

Abstract
Despite the public health challenges associated with the emergence of new pathogenic bacterial strains and/or serotypes, there is a dearth of information regarding the molecular mechanisms that drive this variation. Here, we began to address the mechanisms behind serotype-specific variation between serotype M1 and M3 strains of the human pathogen Streptococcus pyogenes (the group A Streptococcus [GAS]). Spatially diverse contemporary clinical serotype M3 isolates were discovered to contain identical inactivating mutations within genes encoding two regulatory systems that control the expression of important virulence factors, including the thrombolytic agent streptokinase, the protease inhibitor-binding protein-G-related α2-macroglobulin-binding (GRAB) protein, and the antiphagocytic hyaluronic acid capsule. Subsequent analysis of a larger collection of isolates determined that M3 GAS, since at least the 1920s, has harbored a 4-bp deletion in the fasC gene of the fasBCAX regulatory system and an inactivating polymorphism in the rivR regulator-encoding gene. The fasC and rivR mutations in M3 isolates directly affect the virulence factor profile of M3 GAS, as evident by a reduction in streptokinase expression and an enhancement of GRAB expression. Complementation of the fasC mutation in M3 GAS significantly enhanced levels of the small regulatory RNA FasX, which in turn enhanced streptokinase expression. Complementation of the rivR mutation in M3 GAS restored the regulation of grab mRNA abundance but did not alter capsule mRNA levels. While important, the fasC and rivR mutations do not provide a full explanation for why serotype M3 strains are associated with unusually severe invasive infections; thus, further investigation is warranted.
INTRODUCTION
Streptococcus pyogenes (the group A Streptococcus [GAS]) is a human-specific pathogen that causes diseases ranging from pharyngitis or impetigo to a toxic-shock-like syndrome or necrotizing fasciitis (1). GAS strains can be divided into more than 150 serotypes based upon the sequence of the 5′ end of the emm (M protein-encoding) gene (2). Interestingly, for more than half a century it has been known that some GAS serotypes show nonrandom associations with certain disease manifestations (3, 4). For example, M1 strains are the most common serotype isolated in the majority of pharyngeal and invasive case studies (5, 6), M3 strains are associated with unusually severe invasive infections and a high mortality rate (7), and M18 strains are associated with outbreaks of acute rheumatic fever (8). The molecular mechanisms behind GAS serotype disease-phenotype associations are unknown but potentially could be explained by serotype-specific differences in gene content or by differences in the regulation of virulence factor-encoding genes that are common to all serotypes.
No single GAS virulence factor is sufficient to cause any particular disease; rather, the disease potential of GAS is attributable to the coordinated expression of specific subsets of virulence factors (9–12). To this end, GAS uses 13 two-component systems (13), more than 60 “stand-alone” transcriptional regulators (14), and an estimated 40 small regulatory RNAs (sRNAs) (15–17) to regulate gene expression. We and others have described intraserotype variation in GAS regulatory networks (12, 14, 18, 19). For example, in a study of 96 serotype M3 strains, the genes encoding the CovR/S two-component system and the RopB regulator contained statistically significantly more genetic alterations (single nucleotide polymorphisms [SNPs], insertions, deletions) than the genome average, which is indicative of selective pressure acting upon these genes (20). The consequences of covR/S or ropB mutation are an alteration of the virulence factor profile and subsequently an alteration of virulence (21–27). Less is known about the interserotype variation in GAS regulatory networks. However, in one example, serotypes can differ based upon which of two orthologous genes, rofA or nra, is located upstream of the pilus biosynthesis locus (28, 29). While the RofA and Nra proteins are both positive transcriptional regulators of the pilus locus, they are not functionally identical, as their degrees of activation differ (30).
RivR (RofA-like protein IV) is a member of the RofA-like family of regulatory proteins that are conserved across streptococcal species (31, 32). The rivR gene is located upstream of a putative sRNA-encoding gene, rivX. Both RivR and RivX were initially reported to positively regulate the abundance of key virulence factor-encoding mRNAs, including emm (33). However, more recently, we published data showing that the putative rivX gene has no regulatory activity while RivR is a negative regulator of GAS virulence factors (34). RivR-regulated genes include the hyaluronic acid synthesis (hasABC) operon, required for biosynthesis of the antiphagocytic capsule (35), and the protein-G-related α2-macroglobulin-binding (GRAB) protein-encoding gene (grab), required for expression of the GRAB protein, which binds the human protease inhibitor α2-macroglobulin and regulates proteolysis at the GAS cell surface (36).
The fibronectin/fibrinogen-binding/hemolytic activity/streptokinase (SKA) regulatory (Fas) system is encoded by a four-gene locus that encodes three putative proteins (FasBCA) and an sRNA (FasX) (37). The Fas proteins share homology with those of two-component systems, with FasB and FasC having similarity to sensor kinases and FasA having similarity to response regulators. Data indicate that (i) all three Fas proteins are required for activation of fasX transcription, (ii) the regulatory activity of the Fas system occurs through the function of FasX, (iii) FasX enhances the production of the plasminogen activator SKA, and (iv) FasX reduces the abundance of pili on the GAS cell surface (37–39). The reduced expression of adhesins and increased expression of factors that aid bacterial spreading are reminiscent of the role of the Staphylococcus aureus accessory gene regulator (Agr) system (40). Thus, we propose that the Fas system, similar to Agr, plays a central role in the ability of GAS to transition from the colonization to the dissemination phase of infection.
Here, we compared serotype M1 and M3 GAS strains to determine what distinguishes the hypervirulent M3 serotype from other GAS serotypes. We found that M3 isolates harbor inactivating mutations in the rivR and fasC regulatory genes. The deletions within rivR and fasC arose in the M3 population over 80 years ago and have been maintained to the present day. The regulatory consequences of these deletions include alterations of SKA (as a consequence of fasC mutation) and GRAB (as a consequence of rivR mutation) expression. While M3 GAS also produces larger capsules than M1 GAS, this phenotype was not found to be attributable to the rivR mutation, despite the negative regulation of capsule expression by RivR in M1 GAS (34). Insight into the molecular mechanisms controlling serotype emergence in this important human pathogen may enhance public health by facilitating the development of therapeutic approaches targeting conserved regulatory pathways.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
For the clinical GAS isolates used in this study, see Table S2 in the supplemental material. The laboratory-created GAS strains used in this study are listed in Table 1. Routine growth of GAS cultures made use of Todd-Hewitt broth with 0.2% yeast extract (THY broth), and cultures were incubated at 37°C (5% CO2). Chloramphenicol (4 μg/ml) and/or spectinomycin (150 μg/ml) were added when required.
TABLE 1.
Laboratory-constructed GAS strains used in this study
| Strain | Serotype | Information | Reference |
|---|---|---|---|
| 2221ΔfasC | M1 | Derivative of clinical isolate MGAS2221 in which the fasC gene has been replaced with a spectinomycin resistance cassette | This work |
| 2221ΔrivR | M1 | Derivative of clinical isolate MGAS2221 in which the rivR gene has been deleted via homologous recombination | 34 |
| 10870fasCComp | M3 | Derivative of clinical isolate MGAS10870 in which the 4-bp deletion naturally present in the fasC gene has been fixed via homologous recombination | This work |
| 10870ΔrivR | M3 | Derivative of clinical isolate MGAS10870 in which the rivR gene has been deleted via homologous recombination | This work |
| 10870rivR+1bp | M3 | Derivative of clinical isolate MGAS10870 in which the 1-bp deletion naturally present in the rivR gene has been fixed via homologous recombination | This work |
| 10870rivR+1bp fasCComp | M3 | Derivative of 10870fasCComp in which the 1-bp deletion naturally present in the rivR gene has been fixed via homologous recombination (hence has wild-type fasC and rivR alleles) | This work |
| 315fasCComp | M3 | Derivative of clinical isolate MGAS315 in which the mutation naturally present in fasC has been fixed via homologous recombination | This work |
| 315fasCMUT | M3 | Derivative of 315fasCComp in which the fasC gene has been remutated via homologous recombination to look like that present in clinical M3 isolates | This work |
| 2221 + vector | M1 | Derivative of clinical isolate MGAS2221 containing an empty derivative of shuttle vector pDC123 | This work |
| 2221 + pRivR-M1 | M1 | Derivative of clinical isolate MGAS2221 containing the pDC123 derivative pM1rivR, which contains the rivR gene from M1 GAS strain MGAS2221 | This work |
| 2221 + pRivR-M3 | M1 | Derivative of clinical isolate MGAS2221 containing the pDC123 derivative pM3rivR, which contains the rivR gene from M3 GAS strain MGAS10870 | This work |
| 2221ΔrivR + vector | M1 | Derivative of isolate 2221ΔrivR containing an empty derivative of shuttle vector pDC123 | This work |
| 2221ΔrivR + pRivR-M1 | M1 | Derivative of isolate 2221ΔrivR containing the pDC123 derivative pM1rivR, which contains the rivR gene from M1 GAS strain MGAS2221 | This work |
| 2221ΔrivR + pRivR-M3 | M1 | Derivative of isolate 2221ΔrivR containing the pDC123 derivative pM3rivR, which contains the rivR gene from M3 GAS strain MGAS10870 | This work |
| 10870 + vector | M3 | Derivative of clinical isolate MGAS10870 containing an empty derivative of shuttle vector pDC123 | This work |
| 10870 + pRivR-M1 | M3 | Derivative of clinical isolate MGAS10870 containing the pDC123 derivative pM1rivR, which contains the rivR gene from M1 GAS strain MGAS2221 | This work |
| 10870 + pRivR-M3 | M3 | Derivative of clinical isolate MGAS10870 containing the pDC123 derivative pM3rivR, which contains the rivR gene from M3 GAS strain MGAS10870 | This work |
| 10870ΔrivR + vector | M3 | Derivative of isolate 10870ΔrivR containing an empty derivative of shuttle vector pDC123 | This work |
| 10870ΔrivR + pRivR-M1 | M3 | Derivative of isolate 10870ΔrivR containing the pDC123 derivative pM1rivR, which contains the rivR gene from M1 GAS strain MGAS2221 | This work |
| 10870ΔrivR + pRivR-M3 | M3 | Derivative of isolate 10870ΔrivR containing the pDC123 derivative pM3rivR, which contains the rivR gene from M3 GAS strain MGAS10870 | This work |
| 10870 + pFasX | M3 | Derivative of clinical isolate MGAS10870 containing the pDC123 derivative pFasX, which expresses the wild-type fasX gene | This work |
| 315 + vector | M3 | Derivative of clinical isolate MGAS315 containing an empty derivative of shuttle vector pDC123 | This work |
| 315 + pFasX | M3 | Derivative of clinical isolate MGAS315 containing the pDC123 derivative pFasX, which expresses the wild-type fasX gene | This work |
Generation of sequencing data.
The previously determined MGAS315 genome sequence was used as the reference serotype M3 GAS sequence in our studies (41). Internal regions of the fasC and rivR genes containing the identified 4- and 1-bp deletions, respectively, were amplified from 125 serotype M3 GAS stains and 7 serotype M1 strains and sequenced via Sanger sequencing. The GAS strains used were taken from the personal collection of James M. Musser (The Houston Methodist Research Institute). The full rivR gene was also sequenced in 10 M3 strains. For the primers used, see Table S1 in the supplemental material. Generated sequences were compared to the reference sequence by using Sequencher software (Gene Codes Corp.).
Total RNA isolation from GAS.
GAS strains were grown in THY broth to mid-exponential phase, corresponding to an optical density at 600 nm of 0.5. One volume of GAS culture was added to 2 volumes of RNAprotect (Qiagen) and incubated at room temperature for 5 min before centrifugation for 10 min at 4,000 × g. The supernatant was removed, and the pellets were quick frozen in liquid nitrogen and stored at −80°C until ready for use. RNA was isolated from each GAS cell pellet by a mechanical lysis method in conjunction with the miRNeasy kit (Qiagen) (42). Contaminating DNA was removed with TURBO DNase-free (Life Technologies). The quality and quantity of isolated RNA was analyzed with a Bioanalyzer 2100 system (Agilent Technologies).
Northern blot analysis.
Total RNA from exponential-phase GAS cultures was loaded onto a 5% Tris-borate-EDTA-urea gel and separated by electrophoresis. Biotinylated RNA size standards ranging from 100 to 1,000 nucleotides were used to determine the sizes of detected transcripts. RNA was transferred from the gel to nylon membrane via electroblotting, UV cross-linked, and probed overnight with an in vitro-transcribed probe complementary to the FasX sRNA. In vitro-transcribed probes were generated with the Strip-EZ T7 kit (Life Technologies). DNA templates for in vitro transcription reactions were generated by PCR with one primer containing the T7 promoter sequence (see Table S1 in the supplemental material). RNA probes were labeled with biotin prior to hybridization (BrightStar Psoralen-Biotin labeling kit; Life Technologies). After washing, Northern blot assays were developed (BrightStar BioDetect kit; Life Technologies) and exposed to autoradiography film. As loading controls, Northern blot assays were stripped and reprobed with a 5S RNA-specific probe. Note that the probe sequences were selected such that they were fully complementary to FasX and the 5S RNA of both M1 and M3 isolates and hence could be used to compare RNA abundance between serotypes.
Hyaluronic acid capsule assays.
Hyaluronic acid capsule assays were performed by a previously described protocol (34).
Construction of a fasC mutant derivative of serotype M1 isolate MGAS2221.
Strain 2221ΔfasC was created by replacing the fasC gene with a spectinomycin resistance cassette. To replace the fasC gene, 1-kb regions upstream and downstream of fasC were amplified and a spectinomycin resistance cassette was inserted between them via overlap extension PCR. For the PCR primers used, see Table S1 in the supplemental material. The resultant 3-kb PCR product (1-kb left flank, 1-kb spectinomycin resistance cassette, 1-kb right flank) was transformed into competent MGAS2221 cells, which were then plated on THY agar plates containing spectinomycin. Replacement of the fasC gene with the spectinomycin resistance cassette was confirmed via PCR and sequencing.
Construction of fasC- and/or rivR-complemented derivatives of serotype M3 isolates MGAS315 and MGAS10870.
Complementation of the naturally occurring fasC and rivR deletions in M3 GAS isolates was performed by allelic exchange with the suicide vector pBBL740 via a previously described protocol (38). Plasmid pBBL740 was constructed in the laboratory of Benfang Lei (Montana State University) (unpublished data) and is a derivative of plasmid pFW14 in which the Cmr-encoding gene was replaced with the cat gene from plasmid pSET5s (43–45). To briefly describe the complementation protocol, using creation of the fasC-complemented strain 10870fasCComp as an example, 1-kb regions flanking either side of the fasC mutation were amplified by PCR, joined via overlap extension PCR, and cloned into pBBL740. For the PCR primers used, see Table S1 in the supplemental material. The primers located adjacent to the fasC mutation were constructed such that the 2-kb joined PCR product contained the 4 bp missing from contemporary M3 GAS. The resultant plasmid was transformed into MGAS10870, and colonies were selected on THY agar plates containing chloramphenicol (these transformants have the plasmid integrated into the chromosome). To promote excision of the plasmid from the chromosome, which can either leave the mutant (parental) or wild-type (complemented) fasC gene behind, we serially passaged the strain in THY broth without antibiotics. Two 5-h passages, followed by a 15-h passage and two more 5-h passages, were performed (1/100 dilutions of the old culture into the new culture were performed for each of the 5-h passages, with a 1/1,000 dilution for the 15-h passage). The final culture was diluted and plated onto blood agar plates to gain single colonies. Colonies were patched onto THY agar plates, one containing chloramphenicol and one containing no antibiotic. Strains that grew on the THY plate, but not the THY-chloramphenicol plate, were analyzed by PCR and sequencing to identify whether they contained a wild-type or mutant fasC allele.
Remutation of the fasC gene in complemented strain 315fasCComp.
To create strain 315fasCMUT from strain 315fasCComp, we first cloned the mutant fasC allele from MGAS315 into pBBL740. This plasmid was then used to replace the wild-type fasC allele in 315fasCComp with the mutant allele by the same protocol as described above.
Deletion of the rivR gene from serotype M3 isolate MGAS10870.
To create strain 10870ΔrivR, we first performed PCRs to amplify 1-kb regions upstream and downstream of the rivR gene. The two PCR products were joined together via overlap extension PCR and cloned into pBBL740. This plasmid was then used to delete the rivR gene from the MGAS10870 genome by the same protocol as described above.
Creation of RivR-expressing plasmids pRivR-M1 and pRivR-M3.
The rivR genes of MGAS2221 and MGAS10870 were amplified by PCR with primers RIVRCP1 and RIVRCP2 (see Table S1 in the supplemental material). The PCR products were doubly digested with BamHI and NsiI and then cloned into the BglII and NsiI restriction sites of the shuttle vector pDC123. The resultant plasmids were verified by PCR and sequencing. The rivR gene from MGAS2221 is in plasmid pRivR-M1. The rivR gene from MGAS10870 is in plasmid pRivR-M3.
Isolation of secreted protein fractions.
Supernatant proteins from exponential-phase THY broth GAS cultures were concentrated by ethanol precipitation and resuspended in SDS-PAGE buffer at 1/20 of the original volume.
Western blot analysis.
Protein samples were separated on 12% Tris-HCl gels. Primary antibodies against SKA (sheep; Novus Biologicals), streptolysin O (SLO; rabbit; American Research Products), or GRAB (rabbit; custom made by Pacific Immunology) (34) were used at a 1:1,000 dilution. After washing, goat anti-rabbit or rabbit anti-sheep secondary antibodies (horseradish peroxidase conjugated; Thermo Scientific) were used at a 1:10,000 dilution and a signal was generated with the SuperSignal West Pico kit (Thermo Scientific).
qRT-PCR analysis.
To determine relative mRNA abundance, we used quantitative reverse transcription (qRT)-PCR with TaqMan primers and probes. In each experiment, RNA samples from duplicate cultures of the GAS strains to be analyzed were converted into cDNA and used in association with an ABI 7500 Fast System (Life Technologies). For the TaqMan primers and probes for genes of interest and the internal control gene proS, see Table S1 in the supplemental material. Each experiment was performed in triplicate, and mean values ± standard deviations are shown.
RESULTS
Serotype M3 GAS isolates express FasX in lower abundance than M1 isolates.
Previously, we determined that serotype M3 isolate MGAS315 produced FasX sRNA in lower abundance than nine serotype M1 isolates (16). To test whether the low FasX sRNA level of MGAS315 was characteristic of M3 GAS isolates in general, we analyzed FasX expression in four additional isolates via Northern blotting (Fig. 1A). The four additional M3 isolates were isolated from different countries over a 20-year time span (see Table S2 in the supplemental material). The data are consistent with M3 GAS isolates maintaining the FasX sRNA in lower abundance than M1 isolates.
FIG 1.
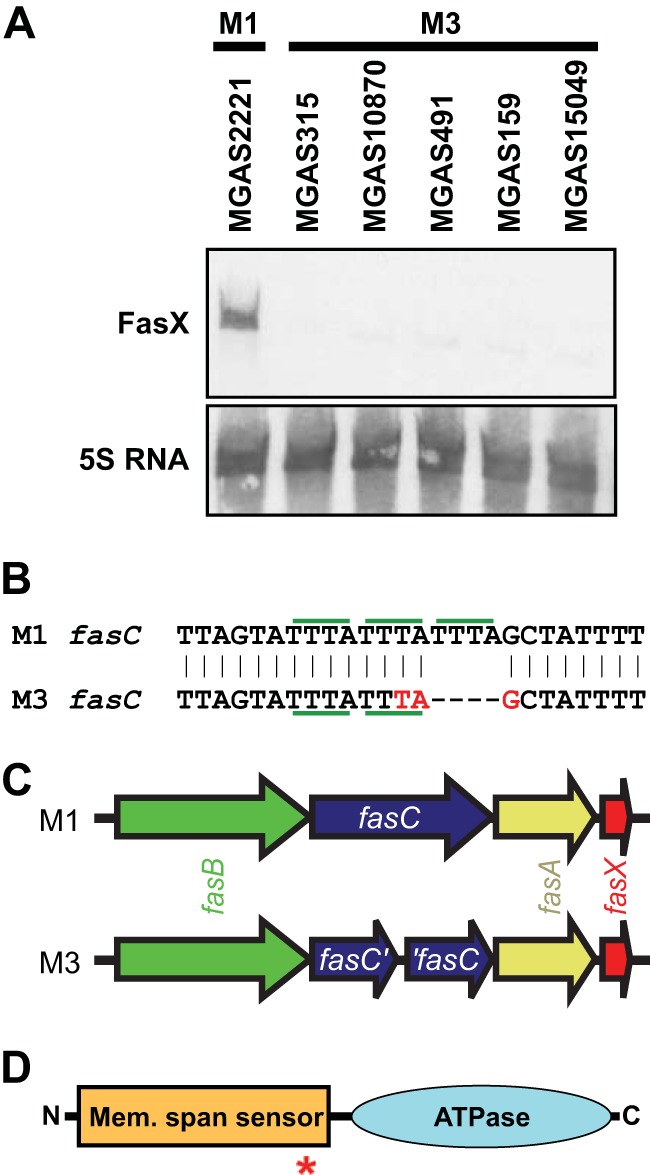
Serotype M3 GAS isolates harbor a 4-bp deletion in fasC leading to a truncated protein. (A) Northern blot analysis of FasX sRNA levels in five representative serotype M3 GAS isolates relative to those in M1 isolate MGAS2221. The 5S RNA was used as a loading control. (B) Schematic of a region of the fasC gene in M1 GAS and also of the corresponding region, which contains a 4-bp deletion, in M3 GAS. Individual tetranucleotide repeats (TTTA) are highlighted with horizontal green bars. The premature stop codon introduced into the fasC gene of serotype M3 GAS by the removal of one tetranucleotide repeat is shown in red (TAG). (C) Comparison of the fas loci between serotype M1 and M3 GAS isolates. Note the truncated fasC gene in M3 GAS. (D) Domain structure of the FasC protein. The N-terminal half of FasC contains multiple putative membrane-spanning regions (Mem. span sensor). The C-terminal half of FasC contains a putative ATPase domain. The red asterisk shows the relative location of the truncation in serotype M3 isolates.
Serotype M3 GAS isolates have harbored a 4-bp deletion in the fasC gene since at least the 1920s.
In serotype M1 GAS strains, the fasBCA genes are all required for high-level FasX transcription (37; P. Sumby, unpublished data). This led us to analyze the fas locus of M3 GAS isolates for mutations that may account for the low FasX levels observed in this serotype. We identified a 4-bp deletion in the middle of the fasC gene in M3 isolate MGAS315. The deleted nucleotides formed a tetranucleotide repeat that is present in three copies in M1 GAS but only two copies in M3 GAS (Fig. 1B). The fasC deletion in M3 GAS introduces a premature stop codon, fragmenting the gene in half (Fig. 1B and C). Given the location of the deletion within fasC, if proteins were translated from the truncated genes, they would not be expected to be active because of the separation of functional domains (Fig. 1D). To identify whether the fasC deletion of MGAS315 was present in additional M3 isolates, we analyzed this gene from 125 isolates that were recovered in a temporally (1920s to 2010) and spatially (Europe, North America, Japan, and Russia) diverse manner. All 125 M3 isolates, but none of 9 M1 isolates, harbored the same 4-bp deletion. A subset of the isolates is shown in Table 2; for all 125 isolates, see Table S2 in the supplemental material. Thus, for more than the last 80 years, a significant number of, if not all, serotype M3 GAS isolates causing disease appear to have been fasC mutants.
TABLE 2.
Subset of the serotype M3 GAS isolates analyzed in this study for fasC and rivR allele status
| Strain | Isolation yr | Isolation location | fasC allele | rivR allele |
|---|---|---|---|---|
| MGAS1251 | 1920s | United Kingdom | 4-bp deletion | SNPs |
| MGAS1254 | 1937 | New York | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS182 | 1940s | Ottawa, Canada | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS1372 | 1969 | Berlin, Germany | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS1428 | 1974 | Cottbus, Germany | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS315 | 1980s | Texas | 4-bp deletion | 1-bp deletion and SNPs |
| SSI-1 | 1994 | Japan | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS9056 | 1998 | Illinois | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS10870 | 2002 | Ontario, Canada | 4-bp deletion | 1-bp deletion and SNPs |
| MGAS22440 | 2010 | Alberta, Canada | 4-bp deletion | 1-bp deletion and SNPs |
The fasC mutation in serotype M3 GAS is responsible for the low abundance of FasX sRNA observed in this serotype.
We reasoned that the disruption of the fasC gene by the 4-bp deletion seen in serotype M3 isolates was responsible for the low level of FasX produced by M3 GAS relative to that produced by M1 GAS (Fig. 1A). To test this, we used allelic exchange to complement the fasC gene in two M3 isolates, MGAS315 and MGAS10870, creating strains 315fasCComp and 10870fasCComp. To ensure that differences between our parental and complemented strains were a consequence of the fasC alleles and not spurious mutations, we also reintroduced the 4-bp deletion into the fasC gene of strain 315fasCComp, creating strain 315fasCMUT. Using Northern blot analysis to compare FasX sRNA abundance, we found that the complemented strains produced FasX at levels similar to that observed in M1 GAS (Fig. 2A). Furthermore, remutation of fasC returned M3 GAS to low FasX expression status. The data are consistent with the idea that M3 isolates have low FasX sRNA levels because they harbor a mutant fasC gene.
FIG 2.
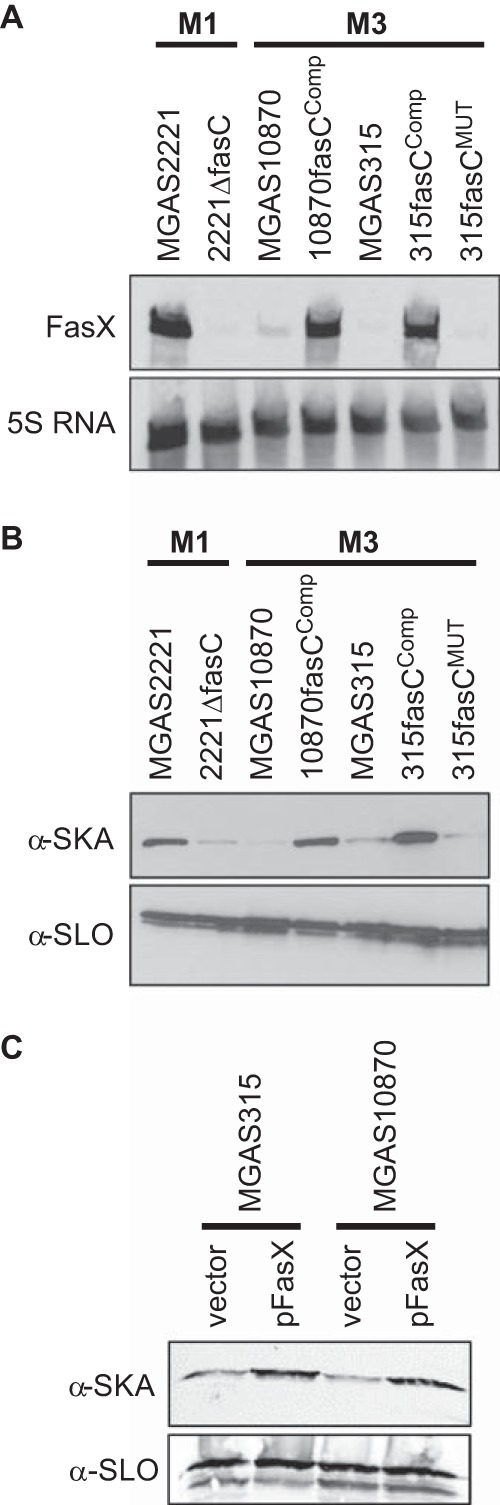
The fasC mutation in serotype M3 GAS isolates is responsible for the low FasX sRNA levels, and subsequently the low level of SKA expression, that are characteristic of this serotype. Parental (MGAS2221) and fasC deletion mutant (2221ΔfasC) M1 isolates, parental (MGAS10870 and MGAS315) and fasC-complemented (10870fasCComp and 315fasCComp) M3 isolates, and a 315fasCComp derivative in which the fasC gene was remutated (315fasCMUT) were compared. (A) Northern blot analysis of FasX sRNA levels. RNA samples from exponential-phase cultures of the indicated strains were analyzed with FasX and 5S RNA-specific probes. The 5S RNA was used as a loading control. (B, C) Western blot analyses of SKA and SLO protein levels. Secreted proteins were isolated from exponential-phase cultures of the indicated strains and interrogated with antibodies against SKA and SLO. The SLO antibody was used as a loading control.
By reducing FasX sRNA abundance, the fasC deletion in M3 GAS results in decreased SKA expression.
FasX positively regulates SKA expression and negatively regulates pilus expression in M1 GAS (37–39). We hypothesized that FasX also regulates these virulence factors in M3 GAS and therefore that the reduced FasX expression in M3 GAS clinical isolates alters their virulence factor profile relative to that of other GAS isolates that have a functional Fas system, for example, M1 GAS. To test our hypothesis, we performed Western blot analysis of SKA expression. Relative to their fasC-complemented derivatives, representative clinical M3 isolates MGAS315 and MGAS10870 produced significantly lower levels of SKA (Fig. 2B). To find out whether the enhanced SKA expression of the fasC-complemented strains was a consequence of greater FasX expression and not some undefined regulatory pathway, we tested whether increased FasX expression alone could enhance SKA expression. The FasX complementation plasmid pFasX was introduced into M3 isolates MGAS315 and MGAS10870, and their SKA expression was compared with that of the same isolates containing the empty vector. Similar to fasC complementation, the presence of plasmid pFasX enhanced SKA expression (Fig. 2C). Our data are consistent with the fasC mutation in M3 isolates contributing to the specific virulence factor profile of this hypervirulent serotype by reducing FasX sRNA expression.
The hyaluronic acid capsule of serotype M3 GAS isolates is an order of magnitude larger than that produced by most serotype M1 isolates.
While working with the serotype M1 and M3 strains, it became apparent that the M3 isolates had mucoid colony morphologies on blood agar plates while the majority of the M1 isolates did not (data not shown). To assess whether the differences in colony morphology were a consequence of greater capsule expression by M3 isolates than by M1 isolates, we determined hyaluronic acid capsule levels. Seven M1 and eight M3 isolates were compared. These isolates were chosen so as to include representatives from different locations (e.g., Europe, North America) and years of isolation (from the 1940s to 2010). In addition, only isolates harboring wild-type covRS genes were selected to prevent this negative regulator of capsule expression from confounding the data (12, 25, 26). In all cases, the M3 isolates produced significantly greater capsule levels than the M1 isolates (Fig. 3).
FIG 3.
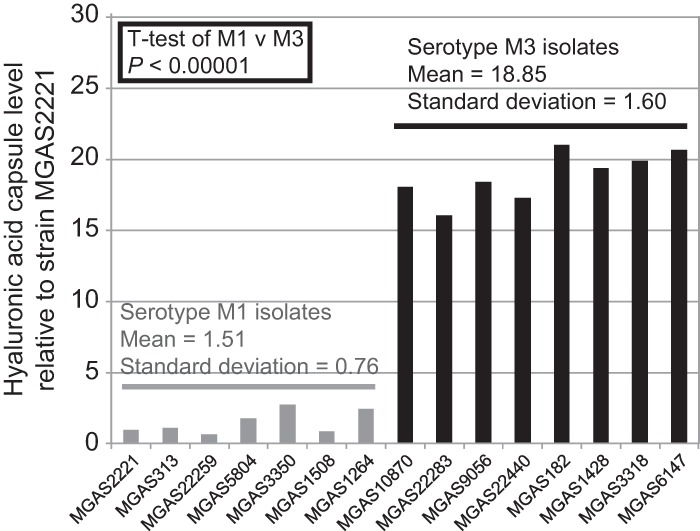
Serotype M3 GAS isolates produce increased levels of hyaluronic acid capsule relative to M1 isolates. Seven serotype M1 (gray bars) and eight serotype M3 (black bars) GAS isolates were compared for hyaluronic acid capsule production.
Serotype M3 GAS isolates since the 1930s have harbored a 1-bp deletion in the rivR gene.
In an attempt to explain the disparity in capsule expression between M1 and M3 GAS strains, we analyzed known capsule regulator genes in the genome of M3 GAS strain MGAS315. The rivR gene, which in M1 GAS strains encodes a negative regulator of capsule expression (34), was found to contain a 1-bp deletion relative to that of M1 GAS (Fig. 4A and B). The 1-bp deletion occurred in a homopolymeric tract of seven T nucleotides, which leads to a premature stop codon occurring early in the gene. As the stop codon occurs within a region of the gene encoding a putative DNA-binding domain (Fig. 4C), neither of the two rivR gene fragments is expected to encode a functional protein. To test whether the same rivR deletion is present in additional M3 GAS isolates, we analyzed this gene in the same 125 isolates used previously in the fasC analysis. All but 1 of the 125 M3 isolates harbored the same 1-bp deletion in rivR (Table 1; see Table S3 in the supplemental material). The single M3 strain not containing the 1-bp deletion was the oldest isolate in our collection and was isolated in the United Kingdom in the 1920s (Table 2). Thus, the data are consistent with M3 isolates developing a 1-bp deletion in rivR during the 1920s or 1930s and maintaining it to the present day.
FIG 4.
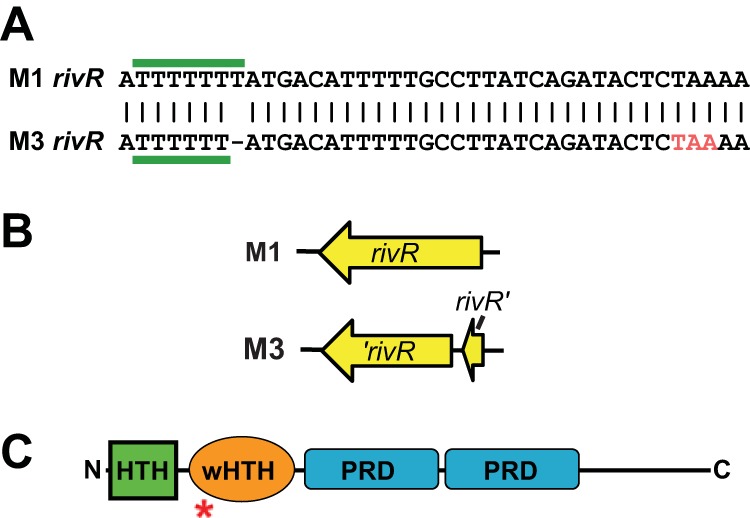
Serotype M3 GAS isolates harbor a 1-bp deletion in rivR that results in a truncated protein. (A) Schematic of a region of the rivR gene in M1 GAS and also of the corresponding region, which contains a 1-bp deletion, in M3 GAS. The homopolymeric tract that contains the 1-bp deletion in M3 GAS is highlighted with horizontal green bars. The premature stop codon introduced into the rivR gene of serotype M3 GAS by the 1-bp deletion is shown in red (TAA). (B) Comparison of the rivR open reading frames in M1 and M3 GAS isolates. Note the truncated rivR gene in M3 GAS. (C) Domain structure of the RivR protein. The locations of putative helix-turn-helix (HTH; green), winged helix-turn-helix (wHTH; orange), and phosphotransferase system regulatory (PRD; blue) domains are shown. The red asterisk shows the relative location of the truncation in serotype M3 isolates.
Reinsertion of the 1 bp deleted from the rivR gene of M3 GAS isolates does not restore the regulation of grab or hasA mRNA abundance.
As RivR is a negative regulator of capsule expression in M1 GAS, we hypothesized that the high-level capsule expression observed in serotype M3 isolates (Fig. 3) was a consequence of the 1-bp deletion in rivR. To test our hypothesis, we reinserted the deleted base pair into the rivR gene of M3 isolate MGAS10870 and compared this strain (10870rivR+1bp) to the parental strain, to the fasC-complemented derivative 10870fasCComp, to the doubly modified derivative 10870rivR+1bpfasCComp, and to the full rivR deletion mutant strain 10870ΔrivR by qRT-PCR. All five strains tested produced hasA mRNA at similar levels (Fig. 5); thus, the 1-bp deletion in rivR is not necessary, at least at the level of hasA mRNA abundance, for the high-level capsule expression observed in M3 GAS. Similar results were obtained with grab mRNA, which is also negatively regulated by RivR in M1 GAS (Fig. 5) (34). Note that while we observed no differences in mRNA abundance in the rivR+1bp strains, we did observe an enhancement of ska mRNA in the fasC-complemented strains, an expected result given the data in Fig. 2B.
FIG 5.
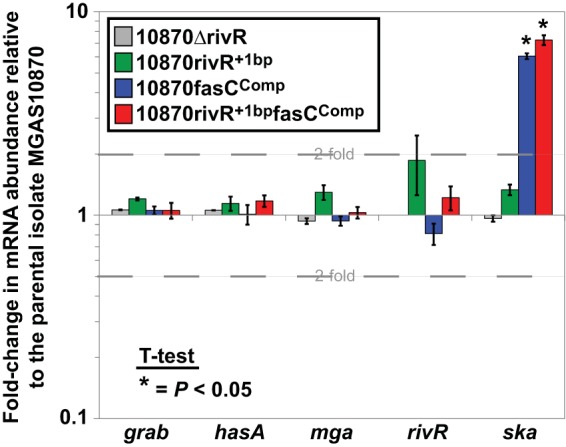
Repair of the 1-bp deletion in the rivR gene of M3 GAS isolate MGAS10870 has no effect on the abundance of hasA or grab mRNA. Parental strain MGAS10870, complete rivR deletion mutant derivative 10870ΔrivR, the rivR-complemented (via a 1-bp insertion) derivative 10870rivRComp, the fasC-complemented (via a 4-bp insertion) derivative 10870fasCComp, and doubly complemented strain 10870rivRCompfasCComp were compared by qRT-PCR. RNAs from exponential-phase cultures of the indicated strains were analyzed for grab, hasA, mga, rivR, and ska mRNA levels. The experiment was performed in triplicate, and means ± standard deviations are shown.
A single nucleotide polymorphism, in addition to the 1-bp deletion, inactivates rivR in serotype M3 GAS strain MGAS10870.
That restoring the 1 bp deleted from the rivR gene of M3 isolate MGAS10870 did not lead to an alteration of grab or hasA mRNA levels, given that they are negatively regulated by RivR in M1 GAS, was unexpected. We hypothesized that this was a consequence of there being rivR mutations in addition to the 1-bp deletion that results in a null mutant allele in M3 GAS. The DNA and amino acid sequences of rivR/RivR from M3 strain 10870rivR+1bp were aligned with those from M1 strain MGAS2221 (see Fig. S1 and S2 in the supplemental material). In addition to the 1-bp deletion, there were four SNPs that distinguished the M1 and M3 GAS rivR alleles. Three of the four SNPs were nonsynonymous, with two of the three amino acid changes being conservative (K to N and K to E) and one being nonconservative (L to P). Proline amino acid substitutions are particularly adept at disrupting protein activity, in part due to the bulky ring structure of this amino acid. To identify whether any of the three nonsynonymous SNPs inactivate RivR activity, we created derivatives of plasmid pRivR-M1, which carries the functional rivR allele from M1 GAS, into which individual M3 rivR SNPs were introduced (creating plasmids pRivR-SNP1 through pRivR-SNP3). To measure the ability of the RivR-SNP plasmids to complement, we introduced them, along with parental M1 and M3 rivR alleles, into strains 2221ΔrivR (a rivR mutant of the M1 clinical isolate MGAS2221) and MGAS10870 (a clinical M3 isolate). In both strain backgrounds, the pRivR-SNP2 and pRivR-SNP3 plasmids complemented as well as the original M1 rivR allele (pRivR-M1), while pRivR-SNP1 failed to complement, similar to pRivR-M3 or the empty vector (Fig. 6). Thus, the leucine-to-proline amino acid change that occurs because of the presence of SNP1 inactivates the activity of the resultant RivR protein.
FIG 6.
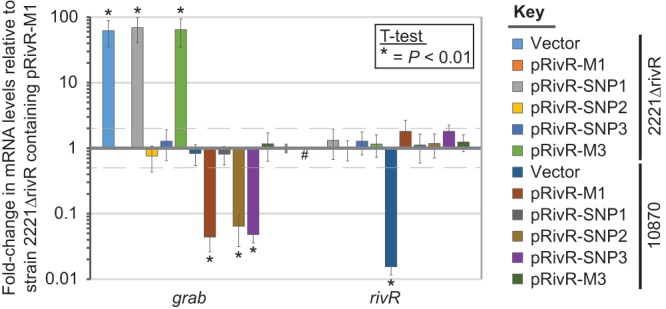
SNP1, but not SNP2 or SNP3, from the rivR gene of serotype M3 isolates results in an inactive RivR protein. Derivatives of serotype M1 strain 2221ΔrivR and serotype M3 clinical isolate MGAS10870 were analyzed by qRT-PCR for grab and rivR mRNA abundance. Derivatives contained either the empty vector or one of five different plasmid-carried rivR alleles. Plasmid pRivR-M1 carries rivR from M1 GAS, plasmid pRivR-M3 carries rivR from M3 GAS, while plasmids pRivR-SNP1 through pRivR-SNP3 are derivatives of pRivR-M1 into which individual SNPs from the M3 rivR allele have been introduced. Analysis of rivR mRNA was performed to ensure that all of the pRivR plasmids transcribed rivR mRNA at similar concentrations. The experiment was performed in triplicate, and means ± standard deviations are shown. Samples in which no transcript was detected are represented by hashtags (#). Asterisks (*) highlight those samples that were statistically significantly different from strain 2221ΔrivR/pRivR-M1 via t test.
M3 isolates going back to at least the 1920s have harbored the same rivR SNPs as MGAS10870.
All but our oldest M3 isolate tested, MGAS1251 from the 1920s, contain the 1-bp deletion in rivR (Table 2). While MGAS1251 lacks a 1-bp deletion, it is possible that this strain is also a rivR mutant, as it may contain the inactivating SNP1 mutation, a possibility that is consistent with the observation that this strain is phenotypically similar to other M3 isolates. To test this, we fully sequenced rivR from MGAS1251 and also from a subset of our other M3 isolates (MGAS491, MGAS159, MGAS15049, MGAS182, MGAS1251, MGAS1254, MGAS1428, MGAS9056, MGAS9507, and MGAS22283). All of the isolates tested contained the same four SNPs as MGAS10870 and no additional SNPs. Therefore, although MGAS1251 does not have the 1-bp deletion in the rivR gene, this strain is nevertheless a rivR mutant because of the presence of SNP1.
M1 GAS RivR regulates grab but not hasA mRNA abundance in M3 GAS, while M3 GAS RivR has dominant negative effects in M1 GAS.
Our data show that M1-RivR, but not M3-RivR, negatively regulates grab mRNA abundance in M1 and M3 GAS (Fig. 6). To test whether the regulation afforded by M1-RivR in M3 GAS extended to hasA mRNA, we performed qRT-PCR analysis. Plasmids expressing M1-RivR or M3-RivR, in addition to the empty vector, were introduced into parental MGAS2221 and MGAS10870 and full rivR deletion mutant derivatives 2221ΔrivR and 10870ΔrivR. The resultant 12 strains were compared to assay grab, hasA, mga, and rivR mRNA abundance. Analysis of mga mRNA levels was used as a negative control, and indeed, none of the plasmid-carried rivR alleles were able to regulate mga mRNA abundance (see Fig. S3 in the supplemental material). Analysis of rivR mRNA levels was performed to ensure that similar levels of transcripts were seen for the two different rivR-carrying plasmids, thus strengthening the hypothesis that any differences observed in our studies were a consequence of the protein products encoded by the rivR alleles (see Fig. S3 in the supplemental material).
The plasmid carrying the rivR gene from M1 GAS (pRivR-M1) reduced grab mRNA abundance not only in the M1 rivR deletion mutant (strain 2221ΔrivR) but also in the parental and rivR deletion mutant M3 strains (MGAS10870 and 10870ΔrivR) (Fig. 7A), consistent with M3 GAS isolates being rivR mutants but maintaining whatever other components (e.g., DNA-binding sites, cofactors) are required for functional RivR protein to regulate grab mRNA levels. In contrast, plasmid pRivR-M1 had no effect on the abundance of hasA mRNA in either M3 isolate tested, despite reducing hasA mRNA abundance in both M1 isolates, consistent with the idea that at least one component (e.g., DNA-binding site, cofactor) required for RivR to regulate hasA mRNA levels is absent from M3 GAS.
FIG 7.
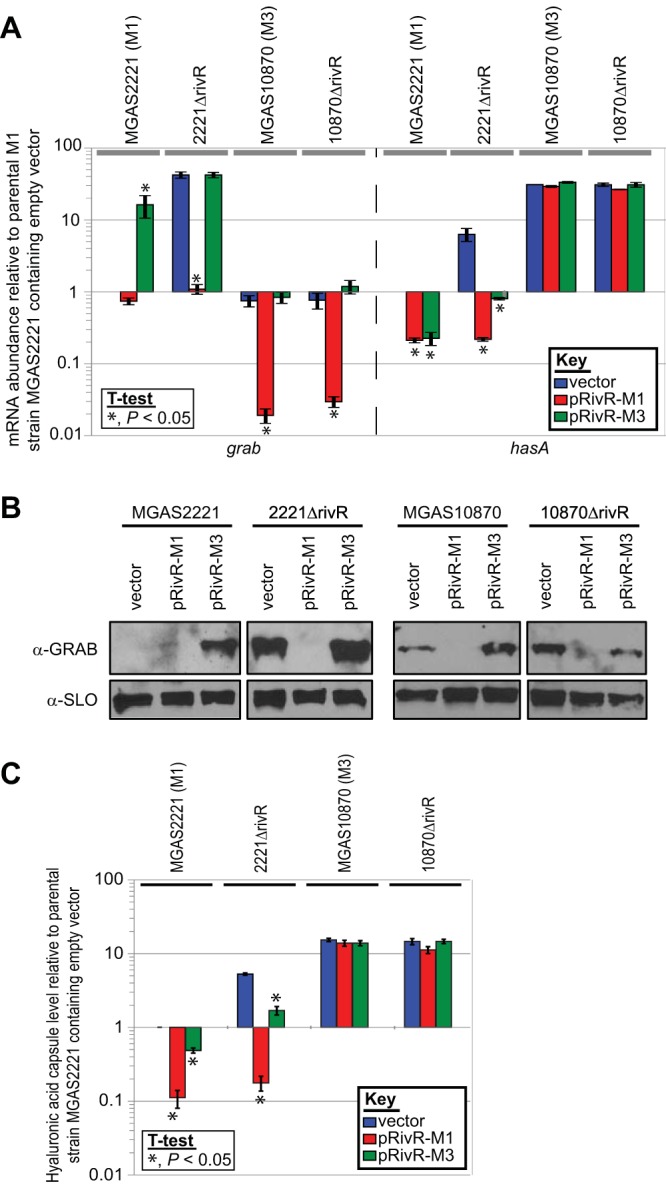
The rivR gene from M1 GAS regulates grab expression in M3 GAS, while the M3 GAS rivR gene has a dominant negative effect on RivR-mediated regulation of grab expression in M1 GAS. Parental (MGAS2221) and full rivR deletion mutant (2221ΔrivR) M1 isolates and parental (MGAS10870) and full rivR deletion mutant (10870ΔrivR) M3 isolates were compared following transformation with the empty vector, a plasmid containing rivR from M1 GAS strain MGAS2221 (pRivR-M1), or a plasmid containing rivR from M3 GAS strain MGAS10870 (pRivR-M3). (A) qRT-PCR analysis. RNA from exponential-phase cultures of the indicated strains were analyzed for grab and hasA mRNA levels. The experiment was performed in triplicate, and means ± standard deviations are shown. Asterisks (*) highlight those samples that were statistically significantly different from the respective empty vector-containing strain via t test. (B) Western blot analysis. Proteins secreted by exponential-phase cultures of the indicated strains were isolated and subjected to Western blot analysis with an anti-GRAB antibody. An antibody against SLO (anti-SLO), a secreted protein that is not RivR regulated, was used as a loading control. (C) Hyaluronic acid capsule assays. Cells present in exponential-phase cultures of the indicated strains were analyzed for hyaluronic acid capsule levels. The experiment was performed twice, with duplicate cultures in each experiment, and means ± standard deviations are shown. Asterisks (*) highlight those samples that were statistically significantly different from the respective empty-vector-containing strain via t test.
The plasmids carrying the rivR gene from M3 GAS (pRivR-M3) had no effect on the abundance of grab or hasA mRNAs produced by M3 isolates MGAS10870 and 10870ΔrivR (Fig. 7A), an expected result given our previous data. However, the M3 rivR plasmid decreased the abundance of hasA in both M1 isolates and, even more surprisingly, increased the abundance of grab mRNA in the parental M1 isolate MGAS2221. With respect to the negative regulation of hasA, the data are consistent with a RivR fragment produced by M3 GAS having some ability to regulate hasA in the M1 background. With respect to the positive regulation of grab mRNA in strain MGAS2221. The data are consistent with a RivR fragment produced by M3 GAS having a dominant negative effect on the functional M1 RivR protein present in this strain. The different grab and hasA mRNA abundances in strain MGAS2221 containing pRivR-M3 are suggestive of RivR regulating these two genes via different mechanisms.
To determine whether the regulation seen at the mRNA level by the two different plasmid-contained rivR alleles resulted in changes at the protein level, we performed Western blot and hyaluronic acid capsule assays. Western analysis of GRAB expression confirmed both that the M1 rivR allele negatively regulates GRAB levels in M3 GAS and that the M3 rivR allele interferes with functioning M1 RivR in MGAS2221, resulting in a dominant negative effect (Fig. 7B). The hyaluronic acid capsule assays confirmed that capsule expression was unchanged in the M3 GAS derivatives regardless of which rivR allele was introduced and that both M1 and M3 rivR alleles could lower the level of capsule in the two M1 isolates, albeit to different degrees (pRivR-M1 > pRivR-M3; Fig. 7C).
DISCUSSION
Significant public health challenges can be associated with the emergence of new bacterial strains and/or serotypes, including the enhanced incidence of treatment failures and/or escape from preventative regimens (46–48). Here, we investigated the molecular mechanisms contributing to serotype-specific variation between serotype M1 GAS strains, which are the most common serotype isolated in the majority of pharyngeal and invasive case studies (5, 6), and serotype M3 GAS strains, which are associated with unusually severe invasive infections and a high mortality rate (7). Our data show that two regulatory genes, encoding the stand-alone transcription factor RivR and the sensor kinase FasC, are disrupted in serotype M3 isolates and that their disruption contributes to differences in virulence factor expression between M1 and M3 isolates. While the rivR and fasC mutations in M3 isolates likely contribute to the association of M3 GAS strains with severe invasive infections, they must do so in combination with other, as yet undefined, regulatory pathways, as the rivR/fasC mutations were not responsible for the high level of capsule expression seen in M3 isolates.
The FasX sRNA both positively (SKA) and negatively (pilus) regulates virulence factor expression in M1 GAS via posttranscriptional mechanisms that require sRNA-mRNA base pairing (38, 39). FasX-ska mRNA interactions lead to enhanced ska mRNA stability, while FasX-pilus mRNA interactions lead to a reduction in pilus mRNA translation. The loss of the Fas system in M3 GAS would be expected to result in a decrease in SKA levels and an increase in cell surface pili relative to M1. A fasC-dependent decrease in M3 GAS SKA expression is observed in the Western blot assay data of Fig. 2B. That pilus levels are increased in M3 GAS because of fasC mutation is expected but has not been tested, in part because of the lack of an appropriate antibody.
The activation of high-level FasX transcription in M1 GAS requires all three Fas proteins (FasBCA) to be functional (Sumby, unpublished) (37). Given that there are no amino acid differences between the FasB and FasA proteins in M1 and M3 GAS (data not shown) and that complementation of the fasC mutation in M3 GAS leads to enhanced FasX sRNA levels (Fig. 2A), we believe that the FasB and FasA proteins are functional in M3 GAS. What regulatory role, if any, FasB and FasA have in M3 GAS strains, given the absence of FasC, is unknown.
The rivR gene from post-1920s M3 isolates contains two separate inactivating mutations, SNP1 and a 1-bp deletion. It should be noted that while our complementation data are consistent with rivR SNP2 and SNP3 not affecting RivR activity, it is possible that subtle differences were missed, in part because of the high level of rivR transcription from the complementation plasmids (Fig. 6). The introduction of RivR from M1 GAS into M3 GAS resulted in the repression of grab mRNA but not repression of mRNA from the has operon (Fig. 7A). If RivR functions as a typical transcriptional repressor, then it would bind to the promoter region of its target genes to inhibit transcription. It is therefore possible that RivR binding sites have been maintained upstream of the grab gene in M3 GAS, allowing for grab repression upon the introduction of RivR from M1 GAS, but have been lost from upstream of the has operon in M3 GAS, preventing repression by RivR. In support of this possibility is the fact that the grab promoter regions are highly similar between representative M1 and M3 strains (only 3 bp differ in the upstream 250 bp; see Fig. S4A in the supplemental material), but the has promoter regions are somewhat divergent (48 bp differ in the upstream 250 bp, including a 29-bp deletion; see Fig. S4B in the supplemental material). Plans to test RivR binding to the grab and hasA promoter regions via electrophoretic mobility assays have not been carried out because of technical issues with the purification of recombinant RivR protein (data not shown).
All of our 125 M3 GAS isolates harbor the same 4-bp deletion in fasC and the same SNP1 in rivR. We favor the notion that, rather than the identical mutations arising independently on multiple occasions, these isolates all have an ancestor in common. Given the small number of older M3 isolates available for analysis, the time when this common M3 ancestor arose and spread globally can only be estimated from the data (Fig. 8). The global spread of the fasC and rivR mutant M3 strains implies that there is a selective advantage to losing the FasBCAX and RivR systems in the M3 background that is not seen in the M1 background (at least no M1 mutants have thus far been identified; see Table S2 in the supplemental material; data not shown).
FIG 8.
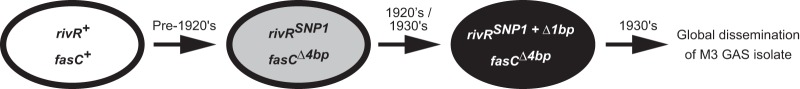
Reconstruction of the molecular evolutionary events resulting in post-1930s M3 GAS isolates. Note that the timing of each event is an approximation based upon the available data.
Loss of RivR activity through rivR mutation appears to have initially been through the gain of SNP1, as the oldest strain in our M3 collection has SNP1 but not the 1-bp deletion, while all subsequent isolates have both SNP1 and the 1-bp deletion. A question arising from this is why the 1-bp deletion has been maintained in the population, given that SNP1 already inactivates RivR activity. That all of the post-1920s M3 isolates tested have the 1-bp deletion in addition to SNP1 indicates that this 1-bp deletion was positively selected for, even in the SNP1 background. Possibly the 1-bp deletion provides a modest fitness advantage by preventing translation of the whole RivR protein, given that a premature stop codon is introduced. Perhaps surprisingly, all 124 post-1920s M3 isolates tested had identical rivR alleles. If the M3 rivR allele provides no activity or function, then we would have expected additional mutations to arise in this gene over time. The observation that M3 GAS rivR, present in plasmid pRivR-M3, has dominant negative effects on M1-RivR and provides some hasA regulatory activity in M1 GAS (Fig. 7A) suggests that some function is maintained by M3-RivR.
The loss of the RivR system in M3 GAS would be expected to result in enhanced GRAB expression relative to serotype M1 GAS. However, GRAB mRNA is in roughly equal abundance in M1 and M3 GAS (compare the vector-containing MGAS2221 and MGAS10870 strains in Fig. 7A). Therefore, either an additional repressor of grab functions in M3 GAS, resulting in lower basal grab transcription, or an additional activator of grab functions in M1 GAS, resulting in higher basal grab transcription. As RivR is unable to regulate hasA expression in M3 GAS, and given that a yet-to-be-determined regulator appears to replace the grab regulatory activity of RivR, this may account for the dispensability of rivR in M3 GAS.
Given the theory that the disease potential of GAS is attributable to the coordinated expression of specific subsets of virulence factors and that the fasC and rivR mutations modify the virulence factor profile of M3 GAS, it is likely that that the disruption of these regulators influences the pathogenicity of M3 isolates. The gain or loss of regulator-encoding genes, along with the potential of changing existing regulatory networks, for example, through the mutation of a regulator binding site upstream of a target gene, provides the opportunity to modulate the regulation of common virulence factor-encoding genes and thus provide for serotype-specific regulation. Only under conditions that favor this new regulatory pattern will these genetic changes be maintained in the population. If a particular combination of regulatory systems proves advantageous, for example, by increasing transmissibility or adherence, then this new strain could arise at greater frequency in the population, possibly resulting in the creation of a new clone or serotype.
An emerging theme in the molecular mechanisms behind strain-specific variation in GAS pathogenicity is the mutation of regulator-encoding genes. We and others have described the enhanced immunomodulatory functions resulting from covR/S mutation and that covR/S mutants are positively selected during invasive GAS infections and negatively selected during pharyngeal infections (12, 23–26). The regulator of protease B (ropB) gene positively regulates the expression of SpeB protease, and this gene was the most polymorphic of all core genome genes in a comparison of serotype M3 GAS strains (20, 22). Mutation of the mtsR (metal transporter of Streptococcus regulator) gene has been identified as contributing to the reduced virulence of some contemporary lineages of serotype M3 isolates (7, 49). Our work presented here adds fasC and rivR as additional regulator-encoding genes that are mutated in certain GAS strains. However, unlike the covR/S, ropB, and mtsR genes, the fasC and rivR mutations appear to be serotype defining; that is, all serotype M3 strains and only serotype M3 strains harbor fasC and rivR mutations (at least no other serotypes have yet been identified with similar mutations). The contribution of the fasC and rivR mutations to M3 GAS virulence factor expression has been shown (Fig. 2 and 8). While the consequences of these mutations with respect to M3 hypervirulence remain to be fully tested, our discovery that this serotype has an altered assortment of functional virulence factor regulatory systems sheds light on mechanisms of serotype-specific variation in GAS, data that may be applicable to strain- or serotype-specific variation in other pathogens.
Supplementary Material
ACKNOWLEDGMENTS
This research was funded in part by grant R01AI087747 from the National Institute of Allergy and Infectious Diseases (to P.S.). Additional support was provided by the Houston Methodist Hospital and the Fondren Foundation.
Footnotes
Published ahead of print 10 February 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01639-13.
REFERENCES
- 1.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin. Microbiol. Rev. 13:470–511. 10.1128/CMR.13.3.470-511.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. 2009. Global emm type distribution of group A streptococci: systematic review and implications for vaccine development. Lancet Infect. Dis. 9:611–616. 10.1016/S1473-3099(09)70178-1 [DOI] [PubMed] [Google Scholar]
- 3.Mitchell ES. 1962. Frequency of serotypes of Streptococcus pyogenes in different diseases. J. Clin. Pathol. 15:231–234. 10.1136/jcp.15.3.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilmers MJ, Cunliffe AC, Williams RE. 1954. Type-12 streptococci associated with acute haemorrhagic nephritis. Lancet 267:17–18 [DOI] [PubMed] [Google Scholar]
- 5.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J. Infect. Dis. 192:771–782. 10.1086/432514 [DOI] [PubMed] [Google Scholar]
- 6.Musser JM, Krause RM. 1998. The revival of group A streptococcal diseases, with a commentary on staphylococcal toxic shock syndrome, p 185–218 In Krause RM. (ed), Emerging infections. Academic Press, New York, NY [Google Scholar]
- 7.Beres SB, Richter EW, Nagiec MJ, Sumby P, Porcella SF, DeLeo FR, Musser JM. 2006. Molecular genetic anatomy of inter- and intraserotype variation in the human bacterial pathogen group A Streptococcus. Proc. Natl. Acad. Sci. U. S. A. 103:7059–7064. 10.1073/pnas.0510279103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smoot JC, Korgenski EK, Daly JA, Veasy LG, Musser JM. 2002. Molecular analysis of group A Streptococcus type emm18 isolates temporally associated with acute rheumatic fever outbreaks in Salt Lake City, Utah. J. Clin. Microbiol. 40:1805–1810. 10.1128/JCM.40.5.1805-1810.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shelburne SA, III, Keith D, Horstmann N, Sumby P, Davenport MT, Graviss EA, Brennan RG, Musser JM. 2008. A direct link between carbohydrate utilization and virulence in the major human pathogen group A Streptococcus. Proc. Natl. Acad. Sci. U. S. A. 105:1698–1703. 10.1073/pnas.0711767105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hondorp ER, McIver KS. 2007. The Mga virulence regulon: infection where the grass is greener. Mol. Microbiol. 66:1056–1065. 10.1111/j.1365-2958.2007.06006.x [DOI] [PubMed] [Google Scholar]
- 11.Kalia A, Bessen DE. 2004. Natural selection and evolution of streptococcal virulence genes involved in tissue-specific adaptations. J. Bacteriol. 186:110–121. 10.1128/JB.186.1.110-121.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog. 2:e5. 10.1371/journal.ppat.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribardo DA, Lambert TJ, McIver KS. 2004. Role of Streptococcus pyogenes two-component response regulators in the temporal control of Mga and the Mga-regulated virulence gene emm. Infect. Immun. 72:3668–3673. 10.1128/IAI.72.6.3668-3673.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIver KS. 2009. Stand-alone response regulators controlling global virulence networks in Streptococcus pyogenes. Contrib. Microbiol. 16:103–119. 10.1159/000219375 [DOI] [PubMed] [Google Scholar]
- 15.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. 10.1038/nature09886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez N, Treviño J, Liu Z, Ho SCM, Babitzke P, Sumby P. 2009. A genome-wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS One 4:e7668. 10.1371/journal.pone.0007668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patenge N, Billion A, Raasch P, Normann J, Wisniewska-Kucper A, Retey J, Boisguerin V, Hartsch T, Hain T, Kreikemeyer B. 2012. Identification of novel growth phase- and media-dependent small non-coding RNAs in Streptococcus pyogenes M49 using intergenic tiling arrays. BMC Genomics 13:550. 10.1186/1471-2164-13-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreikemeyer B, McIver KS, Podbielski A. 2003. Virulence factor regulation and regulatory networks in Streptococcus pyogenes and their impact on pathogen-host interactions. Trends Microbiol. 11:224–232. 10.1016/S0966-842X(03)00098-2 [DOI] [PubMed] [Google Scholar]
- 19.Ikebe T, Ato M, Matsumura T, Hasegawa H, Sata T, Kobayashi K, Watanabe H. 2010. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 6:e1000832. 10.1371/journal.ppat.1000832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. 2010. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc. Natl. Acad. Sci. U. S. A. 107:4371–4376. 10.1073/pnas.0911295107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carroll RK, Shelburne SA, III, Olsen RJ, Suber B, Sahasrabhojane P, Kumaraswami M, Beres SB, Shea PR, Flores AR, Musser JM. 2011. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J. Clin. Invest. 121:1956–1968. 10.1172/JCI45169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olsen RJ, Laucirica DR, Watkins ME, Feske ML, Garcia-Bustillos JR, Vu C, Cantu C, Shelburne SA, III, Fittipaldi N, Kumaraswami M, Shea PR, Flores AR, Beres SB, Lovgren M, Tyrrell GJ, Efstratiou A, Low DE, Van Beneden CA, Musser JM. 2012. Polymorphisms in regulator of protease B (RopB) alter disease phenotype and strain virulence of serotype M3 group A Streptococcus. J. Infect. Dis. 205:1719–1729. 10.1093/infdis/jir825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollands A, Pence MA, Timmer AM, Osvath SR, Turnbull L, Whitchurch CB, Walker MJ, Nizet V. 2010. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A Streptococcus M1T1 clone. J. Infect. Dis. 202:11–19. 10.1086/653124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treviño J, Perez N, Ramirez-Peña E, Liu Z, Shelburne SA, III, Musser JM, Sumby P. 2009. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect. Immun. 77:3141–3149. 10.1128/IAI.01560-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol. Microbiol. 30:209–219. 10.1046/j.1365-2958.1998.01057.x [DOI] [PubMed] [Google Scholar]
- 26.Federle MJ, McIver KS, Scott JR. 1999. A response regulator that represses transcription of several virulence operons in the group A Streptococcus. J. Bacteriol. 181:3649–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kappeler KV, Anbalagan S, Dmitriev AV, McDowell EJ, Neely MN, Chaussee MS. 2009. A naturally occurring Rgg variant in serotype M3 Streptococcus pyogenes does not activate speB expression due to altered specificity of DNA binding. Infect. Immun. 77:5411–5417. 10.1128/IAI.00373-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bessen DE, Manoharan A, Luo F, Wertz JE, Robinson DA. 2005. Evolution of transcription regulatory genes is linked to niche specialization in the bacterial pathogen Streptococcus pyogenes. J. Bacteriol. 187:4163–4172. 10.1128/JB.187.12.4163-4172.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kreikemeyer B, Nakata M, Koller T, Hildisch H, Kourakos V, Standar K, Kawabata S, Glocker MO, Podbielski A. 2007. The Streptococcus pyogenes serotype M49 Nra-Ralp3 transcriptional regulatory network and its control of virulence factor expression from the novel eno ralp3 epf sagA pathogenicity region. Infect. Immun. 75:5698–5710. 10.1128/IAI.00175-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lizano S, Luo F, Tengra FK, Bessen DE. 2008. Impact of orthologous gene replacement on the circuitry governing pilus gene transcription in streptococci. PLoS One 3:e3450. 10.1371/journal.pone.0003450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreikemeyer B, Beckert S, Braun-Kiewnick A, Podbielski A. 2002. Group A streptococcal RofA-type global regulators exhibit a strain-specific genomic presence and regulation pattern. Microbiology 148:1501–1511 [DOI] [PubMed] [Google Scholar]
- 32.Roberts SA, Churchward GG, Scott JR. 2007. Unraveling the regulatory network in Streptococcus pyogenes: the global response regulator CovR represses rivR directly. J. Bacteriol. 189:1459–1463. 10.1128/JB.01026-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts SA, Scott JR. 2007. RivR and the small RNA RivX: the missing links between the CovR regulatory cascade and the Mga regulon. Mol. Microbiol. 66:1506–1522. 10.1111/j.1365-2958.2007.06015.x [DOI] [PubMed] [Google Scholar]
- 34.Treviño J, Liu Z, Cao TN, Ramirez-Peña E, Sumby P. 2013. RivR is a negative regulator of virulence factor expression in group A Streptococcus. Infect. Immun. 81:364–372. 10.1128/IAI.00703-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crater DL, van de Rijn I. 1995. Hyaluronic acid synthesis operon (has) expression in group A streptococci. J. Biol. Chem. 270:18452–18458. 10.1074/jbc.270.31.18452 [DOI] [PubMed] [Google Scholar]
- 36.Rasmussen M, Muller HP, Bjorck L. 1999. Protein GRAB of Streptococcus pyogenes regulates proteolysis at the bacterial surface by binding alpha2-macroglobulin. J. Biol. Chem. 274:15336–15344. 10.1074/jbc.274.22.15336 [DOI] [PubMed] [Google Scholar]
- 37.Kreikemeyer B, Boyle MD, Buttaro BA, Heinemann M, Podbielski A. 2001. Group A streptococcal growth phase-associated virulence factor regulation by a novel operon (Fas) with homologies to two-component-type regulators requires a small RNA molecule. Mol. Microbiol. 39:392–406. 10.1046/j.1365-2958.2001.02226.x [DOI] [PubMed] [Google Scholar]
- 38.Ramirez-Peña E, Treviño J, Liu Z, Perez N, Sumby P. 2010. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol. Microbiol. 78:1332–1347. 10.1111/j.1365-2958.2010.07427.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Treviño J, Ramirez-Peña E, Sumby P. 2012. The small regulatory RNA FasX controls pilus expression and adherence in the human bacterial pathogen group A Streptococcus. Mol. Microbiol. 86:140–154. 10.1111/j.1365-2958.2012.08178.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novick RP. 2003. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 48:1429–1449. 10.1046/j.1365-2958.2003.03526.x [DOI] [PubMed] [Google Scholar]
- 41.Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM. 2002. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. U. S. A. 99:10078–10083. 10.1073/pnas.152298499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treviño J, Perez N, Sumby P. 2010. The 4.5S RNA component of the signal recognition particle is required for group A Streptococcus virulence. Microbiology 156:1342–1350. 10.1099/mic.0.036558-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu M, Hanks TS, Zhang J, McClure MJ, Siemsen DW, Elser JL, Quinn MT, Lei B. 2006. Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152:967–978. 10.1099/mic.0.28706-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takamatsu D, Osaki M, Sekizaki T. 2001. Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid 46:140–148. 10.1006/plas.2001.1532 [DOI] [PubMed] [Google Scholar]
- 45.Podbielski A, Spellerberg B, Woischnik M, Pohl B, Lutticken R. 1996. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS). Gene 177:137–147. 10.1016/0378-1119(96)84178-3 [DOI] [PubMed] [Google Scholar]
- 46.Aracil B, Slack M, Perez-Vazquez M, Roman F, Ramsay M, Campos J. 2006. Molecular epidemiology of Haemophilus influenzae type b causing vaccine failures in the United Kingdom. J. Clin. Microbiol. 44:1645–1649. 10.1128/JCM.44.5.1645-1649.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Croucher NJ, Harris SR, Fraser C, Quail MA, Burton J, van der Linden M, McGee L, von Gottberg A, Song JH, Ko KS, Pichon B, Baker S, Parry CM, Lambertsen LM, Shahinas D, Pillai DR, Mitchell TJ, Dougan G, Tomasz A, Klugman KP, Parkhill J, Hanage WP, Bentley SD. 2011. Rapid pneumococcal evolution in response to clinical interventions. Science 331:430–434. 10.1126/science.1198545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nicol MP, Wilkinson RJ. 2008. The clinical consequences of strain diversity in Mycobacterium tuberculosis. Trans. R. Soc. Trop. Med. Hyg. 102:955–965. 10.1016/j.trstmh.2008.03.025 [DOI] [PubMed] [Google Scholar]
- 49.Olsen RJ, Sitkiewicz I, Ayeras AA, Gonulal VE, Cantu C, Beres SB, Green NM, Lei B, Humbird T, Greaver J, Chang E, Ragasa WP, Montgomery CA, Cartwright J, Jr, McGeer A, Low DE, Whitney AR, Cagle PT, Blasdel TL, DeLeo FR, Musser JM. 2010. Decreased necrotizing fasciitis capacity caused by a single nucleotide mutation that alters a multiple gene virulence axis. Proc. Natl. Acad. Sci. U. S. A. 107:888–893. 10.1073/pnas.0911811107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.