Mas F, Kholodenko BN, Cascante M. Bistability from double phosphorylation in signal transduction. FEBS J. 2006;273:3915–26. doi: 10.1111/j.1742-4658.2006.05394.x. [DOI] [PubMed] [Google Scholar]
- 70.Parker DS, White MA, Ramos AI, Cohen BA, Barolo S. The cis-regulatory logic of Hedgehog gradient responses: key roles for Gli binding affinity, competition and cooperativity. Sci. Signal. 2011;4:ra38. doi: 10.1126/scisignal.2002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prabakaran S, Lippens G, Steen H, Gunawardena J. Post-translational modification: nature's escape from from genetic imprisonment and the basis for cellular information processing. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012 doi: 10.1002/wsbm.1185. to appear. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ptashne M. A Genetic Switch: Phage Lambda Revisited. 3 edition Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- 73.Raveh-Sadka T, Levo M, Segal E. Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res. 2009;19:1480–96. doi: 10.1101/gr.088260.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reich JG, Sel'kov EE. Time hierarchy, equilibrium and non-equilibrium in metabolic systems. BioSystems. 1975:39–50. doi: 10.1016/0303-2647(75)90041-6. [DOI] [PubMed] [Google Scholar]
- 75.Roussel MR, Fraser SJ. Invariant manifold methods for metabolic model reduction. Chaos. 2001;11:196–206. doi: 10.1063/1.1349891. [DOI] [PubMed] [Google Scholar]
- 76.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β2-adrenergic receptor: extending the ternary complex model. J. Biol. Chem. 1993;268:4625–36. [PubMed] [Google Scholar]
- 77.Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patters from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–40. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 78.Segal E, Widom J. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat. Rev. Genetics. 2009;10:443–56. doi: 10.1038/nrg2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Segel IH. Enzyme Kinetics: Behaviour and Analysis of Rapid Equilibrium and Steady-State EnzymeSystems. Wiley-Interscience; 1993. [Google Scholar]
- 80.Segel LA, Slemrod M. The quasi-steady state assumption: a case study in perturbation. SIAM Review. 1989;31:446–77. [Google Scholar]
- 81.Setty Y, Mayo AE, Surette MG, Alon U. Detailed map of a cis-regulatory input function. Proc. Natl. Acad. Sci. USA. 2003;100:7702–7. doi: 10.1073/pnas.1230759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shacter-Noiman E, Chock PB, Stadtman ER. Protein phosphorylation as a regulatory device. Philos. Trans. R. Soc. Lond. B. 1983;302:157–66. doi: 10.1098/rstb.1983.0049. [DOI] [PubMed] [Google Scholar]
- 83.Shea MA, Ackers GK. The OR control system of bacteriophage lambda: a physical-chemical model for gene regulation. J. Mol. Biol. 1985;181:211–30. doi: 10.1016/0022-2836(85)90086-5. [DOI] [PubMed] [Google Scholar]
- 84.Sherman MS, Cohen BA. Thermodynamic state ensemble models of cis-regulation. PLoS Comp. 2012;8:e1002407. doi: 10.1371/journal.pcbi.1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thomson M, Gunawardena J. The rational parameterisation theorem for multisite post-translational modification systems. J. Theor. Biol. 2009;261:626–36. doi: 10.1016/j.jtbi.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomson M, Gunawardena J. Unlimited multistability in multisite phosphorylation systems. Nature. 2009;460:274–7. doi: 10.1038/nature08102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tutte WT. The dissection of equilateral triangles into equilateral triangles. Proc. Camb. Phil. Soc. 1948;44:463–82. [Google Scholar]
- 88.Walsh CT. Posttranslational Modification of Proteins. Roberts and Company; Englewood, Colorado: 2006. [Google Scholar]
- 89.Xu Y, Gunawardena J. Realistic enzymology for post-translational modification: zero-order ultrasensitivity revisited. J. Theor. Biol. 2012;311:139–52. doi: 10.1016/j.jtbi.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zinzen RP, Senger K, Levine M, Papatsenko D. Computational models for neurogenic gene expression in the Drosophila embryo. Current Biol. 2006;16:1358–65. doi: 10.1016/j.cub.2006.05.044. [DOI] [PubMed] [Google Scholar]