

Abstract
The atypical two-component system (TCS) AbrC1/C2/C3 (encoded by SCO4598, SCO4597, and SCO4596), comprising two histidine kinases (HKs) and a response regulator (RR), is crucial for antibiotic production in Streptomyces coelicolor and for morphological differentiation under certain nutritional conditions. In this study, we demonstrate that deletion of the RR-encoding gene, abrC3 (SCO4596), results in a dramatic decrease in actinorhodin (ACT) and undecylprodiginine (RED) production and delays morphological development. In contrast, the overexpression of abrC3 in the parent strain leads to a 33% increase in ACT production in liquid medium. Transcriptomic analysis and chromatin immunoprecipitation with microarray technology (ChIP-chip) analysis of the ΔabrC3 mutant and the parent strain revealed that AbrC3 directly controls ACT production by binding to the actII-ORF4 promoter region; this was independently verified by in vitro DNA-binding assays. This binding is dependent on the sequence 5′-GAASGSGRMS-3′. In contrast, the regulation of RED production is not due to direct binding of AbrC3 to either the redZ or redD promoter region. This study also revealed other members of the AbrC3 regulon: AbrC3 is a positive autoregulator which also binds to the promoter regions of SCO0736, bdtA (SCO3328), absR1 (SCO6992), and SCO6809. The direct targets share the 10-base consensus binding sequence and may be responsible for some of the phenotypes of the ΔabrC3 mutant. The identification of the AbrC3 regulon as part of the complex regulatory network governing antibiotic production widens our knowledge regarding TCS involvement in control of antibiotic synthesis and may contribute to the rational design of new hyperproducer host strains through genetic manipulation of such systems.
INTRODUCTION
Antibiotic production in Streptomyces species is a complex process controlled by multiple factors through intricate genetic networks (1–4). These organisms inhabit the soil, where the environmental conditions are extremely variable, and must therefore compete with other organisms for scarce nutrients. Secretion of antibiotics provides a selective advantage to survive in this adverse environment (5), where it is important to respond quickly to different stimuli. Two-component systems (TCSs) are perhaps the most important signal transduction mechanism in bacteria (6, 7; http://mistdb.com/; http://www.p2cs.org/index.php?PHPSESSID=4c2f13770410fa0fab046d10e6cf90d1). Their relative abundance in a genome reflects an organism's ability to adapt to different conditions, with less abundance, for example, in pathogens such as Chlamydia and Rickettsia than in free-living organisms, such as Streptomyces or Myxococcus, where more than 60 TCSs are present in their respective genomes (8–10). The only bacterial species lacking TCSs are intracellular pathogens (e.g., Mycoplasma species) and endosymbionts (e.g., Amoebophilus species) with severely reduced genomes (11).
The Streptomyces AbrC1/C2/C3 gene cluster (SCO4598, SCO4597, and SCO4596) constitutes an atypical TCS in that it encodes two histidine kinases (HKs) (AbrC1 and AbrC2) of group II and one response regulator (RR) (AbrC3) belonging to the NarL group of the helix-turn-helix (HTH) LuxR family regulators (12). It is conserved across all Streptomyces species sequenced to date and has been found to be critical for antibiotic production and morphological differentiation in Streptomyces coelicolor under certain culture conditions (13). Both kinases of the system (AbrC1 and AbrC2) share high similarity in their kinase domains (85%), although AbrC1 lacks an internal domain of 50 amino acid residues of AbrC2. Independent promoters control the three genes, even though the genes for HKs and cognate RRs of TCSs are usually arranged in operons (14); these features are conserved in all sequenced Streptomyces species, suggesting the importance of regulation of these genes. The RR, AbrC3, has orthologues in more than 20 Streptomyces species sequenced, sharing more than 90% amino acid identity among them. The deletion of these three genes correlates with delayed morphological differentiation and a marked reduction in the production of the following antibiotics in S. coelicolor M145: actinorhodin (ACT), undecylprodiginine (RED), and the calcium-dependent antibiotic (CDA) (13).
In this work, we present a detailed genetic, physiological, and biochemical characterization of this important regulatory system. We show that deletion of abrC3 alone leads to a dramatic reduction in ACT and RED production and in morphological development in S. coelicolor. Moreover, overexpression of the AbrC3 regulator yields high ACT production in both S. coelicolor and S. lividans. To better characterize the pleiotropic effects exhibited by the ΔabrC3 mutant, we conducted a global transcriptome analysis comparing the ΔabrC3 mutant and the parental strain and showed that the low-level production of ACT observed in the abrC3 mutant was due to a general downregulation of transcription of the act gene cluster. Other regulatory genes previously associated with antibiotic production, such as the sIHF gene (SCO1480) (15, 16), afsS (SCO4425) (17, 18), and absR1 (SCO6992) (19), and with morphological development, for example, wblE (SCO5240) (20, 21) and bdtA (SCO3328) (22, 23), were also downregulated in this mutant. Furthermore, through chromatin immunoprecipitation with microarray technology (ChIP-chip) analyses, we demonstrated a direct regulation of act gene expression through AbrC3, and we provided independent verification with in vitro DNA-binding studies. This genomewide study also revealed additional direct DNA targets of AbrC3. All of these targets share a 10-bp sequence (5′-GAASGSGRMS-3′; S = G or C, R = A or G, and M = A or C), and we have shown, by mutagenesis, that it is indispensable for the binding of AbrC3 to the actII-ORF4 promoter region. Therefore, this sequence defines the putative consensus AbrC3 binding site.
MATERIALS AND METHODS
Strains, media, and culture conditions.
Escherichia coli strain BW25113(pIJ790) (containing the λRed system) is an E. coli K-12 (ΔaraBAD ΔrhaBAD) derivative (24), and nonmethylating ET12567(pUZ8002) is a dam dcm hsdS cat tet strain harboring the tra genes in the nontransmissible RP4-derivative plasmid pUZ8002 (25). Both strains were grown in Luria-Bertani (LB) liquid broth or on LB agar (26). For CDA bioassays, a parent strain of Bacillus subtilis (CECT 4522) was grown as an overlay on NA medium (27). S. lividans 66, S. coelicolor M145 (prototroph; SCP1− SCP2−), and mutant strains were grown on R2YE and MS solid media for transformation and sporulation, respectively (28). The liquid cultures used for transcriptional profiling and ChIP-chip analyses were grown in nutrient broth (NB) at 30°C in 500-ml baffled flasks containing 160 ml of medium (each) in a shaking orbital incubator (250 rpm). Where necessary, the medium was supplemented with antibiotics (for E. coli media, 100 μg ml−1 for ampicillin, 50 μg ml−1 for apramycin, 50 μg ml−1 for kanamycin, 34 μg ml−1 for chloramphenicol, and 25 μg ml−1 for nalidixic acid; and for S. coelicolor media, 20 μg ml−1 for neomycin, 50 μg ml−1 for apramycin, and 20 μg ml−1 for hygromycin).
Nucleic acid manipulations.
Plasmid isolation, restriction enzyme digestion, ligation, and transformation of E. coli and S. coelicolor were carried out as described by Sambrook et al. (26) and Kieser et al. (28), respectively. The plasmids and cosmids used are listed in Table S1 in the supplemental material. Total genomic DNA (gDNA) from S. coelicolor was isolated from 24- to 36-h cultures in Trypticase soy broth (TSB), following the procedure described by Hopwood et al. (27), but scaled to 1 to 2 g of mycelium.
Where needed, the DNA was sequenced on both strands by using a PerkinElmer ABI Prism 377 DNA sequencer. In silico plasmid designs were obtained with Gene Construction kit software (GCK; Texco).
Plasmid construction.
The integrative plasmid pSETabrC3 was constructed by cloning the abrC3 gene under the regulation of its own promoter into the Streptomyces integrative shuttle plasmid pSET152t, using NdeI and BamHI restriction sites. This plasmid was used for complementation of the ΔabrC3 mutant strain (see Table S1 in the supplemental material).
The low-copy-number pHJLAbrC3 plasmid, derived from pHJL401 (29), was obtained by cloning the abrC3 gene with its own promoter into pHJL401, using BamHI and EcoRI sites.
The new plasmids were introduced into the corresponding Streptomyces strains by protoplast transformation as previously described (28).
The E. coli expression vector pETabrC3 was a derivative of the pET22(b) plasmid obtained by cloning the coding sequence for AbrC3, previously amplified by PCR with the corresponding oligonucleotides SRG-001 and SRG-002 (see Table S2 in the supplemental material), into the NdeI and XhoI sites of the polylinker.
AbrC3 overexpression in E. coli.
Cultures of E. coli BL21(DE3) transformed with pETabrC3 were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to overproduce AbrC3-6His, subsequently purified with Ni-nitrilotriacetic acid (Ni-NTA) (Qiagen) as described before (30), and used to immunize two rabbits in order to obtain anti-AbrC3 antibodies.
Mutant construction (abrC3 knockout).
The PCR-targeted system established by Gust et al. (31) was used to replace the complete coding sequence of abrC3 (SCO4596) in cosmid SCD20 (http://streptomyces.org.uk/) with the apramycin resistance gene aac(3)IV. The primers used to amplify the mutagenesis cassette, with pIJ773 as the template, are listed in Table S2 in the supplemental material. The mutated cosmid SCD20 ΔabrC3::acc(3)IV, obtained in E. coli BW25113(pIJ790), was demethylated in E. coli ET12567(pUZ8002) and transferred by conjugation to S. coelicolor M145. The desired double recombinants, carrying apramycin resistance while being sensitive to kanamycin (the selection marker for the vector sequences), were selected. Southern blotting and PCR assays confirmed the deletion of the abrC3 gene in S. coelicolor M145.
RNA isolation and DNA microarray analysis.
For RNA extraction from the S. coelicolor parent strain and ΔabrC3 mutant strains, 160 ml of NB medium was inoculated into 500-ml baffled flasks with 4 × 106 spores/ml and incubated at 30°C for 36, 48, and 60 h. Two biological replicates and three technical replicates were used for each time point. Prior to RNA isolation using a modified version of the mirVana miRNA isolation kit protocol (Life Technologies), cells from 20 ml of culture were harvested and suspended in RNA-protect bacterial reagent (Qiagen). Following mycelial lysis with lysozyme (15 mg/ml in Tris-EDTA [TE] at room temperature), 3 volumes of RLT buffer from an RNeasy Plus minikit (Qiagen) was added, and mycelia were disrupted using a TissueLyser (Qiagen) for 4 min. The lysate was clarified by centrifugation, extracted twice with phenol-chloroform and once with chloroform, and applied to a gDNA eliminator filter (RNeasy Plus minikit; Qiagen) to remove any contaminating DNA. After this step, 1.25 volumes of 100% ethanol was added, and the total RNAs (including small RNAs) were purified through mirVana columns following the manufacturer's recommendations. The quality and concentration of RNA were assayed using a Bioanalyzer 2100 system (Agilent) and spectrophotometric assays (model ND1000 Nanodrop instrument).
The S. coelicolor IJISS 105K microarrays used for ChIP-chip and high-resolution transcriptome analysis were designed by the Functional Genomics Laboratory of Surrey University (United Kingdom) in collaboration with Oxford Gene Technology (United Kingdom), and they were manufactured by Agilent. Each microarray comprised almost 105,000 unique 60-mers, with an average genome spacing of 35 nucleotides. The 105K probes were designed to cover the coding strands of all known protein coding genes and both strands of all intergenic regions. A cDNA versus reference gDNA microarray two-color experimental design was utilized for the transcriptomic comparison of S. coelicolor M145 and the ΔabrC3 mutant. All Cy3-cDNA and Cy5-gDNA labeling reactions and hybridization assays were performed according to the recommendations described at http://www.surrey.ac.uk/fhms/microarrays/Downloads/Protocols/index.htm.
All hybridized arrays (expression and ChIP-chip arrays) were scanned using an Agilent Technologies microarray scanner, and resultant images were analyzed using Agilent Technologies Feature Extraction software (version 9.1.3.1.), with local background correction.
Raw gene expression data were normalized within the statistical computing environment R by using the LIMMA package (32–34) global median within-array normalization, followed by “scale” across-array normalization. Probes that were flagged as poor quality by Agilent Feature Extraction were removed from the analysis. A gene's expression was calculated by averaging across all remaining good-quality probes that targeted the annotated coding sequence. Expression data sets were analyzed using rank product analysis via RankProdIt (http://strep-microarray.sbs.surrey.ac.uk/RankProducts), a Web-based interactive analysis tool (35). Differentially expressed genes were identified as having a PFP (probability of false prediction) value of ≤0.15, equal to a false discovery rate of approximately 15% and at a level that has been validated experimentally in this study and others (e.g., see reference 36). A BED-formatted track with the average log2 expression data to be visualized across the chromosome, using the UCSC microbial gene browser (http://microbes.ucsc.edu/), was generated using in-house Perl scripts.
qRT-PCR.
Real-time quantitative reverse transcription-PCR (qRT-PCR) was used to validate the microarray data. Specific primers and probes for the tested genes were designed using the Primer3 Web-based tool (see Table S2 in the supplemental material). Five-microgram RNA samples were treated with RNase-free DNase I (Promega) according to the manufacturer's instructions. One microgram of the resulting RNA was used as the template for cDNA synthesis using iScript reverse transcription supermix for RT-qPCR (Bio-Rad) in 20-μl reaction volumes. The samples were diluted 1:1 with distilled water, and 5 μl was used in the quantitative PCR, with 10 pmol (each) of forward and reverse primers, 2.5 pmol of 6-carboxyfluorescein (FAM)- and 6-carboxytetramethylrhodamine (TAMRA)-dual-labeled specific probe, and 12.5 μl SsoFast Probes supermix with ROX master mix (Bio-Rad) in a final volume of 25 μl. Each assay was performed in duplicate, using a CFX96 Touch real-time PCR detection system (Bio-Rad). Control PCRs were included to verify that there was no DNA contamination (mixtures without reverse transcriptase as an enzyme negative control). Relative quantification of gene expression was performed by the ΔΔCT method, a comparative qPCR data analysis (37) in which the threshold cycle (CT) values of the genes of interest obtained from two different experimental RNA samples (parent strain and mutant) are directly normalized to the CT values of a housekeeping gene (gene control) in the same samples. First, the difference between the CT values of the gene of interest between parent and mutant strains (ΔCT1) and the difference between the CT values of the housekeeping gene between both strains (ΔCT2) are calculated for each experimental sample. Next, the difference in the ΔCT values between the gene of interest and the control gene (ΔΔCT = ΔCT1 − ΔCT2) is calculated. The fold change in expression of the gene of interest between the two experimental samples (parent strain and mutant) is then equal to 2−ΔΔCT. The hrdB (SCO5820) gene was the internal control used to quantify the relative expression of the target genes, since its expression level remained constant under all the conditions analyzed by the microarray. The expression ratios measured by microarrays and by qRT-PCR assays were plotted, and the linear correlation coefficient was calculated (y = 1.60x − 0.09; R2 = 0.93) (see Table 3).
TABLE 3.
qRT-PCR validation of five differentially expressed genes identified from microarray dataa
| Gene | Function | Expression ratio (fold change)b |
|
|---|---|---|---|
| Microarray | qRT-PCR | ||
| SCO2779/acdH | Acyl-CoA dehydrogenase | −1.27 | −2.13 |
| SCO4425/afsS | Sigma-like protein | −1.83 | −3.06 |
| SCO4597/abrC2 | Sensor kinase | −1.085 | −1.12 |
| SCO4598/abrC1 | Sensor kinase | −1.04 | −1.08 |
| SCO5085/actII-ORF4 | ACT cluster activation protein | −2.85 | −3.90 |
| SCO6992/absR1 | Regulatory protein | −1.57 | −3.65 |
| SCO4709/rplP | 50S ribosomal protein L16 | 1.16 | 2.11 |
Linear regression fit analysis was applied, and a correlation coefficient was used to validate the data (y = 1.60x − 0.09; R2 = 0.93).
Parent strain M145 versus ΔabrC3 mutant.
ChIP-chip analysis.
For ChIP, three 20-ml aliquots from the same cultures used in the microarray gene expression analysis were taken (S. coelicolor M145 and ΔabrC3 mutant strains, incubated at 30°C for 36, 48, and 60 h). These samples were treated with formaldehyde (Sigma) at a final concentration of 1%. Cross-linking was allowed to proceed for 30 min at 30°C. Glycine, at a final concentration of 0.5 M, was added to stop the cross-linking for a further 5 min at 30°C. Mycelium was harvested by centrifugation and frozen at −20°C. Chromatins with an average size range of 300 to 600 bp was obtained as described by Bucca et al. (38). ChIP was carried out as described before (38), using purified rabbit polyclonal antibodies against AbrC3 obtained in our laboratory. The IgG fraction containing anti-AbrC3 polyclonal antibodies and the fraction from preimmune serum from the same rabbit used for immunization were purified by affinity chromatography using CNBr-activated Sepharose 4B (Sigma). The chromatins purified from S. coelicolor M145 and the ΔabrC3 mutant strain were sonicated to obtain an average size range of 300 to 600 bp. DNA samples were immunoprecipitated with either AbrC3 antibody (AbIP) or a mock control (NoIP), purified, labeled with Cy3- or Cy5-dCTP, respectively, and hybridized to 105K arrays as described above. Two biological replicates were hybridized such that the AbIP/NoIP samples from each of the two replicates were labeled in opposite dye orientations to control for dye bias. Arrays were scanned and processed as described above.
The raw data were normalized within R (as described above), using the across-array scale function of the LIMMA package only. Again, poor-quality probes were filtered out from downstream analysis. The data analysis was carried out essentially as described by Allenby et al. (39). Values for significant probes (P < 0.05), defined on the basis of a nonparametric t test run with multtest and pnorm (within R), were plotted as ratios [(parent strain AbIP value/parent strain NoIP value)/(ΔabrC3 AbIP value/ΔabrC3 NoIP value)]. The probes most differentially enriched for AbrC3 binding between the parent strain and the mutant were selected from the right tail of the distribution. Probes within 3 kb (based on their annotated genomic locations) of each other were grouped, and genomic regions of interest were those in which at least two enriched probes were found. Finally, tracks (BED files) to visualize the data across the chromosome, using the UCSC microbial genome browser (http://microbes.ucsc.edu/), were generated using in-house Perl scripts.
Western blot assays.
Total protein extracts were obtained from 5-ml samples (in triplicate) of the cultures used for the global analysis described above. The proteins were resolved by SDS-PAGE (15% polyacrylamide in a MiniProtean II system; Bio-Rad). After transfer to Immobilon-P membranes (Millipore), the proteins were incubated with prepurified polyclonal anti-AbrC3 at a 1:100,000 dilution. AbrC3 was detected by chemiluminescence with ECL Western blotting detection reagents following the manufacturer's instructions (GE Healthcare), using horseradish peroxidase-coupled anti-rabbit secondary antibody.
In vitro DNA-binding assays.
Gel shift assays were conducted using DNA fragments containing the intergenic region (up to −400 bp from the translational start point) plus another 100 bp (+100 bp) inside the open reading frame (ORF), amplified using the corresponding primers in a PCR (see Table S2 in the supplemental material). These fragments were 5′ radiolabeled with T4 polynucleotide kinase in the presence of [γ-32P]dATP, and the unreacted [γ-32P]dATP was removed using Illustra ProbeQuant G-50 microcolumns (GE Healthcare). The purified proteins were incubated in 50 mM Tris-HCl, pH 7, 25 mM MgCl2, 0.1 M NaCl at 30°C for 30 min prior to binding. Different concentrations of the proteins were then incubated with the labeled DNA probes (10 fmol) in binding buffer (10 mM Tris-HCl, pH 7.8, 2 mM dithiothreitol [DTT], 150 mM NaCl, 10% glycerol, and 45 ng of sheared salmon sperm DNA) for 30 min at 30°C. Protein-bound DNA and free DNA were resolved in a 5% acrylamide gel in 0.5× TBE buffer (44.5 mM Tris, 44.5 mM boric acid, 1 mM EDTA, pH 8) at 4°C. The gel was exposed overnight and bands revealed by autoradiography. As a control, an unlabeled specific probe (S) (100-fold molar excess) or nonspecific competitor DNA (N) (100-fold molar excess of sheared salmon sperm DNA) was added to the reaction mixture. Also, a bovine serum albumin (BSA) (4 μM) protein-binding reaction was conducted as a control to demonstrate the specificity of the electrophoretic mobility shift assays (EMSAs).
Identification of the putative AbrC3 binding site.
The Regulatory Sequence Analysis Tools (RSAT) “consensus” alignment (http://rsat.ulb.ac.be/) was used to identify putative conserved DNA sequences in the upstream regions (with respect to the annotated translational start codon) of the AbrC3 target genes (including 100 bp into the respective ORF) identified from the ChIP-chip experiment. The functionality of the conserved 10-base sequence identified with RSAT consensus alignment was demonstrated by eliminating it from actII-ORF4p. The elimination of this sequence was achieved using overlap PCR. As the first step, PCR amplicons were made using primers SRG95-SRG97 and SRG96-SRG90 (see Table S2 in the supplemental material). Using both resulting fragments as templates, a second PCR was conducted using oligonucleotides SRG95 and SRG90. The final fragment, with EcoRI and PstI sites in the ends, was cloned into the pGEM-T Easy plasmid. The elimination of the consensus binding sequence from absR1p was performed by PCR amplification of the region with the oligonucleotides SRG-116 and SRG073 (see Table S2).
Antibiotic production measurements.
Production of ACT in liquid cultures was determined by measuring the A640 (ε640 = 25,320) in the supernatants of samples treated with 1 N KOH overnight at 4°C and then centrifuged (15,000 × g, 10 min). The experiments were all performed in triplicate, and the dry weights of samples at different times were measured to monitor culture growth.
Chromatographic analysis.
Liquid cultures were centrifuged, and supernatants and cell pellets were extracted separately. The supernatants were extracted twice with an equal volume of ethyl acetate containing 1% formic acid. The cell pellets were extracted with acetonitrile. In each case, the solvent was evaporated and the residue redissolved in a small volume of a mixture of dimethyl sulfoxide and methanol (50:50). These samples were chromatographed as previously described (40).
Microarray data accession numbers.
All microarray data are MIAME compliant, and the raw and normalized data have been deposited in ArrayExpress under accession number E-MTAB-1453. The raw and normalized ChIP-chip data have been deposited in ArrayExpress under accession number E-MTAB-1451.
RESULTS
Influence of the AbrC3 RR in antibiotic production in Streptomyces.
The S. coelicolor ΔabrC3 mutant strain was generated using a PCR-targeting approach described by Gust et al. (31), which replaced the entire coding region of the gene with an apramycin resistance cassette (see Materials and Methods). Phenotypic analysis of growth and antibiotic production of the parent strain (strain M145), the ΔabrC3 mutant, and the ΔabrC1C2C3 triple mutant, which was described previously (13), revealed significant differences among the three strains (Fig. 1A). ACT and RED production was highly diminished in both the triple and single mutants compared to the parent strain on nutrient agar medium. In contrast, CDA production in the mutant lacking only the response regulator (ΔabrC3 mutant) was unaffected, while it was clearly diminished in the triple mutant. A delay in morphological differentiation on solid medium was also observed in both mutants. When the strains were grown in liquid NB, the ΔabrC3 mutant produced almost no ACT. Complementation of the ΔabrC3 mutant with the integrative plasmid pSET152-abrC3 restored ACT production to almost the parent strain levels (Fig. 1B). Both mutants and the parent strain produced similar growth curves (e.g., see Fig. S1A in the supplemental material), indicating that growth per se was not compromised in the mutant strains.
FIG 1.

Phenotypic study of the mutants and complementation. (A) Production of the antibiotics ACT (72 h), CDA (bioassay against B. subtilis at 48 h), and RED (48 h) by the parent strain (M145) and the ΔabrC1C2C3 and ΔabrC3 mutants, as well as their morphological differentiation (MD) on NA medium. (B) Complementation of ACT production by integration of plasmid pSETabrC3 in the ΔabrC3 mutant strain in liquid NB medium (ACT liquid), ACT (72 h) and RED (48 h) production, and their morphological differentiation (MD) on solid NA medium.
Complementation with pSET152-abrC3 was also tested on solid medium (NA). ACT production and morphological differentiation were complemented, and there was a partial complementation of RED production (Fig. 1B). It was noted that ACT production on solid medium was found to be higher in the complemented strain than in the parent strain harboring the empty plasmid; pSET152 derivatives tend to exist as tandem duplications in the genome (C. P. Smith, unpublished data), and it is therefore conceivable that an enhanced level of AbrC3 was responsible for the elevated ACT production.
It is noteworthy that quantification of ACT production from M145 cells transformed with abrC3 moderately overexpressed from the low-copy-number plasmid pHJLabrC3 showed 33% more ACT production at 5 days than that of the plasmid control, pHJL401 (Fig. 2). This result was consistent with AbrC3 acting as a positive regulator of the ACT gene cluster and offers the possibility of using this low-copy-number plasmid to enhance antibiotic production. Indeed, when pHJLabrC3 was introduced into the S. lividans 66 strain, the normally silent ACT pathway was induced to even higher levels than those in S. coelicolor (Fig. 2).
FIG 2.
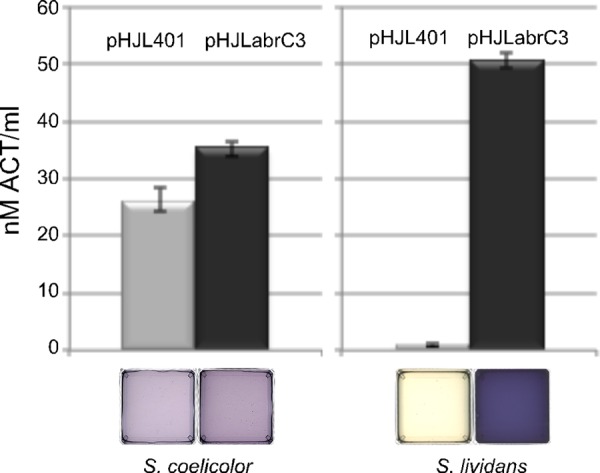
Overexpression of AbrC3 in S. coelicolor and S. lividans. ACT production is shown for S. coelicolor M145 (left) and S. lividans 66 (right) transformed with the low-copy-number plasmid pHJL401 (control) and its derivative, pHJLabrC3, in NB medium at 5 days. Error bars correspond to three independent experiments. The bottom panels show photographs of the corresponding supernatants.
Effects of abrC3 deletion on the S. coelicolor transcriptome.
Comparison of genomewide mRNA abundances between the S. coelicolor M145 parent strain and its congenic ΔabrC3 mutant was performed. Samples were taken after 36, 48, and 60 h of growth (see Fig. S1A in the supplemental material), and antibiotic production at these time points was assessed by high-pressure liquid chromatography (HPLC). As shown in Fig. S1B, at 60 h of growth there was an ACT production peak in the parent strain but not the mutant strain, in both the supernatant and cell extract fractions. Likewise, RED production was observed only in parent strain cell extracts. In addition, the level of AbrC3 protein in the parent strain and its absence in the ΔabrC3 mutant strain at the three sampling points used in this study were confirmed by Western blotting using anti-AbrC3 antibodies (see Fig. S1C).
Total RNA samples from the ΔabrC3 mutant and the parent strain, cultivated in NB medium for 36, 48, and 60 h, were labeled and hybridized to 60-mer, high-density, whole-genome S. coelicolor IJSS 105K DNA microarrays as described by Lewis et al. (41) (see Materials and Methods). Analysis of the DNA microarray data from two independent experiments revealed that only 32 genes were downregulated and 8 genes were upregulated at statistically significant levels (Benjamini and Hochberg-corrected P values of <0.005 and PFP values of <0.15) (Tables 1 and 2). Of the 32 genes that showed significantly reduced expression in the mutant strain, 16 corresponded to the entire act cluster, including actII-ORF4 (SCO5085), which encodes the SARP pathway-specific regulator of the act cluster (Table 1). Three other significantly downregulated genes, i.e., the sIHF gene (SCO1480) (15, 16), afsS (SCO4425) (17, 18), and absR1 (SCO6992) (19), encode regulatory proteins and were previously related to antibiotic regulation. An additional gene, SCO1839, encoding a putative transcriptional regulator with a helix-turn-helix motif, and two genes involved in morphological development, i.e., wblE (SCO5240; encodes an extracytoplasmic function [ECF] sigma factor [20, 21]) and bdtA (SCO3328; encodes a putative transcription factor regulated by BldD and forms an operon with SCO3327 [22, 23]), were also significantly downregulated. The acdH gene (SCO2779), encoding an acyl-coenzyme A (acyl-CoA) dehydrogenase involved in branched-amino-acid catabolism in S. coelicolor and S. avermitilis (42), and SCO3218, a gene of unknown function located in the cda gene cluster, were also significantly downregulated in the mutant (Table 1). Among the eight significantly upregulated genes were the ribosomal protein gene rplP (SCO4709) and the chpE gene (SCO1800) (43) (Table 2).
TABLE 1.
Selected genes significantly downregulated in the ΔabrC3 mutant relative to the parent strain M145 (P < 0.005 and/or PFP < 0.15)
| Gene | Function | P value | PFP | Fold change | Time (h) |
|---|---|---|---|---|---|
| SCO5071 | Hydroxylacyl-CoA dehydrogenase | 0 | 0.002 | −1.77 | 48 |
| SCO5072 | Hydroxylacyl-CoA dehydrogenase | 0 | 0 | −2.13 | 48 |
| SCO5073 | Putative oxidoreductase | 0 | 0 | −2.09 | 48 |
| SCO5074 | Putative dehydratase | 0 | 0 | −3.25 | 48 |
| SCO5075/ORF4 | Putative oxidoreductase | 0 | 0 | −2.23 | 48 |
| SCO5077/actVA2 | Hypothetical protein | 0 | 0.001 | −1.99 | 48 |
| SCO5078/actVA3 | Hypothetical protein | 0 | 0 | −2.28 | 48 |
| SCO5079/actVA4 | Conserved hypothetical protein | 0 | 0 | −2.28 | 48 |
| SCO5080/actVA5 | Putative hydrolase | 0 | 0 | −2.25 | 48 |
| SCO5081/actVA6 | Hypothetical protein | 0 | 0 | −2.59 | 48 |
| SCO5085/actII-ORF4 | Actinorhodin cluster activator protein | 0 | 0 | −2.85 | 48 |
| SCO5086/actIII | Ketoacyl reductase | 0 | 0 | −2.80 | 48 |
| SCO5087/actI-ORF1 | Actinorhodin polyketide beta-ketoacyl synthase alpha subunit | 0 | 0.008 | −1.83 | 48 |
| SCO5088/actI-ORF2 | Actinorhodin polyketide beta-ketoacyl synthase beta subunit | 0 | 0.003 | −1.92 | 48 |
| SCO5089/actI-ORF3 | Actinorhodin polyketide synthase acyl carrier protein | 0 | 0 | −3.11 | 48 |
| SCO5090/actVII | Actinorhodin polyketide synthase bifunctional cyclase/dehydratase | 0.0002 | 0.060 | −1.63 | 48 |
| SCO0045 | Hypothetical protein | 0.0005 | 0.127 | −1.23 | 48 |
| SCO0682 | Hypothetical protein SCF15.03c | 0.0001 | 0.053 | −1.25 | 48 |
| SCO0736 | Putative secreted protein | 0 | 0.019 | −1.45 | 60 |
| SCO1480/sIHF gene | Streptomyces integration host factor | 0.0003 | 0.090 | −1.28 | 48 |
| SCO1550 | Putative small membrane protein | 0.0002 | 0.070 | −1.35 | 36 |
| SCO1839 | Putative transcriptional regulator | 0.0005 | 0.120 | −1.40 | 36 |
| SCO2113/bfr | Putative bacterioferritin | 0.0001 | 0.050 | −1.24 | 48 |
| SCO2779/acdH | Acyl-CoA dehydrogenase | 0.0006 | 0.145 | −1.27 | 36 |
| SCO3218 | Putative small conserved hypothetical protein | 0 | 0.018 | −1.39 | 36 |
| SCO3327 | Hypothetical protein | 0.0002 | 0.051 | −1.41 | 60 |
| SCO3328/bdtA | Hypothetical protein | 0.0002 | 0.055 | −1.47 | 60 |
| SCO4425/afsS | Sigma-like protein | 0 | 0.001 | −1.83 | 48 |
| SCO5240/wblE | Hypothetical protein | 0.0007 | 0.158 | −1.36 | 48 |
| SCO5915 | Fatty acid desaturase | 0.0003 | 0.081 | −1.39 | 36 |
| SCO6205 | Putative threonine dehydrogenase | 0.0007 | 0.158 | −1.43 | 48 |
| SCO6992/absR1 | Regulatory protein | 0.0001 | 0.056 | −1.57 | 60 |
TABLE 2.
Selected genes upregulated in the ΔabrC3 mutant relative to the parent strain M145 (P < 0.005 and/or PFP < 0.15)
| Gene | Function | P value | PFP | Fold change | Time (h) |
|---|---|---|---|---|---|
| SCO1800/chpE | Putative small secreted protein | 0.0001 | 0.145 | 1.12 | 48 |
| SCO2605 | Hypothetical protein SCC88.16 | 0.0002 | 0.140 | 1.36 | 36 |
| SCO4187 | Putative membrane protein | 0 | 0.040 | 1.52 | 36 |
| SCO4709/rplP | 50S ribosomal protein L16 | 0.0001 | 0.119 | 1.16 | 36 |
| SCO6329 | Hypothetical protein SC10H5.05 | 0.0001 | 0.115 | 1.38 | 60 |
| SCO6809 | Putative integral membrane transport protein | 0.0001 | 0.124 | 1.41 | 60 |
| SCO7665 | Hypothetical protein | 0.0002 | 0.131 | 1.07 | 36 |
| SCO7801 | Putative membrane protein | 0.0002 | 0.131 | 1.39 | 60 |
Several of the differentially expressed genes were selected for independent validation by qRT-PCR (Table 3). These included the downregulated genes acdH (SCO2779), encoding acyl-CoA dehydrogenase; absR1 (SCO6992), encoding an activator of secondary metabolism; afsS (SCO4425), a sigma-like protein gene; and the specific regulator of the ACT cluster, actII-ORF4 (SCO5085). We also analyzed the expression levels of one of the genes that was differentially upregulated in the mutant and encodes the 50S ribosomal protein L16 (rplP; SCO4709). Moreover, we included both histidine kinase genes of the TCS, abrC1 and abrC2 (SCO4598 and SCO4597), in order to confirm their nondifferential expression levels. A good correlation of transcript measurements from the microarray data and the qRT-PCR data (yielding a correlation regression coefficient of 0.93) was obtained (Table 3). Note that the validation of differential expression for SCO4709 confirms the use of 0.15 as a suitable PFP threshold for determining significance, similar to that found in other expression-based studies (e.g., see reference 36).
In vivo identification of AbrC3 targets on the S. coelicolor genome.
The genes exhibiting marked AbrC3-dependent changes in expression could be regulated directly by AbrC3 or could be regulated by transcription factors downstream of AbrC3, in a possible regulatory cascade. Hence, to identify genes directly regulated by AbrC3, by interaction with their respective promoter regions, a genomewide ChIP-chip approach was undertaken. Samples from the same cultures and time points used for the transcriptomic analysis (described above) were treated with formaldehyde to cross-link proteins to DNA and then were subjected to chromatin immunoprecipitation with either purified anti-AbrC3 antibodies or mock controls (see Materials and Methods). The immunoprecipitated DNA from each strain was labeled with Cy3 or Cy5 in a dye-balanced direct comparison experimental design and hybridized to the above-mentioned 105K S. coelicolor microarrays (see Materials and Methods). Two biological replicate experiments were conducted, and significantly enriched genomic binding regions in the parent strain relative to the ΔabrC3 mutant were identified. We identified 617, 520, and 458 AbrC-enriched (AbrC3 DNA targets bound at significantly higher levels in the parent strain than in the mutant) regions from chromatins isolated at 36 h, 48 h, and 60 h of growth, respectively. The genes with high enrichment ratios are summarized in Table 4, and the overlapping targets in ChIP-chip and transcriptomic analyses are shown in Table 5.
TABLE 4.
AbrC3 ChIP-chip targets selected by high enrichment ratiose
| Regulated genea | Enrichment ratiob | Predicted function of regulated gene | Time (h)c | Predicted binding site sequenced |
|---|---|---|---|---|
| SCO0167 | 2.45 | Conserved hypothetical protein SCJ1.16c | 36 | 354-GAAGGGGGCG-363 |
| SCO0951 | 2.22 | Putative transport system permease protein | 36 | −329-GGTCGCCTTC-−318 |
| SCO1391 | 5.44 | Crr, phosphoenolpyruvate-protein phosphotransferase | 48 | 1113- GAAGGCGGCG-1122 |
| SCO3020 | 4.22 | Putative integral membrane protein | 48 | Not found |
| SCO3263 | 2.60 | Conserved hypothetical protein | 36 | −240-GTTCGCCTTC-−231 |
| SCO3334 | 5.70 | TrpS, tryptophanyl tRNA synthetase | 48 | −387-GGCCGCGTTC-−378 |
| SCO3590 | 2.54 | VanR, putative TCS response regulator | 36 | Not found |
| SCO3992 | 2.45 | Hypothetical protein | 36 | Not found |
| SCO4075 | 6.32 | RagA, ABC transport protein, ATP-binding subunit | 48 | Not found |
| SCO4123 | 5.02 | Putative TCS response regulator | 48 | −674-GAACGCGACC-−665 |
| SCO4596 | 2.24 | AbrC3, TCS response regulator | 60 | −137-CTTCCCGTTC-−128 |
| SCO4630 | 2.78 | Hypothetical protein | 36 | Not found |
| SCO5085 | 2.23 | ActII-ORF4, ACT cluster activator protein | 36 | 72-GAAGGCGACC-81 |
| SCO5236 | 2.86 | NagB, putative glucosamine phosphate isomerase | 48 | −213-GGTCGCGTTC-−204 |
| SCO5330 | 3.26 | Hypothetical protein SC6G9.03c | 36 | −975-GAAGGGGGCG-−966 |
| SCO5332 | 2.21 | Hypothetical protein | 36 | 1134-CTTCGCCTTC-1143 |
| SCO5632 | 2.36 | Hypothetical protein SC6A9.3 | 36 | −1261-GAACGGGACG-−1252 |
| SCO5638 | 2.76 | Integral membrane protein | 36 | Not found |
| SCO5691 | 2.24 | Putative secreted sugar hydrolase | 48 | −777-GAACGCGGCG-−768 |
| SCO5729 | 2.33 | Conserved hypothetical protein SC3C3.15c | 36 | Not found |
| SCO5912 | 4.30 | Probable secreted protease | 48 | −54-CTCCCCCTTC-−45 |
| SCO5966 | 2.24 | Putative oxidase | 48 | −803-GAAGGCGGAC-−794 |
| SCO6273 | 2.18 | CpkC, putative type I polyketide synthase | 48 | 3138-CGCCCCGTTC-3147 |
| SCO6732 | 2.29 | Putative fatty acid oxidative multifunctional enzyme | 36 | 1140-GAAGGCGGCC-1149 |
| SCO6820 | 2.94 | Putative oxidoreductase | 48 | −519-GAACGCGGCC-−510 |
| SCO6941 | 2.55 | CvnC8, hypothetical protein SC1G8.13c | 36 | −306-GGCCGCCTTC-−297 |
| SCO6951 | 2.55 | Conserved hypothetical protein | 36 | −411-CGCCGCGTTC-−402 |
| SCO6992 | 2.78 | AbsR1, regulatory protein | 48 | −321-GAAGGGGACG-−312 |
| SCO7077 | 2.24 | Putative integral membrane protein | 36 | −294-GAAGGCGGCG-−285 |
| SCO7545 | 2.45 | Putative ABC transport protein, ATP-binding component | 48 | −369-GGCCGCGTTC-−360 |
Putative AbrC3 target genes indicated by the ChIP-chip analysis.
AbrC3-DNA interaction affinity.
Time in the ChIP-chip assay at which the shown enrichment ratio was found.
The consensus binding site sequence is 5′-GAASGSGRMS-3′. Numbers indicate the position of the sequence relative to the start codon of the corresponding gene.
Genes that also had differential transcription profiles are shown in bold.
TABLE 5.
AbrC3 overlapping targets in ChIP-chip and transcriptome analysesd
| Gene | Enrichment ratioa | Predicted function of regulated gene | Time (h)b | Predicted binding site sequencec |
|---|---|---|---|---|
| SCO0736 | 1.50 | Putative secreted protein | 36 | −53-GAAGGCGACC-−44 |
| SCO2113 | 1.59 | Bfr, putative bacterioferritin | 48 | −26-GTTCCCGTTC-−17 |
| SCO3328 | 1.48 | BdtA, hypothetical protein | 48 | −46-GAAGGGGAAG-−37 |
| SCO4596 | 2.24 | AbrC3, TCS response regulator | 60 | −137-CTTCCCGTTC-−128 |
| SCO4709 | 1.89 | RplP, 50S ribosomal protein L16 | 36 | Not found |
| SCO5085 | 2.23 | ActII-ORF4, ACT cluster activator protein | 36 | 72-GAAGGCGACC-81 |
| SCO6809 | 1.77 | Putative integral membrane transport protein | 48 | −112-GAAGGCGGAC-−103 |
| SCO6992 | 2.78 | AbsR1, regulatory protein | 48 | −321-GAAGGGGACG-−312 |
AbrC3-DNA interaction affinity by ChIP-chip analysis.
Time in the ChIP-chip assay at which the shown enrichment ratio was found.
The consensus binding site sequence is 5′-GAASGSGRMS-3′. Numbers indicate the position of the sequence related to the start codon of the corresponding gene.
The genes shown in bold are upregulated. The other genes in the table are downregulated.
This analysis demonstrated that the AbrC3 protein was able to bind in vivo, with high efficiency, to the promoter of the pathway-specific regulator gene of the ACT cluster, actII-ORF4p (SCO5085) (Tables 4 and 5), at 36 h. No binding was detectable at 48 h or 60 h. This result suggests that the downregulation of the act gene cluster (Table 1) was due to a direct regulation of actII-ORF4 exerted by AbrC3 at around 36 h of culture.
Efficient binding of AbrC3 to its own promoter, abrC3p (SCO4596), was also observed at 60 h (Table 5), indicating that AbrC3 is an autoregulator. No binding to the promoters of the histidine kinase-encoding genes of the AbrC system (abrC1 and abrC2) or differential expression of these two genes under the experimental conditions used was observed (Table 3).
In vivo binding of AbrC3 to several of the promoters of the genes previously identified as downregulated was observed, and therefore their expression is very likely directly controlled by AbrC3 (Table 5). Weaker AbrC3 enrichment was observed upstream of SCO1480 (sIHF gene), SCO1550, the regulator gene SCO1839, the acyl-CoA dehydrogenase AcdH gene (SCO2779), and the transcription factor WblE gene (SCO5240), and no binding at all was observed for three of the differentially expressed genes identified in our transcriptome analysis: SCO0682, SCO3218, and afsS (SCO4425) (data not shown). We hypothesize that these genes could be regulated by factors downstream of AbrC3, in a possible regulatory cascade, or that additional transcription factors/cofactors bound to the promoter regions blocked access of the anti-AbrC3 antibody, yielding false-negative results (for example, see reference 39).
Among the significantly upregulated genes, we detected binding of AbrC3 only to the putative promoters of the rplP (SCO4709) and SCO6809 genes (Table 5), suggesting that the other six upregulated genes may be regulated by downstream factors.
EMSAs were conducted to test the in vitro binding capacity of the protein for the DNA sequences upstream of the corresponding genes that showed AbrC3 binding in vivo. DNA fragments comprising the intergenic region (up to −400 bp from the translational start point) plus 100 bp inside the ORF (+100) were used to perform the experiments and are referred to henceforth as “promoters*” (SCOXXXXp). Most of the identified targets that were bound in vivo by the AbrC3 protein (Table 5) were also able to be bound in vitro with a high specificity, including actII-ORF4p, abrC3p, SCO0736p, absR1p, and SCO6809p (Fig. 3A to D and H). The promoters* of SCO2113 and bdtA were bound with less specificity (Fig. 3E and F), and some promoters*, such as SCO4709p, did not yield mobility shifts in vitro (Fig. 3G). Competition experiments to verify the specificity of AbrC3 binding to each promoter* were conducted by adding different amounts of the corresponding unlabeled DNA or different amounts of sheared salmon sperm DNA to the binding reaction mixtures.
FIG 3.

EMSAs of different concentrations of AbrC3 protein with ChIP-chip-selected promoters. (A) actII-ORF4p; (B) abrC3p; (C) SCO0736p; (D) absR1p (SCO6992p); (E) SCO2113p; (F) bdtAp (SCO3328p); (G) SCO4709p; (H) SCO6809p. S, specific competition control (adding the corresponding unlabeled specific probe); N, nonspecific competitor DNA (100-fold molar excess of sheared sperm salmon DNA); BSA, specific protein control (4 μM BSA).
Identification of a putative consensus AbrC3-binding site.
Aligning the 500-bp upstream sequences (including the first 100 bp of the respective ORFs) of genes enriched for AbrC3 in the ChIP-chip and EMSA analyses by using the RSAT consensus alignment tool (http://rsat.ulb.ac.be/) identified a minimal consensus binding site for AbrC3. A common 10-bp sequence (5′-GAASGSGRMS-3′) was present in all of the putative promoters of the identified target genes and was absent in the ones that did not yield mobility shifts in vitro, such as SCO4709p (Fig. 4A). This putative AbrC3-binding sequence was present within the ORF of actII-ORF4, at positions +72 to +81 (from the translational start codon), near one of the described target sequences of the regulator protein AtrA (44) (Fig. 5). This sequence was also found in 23 of 30 of the genes identified by ChIP-chip analysis (Table 4).
FIG 4.

Identification of a putative AbrC3-binding sequence in different genes. (A) Consensus sequence identified by RSAT consensus alignment of putative promoters* (upstream sequences up to −400 bp plus +100 bp from the translational start point) of genes identified by ChIP-chip and EMSAs. (B) EMSAs of different fragments of the actII-ORF4 promoter* region with 4 μM AbrC3 (the top panel shows the amplified fragment locations in the promoter*). (C) EMSA of different concentrations of the AbrC3 protein with the actII-ORF4 fragment 4 promoter*. S, specific competition control (adding 100-fold molar excess of unlabeled specific probe); N, nonspecific competitor DNA (adding 100-fold excess of sheared sperm salmon DNA); BSA, specific control 2 (adding 4 μM BSA protein).
FIG 5.
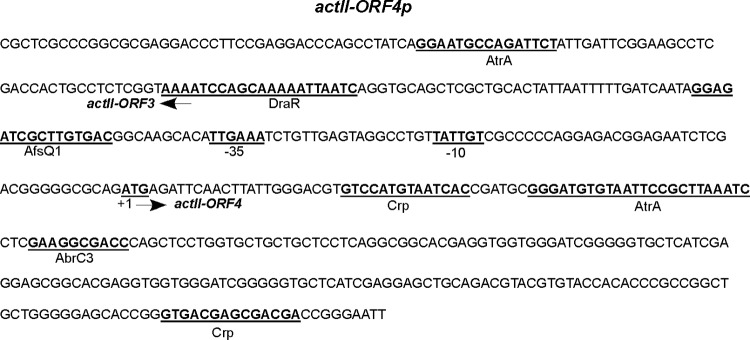
Locations of regulatory protein-binding sites in the actII-ORF4 promoter* region. Locations are shown for the binding sequences in the actII-ORF4 promoter* described in the literature for the AtrA, DraR, and AfsQ1 proteins, as well as the position of the deduced target sequence for AbrC3 identified in this work.
The functionality of this putative AbrC3-binding sequence was demonstrated by EMSAs using different fragments of actII-ORF4p or deleting the putative binding sequence (Fig. 4B). Only the whole promoter* region (fragment 1, also used for Fig. 3) or a 3′ part of the promoter* that included the 10-base sequence generated a mobility shift in the presence of the AbrC3 protein (Fig. 4B, panels 1 and 4). Conversely, the mobility shift did not take place when the 10-base sequence was removed (see Materials and Methods) (Fig. 4B, panel 5); controls used to demonstrate the specificity of AbrC3 binding to the 3′ segment of the promoter* (fragment 4) are shown in Fig. 4C. This result clearly indicated that the sequence (5′-GAAGGCGACC-3′) was essential in the direct regulation of actII-ORF4 expression by the AbrC3 regulator.
The importance of this sequence in the binding of AbrC3 was also demonstrated by an EMSA with absR1p, another direct target of this regulator. The elimination of the proposed binding sequence (5′-GAAGGGGACG-3′) originated the sequence absR1pDel, which was unable to bind AbrC3, and therefore the shifted band was not observed (see Fig. S2 in the supplemental material).
Moreover, EMSAs were performed using shorter DNA probes consisting of a 50-bp labeled oligonucleotide (resulting from annealing two long, complementary oligonucleotides, SRG119 and SRG120 [see Table S2 in the supplemental material]) corresponding to a small fragment from actIIORF4p and containing the predicted binding sequence (probe 1), and also another artificially labeled oligonucleotide of the same size, containing this sequence but surrounded by foreign DNA (probe 2, resulting from annealing of oligonucleotides SRG121 and SRG122 [see Table S2]). The results (see Fig. S3) showed the ability of AbrC3 to bind specifically to probe 1 and also to the artificial probe 2. Therefore, these 10 nucleotides (5′-GAAGGCGACC-3′) were enough to recruit the protein.
DISCUSSION
AbrC3 directly regulates actII-ORF4 expression.
An integrated global ChIP-chip and transcriptome analysis of an S. coelicolor parent strain and an abrC3 mutant derivative has identified the regulon of the response regulator AbrC3. actII-ORF4, the pathway-specific regulator of ACT, is one of its direct targets, as corroborated by EMSA in vitro. A 10-bp sequence, 5′-GAASGSGRMS-3′, identified by RSAT consensus alignment of the putative promoters* targeted by AbrC3, defines the AbrC3-binding site. In the case of actII-ORF4, the binding sequence (5′-GAAGGCGACC-3′) is found within the ORF, at positions +72 to +81 (from the translational ATG start codon) (Fig. 5). This is not the first time that a transcriptional regulator-binding site has been found within the actII-ORF4 coding sequence; there are two other regulators, AtrA and Crp, whose binding sites are at positions +45 to +69 (44) and +24 to +38 (45), respectively. DNase footprinting assays using selected AbrC3-associated sequences failed to yield a DNase-protected region (data not shown); we consider that the observed poor solubility of AbrC3 in vitro may account for the incomplete shifting of template DNA obtained in the EMSAs and for our failure to observe a footprint. Nevertheless, this consensus sequence has been shown to be indispensable for AbrC3 binding to actII-ORF4p (Fig. 4) and to another of its targets (absR1p) (see Fig. S2 in the supplemental material). We speculate that binding of AbrC3 to actII-ORF4p at 36 h may be necessary for the recruitment of other regulators and that the binding of these regulators is necessary for the first steps of actII-ORF4 expression: after this initial recruitment, the regulators are subsequently released from the promoter* to allow expression of downstream genes.
The expression of the ACT-specific pathway regulator actII-ORF4 is under the control of different regulatory proteins that respond to diverse signaling routes in S. coelicolor, such as PhoPR (46), RapA1A2 (47), the LAL regulators SCO0877 and SCO7173 (48), and the ECF SigT (49), among others (reviewed by van Wezel and McDowall [3]). To date, demonstration of the direct binding of transcription factors to the actII-ORF4 promoter region has been limited (Fig. 5). In vitro binding assays (EMSAs) have shown that the AtrA protein binds to the actII-ORF4 promoter (44). More recently, DNA affinity capture assays have shown the capability of four more proteins (SCO0310, SCO3932, SCO5405, and AdpA) to bind to the actII-ORF4 promoter (15). Among the TCSs implicated in antibiotic production, thus far, only AbsA2 has been shown to bind to the actII-ORF4 promoter in vivo (50). Very recently, EMSAs demonstrated in vitro binding of the TCSs DraRK and AfsQ1Q2 to the actII-ORF4 promoter (51, 52). Control of secondary metabolism by the global regulator Crp was also recently described, and Crp binding to actII-ORF4 was detected by ChIP-chip analysis, with both of the described binding sites located within the ORF, at positions +24 to +38 and +234 to +248 (Fig. 5) (45).
AbrC3 regulon.
The putative binding sequence (5′-GAASGSGRMS-3′) defined in this work is present in 23 of the 30 genes with the highest binding enrichment levels (wild-type AbIP/NoIP versus ΔabrC3 AbIP/NoIP enrichment ratios obtained in the ChIP-chip assay) (Table 4). The position of the putative binding sequence is typically upstream of the respective ORF, suggesting regulation of the gene downstream of the binding site. However, in some cases, the target sequences are within the coding sequences. This feature is coincident with data described previously in the study of the regulation exerted by the protein Crp, where more than 50% of all Crp-associated sequences were mapped within ORFs (45). The regulatory role of these sequences located inside the ORFs is not clear, and in a recent study using ChIP-sequencing (ChIP-seq) and chromatin affinity precipitation sequencing (ChAP-seq) technologies to characterize the AdpA regulon, it was found that approximately 75% of the in vivo AdpA-binding sites apparently have no function in the regulation of gene expression (53).
Conversely, it is also possible that some bona fide AbrC3 targets were missed in this study, because the antibody does not recognize its epitopes when AbrC3 is one of several transcription factors bound to the promoter regions. It is known that false-negative results can occur in ChIP-chip studies (38).
Pleiotropic regulation by the abrC TCS.
The pleiotropic phenotype of the triple ΔabrC1C2C3 mutant (13) is different (decreased CDA production) from the phenotype of the RR gene deletion mutant, i.e., the ΔabrC3 strain. This result suggests that other RRs may be controlled by the kinases of this TCS, such as the orphan RR SCO2281, proposed by Wang et al. to be recognized by AbrC2 (54). It is known that the biosynthetic RED genes are expressed early, so the existence of direct AbrC3 regulation of the promoter regions of redD or redZ cannot yet be excluded for the experimental conditions tested. If the RED cluster regulation by AbrC3 is indirect, this could be exerted through the regulatory cascade triggered by AfsS, which is one of the indirect AbrC3 targets (17, 18), or by sIHF, which is downregulated in the ΔabrC3 mutant and is able to bind to the redD and redZ promoters (15, 16).
Several transcriptional regulators whose genes were downregulated in our study could be responsible for the morphological phenotype observed in the ΔabrC3 mutant, including sIHF, which also binds to the bldD promoter, a key regulator of development in S. coelicolor (16); the extracytoplasmic function sigma factor WblE (20, 21); and BdtA, a putative transcription factor regulated by BldD and involved in morphological differentiation (22, 23).
ChIP-chip assays demonstrated that AbrC3 is an autoregulator. No differential regulation between the wild-type strain and the AbrC3 mutant of the two kinase-encoding genes of the AbrC TCS system was identified in our ChIP-chip or transcriptome analysis.
A summary of the main interactions within the AbrC3 regulon deduced from our global in vivo analyses (ChIP-chip and transcriptome analyses) is illustrated in the model depicted in Fig. 6. We observed a global positive effect on secondary metabolism, mainly on the ACT cluster, and morphological differentiation through the direct interaction of AbrC3 with the different genes mentioned above. This activation could also be facilitated by negative control of primary metabolism by AbrC3, through ribosomal proteins such as RplP. Indeed, another gene involved in primary metabolism, acdH, was downregulated in the ΔabrC3 mutant; this gene encodes an acyl-CoA dehydrogenase involved in biosynthesis of precursors of ACT and RED, which could explain the positive regulation of this gene by AbrC3. In addition, Wang et al. have just reported in vitro evidence that abrC3 is a target of AfsQ1 and that it positively regulates its expression. This suggests that the function of AfsQ1Q2 is mediated at least partly via the AbrC1/C2/C3 system, further illustrating the complexity of the regulatory network governing antibiotic production (Fig. 6) (51, 52).
FIG 6.
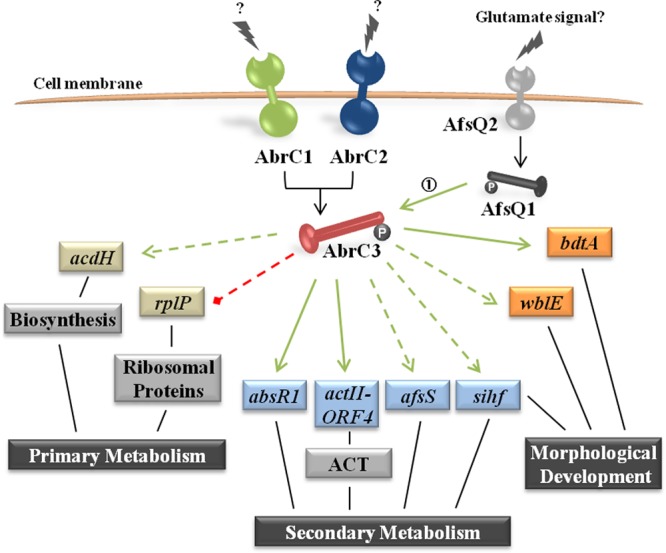
Scheme of the AbrC3 regulon of S. coelicolor. The schematic model shows the pleiotropic regulatory roles of AbrC3 in primary and secondary metabolism, morphological differentiation, and the putative gene network governed by AbrC3. Green arrows, positive control; red dashed line, negative control; solid arrows, direct control; dashed arrows, indirect control. The data for arrow 1 are from the work of Wang et al. (51).
The emergence of new antibiotic-resistant isolates makes new antibiotic discovery and the improvement of their production major challenges for microbial biotechnology. Widening our knowledge regarding TCS involvement in antibiotic synthesis regulation can contribute to the rational design of new hyperproducer host strains through genetic manipulation of those complex systems. Moreover, strategies involving TCSs could be used to unveil new antimicrobial molecules that are not normally produced under laboratory culture conditions.
Supplementary Material
ACKNOWLEDGMENTS
Thanks are due to M. J. Jiménez Rufo for her excellent technical work and to A. Braña for his chromatographic analysis.
This work was supported by the Spanish Comisión Interministerial de Ciencia y Tecnología (CICYT) (grant GEN2003-20245-C09-02), the Junta de Castilla y León (JCyL) (grants SA072A07 and CSI099A12-1), the Spanish Ministerio de Ciencia e Innovación (MICINN) (grant BFU2010-17551), and Fundación Ramón Areces institutional funding to the Instituto de Biología Funcional y Genómica (IBFG). S.R. had a JAE predoctoral grant from the CSIC. H.R. had a postdoctoral fellowship from the Botín Foundation.
Footnotes
Published ahead of print 7 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03378-13.
REFERENCES
- 1.Bibb M, Hesketh A. 2009. Chapter 4. Analyzing the regulation of antibiotic production in streptomycetes. Methods Enzymol. 458:93–116. 10.1016/S0076-6879(09)04804-6 [DOI] [PubMed] [Google Scholar]
- 2.Huang J, Shi J, Molle V, Sohlberg B, Weaver D, Bibb MJ, Karoonuthaisiri N, Lih CJ, Kao CM, Buttner MJ, Cohen SN. 2005. Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol. Microbiol. 58:1276–1287. 10.1111/j.1365-2958.2005.04879.x [DOI] [PubMed] [Google Scholar]
- 3.van Wezel GP, McDowall KJ. 2011. The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat. Prod. Rep. 28:1311–1333. 10.1039/c1np00003a [DOI] [PubMed] [Google Scholar]
- 4.Liu G, Chater KF, Chandra G, Niu G, Tan H. 2013. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 77:112–143. 10.1128/MMBR.00054-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hopwood DA. 2007. Streptomyces in nature and medicine. The antibiotic makers. Oxford University Press Inc, New York, NY [Google Scholar]
- 6.Barakat M, Ortet P, Jourlin-Castelli C, Ansaldi M, Mejean V, Whitworth DE. 2009. P2CS: a two-component system resource for prokaryotic signal transduction research. BMC Genomics 10:315. 10.1186/1471-2164-10-315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulrich LE, Zhulin IB. 2010. The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38:D401–D407. 10.1093/nar/gkp940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee B, Schramm A, Jagadeesan S, Higgs PI. 2010. Two-component systems and regulation of developmental progression in Myxococcus xanthus. Methods Enzymol. 471:253–278. 10.1016/S0076-6879(10)71014-4 [DOI] [PubMed] [Google Scholar]
- 9.Kim D, Forst S. 2001. Genomic analysis of the histidine kinase family in bacteria and archaea. Microbiology 147:1197–1212 [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Hirakawa H, Muta S, Kuhara S. 2010. Identification and classification of a two-component system based on domain structures in bacteria and differences in domain structure between Gram-positive and Gram-negative bacteria. Biosci. Biotechnol. Biochem. 74:716–720. 10.1271/bbb.90746 [DOI] [PubMed] [Google Scholar]
- 11.Wuichet K, Cantwell BJ, Zhulin IB. 2010. Evolution and phyletic distribution of two-component signal transduction systems. Curr. Opin. Microbiol. 13:219–225. 10.1016/j.mib.2009.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutchings MI, Hoskisson PA, Chandra G, Buttner MJ. 2004. Sensing and responding to diverse extracellular signals? Analysis of the sensor kinases and response regulators of Streptomyces coelicolor A3(2). Microbiology 150:2795–2806. 10.1099/mic.0.27181-0 [DOI] [PubMed] [Google Scholar]
- 13.Yepes A, Rico S, Rodriguez-Garcia A, Santamaria RI, Diaz M. 2011. Novel two-component systems implied in antibiotic production in Streptomyces coelicolor. PLoS One 6:e19980. 10.1371/journal.pone.0019980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castro-Melchor M, Charaniya S, Karypis G, Takano E, Hu WS. 2010. Genome-wide inference of regulatory networks in Streptomyces coelicolor. BMC Genomics 11:578. 10.1186/1471-2164-11-578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park SS, Yang YH, Song E, Kim EJ, Kim WS, Sohng JK, Lee HC, Liou KK, Kim BG. 2009. Mass spectrometric screening of transcriptional regulators involved in antibiotic biosynthesis in Streptomyces coelicolor A3(2). J. Ind. Microbiol. Biotechnol. 36:1073–1083. 10.1007/s10295-009-0591-2 [DOI] [PubMed] [Google Scholar]
- 16.Yang YH, Song E, Willemse J, Park SH, Kim WS, Kim EJ, Lee BR, Kim JN, van Wezel GP, Kim BG. 2012. A novel function of Streptomyces integration host factor (sIHF) in the control of antibiotic production and sporulation in Streptomyces coelicolor. Antonie Van Leeuwenhoek 101:479–492. 10.1007/s10482-011-9657-z [DOI] [PubMed] [Google Scholar]
- 17.Lee PC, Umeyama T, Horinouchi S. 2002. afsS is a target of AfsR, a transcriptional factor with ATPase activity that globally controls secondary metabolism in Streptomyces coelicolor A3(2). Mol. Microbiol. 43:1413–1430. 10.1046/j.1365-2958.2002.02840.x [DOI] [PubMed] [Google Scholar]
- 18.Lian W, Jayapal KP, Charaniya S, Mehra S, Glod F, Kyung YS, Sherman DH, Hu WS. 2008. Genome-wide transcriptome analysis reveals that a pleiotropic antibiotic regulator, AfsS, modulates nutritional stress response in Streptomyces coelicolor A3(2). BMC Genomics 9:56. 10.1186/1471-2164-9-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park U, Suh J, Hong S. 2000. Genetic analysis of absR, a new abs locus of Streptomyces coelicolor. J. Microbiol. Biotechnol. 10:169–175 [Google Scholar]
- 20.Fowler-Goldsworthy K, Gust B, Mouz S, Chandra G, Findlay KC, Chater KF. 2011. The actinobacteria-specific gene wblA controls major developmental transitions in Streptomyces coelicolor A3(2). Microbiology 157:1312–1328. 10.1099/mic.0.047555-0 [DOI] [PubMed] [Google Scholar]
- 21.Homerova D, Sevcikova J, Kormanec J. 2003. Characterization of the Streptomyces coelicolor A3(2) wblE gene, encoding a homologue of the sporulation transcription factor. Folia Microbiol. 48:489–495. 10.1007/BF02931330 [DOI] [PubMed] [Google Scholar]
- 22.den Hengst CD, Tran NT, Bibb MJ, Chandra G, Leskiw BK, Buttner MJ. 2010. Genes essential for morphological development and antibiotic production in Streptomyces coelicolor are targets of BldD during vegetative growth. Mol. Microbiol. 78:361–379. 10.1111/j.1365-2958.2010.07338.x [DOI] [PubMed] [Google Scholar]
- 23.Elliot MA, Bibb MJ, Buttner MJ, Leskiw BK. 2001. BldD is a direct regulator of key developmental genes in Streptomyces coelicolor A3(2). Mol. Microbiol. 40:257–269. 10.1046/j.1365-2958.2001.02387.x [DOI] [PubMed] [Google Scholar]
- 24.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, MacNeil T. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111:61–68. 10.1016/0378-1119(92)90603-M [DOI] [PubMed] [Google Scholar]
- 26.Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 27.Hopwood DA, Bibb JM, Chater KF, Kieser T, Bruton CJ, Kieser HM, Lydiate DJ, Smith CP, Ward JM, Schrempf H. 1985. Genetic manipulation of Streptomyces: a laboratory manual. John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 28.Kieser T, Hopwood DA, Bibb JM, Chater KF, Buttner MJ. 2000. Practical Streptomyces genetics. John Innes Foundation, Norwich, United Kingdom [Google Scholar]
- 29.Larson JL, Hershberger CL. 1986. The minimal replicon of a streptomycete plasmid produces an ultrahigh level of plasmid DNA. Plasmid 15:199–209. 10.1016/0147-619X(86)90038-7 [DOI] [PubMed] [Google Scholar]
- 30.Sevillano L, Diaz M, Yamaguchi Y, Inouye M, Santamaria RI. 2012. Identification of the first functional toxin-antitoxin system in Streptomyces. PLoS One 7:e32977. 10.1371/journal.pone.0032977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. 2003. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 100:1541–1546. 10.1073/pnas.0337542100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5:R80. 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Core Team. 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria [Google Scholar]
- 34.Smyth GK, Speed TP. 2003. Normalization of cDNA microarray data. Methods 31:265–273. 10.1016/S1046-2023(03)00155-5 [DOI] [PubMed] [Google Scholar]
- 35.Laing E, Smith CP. 2010. RankProdIt: a web-interactive rank products analysis tool. BMC Res. Notes 3:221. 10.1186/1756-0500-3-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hesketh A, Bucca G, Laing E, Flett F, Hotchkiss G, Smith CP, Chater KF. 2007. New pleiotropic effects of eliminating a rare tRNA from Streptomyces coelicolor, revealed by combined proteomic and transcriptomic analysis of liquid cultures. BMC Genomics 8:261. 10.1186/1471-2164-8-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 38.Bucca G, Laing E, Mersinias V, Allenby N, Hurd D, Holdstock J, Brenner V, Harrison M, Smith CP. 2009. Development and application of versatile high density microarrays for genome-wide analysis of Streptomyces coelicolor: characterization of the HspR regulon. Genome Biol. 10:R5. 10.1186/gb-2009-10-1-r5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allenby NE, Laing E, Bucca G, Kierzek AM, Smith CP. 2012. Diverse control of metabolism and other cellular processes in Streptomyces coelicolor by the PhoP transcription factor: genome-wide identification of in vivo targets. Nucleic Acids Res. 40:9543–9556. 10.1093/nar/gks766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pérez J, Muñoz-Dorado J, Braña AF, Shimkets LJ, Sevillano L, Santamaría RI. 2011. Myxococcus xanthus induces actinorhodin overproduction and aerial mycelium formation by Streptomyces coelicolor. Microb. Biotechnol. 4:175–183. 10.1111/j.1751-7915.2010.00208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis RA, Laing E, Allenby N, Bucca G, Brenner V, Harrison M, Kierzek AM, Smith CP. 2010. Metabolic and evolutionary insights into the closely-related species Streptomyces coelicolor and Streptomyces lividans deduced from high-resolution comparative genomic hybridization. BMC Genomics 11:682. 10.1186/1471-2164-11-682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YX, Denoya CD, Skinner DD, Fedechko RW, McArthur HA, Morgenstern MR, Davies RA, Lobo S, Reynolds KA, Hutchinson CR. 1999. Genes encoding acyl-CoA dehydrogenase (AcdH) homologues from Streptomyces coelicolor and Streptomyces avermitilis provide insights into the metabolism of small branched-chain fatty acids and macrolide antibiotic production. Microbiology 145:2323–2334 [DOI] [PubMed] [Google Scholar]
- 43.Capstick DS, Jomaa A, Hanke C, Ortega J, Elliot MA. 2011. Dual amyloid domains promote differential functioning of the chaplin proteins during Streptomyces aerial morphogenesis. Proc. Natl. Acad. Sci. U. S. A. 108:9821–9826. 10.1073/pnas.1018715108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uguru GC, Stephens KE, Stead JA, Towle JE, Baumberg S, McDowall KJ. 2005. Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol. Microbiol. 58:131–150. 10.1111/j.1365-2958.2005.04817.x [DOI] [PubMed] [Google Scholar]
- 45.Gao C, Hindra, Mulder D, Yin C, Elliot MA. 2012. Crp is a global regulator of antibiotic production in Streptomyces. mBio 3:e00407-12. 10.1128/mBio.00407-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sola-Landa A, Moura RS, Martin JF. 2003. The two-component PhoR-PhoP system controls both primary metabolism and secondary metabolite biosynthesis in Streptomyces lividans. Proc. Natl. Acad. Sci. U. S. A. 100:6133–6138. 10.1073/pnas.0931429100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Y, Wang W, Shu D, Zhang W, Chen L, Qin Z, Yang S, Jiang W. 2007. Characterization of a novel two-component regulatory system involved in the regulation of both actinorhodin and a type I polyketide in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 77:625–635. 10.1007/s00253-007-1184-5 [DOI] [PubMed] [Google Scholar]
- 48.Guerra SM, Rodriguez-Garcia A, Santos-Aberturas J, Vicente CM, Payero TD, Martin JF, Aparicio JF. 2012. LAL regulators SCO0877 and SCO7173 as pleiotropic modulators of phosphate starvation response and actinorhodin biosynthesis in Streptomyces coelicolor. PLoS One 7:e31475. 10.1371/journal.pone.0031475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng WH, Mao XM, Liu ZH, Li YQ. 2011. The ECF sigma factor SigT regulates actinorhodin production in response to nitrogen stress in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 92:1009–1021. 10.1007/s00253-011-3619-2 [DOI] [PubMed] [Google Scholar]
- 50.McKenzie NL, Nodwell JR. 2007. Phosphorylated AbsA2 negatively regulates antibiotic production in Streptomyces coelicolor through interactions with pathway-specific regulatory gene promoters. J. Bacteriol. 189:5284–5292. 10.1128/JB.00305-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R, Mast Y, Wang J, Zhang W, Zhao G, Wohlleben W, Lu Y, Jiang W. 2013. Identification of two-component system AfsQ1/Q2 regulon and its cross-regulation with GlnR in Streptomyces coelicolor. Mol. Microbiol. 87:30–48. 10.1111/mmi.12080 [DOI] [PubMed] [Google Scholar]
- 52.Yu Z, Zhu H, Dang F, Zhang W, Qin Z, Yang S, Tan H, Lu Y, Jiang W. 2012. Differential regulation of antibiotic biosynthesis by DraR-K, a novel two-component system in Streptomyces coelicolor. Mol. Microbiol. 85:535–556. 10.1111/j.1365-2958.2012.08126.x [DOI] [PubMed] [Google Scholar]
- 53.Higo A, Hara H, Horinouchi S, Ohnishi Y. 2012. Genome-wide distribution of AdpA, a global regulator for secondary metabolism and morphological differentiation in Streptomyces, revealed the extent and complexity of the AdpA regulatory network. DNA Res. 19:259–274. 10.1093/dnares/dss010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang W, Shu D, Chen L, Jiang W, Lu Y. 2009. Cross-talk between an orphan response regulator and a noncognate histidine kinase in Streptomyces coelicolor. FEMS Microbiol. Lett. 294:150–156. 10.1111/j.1574-6968.2009.01563.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.