

Significance
Many genetic disorders, including Duchenne muscular dystrophy and cystic fibrosis, are caused by a defective protein resulting from a premature termination codon (PTC) in the mutant gene. Aminoglycosides have been proposed as therapies for these disorders because they increase the frequency of translational read-through of PTCs, permitting expression of full-length protein. We consider the possibility that this approach may prompt an autoimmune response to HLA-presented epitopes encoded downstream of the PTC or other stop codons. We demonstrate that gentamicin induces immunologically relevant levels of an epitope derived from PTC read-through. Furthermore, we identify multiple HLA class I-binding peptides derived from read-through of conventional stop codons in gentamicin-treated cells. These results substantiate the possibility of immune autoreactivity from read-through therapies.
Keywords: recoding, antigen presentation, autoimmunity
Abstract
Aminoglycosides have been proposed as therapies for genetic disorders caused by nonsense mutations, because of their capacity to enhance translational read-through of premature termination codons (PTCs), thereby permitting expression of functional full-length protein. However, a potential consequence of this strategy is the development of an autoimmune response to HLA-presented epitopes encoded downstream of the PTC or other stop codons. Using a recombinant virus-expression system in tissue culture and in mice, we demonstrate that gentamicin can induce expression and MHC class I presentation of a model epitope encoded downstream of a PTC at levels sufficient to activate CD8+ T cells. The degree of read-through–derived peptide presentation varies with the sequence of the stop codon and +1 nucleotide. Additionally, we applied a mass spectrometry exploration of the HLA class I peptide repertoire of gentamicin-treated cells and identified multiple peptides derived from read-through of conventional stop codons. These results substantiate the possibility of self-reactivity to cryptic epitopes revealed by stop codon read-through therapies and potentially other therapeutic approaches involving compounds that alter translational fidelity.
CD8+ T cells (TCD8+) of the adaptive immune system recognize antigens via specific interaction of their distinct T-cell receptors with peptide fragments, termed “epitopes,” that are bound to MHC class I molecules at the surface of target cells (1). Relatively few peptide–MHC complexes, on the order of tens to hundreds, are required for TCD8+ activation (2). Indeed, immunologically relevant levels of peptide can be derived from amounts of protein that are undetectable by standard biochemical methods (3), such as minor protein species generated by nonstandard or aberrant translation (4).
Nonstandard translation, or translational recoding, can produce functional or nonfunctional proteins by such mechanisms as initiation at an internal AUG (5) or non-AUG start codon (6), ribosome frame-shifting (3, 7), or continuing past a stop codon. Stop codon read-through, which occurs when the ribosome inserts an amino acid when reading a termination codon and then continues translation into the 3′ UTR, is generally rare but may occur at an increased rate under certain circumstances. In yeast, a prion conformation of the termination factor eRF3 increases read-through of termination codons (8), and recent evidence from ribosome profiling in Drosophila suggests that a subset of mRNAs may be regulated by stop codon read-through to control gene expression and protein function (9). Additionally, and central to the studies reported here, stop codon read-through is enhanced by certain ribosome-targeting pharmaceutical compounds, such as aminoglycoside antibiotics (10) or the small molecule ataluren (PTC124) (11).
Aminoglycosides, such as gentamicin, disrupt prokaryotic protein synthesis by binding to 16S ribosomal RNA, inducing a conformational change in the ribosome-RNA complex (12) and increasing the rate of translational error and premature termination. This disruption of translational fidelity can also allow for pairing of an aminoacylated near-cognate tRNA with a stop codon to permit continued translation (10). Interaction of certain aminoglycosides with the eukaryotic ribosome similarly promotes stop codon read-through in mammalian cells, with an efficiency that is determined by multiple factors, including the identity of the stop codon (in humans, UGA > UAG > UAA) and the nucleotide immediately downstream (C > U > G ≥ A), as well as the larger sequence context in which they are embedded (13, 14).
Premature termination codons (PTCs), caused by nonsense mutations, frame-shift mutations, or aberrant splicing, can result in truncated, nonfunctional proteins, such as those implicated in many genetic diseases, including Duchenne/Becker muscular dystrophy, lysosomal storage disorders, cancer syndromes, and cystic fibrosis (15). PTCs show greater susceptibility than conventional stop codons to drug-induced read-through (11), likely because of their lack of the termination regulatory factors that are present in the conventional 3′ UTR and the poly-A tail (16). Consequently, aminoglycosides and ataluren have been explored as therapies for multiple genetic diseases caused by nonsense mutations, in which even a small amount of full-length functional protein may be sufficient to achieve a therapeutic effect. Clinical trials of these drugs have demonstrated some promise in Duchenne muscular dystrophy (DMD) and cystic fibrosis patients, although results have been inconsistent among trials (17–19).
A potential consequence of stop codon read-through therapy is the induction of autoimmunity because of production of epitopes from 3′ regions that are normally untranslated. Indeed, CD4+ T-cell (TCD4+) responses to a dystrophin epitope downstream of the PTC were detected in a recent clinical trial of gentamicin therapy for DMD (17). Furthermore, despite the lower frequency of read-through at native stop codons, the high sensitivity of T cells may permit reactivity to cryptic epitopes encoded downstream of conventional stop codons as well. Any novel peptide–MHC complex generated by induced nonstandard translation carries the potential hazard of autoimmunity, because responsive T cells might not have been subject to negative selection or peripheral tolerance mechanisms.
We investigated the possibility of aminoglycoside-induced generation of MHC class I-restricted epitopes using a recombinant virus-expression system in tissue culture and in mice, as well as exploration by peptide mass spectrometry for 3′ UTR-derived MHC epitopes presented on gentamicin-treated human cells. We demonstrate that a model epitope downstream of a stop codon can be functionally presented by MHC class I to T cells in the presence of gentamicin, with varying levels of presentation depending on the identity of the stop codon and +1 nucleotide. Additionally, we identify several cryptic class I-binding peptides generated by gentamicin-induced translational read-through of native stop codons in human cells. Our results suggest that aminoglycoside treatment can induce the generation of cryptic self-epitopes via stop codon read-through and may therefore pose a risk for autoimmunity.
Results
Stop Codon Construct Design.
We designed constructs to encode the MHC class I epitope Ova257–264 (SIINFEKL) downstream of various stop codons within a model protein, using influenza nucleoprotein (NP) as the backbone sequence (Fig. 1). Thus, expression of SIINFEKL would require read-through of the PTC, whereas the endogenous epitope NP50–57, encoded upstream of the stop codon, serves as a control for construct expression. A total of seven different stop codon contexts (Table S1), representing a range of stop codon read-through efficiencies (14), were included in the study. The positive control construct contains no stop codon. In the negative control construct, the downstream coding sequence was placed in the +1 reading frame, preventing production of the SIINFEKL epitope even if the stop codon were read through. All constructs were recombined into the vaccinia virus (VV) genome for expression in cells (20). Virus nomenclature indicates the identity of the stop codon and the +1 nucleotide (immediately downstream of the stop codon).
Fig. 1.
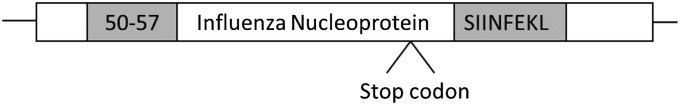
Stop codon construct generation. Stop codon sequences (Table S1) were cloned into an engineered SphI site in influenza NP, such that the stop codon would lie downstream of the endogenous MHC class I epitope NP50–57 and upstream of the inserted Ova257–264 (SIINFEKL) epitope. Constructs were recombined into VV for expression in cells.
Read-Through of Premature Stop Codons Is a Low-Frequency Event.
To determine whether aminoglycoside treatment can generate biochemically appreciable levels of full-length protein via read-through of PTCs, we initially used radio-immunoprecipitation of NP. L929 cells were infected, in the presence or absence of gentamicin, with virus-encoding NP constructs representing three distinct premature stop codons. The samples were then radio-labeled with [35S]methionine, and NP was immunoprecipitated and separated by gel electrophoresis. Termination at the PTC would yield a truncated protein product of 36 kDa, whereas stop codon read-through would generate a full-length protein of 56 kDa. In cells infected with virus encoding the positive control construct, the 56-kDa full-length protein is clearly expressed, and the 36-kDa protein fragment is evident with expression of the negative control construct (Fig. 2A). However, no full-length product was appreciable in cells infected with any of the stop constructs. The absence of observable full-length protein was further confirmed by Western blot, which failed to detect 56-kDa NP in cells infected with any of the seven stop codon constructs in the presence or absence of 900 μg/mL of gentamicin (Fig. 2B). We selected 900 μg/mL as the maximum gentamicin concentration because of the potential for a higher dose to cause nephrotoxicity in subsequent in vivo experiments (21).
Fig. 2.

Read-through of a premature stop codon is undetectable by radio-immunoprecipitation and Western blot. (A) LKb cells infected with the indicated VV were 35S-labeled and incubated ± 300 μg/mL of gentamicin. Autoradiography of immunoprecipitated NP demonstrates 36-kDa truncated NP, resulting from termination at the PTC, in the negative control (NEG) and 56-kDa full-length NP in the positive control (POS). No full-length protein was detectable from any of the test constructs using this approach with a range of gentamicin concentrations. (B) LKb cells were cultured ± 900 μg/mL of gentamicin and infected with the indicated VV before assessing the presence of 56-kDa NP by anti-NP Western blot. Expression of full-length NP was not observed from any stop codon construct.
Given the insufficient sensitivity of radio-immunoprecipitation and Western blot for detection of such minor species, we applied a more sensitive dual luciferase reporter assay that was developed for detection of unconventional translation events (14). Plasmids encoding Renilla and firefly luciferases, separated by the selected stop codons, were expressed in LKb cells cultured with 0, 300, or 900 μg/mL gentamicin. Stop codon read-through was quantified as luminescence from firefly luciferase normalized to Renilla luciferase luminescence. Using this assay, gentamicin-induced stop codon read-through was readily demonstrated from multiple stop codon constructs in a concentration-dependent manner, with the highest levels detected from TAAA, TAAC, and TGAC sequences (Fig. 3).
Fig. 3.

Stop codon read-through is detectable by luciferase assay. LKb cells were transfected with plasmids expressing Renilla luciferase upstream and firefly luciferase downstream of the indicated stop codon sequences. After 6 h of incubation with the indicated concentration of gentamicin, the respective luciferase luminescences were quantified. Stop codon read-through is evident as luminescence from firefly luciferase, normalized to permit comparison across samples: (luminescence firefly luciferase)/(luminescence Renilla luciferase) × 100%. Each sample was analyzed in triplicate in three independent assays. Average values + SEM are shown.
These results demonstrate that aminoglycoside treatment induces stop codon read-through at a level insufficient for protein visualization by radio-immunoprecipitation or Western blot but observable by a more sensitive enzymatic assay. The predicted permissiveness of particular stop codon contexts was largely upheld in the results of the luciferase assay, with increased levels of read-through from the “leaky” TGA termination codon and from constructs with a C in the +1 position after the stop codon, in general agreement with the hierarchy described in earlier reports (14, 22). Indeed, some read-through of TGAC is evident even in the absence of gentamicin. A notable exception was seen with the increased level of read-through from TAAA, the stop codon correlating to the mutation in the mdx mouse model for DMD, which has previously been observed to demonstrate high termination fidelity.
Epitopes Generated by Stop Codon Read-Through Are Sufficient for T-Cell Activation.
Because T cells are sensitive to even low numbers of MHC-peptide complexes, we hypothesized that gentamicin-induced stop codon read-through can generate epitopes in amounts sufficient for T-cell activation. We initially assessed in vitro T-cell responses to the SIINFEKL epitope in our NP constructs, using the TCD8+ hybridoma B3Z, which produces β-galactosidase upon recognition of the SIINFEKL/Kb complex at a level that is proportional to epitope density (23). LKb cells were infected, in the presence of 0, 300, or 900 μg/mL of gentamicin, with viruses encoding the NP stop codon constructs. The infected cells were cultured overnight with B3Z cells, and SIINFEKL-specific TCD8+ activation was quantified using a fluorogenic β-galactosidase substrate. A marked gentamicin dose-dependent increase in T-cell activation was observed for several of the stop codon constructs (Fig. 4A), with TGAC, TAGC, and TAAA demonstrating the highest read-through efficiencies. These results suggest that gentamicin can induce generation of cryptic epitopes at levels sufficient for T-cell activation despite a very small amount of the read-through–generated polypeptide.
Fig. 4.

T-cell activation in vitro and in vivo by read-through–derived epitopes. (A) LKb cells were infected with the indicated VV and cocultured ± gentamicin with the SIINFEKL-specific B3Z T-cell hybridoma, which produces β-galactosidase upon activation, measurable as fluorescence from MUG product. To account for the effects of gentamicin on B3Z, the values obtained with the stop constructs were normalized to those of the controls: (sample − negative)/(positive − negative) × 100%. Each sample was analyzed in triplicate in three independent assays. Average values + normalized SEM are shown. (B) Splenocytes primed with the indicated VV in mice treated ± gentamicin were assessed by ELISpot assay for IFN-γ production in response to target cells pulsed with NP50–57 or SIINFEKL peptide. NP50–57-specific and SIINFEKL-specific responses from four mice are shown, as well as normalized values (SIINFEKL spots/NP50–57 spots) to control for gentamicin effects and differences in VV priming. Data are shown as the range per condition with second and third quartile boxes.
A subset of the recombinant VV panel was used to confirm read-through–derived epitope presentation in vivo. Mice were pretreated for 2 d with subcutaneous injections of PBS ± 12.75 mg/kg gentamicin twice daily, corresponding to 150% of the allometric dose equivalent (24). The TGAG, TGAC, TAAA, and negative and positive control viruses were used to infect mice with a single intraperitoneal inoculation, and PBS/gentamicin treatments were continued for 2 wk. Splenocytes were then harvested and analyzed for IFN-γ activity in an ELISpot assay using target cells presenting either the NP50–57 (prestop codon) or SIINFEKL (poststop codon) epitope. Splenocytes from mice treated with gentamicin demonstrated a depressed NP50–57 response (Fig. 4B), possibly as a consequence of immunosuppression secondary to gentamicin toxicity (25). Nevertheless, the SIINFEKL-specific responses to all three stop codon constructs were significantly higher in gentamicin-treated animals. These studies demonstrate that a robust in vivo TCD8+ immune response can be provoked from the low amounts of epitope that are generated after aminoglycoside-induced stop codon read-through.
Gentamicin-Induced Read-Through of Native Stop Codons Uncovers Cryptic HLA Class I Epitopes.
Having demonstrated that gentamicin-induced read-through of PTCs can generate immunologically relevant levels of a model epitope, we proceeded to explore the possibility of self-epitope generation from read-through of conventional stop codons. We cultured human HeLa S3 cells in the presence or absence of 900 μg/mL gentamicin, immunoprecipitated HLA (human MHC) class I, and analyzed the associated peptides by mass spectrometry. From untreated and gentamicin-treated cells, respectively, we observed 8,435 and 7,654 peptides matching 8–11mer sequences of the RefSeq human proteome at 90% confidence. The mass results were then searched against a custom RefSeq database of human proteome sequences that include sequences at and after the natural stop codon of each gene, to identify read-through–derived peptides while excluding masses that had matched within conventional coding regions.
We identified 17 read-through–derived 8–11mer peptides that appear on gentamicin-treated but not untreated cells, as well as 2 that appear on both untreated and gentamicin-treated cells (Table 1). Several of these peptides derive from the same parent proteins, in variations of the amino acid inserted at the stop codon site or overlapping sequences with varying lengths. Interestingly, read-through of the TGA stop codon of d-dopachrome tautomerase yielded an HLA-binding peptide presented on untreated cells; additional peptides including sequence further downstream appeared with gentamicin treatment. Both 3′ UTR peptides identified on untreated cells arise from read-through of TGA stop codons; however, the stop codons yielding 3′ UTR peptides in gentamicin-treated cells are varied, and no clear preference for +1 nucleotide is seen.
Table 1.
HLA-associated 3′ UTR peptides from HeLa S3 cells
| Sample | Sequence* | Parent protein | Stop codon |
| Gent | IERMYGRKDLF | Acyl-CoA synthetase long-chain family member 3 | A TAA T |
| Gent | TAQEPILGSR | ARAP1 | C TGA G |
| Gent | AFGARGGCSRS | BTBD6 | C TGA G |
| Gent | VIGATAPLCSS | Claudin 4 | G TAA G |
| Gent | VLGATAPLCSS | Claudin 4 | G TAA G |
| Untreated + Gent | VMTFLMLG | d-dopachrome tautomerase | A TGA T |
| Gent | MTFLNLGTE | d-dopachrome tautomerase | A TGA T |
| Gent | VMTFLTLGTEG | d-dopachrome tautomerase | A TGA T |
| Gent | LKLGTEGSR | d-dopachrome tautomerase | A TGA T |
| Untreated + Gent | MNFVGLLIL | Defender against cell death 1 | C TGA C |
| Gent | MNFVGILIL | Defender against cell death 1 | C TGA C |
| Gent | PCKIFFLFY | Heterogeneous nuclear ribonucleoprotein K | C TAA T |
| Gent | GFSLFHKNF | Microtubule associated serine/threonine kinase-like | G TAG C |
| Gent | CAVPLPQGY | Sialidase 1 (lysosomal sialidase) | C TGA G |
| Gent | SQLIGEMLY | Sortilin-related VPS10 domain containing receptor 1 | G TAG C |
| Gent | DSIKRQDITY | Translocase of outer mitochondrial membrane 5 | A TGA A |
| Gent | IWRQDITY | Translocase of outer mitochondrial membrane 5 | A TGA A |
| Gent | AVVNVHSTAW | Transmembrane protein 147 | C TAG G |
| Gent | VGLALELEAY | Voltage-dependent anion channel 2 | T TAA T |
Gent, gentamicin.
The amino acid inserted at the stop codon for each peptide is in bold.
To confirm that these peptides were derived from HLA class I molecules, we selected one from each parent protein to include in a binding analysis adapted from the traditional RMA-S binding assay (26). In essence, cells lacking the transporter associated with antigen processing (TAP) deliver unloaded, unstable HLA class I molecules to the cell surface. Introducing an HLA class I-binding peptide into the cell media stabilizes the formerly empty HLA molecule, increasing surface half-life, thus increasing surface HLA concentration. To inhibit TAP in HeLa S3 cells, we used a recombinant VV encoding ICP47, a herpes simplex virus protein that binds human TAP and blocks its binding of antigenic peptides (27). Using this approach, we confirmed that all of the tested peptides can bind to at least one of the HLA molecules of HeLa S3 cells (Fig. 5), increasing surface HLA class I concentration by 25–70%, comparable to two positive controls that were included in the assay. Some patterns are evident among these HeLa S3 epitopes, including a C-terminal tyrosine, but the HLA allotypes are not specified in this assay. Taken together, these results indicate that gentamicin-induced read-through of native stop codons can introduce cryptic peptides into the HLA repertoire of human cells.
Fig. 5.

Confirmed HLA binding of identified read-through peptides. ICP47-expressing HeLaS3 cells were incubated with test peptides before quantification of surface HLA class I by flow cytometry. SIINFEKL peptide was included as a negative control. Two peptides, YKIGDLTGY and VKVDELSLY, were selected as positive controls, having been consistently observed among the RefSeq human proteome matches of HeLa S3 HLA class I-associated peptides. Average percent increase in surface HLA class I from three independent assays is shown + SEM.
Discussion
The ability of aminoglycosides to reduce the fidelity of translation termination (10) provides the rationale for the current interest in using ribosome-binding pharmaceutical agents to treat human diseases caused by PTCs. Clinical trials of gentamicin and ataluren have been encouraging, although the lack of response in some patients and only partial restoration of protein expression has compelled efforts to identify new drugs with increased activity (28). Similarly, the ability of some antimicrobial compounds to induce ribosomal frame-shifting has raised the prospect that these compounds could be useful in treating frame-shift mutations (29). The utility of such approaches will likely depend not only on the amount of functional protein that can be restored but also on the degree to which a reduction in translational fidelity affects global protein synthesis. Even small changes in translational fidelity may result in the widespread production of previously unexpressed polypeptides because of aberrant translation of “off-target” mRNAs.
A major concern in any effort to reconstitute functional gene expression for patients with genetic mutations is the possibility of immune reactivity to new protein products, which in many cases will appear essentially foreign to the patient’s immune system. In the case of stop codon read-through therapies, the generation of neoepitopes arising either from restored expression of the desired protein product or by read-through of native stop codons may contribute to potential self-reactive immune responses. Indeed, a TCD4+ response specific for an epitope downstream of the dystrophin PTC has been detected in a DMD patient treated with gentamicin in a recent clinical trial (17), whereas a different downstream TCD4+ epitope has been reported in an untreated patient (30). If basal levels of read-through are insufficient to induce immune tolerance to cryptic epitopes, it is possible that aminoglycoside-induced read-through could introduce autoimmune effects, potentially in a dose-dependent manner.
In this report, we demonstrated in vitro and in vivo TCD8+ responses to an epitope expressed downstream of a stop codon following treatment with gentamicin. These results add to the list of nonstandard translation events that have been shown to reveal “cryptic” T-cell epitopes (3, 4, 7, 17). Additionally, activating levels of a cryptic epitope can be presented despite minimal levels of translation. Indeed, translation past the inserted PTC in influenza nucleoprotein was not visible by radio-immunoprecipitation nor by Western blot, but the more sensitive readouts of luciferase expression and TCD8+ activation confirmed translation downstream of the PTC in the presence of gentamicin. Both the dual luciferase assay and TCD8+ activation assay revealed variable read-through levels in vitro, depending on the identity of the stop codon and its surrounding sequence context. Of the seven sequences of stop codons and +1 nucleotides examined in this study, UGAC, UAGC, UAAC, and UAAA revealed the highest levels of stop codon read-through. These results are in general agreement with previous findings (14, 22), with the exception of UAAA, for which higher gentamicin-induced read-through levels were revealed in our experiments than have been previously reported (14). The sequence-specific levels of stop codon suppression observed here and in other studies (14, 17, 31) suggest that the effectiveness of aminoglycoside treatment may vary for different nonsense mutations, a finding supported by at least one clinical trial of DMD patients (32).
Although aminoglycosides preferentially overcome PTCs, lower frequency read-through events at conventional stop codons may also generate adequate presentable peptides to activate T cells because of their high sensitivity (3). Using direct mass spectrometry peptide analysis, we identified several HLA class I peptides derived from stop codon read-through in gentamicin-treated but not untreated human HeLa S3 cells. Two of these 3′ UTR-derived HLA-binding peptides were also observed in the absence of gentamicin, indicating that a basal level of stop codon read-through can contribute peptides to the HLA repertoire of untreated cells. With gentamicin treatment, additional nearby or overlapping peptides from these proteins were identified, likely as a consequence of an increased concentration of these HLA-binding peptides because of an amplified level of stop codon read-through of these genes. Moreover, four genes each contributed two or more peptides to the list of 19 read-through–derived peptides identified here, with variations in length and the amino acid inserted at the stop codon. The variability permitted for the tRNA accepted at the stop codon site during read-through may further increase the array of peptides available for HLA presentation, varying anchor residues available to interact with HLA peptide-binding grooves and residues available to interact with T-cell receptors.
Three of the four genes contributing multiple peptides, including the two observed from untreated cells, terminate at a TGA stop codon, suggesting that basal and possibly gentamicin-induced read-through preferentially occur at this stop codon sequence. In contrast to the sequence hierarchy seen with our PTC constructs, the stop codons and nucleotides immediately downstream or upstream show no clear pattern in the small sample of 3′ UTR peptides identified on gentamicin-treated cells. It is possible that selectivity in read-through for conventional stop codons differs from that of PTCs. Moreover, numerous factors may influence translation fidelity, including nucleotides up to six bases away from the stop codon (33, 34), translation factors (35), and local mRNA tertiary structure (36).
In summary, we report that robust TCD8+ responses can be generated against epitopes produced by aminoglycoside-induced read-through of premature termination codons, and we demonstrate that novel HLA-binding peptides are generated by aminoglycoside-induced read-through of native stop codons. Given the wide range of peptide binding specificities among hundreds of distinct human class I HLA alleles, it is likely that stop codon read-through can drive a broad array of foreign and self-neoepitope formation, and levels of immune activation by cryptic epitopes from 3′ UTRs may vary by HLA type. Although the immunogenicity of the given assortment of HLA peptides has not been determined, the potential generation and presentation of diverse self-antigens as a consequence of stop codon read-through, or other therapeutic compounds that affect ribosome fidelity, invokes a concern for autoimmune pathology.
Materials and Methods
Mice, Cell Lines, Antibodies, and Chemicals.
C3FeB6F1/J (H-2b, H-2k) mice were purchased from the Taconic or Jackson Laboratories and maintained in the Thomas Jefferson University Animal Facilities. All experimental protocols were preapproved by the Thomas Jefferson University Institutional Animal Care and Use Committee. LKb cells [L929 (H-2k) stably transfected with H-2Kb] were cultured in DMEM supplemented with 5% (vol/vol) FCS (D5). HeLa S3 cells were maintained in spinner flasks in DMEM supplemented with 10% (vol/vol) FCS (D10) + 2 mM l-glutamine. B3Z hybridomas were cultured in RPMI 1640 supplemented with 10% (vol/vol) FCS. Anti-NP monoclonal antibodies were generated by harvesting supernatant of mouse hybridomas HB-65 and H19-S24, cultured in D10. Anti-HLA monoclonal antibody (W6/32) was generated by Protein A (Pierce) purification from supernatant of mouse hybridoma HB-95 cultured in CELLine bioreactors (Integra) in D10. General chemical supplies were obtained from Sigma-Aldrich unless otherwise noted.
Molecular Constructs and Viruses.
Construction of the NP/SIINFEKL gene (NP/S) has been described previously (23). Annealed complimentary oligonucleotides (sense strand sequences shown in Table S1) with SphI-compatible ends were ligated into an engineered SphI site in NP/S within a modified pSC11 plasmid for recombination into the wild-type CR19 VV genome by standard methods (37).
Radio-Immunoprecipitation.
For radio-immunoprecipitaton, 1 × 106 LKb cells were infected with 5 pfu per cell stop codon VV for 3 h ± 300 μg/mL of gentamicin. Medium lacking methionine and cysteine was substituted for 1 h before addition of 50 μCi 35S Met/Cys (Amersham) for 2–3 h ± gentamicin. Cells were lysed in 0.5% Nonidet P-40 buffer with 1 mM PMSF, and NP/S was immunoprecipitated using Protein A-bound anti-NP monoclonal antibodies (1:1 HB-65 and H19-S24). NP/S was separated by SDS/PAGE, and gels were dried (Hoefer GD2000; Amersham Pharmacia Biotech) and exposed to film (X-Omat LS, Kodak) for 24 h.
Western Blot.
For Western blot, 1 × 106 l-Kb cells pretreated for 18–24 h ± 900 μg/mL of gentamicin were infected with 5 pfu per cell stop codon VV for 16 h ± gentamicin. Cells were lysed in 1% Triton-X buffer with protease inhibitor mixture (Roche) and lysates separated by SDS/PAGE before transfer to nitrocellulose membrane. Protein bands were detected using mouse anti-NP (1:5 dilution H19-S24 supernatant) or rabbit anti-actin (I-19; Santa Cruz Biotechnology), peroxidase-labeled secondary Abs (Jackson Immunoresearch Laboratories), and chemiluminescence detection (SuperSignal West Pico; Thermo Scientific) by autoradiography.
Dual Luciferase Assay.
Dual luciferase plasmids for our assay were constructed by cloning stop codon sequences (Table S1) into an Sph1 restriction site between Renilla and firefly luciferases in the T7 promoter-driven plasmid (14). In a 96-well plate, 5 × 104 LKb cells per well were infected for 1 h at 5 pfu per cell with recombinant VV expressing the T7 polymerase before 1 h of transfection with 1 μg of the indicated dual luciferase plasmid using Lipofectamine 2000 (Invitrogen). Infected tranfected cells were incubated in media with 0, 300, or 900 μg/mL gentamicin for 6 h, and luminescence from the respective Renilla and firefly luciferase genes was then quantified using the Dual Luciferase Reporter assay (Promega) with a Wallac Victor2 1420 multilabel plate reader.
T-Cell Hybridoma Assays.
Assays for presentation of the SIINFEKL epitope using B3Z hybridomas have been described previously (23). Briefly, LKb cells pretreated for 18–24 h ± gentamicin were infected with 5 pfu per cell stop codon VV for 2 h. Infected cells were washed with PBS and cocultured 1:1 with B3Z cells at 5 × 104 per well in a 96-well plate in media + 100 μg/mL Ara-C ± gentamicin for 16 h. β-Galactosidase production was assessed by addition of the fluorogenic substrate 4-methylumbelliferyl β-d-glucuronide (MUG) at 33 μg/mL.
ELISpot.
Mice were injected subcutaneously every 12 h with 50 μL PBS ± 12.75 mg/kg of gentamicin for 16 d. On day 3, mice were immunized intraperitoneally with 1 × 107 pfu of recombinant stop codon VV in 250 μL BSS/BSA. Splenocytes harvested 14 d postinfection were assessed by ELISpot, as described previously (3), for IFN-γ production in response to LKb cells either untreated, pulsed with 10−8 M NP50–57 or SIINFEKL synthetic peptide, or infected with wild-type VV.
HLA Immunoprecipitation.
For HLA immunoprecipitation, 3 × 108 HeLa S3 cells, cultured ± 900 μg/mL gentamicin, were lysed in 1% Triton X buffer + protease inhibitor mixture (Roche). HLA class I was purified from lysate supernatant using W6/32-conjugated protein A agarose, and peptides were eluted in 10% (vol/vol) acetic acid, filtered through a 10-kD size-exclusion column (Microcon Ultracel YM-10), and purified on a C18 column (Thermo Scientific Hypersep). Purified peptides were dried by cold-trap centrifugation (Labconco Centrivap) and resuspended in 0.1% TFA for LC-MS-MS. Triplicate samples of purified HLA-associated peptides were isolated per condition.
Mass Spectrometry.
Peptides were analyzed by nanoLC-MS/MS at the Rutgers Biological Mass Spectrometry Facility using a RSLC system (Dionex) interfaced with a Velos-LTQ-Orbitrap (ThermoFisher). Samples were loaded onto a 100-μm × 2-cm trap packed with Magic C18AQ, 5-μm 200 A (Michrom Bioresources), and washed with Buffer A (0.2% formic acid) for 5 min with a flowrate of 10 μL/min. The trap was brought in-line with a 75-µm × 50-cm analytical column (Magic C18AQ; 3-µm 200 A) and peptides fractionated at 300 nL/min with a multistep gradient of 4–15% (vol/vol) Buffer B [80% (vol/vol) acetonitrile + 0.16% formic acid] in 5 min and 15–50% (vol/vol) B in 45 min. Data were acquired from a cyclic series of Orbitrap scans at 60,000 resolution followed by linear ion trap MS-MS scans of the 20 most intense ions with a repeat count of two and the dynamic exclusion duration of 60 s.
Peptide Informatics.
The LC-MS-MS data were converted to mascot generic file format using Proteome Discoverer v1.3 (Thermofisher) and searched against custom FASTA databases using the MASCOT search engine (Matrixscience) with no modifications and protease set as “none.” The “ORF” FASTA database contains the total RefSeq human proteome sequences. The “ORF+3′UTR” FASTA database includes the RefSeq human proteome as well as sequences containing the last 15 amino acids of RefSeq annotated ORFs of the human genome and extending to the next stop codon, while inserting each of the possible 20 amino acids at the stop codon. To identify peptide masses specifically representing 3′ UTR sequences, the LC-MS-MS data were searched against both databases at a confidence threshold of 90%, and sequences matching only the “ORF+3′UTR” database were considered. The resulting spectra were confirmed by manual inspection.
Peptide Binding Assay.
HeLa S3 cells were infected with 2 pfu per cell VV expressing ICP47. At 12 h of infection, cells were stripped of surface HLA molecules by exposure to cold citrate phosphate buffer, pH 3, for 2 min before replating ± 0.5 mM test peptide. After 4 h, cells were stained for HLA class I using the LY5.1 mouse pan anti-HLA antibody (Santa Cruz Biotechnology) and FITC-conjugated anti-mouse IgG (eBioscience). Flow cytometry was performed using a Becton Dickinson FACSCalibur cytometer, and CellQuest software (Becton Dickinson) was used for acquisition and analysis. The percent increase in surface HLA was calculated from median fluorescence intensity (MFI) values: [(MFI test peptide – MFI no peptide)/MFI no peptide] × 100%.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants R01AI039501 (to L.C.E.), R01AI100561 (to L.C.E.), and R21NS083884 (to M.T.H.); and Science Foundation Ireland Grant 12/IP/1492 (to J.F.A.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. B.J.V.d.E. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1402670111/-/DCSupplemental.
References
- 1.Neefjes J, Jongsma MLM, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 2.Kageyama S, Tsomides TJ, Sykulev Y, Eisen HN. Variations in the number of peptide-MHC class I complexes required to activate cytotoxic T cell responses. J Immunol. 1995;154(2):567–576. [PubMed] [Google Scholar]
- 3.Zook MB, Howard MT, Sinnathamby G, Atkins JF, Eisenlohr LC. Epitopes derived by incidental translational frameshifting give rise to a protective CTL response. J Immunol. 2006;176(11):6928–6934. doi: 10.4049/jimmunol.176.11.6928. [DOI] [PubMed] [Google Scholar]
- 4.Starck SR, Shastri N. Non-conventional sources of peptides presented by MHC class I. Cell Mol Life Sci. 2011;68(9):1471–1479. doi: 10.1007/s00018-011-0655-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malarkannan S, Horng T, Shih PP, Schwab S, Shastri N. Presentation of out-of-frame peptide/MHC class I complexes by a novel translation initiation mechanism. Immunity. 1999;10(6):681–690. doi: 10.1016/s1074-7613(00)80067-9. [DOI] [PubMed] [Google Scholar]
- 6.Starck SR, et al. A distinct translation initiation mechanism generates cryptic peptides for immune surveillance. PLoS ONE. 2008;3(10):e3460. doi: 10.1371/journal.pone.0003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS ONE. 2011;6(11):e26517. doi: 10.1371/journal.pone.0026517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urakov VN, et al. N-terminal region of Saccharomyces cerevisiae eRF3 is essential for the functioning of the eRF1/eRF3 complex beyond translation termination. BMC Mol Biol. 2006;7:34. doi: 10.1186/1471-2199-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS (2013) Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2. Available at: http://elife.elifesciences.org/content/2/e01179 [(Accessed February 9, 2014)
- 10.Palmer E, Wilhelm JM, Sherman F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature. 1979;277(5692):148–150. doi: 10.1038/277148a0. [DOI] [PubMed] [Google Scholar]
- 11.Welch EM, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 12.Carter AP, et al. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407(6802):340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 13.Bidou L, et al. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11(7):619–627. doi: 10.1038/sj.gt.3302211. [DOI] [PubMed] [Google Scholar]
- 14.Howard MT, et al. Sequence specificity of aminoglycoside-induced stop condon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48(2):164–169. [PubMed] [Google Scholar]
- 15.Bidou L, Allamand V, Rousset J-P, Namy O. Sense from nonsense: Therapies for premature stop codon diseases. Trends Mol Med. 2012;18(11):679–688. doi: 10.1016/j.molmed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 16.Amrani N, et al. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature. 2004;432(7013):112–118. doi: 10.1038/nature03060. [DOI] [PubMed] [Google Scholar]
- 17.Malik V, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67(6):771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 18.Wilschanski M, et al. Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J. 2011;38(1):59–69. doi: 10.1183/09031936.00120910. [DOI] [PubMed] [Google Scholar]
- 19.Finkel RS, et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE. 2013;8(12):e81302. doi: 10.1371/journal.pone.0081302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tatsis N, Sinnathamby G, Eisenlohr LC. Vaccinia virus as a tool for immunologic studies. Methods Mol Biol. 2004;269:267–288. doi: 10.1385/1-59259-789-0:267. [DOI] [PubMed] [Google Scholar]
- 21.Carver MP, Monteiro-Riviere NA, Brown TT, Riviere JE. Dose-response studies of gentamicin nephrotoxicity in rats with experimental renal dysfunction. II. Polyvinyl alcohol glomerulopathy. Toxicol Appl Pharmacol. 1985;80(2):264–273. doi: 10.1016/0041-008x(85)90083-3. [DOI] [PubMed] [Google Scholar]
- 22.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6(7):1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wherry EJ, McElhaugh MJ, Eisenlohr LC. Generation of CD8(+) T cell memory in response to low, high, and excessive levels of epitope. J Immunol. 2002;168(9):4455–4461. doi: 10.4049/jimmunol.168.9.4455. [DOI] [PubMed] [Google Scholar]
- 24.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104(4):375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Exon JH, Stevens MG, Koller LD, Mather GG. Immunotoxicity assessment of gentamycin and liquamycin. Vet Hum Toxicol. 1989;31(5):427–430. [PubMed] [Google Scholar]
- 26.Schumacher TN, et al. Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell. 1990;62(3):563–567. doi: 10.1016/0092-8674(90)90020-f. [DOI] [PubMed] [Google Scholar]
- 27.Hill A, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375(6530):411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- 28.Keeling KM, Wang D, Conard SE, Bedwell DM. Suppression of premature termination codons as a therapeutic approach. Crit Rev Biochem Mol Biol. 2012;47(5):444–463. doi: 10.3109/10409238.2012.694846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta P, Kannan K, Mankin AS, Vázquez-Laslop N. Regulation of gene expression by macrolide-induced ribosomal frameshifting. Mol Cell. 2013;52(5):629–642. doi: 10.1016/j.molcel.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanigan KM, et al. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: Prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24(9):797–806. doi: 10.1089/hum.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Floquet C, Rousset J-P, Bidou L. Readthrough of premature termination codons in the adenomatous polyposis coli gene restores its biological activity in human cancer cells. PLoS ONE. 2011;6(8):e24125. doi: 10.1371/journal.pone.0024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Politano L, et al. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003;22(1):15–21. [PubMed] [Google Scholar]
- 33.Namy O, Hatin I, Rousset JP. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001;2(9):787–793. doi: 10.1093/embo-reports/kve176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Floquet C, Hatin I, Rousset J-P, Bidou L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012;8(3):e1002608. doi: 10.1371/journal.pgen.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orlova M, Yueh A, Leung J, Goff SP. Reverse transcriptase of Moloney murine leukemia virus binds to eukaryotic release factor 1 to modulate suppression of translational termination. Cell. 2003;115(3):319–331. doi: 10.1016/s0092-8674(03)00805-5. [DOI] [PubMed] [Google Scholar]
- 36.Houck-Loomis B, et al. An equilibrium-dependent retroviral mRNA switch regulates translational recoding. Nature. 2011;480(7378):561–564. doi: 10.1038/nature10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eisenlohr LC, Bacik I, Bennink JR, Bernstein K, Yewdell JW. Expression of a membrane protease enhances presentation of endogenous antigens to MHC class I-restricted T lymphocytes. Cell. 1992;71(6):963–972. doi: 10.1016/0092-8674(92)90392-p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.