

Abstract
The spindle assembly checkpoint (SAC) monitors defects in kinetochore-microtubule attachment or lack of tension at kinetochores and arrests cells at prometaphase. In fission yeast, the double mutant between pot1Δ and the helicase-dead point mutant of the RecQ helicase Rqh1 gene (rqh1-hd) accumulates Rad51-dependent recombination intermediates at telomeres and enters mitosis with those intermediates. Here, we found that SAC-dependent prometaphase arrest occurred more frequently in pot1Δ rqh1-hd double mutants than in rqh1-hd single mutants. SAC-dependent prometaphase arrest also occurred more frequently in rqh1-hd single mutants after cells were released from DNA replication block compared to the rqh1-hd single mutant in the absence of exogenous insult to the DNA. In both cases, Mad2 foci persisted longer than usual at kinetochores, suggesting a defect in kinetochore-microtubule attachment. In pot1Δ rqh1-hd double mutants and rqh1-hd single mutants released from DNA replication block, SAC-dependent prometaphase arrest was suppressed by the removal of the recombination or replication intermediates. Our results indicate that the accumulation of recombination or replication intermediates induces SAC-dependent prometaphase arrest, possibly by affecting kinetochore-microtubule attachment.
INTRODUCTION
The spindle assembly checkpoint (SAC) monitors defects in kinetochore-spindle interactions (1). Proteins involved in the SAC, such as Bub1 and Mad2, are conserved from yeast to humans (2). Bub1 binds to kinetochores that are not under tension (3). In contrast, Mad2 binds to unattached kinetochores (4). When proper kinetochore-spindle attachment is achieved, the anaphase-promoting complex/cyclosome (APC/C) is activated, which degrades APC substrates such as securin (Cut2 in Schizosaccharomyces pombe), allowing anaphase to proceed. The DNA damage checkpoint detects DNA damage and arrests the cell cycle, which provides time for cells to repair DNA before they enter mitosis. In S. pombe, the DNA damage checkpoint is activated by the recruitment of Rad3 and other proteins to sites of DNA double-strand breaks (5, 6). Rad3 phosphorylates and activates the downstream effecter kinase Chk1. When the DNA damage checkpoint is activated, Chk1 phosphorylates Cdc25 and Wee1, which maintain Cdc2 in an inactive state, resulting in cell cycle arrest at the G2/M transition (7–10). A link between DNA damage and the SAC in many organisms, including Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila, and humans (11–18), has been suggested. However, the molecular details of this link are not well understood.
Chromosome ends are protected by several telomere-binding proteins. In S. pombe, the double-stranded telomere-binding protein Taz1 and the single-stranded telomere-binding protein Pot1 play important roles in telomere maintenance (19). Deletion of taz1+ causes massive telomere elongation (20). Although cell viability is unaffected by taz1 deletion under standard growth conditions (30 to 32°C), telomere entanglement in taz1 disruptants makes the cells sensitive to low temperatures (20°C) (21). At 20°C, mitosis is delayed in taz1 disruptants, and Bub1, but not Mad2, is required for cell survival. This suggests that telomere entanglement disrupts chromosome segregation and that Bub1 helps maintain viability at 20°C (21). Deletion of pot1+ causes rapid telomere loss and chromosome circularization (22). Rqh1 suppresses recombination and promotes the resolution of recombination intermediates (23–28). Double mutants between pot1Δ and the Rqh1 helicase-dead (rqh1-hd) point mutant, in which lysine 547 is mutated to alanine, maintain chromosome ends by homologous recombination (HR) (29). The recombination intermediates exist at chromosome ends even in M phase, which makes cells sensitive to the antimicrotubule drug thiabendazole (TBZ). One study has shown that telomere dysfunction activates the SAC in Drosophila (30). However, fly telomeres are maintained by transposons and do not have telomeric sequences. It remains unclear whether telomere dysfunction activates the SAC in organisms that have telomeric sequences.
In this study, we sought to determine whether the accumulation of recombination intermediates at telomeres in the pot1Δ rqh1-hd double mutant caused defects in M-phase progression. We found that the SAC was activated in the pot1Δ rqh1-hd double mutant. Bub1 and Mad2 foci persisted longer than usual in the prometaphase-arrested pot1Δ rqh1-hd double mutant. Moreover, the accumulation of replication intermediates at ribosomal DNA (rDNA) also arrested the cell cycle at prometaphase and activated the SAC. Based on these and other data, we propose that the accumulation of recombination or replication intermediates activates the SAC, possibly by affecting kinetochore-microtubule attachment.
MATERIALS AND METHODS
Strain construction and growth media.
The strains used in this report are listed in Table 1. Strains were constructed by mating or transformation according to previously described procedures (29, 31). To tag the Atb2 protein in pot1Δ rqh1-hd-related strains with mCherry at the N terminus, pNATZA13-mCherry-atb2+ (a gift from Y. Watanabe and T. Sakuno) was linearized with ApaI and used for transformation (32). Cells were grown in YEA medium (0.5% yeast extract, 3% glucose, and 40 μg of adenine/ml) or Edinburgh minimal medium with the required supplements at the indicated temperature (33). For spot assays, cells were grown to 107 cells/ml in YEA. Serial dilutions (1:10) were prepared, and 4-μl aliquots were spotted onto plates and incubated at 30°C.
TABLE 1.
S. pombe strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| JY741 | h− leu1-32 ura4-D18 ade6-M216 | M. Yamamoto |
| JY746 | h+ leu1-32 ura4-D18 ade6-M210 | M. Yamamoto |
| GT000 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A pPC27-pot1+-HA | K. Takahashi et al. |
| FY18585 | h− leu1 mad2::kanMX6 | NBRPa |
| YK002 | h+ leu1-32 ura4-D18 ade6-M210 rqh1-K547A | K. Takahashi et al. |
| GT002 | h− leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A | K. Takahashi et al. |
| TH025 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A (pPC27-Leu-pot1+-HA) | This study |
| FY10134 | h− leu1 cut2-6×GFP::LEU2 | NBRP |
| NN439 | h− leu1 mis12-GFP::LEU2 | M. Yanagida |
| Sp635 | h− leu1 bub1-GFP::kanMX6 | S. Saito |
| TH002 | h− leu1 bub1-GFP::hphMX6 | This study |
| FY18581 | h− leu1 bub1::kan MX6 | NBRP |
| TH026 | h+ leu1-32 ura4-D18 ade6-M210 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH003 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+ (pPC27-pot1+-HA) | This study |
| pw233 | h− leu1-32 urad4-D18 ade6-M216 kanr≪Padh1-rec8-HA mad2::hphMX6 | Y. Watanabe |
| TH004 | h+ leu1-32 ura4-D18 ade6-M210 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH005 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH006 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A mad2::hphMX6 Z:natMX ≪p adh13-mCherry-atb2+ (pPC27-pot1+-HA) | This study |
| TH000 | h− bub1::LEU2 ade6-210 leu1-32 ura4-DSE his1-102 | S. W.Wang |
| TH007 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A mad2::hphMX6 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH008 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1::LEU2 Z:natMX ≪p adh13-mCherry-atb2+ (pPC27-pot1+-HA) | This study |
| TH009 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1::LEU2 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH011 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A mad2::hphMX6 (pPC27-pot1+-HA) | This study |
| TH012 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A mad2::hphMX6 | This study |
| TH013 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1::LEU2 (pPC27-pot1+-HA) | This study |
| TH014 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1::LEU2 | This study |
| TH015 | h− leu1 ade6-M210 pot1::kanMX6 rqh1-K547A cut2-6×GFP::LEU2 Z:natMX ≪p adh13-mCherry-atb2+ (pPC27-pot1+-HA) | This study |
| TH016 | h− leu1 ade6-M210 pot1::kanMX6 rqh1-K547A cut2-6×GFP::LEU2 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH019 | h− leu1 pot1::kanMX6 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+ mis12-GFP::LEU2 (pPC27-pot1+-HA) | This study |
| TH020 | h− leu1 pot1::kanMX6 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+mis12-GFP::LEU2 | This study |
| TH021 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1-GFP::hphMX6 Z:natMX ≪p adh13-mCherry-atb2+ (pPC27-pot1+-HA) | This study |
| TH022 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX6 rqh1-K547A bub1-GFP::hphMX6 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| TH023 | h− leu1 Z:natMX ≪p adh13-mCherry-atb2+ bub1-GFP::kanMX6 | This study |
| TH024 | h+ leu1 rqh1-K547A Z:natMX ≪p adh13-mCherry-atb2+ bub1-GFP::kanMX6 | This study |
| MT25 | h+ mal3::kanMX6 mad2-GFP-LUE2 bub1-mRFP::kanMx6 his2 | T. Toda |
| FY18537 | h− leu1-32 ura4-D18 rad51::hphMX6 | NBRP |
| MBY1747-2 | h− leu1-32 ura4-D18 bub1::ura4 | D. Hirata |
| AE148 | h− leu1-32 ura4-D18 mad2::ura4 | D. Hirata |
| AN057 | h− leu1-32 ura4-D18 mad3-GFP:kanMX Z:natMX ≪p adh13-mCherry-atb2+ pot::kanMX rqh1K547A | This study |
| AN021 | h− leu1-32 ura4-D18 mad3-GFP:kanMX Z:natMX ≪p adh13-mCherry-atb2+ pot::kanMX rqh1K547A pPC27 pot1+-HA | This study |
| AN076 | h− ade6-M210 leu1-32 mad3-GFP:kanMX Z:natMX ≪p adh13-mCherry-atb2+ rqh1K547A | This study |
| AN092 | h− ade6-M210 leu1-32 ura4-D18 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ pot::kanMX rqh1K547A | This study |
| AN077 | h− ade6-M210 ura4-D18 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ pot::kanMX rqh1K547A pPC27 pot1+-HA | This study |
| AN065 | h− ade6-M210 leu1-32 ura4-D18 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ rqh1K547A | This study |
| AN105 | h− ade6-M210 leu1-32 ura4-D18 bub1::ura4 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ rqh1-K547A | This study |
| AN122 | h− ura4-D18 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ rqh1K547A rad51::hph | This study |
| AN106 | h+ ade6-D18 leu1-32 cut2-6×GFP::LEU2 Z:natMX ≪p adh13-mCherry-atb2+ rqh1-K547A | This study |
| AN131 | h− ade6-M210 leu1-32 ura4-D18 Chk1::hph rad11-GFP:kanMX rqh1-K547A | This study |
| AN126 | h− ura4-D18 mad2-GFP:LEU2 Z:natMX ≪p adh13-mCherry-atb2+ chk1::hph rqh1K547A | This study |
| 738 | h+ chk1d155A:ep ade6-216 leu1-32 ura4-d18 | N. C. Walworth |
| KTA014 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX rqh1-K547A chk1::ura4 | This study |
| TH010 | h+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX rqh1-K547A chk1::ura4 Z:natMX ≪p adh13-mCherry-atb2+ | This study |
| KM005 | H+ leu1-32 ura4-D18 ade6-M210 pot1::kanMX rqh1-K547A chk1::ura4 rad11-mRFP::natMX6 | This study |
| KM003 | h− ura4-D18 pot1::kanMX rqh1-K547A chk1::D155A:ep(HA) | This study |
| KM004 | h− ura4-D18 pot1::kanMX rqh1-K547A chk1::D155A:ep(HA) rad11-mRFP::natMX6 | This study |
| AN216 | h− leu1 pot1::kanMX rqh1-K547A rad51::hph Z:natMX≪p adh13-mCherry-atb2+ mis12-GFP::LEU2+ | This study |
| AN200 | h+ leu1-32 ura4-D18 ade6-M210 rqh1-K547A Z:natMX ≪p adh13-mcherry-atb2+ reb1::kanMX | This study |
| D7 | h− leu1-32 ura4-d18 ade6-M210 reb1Δ::kanMX6 | P. Hernandez |
| AN215 | h? ade6 ura4-D18 leu1 pot1::kanMX rqh1-K547A mis6-2mRFP-hph mad2-GFP-LEU2 | This study |
| AN207 | h− ade6 ura4-D18 mis6-2mRFP-hph mad2-GFP-LEU2 cut12-cfp-nat rqh1-K547A | This study |
NBRP, National Bio Resource Project.
Microscopy.
Microscopy images were obtained using an AxioCam digital camera (Zeiss) connected to an Axio Observer.Z1 microscope (Zeiss) with a Plan-Apochromat 63×, numerical aperture (NA) 1.4 objective lens or an αPlan-FLUAR 100×, NA 1.45 objective lens (see Fig. 1A, 1C, 2A, 4B, 5B, and 5C). Pictures were captured and analyzed using AxioVision Rel. 4.8.2 Software (Zeiss). For Fig. 2C, 3A, and 5D, microscopy images were obtained using an iXon3 897 EMCCD camera (Andor) connected to a Yokokawa CSU-W1 spinning-disc scan head (Yokokawa Electric Corp.) and an Olympus IX83 microscope (Olympus) with a UPlanSApo 100× NA 1.4 objective lens (Olympus). Pictures were captured and analyzed using MetaMorph Software (Molecular Devices). A glass-bottom dish (Iwaki) was coated with 5 mg of lectin/ml from Bandeiraea simplicifolia BS-I (Sigma). For Fig. 1A, 1C, 2A, 4B, 5B, and 5C, optical section data (3 focal planes with 0.5-μm spacing) were collected, and the best focus plane was used. For Fig. 2C, 3A, and 5D, optical section data (10 focal planes with 0.3-μm spacing, every 30 s) were collected, and the time-lapse sequences were deconvolved using the Huygens image analysis software (Scientific Volume Imaging). The z-stack was then projected using the maximum intensity algorithm of the Huygens image analysis software.
FIG 1.
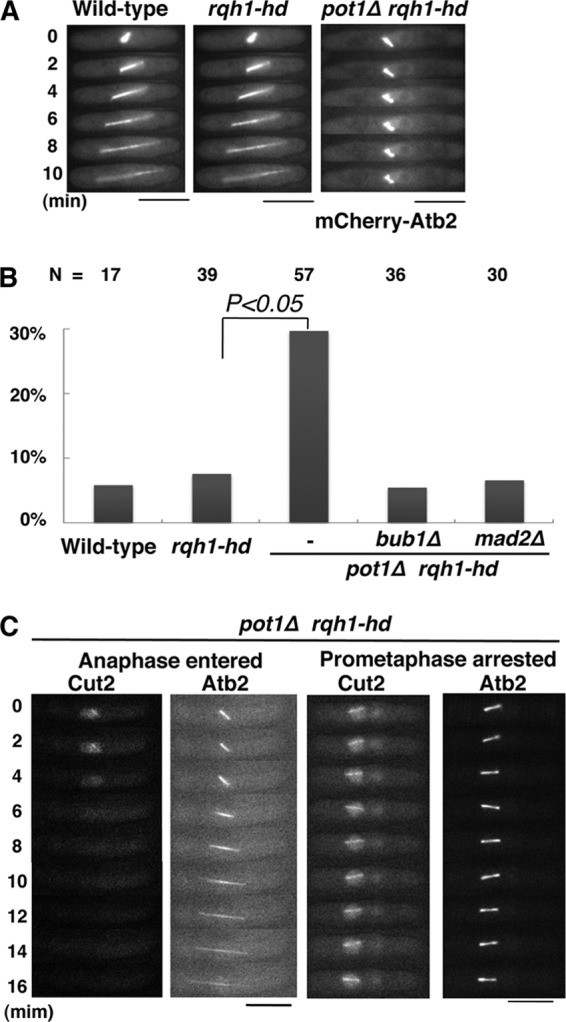
The pot1Δ rqh1-hd double mutant arrests at prometaphase in a Mad2- and Bub1-dependent manner. (A) Representative time-lapse fluorescence images of the spindle microtubule (mCherry-Atb2) in mCherry-Atb2-expressing wild-type, rqh1-hd, and pot1Δ rqh1-hd cells. Bars, 5 μm. (B) Percentage of prometaphase-arrested cells among mCherry-Atb2-expressing wild-type, rqh1-hd, pot1Δ rqh1-hd, pot1Δ rqh1-hd bub1Δ, and pot1Δ rqh1-hd mad2Δ cells. Cells in which spindle elongation arrested for more than 7.5 min were counted as prometaphase-arrested cells. The total cell number examined (N) is shown at the top. (C) Time-lapse fluorescence images of Cut2-GFP and mCherry-Atb2 in pot1Δ rqh1-hd cells. Left side, pot1Δ rqh1-hd cells in anaphase; right side, pot1Δ rqh1-hd cells arrested in prometaphase. Bars, 5 μm.
FIG 2.
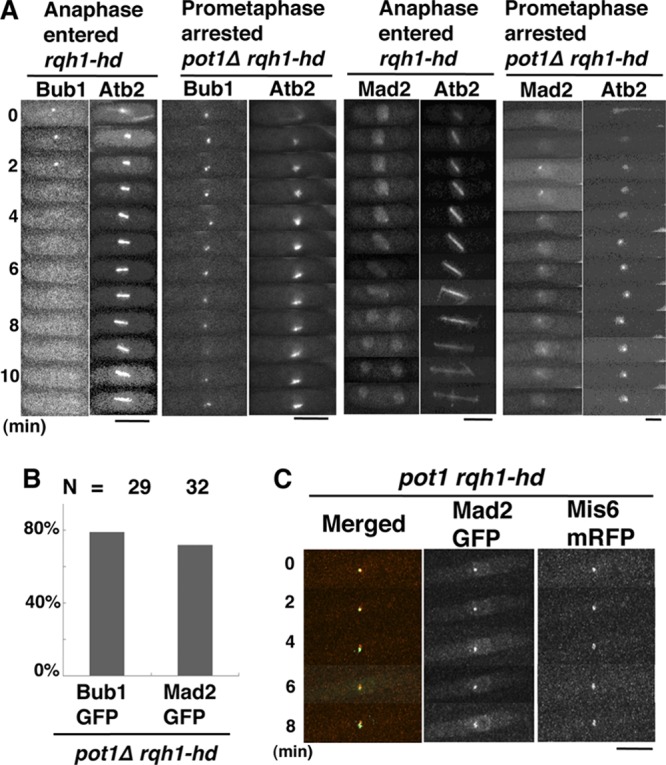
Bub1 and Mad2 foci persist longer than usual in prometaphase-arrested pot1Δ rqh1-hd cells. (A) Time-lapse fluorescence images of Bub1-GFP or Mad2-GFP and mCherry-Atb2 in rqh1-hd cells in anaphase and pot1Δ rqh1-hd cells arrested in prometaphase. Bars, 5 μm. (B) The percentage of prometaphase-arrested pot1Δ rqh1-hd cells in which Bub1 or Mad2 foci were present for >8 min. Cells in which spindle elongation was arrested for >7.5 min were counted as prometaphase-arrested cells. The number of prometaphase-arrested pot1Δ rqh1-hd cells examined (N) is shown at the top. (C) Merged images of fluorescence micrographs showing Mad2-GFP (green) and Mis6-mRFP (red) in prometaphase-arrested pot1Δ rqh1-hd cells.
FIG 4.
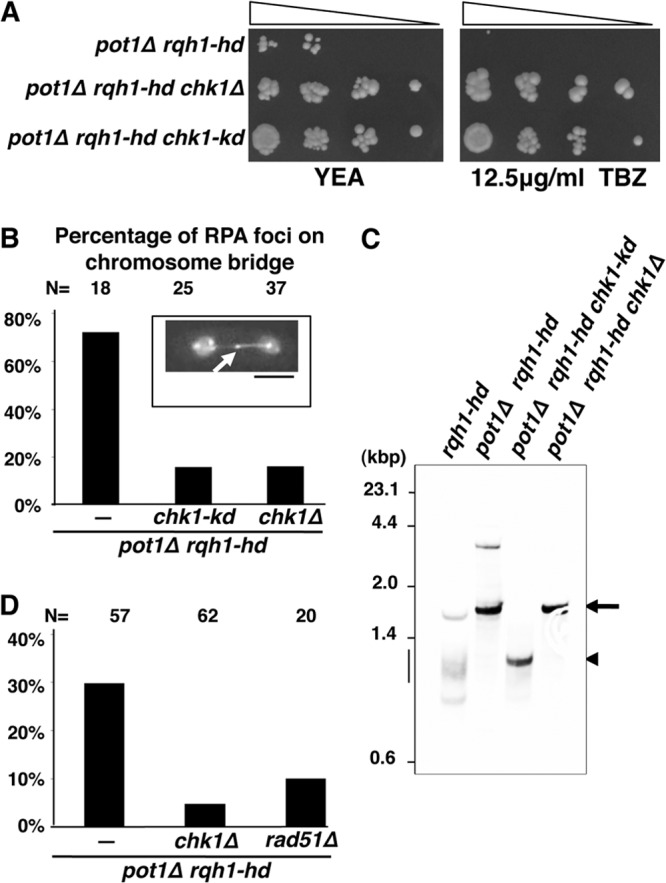
Deletion or mutation of the kinase domain in chk1+ suppresses both the accumulation of recombination intermediates at telomeres and the prometaphase arrest of pot1Δ rqh1-hd double mutants. (A) Deletion or mutation of the kinase domain in chk1+ suppressed TBZ sensitivity. A spotting assay using 10-fold serial dilutions of cells was performed. Sensitivity to TBZ was assessed by spotting pot1Δ rqh1-hd, pot1Δ rqh1-hd chk1Δ, and pot1Δ rqh1-hd chk1-kd cells onto YEA plates in the absence or presence of the indicated concentrations of TBZ at 30°C. (B) Percentage of cells in which RPA foci appeared on chromosome bridges (arrow in the box). A representative fluorescence image of a Rad11-mRFP-expressing pot1Δ rqh1-hd cell, with RPA foci on the chromosome bridge, is shown in the box. Bar, 5 μm. Rad11-mRFP-expressing pot1Δ rqh1-hd, pot1Δ rqh1-hd chk1Δ, and pot1Δ rqh1-hd chk1-kd cells were analyzed. The total cell number examined (N) is shown at the top. (C) Telomere length in rqh1-hd, pot1Δ rqh1-hd, pot1Δ rqh1-hd chk1-kd, and pot1Δ rqh1-hd chk1Δ cells was analyzed by Southern hybridization. Genomic DNA was digested with EcoRI, separated by 1.5% agarose gel electrophoresis, and hybridized to a probe containing 300 bp of telomeric DNA and 700 bp of subtelomeric DNA. The bands corresponding to telomeres are indicated by an arrow (for pot1Δ rqh1-hd and pot1Δ rqh1-hd chk1Δ), arrowhead (for pot1Δ rqh1-hd chk1-kd), or bar (for rqh1-hd). (D) The percentage of prometaphase-arrested cells in mCherry-Atb2-expressing pot1Δ rqh1-hd, pot1Δ rqh1-hd chk1Δ, and pot1Δ rqh1-hd rad51Δ cells. Cells in which spindle elongation was arrested for >7.5 min were counted as prometaphase-arrested cells. The total cell number examined (N) is shown at the top.
FIG 5.
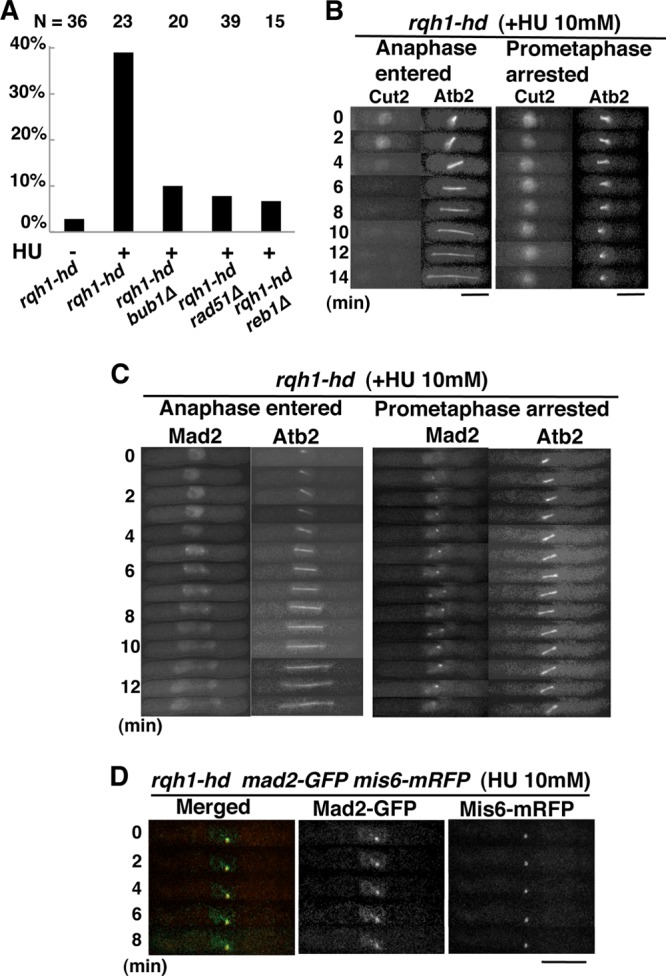
The SAC is activated in rqh1-hd cells released from S-phase arrest. (A) Percentage of prometaphase-arrested cells among mCherry-Atb2-expressing rqh1-hd, rqh1-hd bub1Δ, rqh1-hd rad51Δ, and rqh1-hd reb1Δ cells released from S-phase arrest. Cells in which spindle elongation was arrested for >7.5 min were counted as prometaphase-arrested cells. The total cell number examined (N) is shown at the top. We added 10 mM HU to asynchronous cultures in YEA medium. After exposure to HU for 4 h, cells were washed and transferred to YEA medium without HU and cultured for a further 2 h. (B) Time-lapse fluorescence images of Cut2-GFP and mCherry-Atb2 in rqh1-hd cells released from S-phase arrest. Left, rqh1-hd cells in anaphase; right, rqh1-hd cells arrested in prometaphase. Bars, 5 μm. (C) Time-lapse fluorescence images of Mad2-GFP and mCherry-Atb2 in rqh1-hd cells released from S-phase arrest. Left, rqh1-hd cells in anaphase; right, rqh1-hd cells arrested in prometaphase. Bar, 5 μm. (D) Merged images of fluorescence micrographs showing Mad2-GFP (green) and Mis6-mRFP (red) in prometaphase-arrested rqh1-hd cells released from S-phase arrest.
FIG 3.
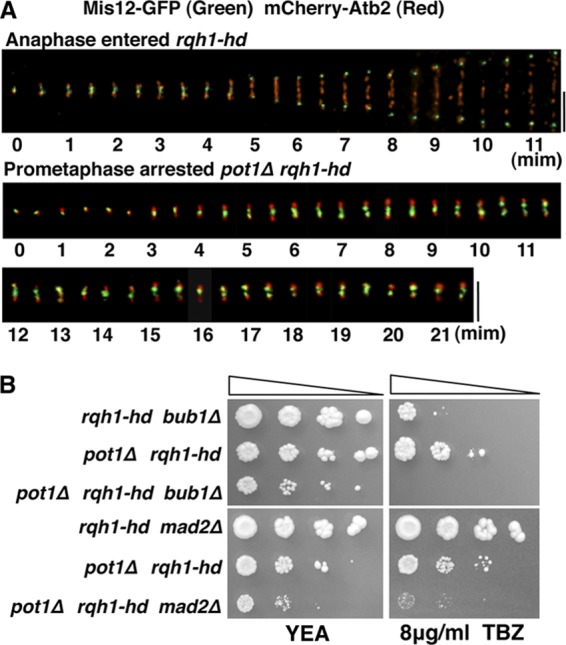
Kinetochores continue to move between the two SPBs in prometaphase-arrested pot1Δ rqh1-hd cells (A) Merged time-lapse fluorescence images of Mis12-GFP (green) and mCherry-Atb2 (red) in rqh1-hd and pot1Δ rqh1-hd cells. Images were captured every 30 s. Top, rqh1-hd cells in anaphase; bottom, pot1Δ rqh1-hd cells arrested in prometaphase. Bars, 5 μm. (B) Spotting assay using 10-fold serial dilutions of cells. Sensitivity to TBZ was assessed by spotting pot1Δ rqh1-hd, pot1Δ rqh1-hd bub1Δ, rqh1-hd bub1Δ, pot1Δ rqh1-hd mad2Δ, and rqh1-hd mad2Δ cells onto YEA platesin the absence or presence of the indicated concentrations of TBZ at 30°C.
Measurement of telomere length.
Telomere length was measured by Southern hybridization according to a previously described procedure (20) with an AlkPhos Direct Labeling and Detection System (GE Healthcare). A telomere associated sequence (TAS1) plus telomere fragment derived from pNSU70 (34) was used as a probe. Genomic DNA was digested with EcoRI, fractioned by 1.5% agarose gel electrophoresis and hybridized to a probe containing the TAS1 plus telomere fragment.
Release from HU arrest.
HU (10 mM) was added to an asynchronous culture of mCherry-Atb2-expressing cells in YEA medium at 30°C. After 4 h in HU, the cells were washed and transferred to YEA medium without HU and cultured for a further 2 h at 30°C. The cells were then observed by microscopy.
RESULTS
The SAC is activated in the pot1Δ rqh1-hd double mutant.
The pot1Δ rqh1-hd double mutant has recombination intermediates at telomeres even in M phase, which may affect the progression of M phase. To examine the phenotype of the pot1Δ rqh1-hd double mutant in M phase, we analyzed M-phase progression by monitoring the elongation of mitotic spindles in mCherry-Atb2-expressing cells (32). Mitotic spindles elongated normally in >90% of wild-type cells and rqh1-hd single mutants (Fig. 1A and B). In contrast, arrest of spindle elongation was detected in ca. 30% of pot1Δ rqh1-hd double mutants (Fig. 1A and B).
We hypothesized that the arrest of spindle elongation was the result of SAC activation. To test this, we assessed whether the arrest of spindle elongation was Mad2 or Bub1 dependent. Among pot1Δ rqh1-hd mad2Δ and pot1Δ rqh1-hd bub1Δ triple mutants, the percentage of spindle elongation-arrested cells was reduced to the wild-type level (Fig. 1B). This suggests that arrest in the pot1Δ rqh1-hd double mutant is SAC dependent. In fission yeast, securin (Cut2) localizes to the nucleus and mitotic spindles when the SAC is activated, and it is degraded by the APC/C after the SAC is satisfied (35, 36). To confirm that the SAC was activated in prometaphase-arrested pot1Δ rqh1-hd double mutants, we monitored the Cut2-GFP signal in living cells. In pot1Δ rqh1-hd double mutants, the Cut2-GFP signal disappeared in cells in which the spindle microtubules elongated normally, whereas the Cut2-GFP signal did not disappear in cells in which spindle microtubule elongation was arrested (Fig. 1C). These data further confirm that the SAC is activated in the pot1Δ rqh1-hd double mutant.
Prolonged Bub1 and Mad2 focus formation in the pot1Δ rqh1-hd double mutant.
To understand the mechanism of SAC activation in the pot1Δ rqh1-hd double mutant, the localization of Bub1 and Mad2 was examined. An intense Bub1-GFP kinetochore signal was visible for 2 to 3 min during the very early stage of mitosis (prometaphase) in wild-type cells (37). Similarly, Bub1 foci were visible for 2 to 3 min during prometaphase in the rqh1-hd single mutant. In contrast, 79% of the prometaphase-arrested pot1Δ rqh1-hd double mutants exhibited Bub1 foci for more than 8 min (Fig. 2A and B). Likewise, 72% of the prometaphase-arrested pot1Δ rqh1-hd double mutants exhibited Mad2 foci for more than 8 min. The Mad2 foci colocalized with Mis6, a kinetochore protein (38), indicating that the Mad2 detected in pot1Δ rqh1-hd cells localized to kinetochores (Fig. 2C). These data suggest that the pot1Δ rqh1-hd double mutant has a defect in kinetochore-microtubule attachment.
Kinetochore movement between the two spindle pole bodies (SPBs) in the pot1Δ rqh1-hd double mutant is prolonged, suggesting a defect in kinetochore-microtubule attachment.
Our results suggest there is a defect in kinetochore-microtubule attachment in the prometaphase-arrested pot1Δ rqh1-hd double mutant. If this is true, kinetochores should continue to move between the two SPBs (39). To test this, we examined the localization of the kinetochore in pot1Δ rqh1-hd double mutants expressing Mis12-GFP (a kinetochore marker [40]) and mCherry-Atb2. In the rqh1-hd single mutant, Mis12-GFP signals oscillated between the two ends of the mitotic spindle (SPBs) until all of the kinetochores were attached to microtubules; Mis12-GFP signals then moved rapidly to the SPBs (Fig. 3A). In contrast, Mis12-GFP signals continued to move between the two SPBs in 71% of the prometaphase-arrested pot1Δ rqh1-hd double mutants (n = 14), supporting the conclusion that the double mutant is defective in kinetochore-microtubule attachment (Fig. 3A).
Bub1 and Mad2 are required for the viability of pot1Δ rqh1-hd cells in the presence or absence of TBZ.
The pot1Δ rqh1-hd double mutant causes prometaphase arrest in Bub1- and Mad2-dependent manners. We next sought to determine whether Bub1 and Mad2 contributed to the viability of the pot1Δ rqh1-hd double mutant in the presence or absence of TBZ. The pot1Δ rqh1-hd bub1Δ triple mutant was more sensitive to TBZ than the pot1Δ rqh1-hd and rqh1-hd bub1Δ double mutants (Fig. 3B). Similarly, the pot1Δ rqh1-hd mad2Δ triple mutant was more sensitive to TBZ than the pot1Δ rqh1-hd and rqh1-hd mad2Δ double mutants (Fig. 3B). Importantly, the growth of the pot1Δ rqh1-hd mad2Δ and pot1Δ rqh1-hd bub1Δ triple mutants was less than that of the pot1Δ rqh1-hd double mutant in the absence of TBZ. These results show that Bub1 and Mad2 are important for the viability of pot1Δ rqh1-hd cells in the presence or absence of TBZ.
Deletion of chk1+ or mutation of the kinase domain in chk1+ suppresses TBZ sensitivity and the accumulation of recombination intermediates at telomeres.
Our results suggest that recombination intermediates underlie SAC activation in the pot1Δ rqh1-hd double mutant. To substantiate this hypothesis, we searched for a mutant that suppressed the accumulation of recombination intermediates at telomeres in the pot1Δ rqh1-hd double mutant. Specifically, because the accumulation of recombination intermediates may underlie the TBZ sensitivity of the pot1Δ rqh1-hd double mutant (29), we searched for a mutant that suppressed TBZ sensitivity. We found that deletion of chk1+ suppressed the TBZ sensitivity of the pot1Δ rqh1-hd double mutant (Fig. 4A). We then examined the importance of Chk1 kinase activity. We used a chk1-kd (kinase dead) point mutant, in which aspartic acid 155 is mutated to alanine, which has no kinase activity in vitro (41). The TBZ sensitivity of the pot1Δ rqh1-hd chk1-kd triple mutant was significantly lower than that of the pot1Δ rqh1-hd double mutant, demonstrating that the kinase domain of Chk1 is important for the suppression of TBZ sensitivity. Next, we sought to determine whether the deletion of chk1+ or mutation of the kinase domain in chk1+ suppressed the accumulation of recombination intermediates at telomeres. Foci containing Rad11, a large subunit of replication protein A (RPA), were detected on the chromosome bridge during M phase in ca. 80% of pot1Δ rqh1-hd double mutants (Fig. 4B, arrow), suggesting that recombination intermediates accumulate at telomeres even during M phase (29). Deletion or mutation of the kinase domain in chk1+ in the pot1Δ rqh1-hd double mutant significantly reduced the percentage of cells with Rad11 foci on the chromosome bridge during M phase to ca. 20% (Fig. 4B). The reduction in Rad11 foci was not due to a reduction in the amount of single-stranded DNA, at unreplicated regions or single-stranded telomere overhangs, for example, because the percentage of asynchronous pot1Δ rqh1-hd cells containing Rad11 foci was not affected by deletion of chk1+ (data not shown). These results suggest that deletion or mutation of the kinase domain in chk1+ suppresses the accumulation of recombination intermediates at telomeres.
Telomeres in rqh1-hd cells, which are maintained by telomerase, were detected as broad bands of approximately 1 kbp when chromosomes were digested by EcoRI (Fig. 4C, bar). In contrast, chromosome ends in the pot1Δ rqh1-hd double mutant are maintained by recombination, and the EcoRI site-containing chromosome ends in the pot1Δ rqh1-hd double mutant were highly amplified (Fig. 4C, arrow) (29). A similar band pattern was detected for pot1Δ rqh1-hd chk1Δ cells (Fig. 4C, arrow), suggesting that the chromosome ends are maintained by recombination (Fig. 4C). Although the band size of the pot1Δ rqh1-hd chk1-kd triple mutant (Fig. 4C, arrowhead) was different from that of the pot1Δ rqh1-hd and pot1Δ rqh1-hd chk1Δ cells, a sharp band was detected, implying that the EcoRI site-containing chromosome end fragments were amplified by recombination. Moreover, pulsed-field gel electrophoresis demonstrated that the chromosomes in pot1Δ rqh1-hd chk1-kd cells were linear (data not shown). Telomeres should be maintained by recombination in the absence of Pot1, because Pot1 is essential for telomerase-dependent telomere maintenance (22, 29). These facts suggest that the chromosome ends in pot1Δ rqh1-hd chk1-kd cells are still maintained by recombination, and yet no recombination intermediates are accumulated.
Deletion of chk1+ or rad51+ suppresses the prometaphase arrest of the pot1Δ rqh1-hd double mutant.
We next sought to determine whether the prometaphase arrest of the pot1Δ rqh1-hd double mutant was suppressed by deletion of chk1+. The percentage pot1Δ rqh1-hd double mutants arrested in prometaphase was significantly reduced by the deletion of chk1+ (Fig. 4D), suggesting a link between the accumulation of recombination intermediates and SAC activation.
Because the pot1Δ rqh1-hd rad51Δ triple mutant has circular chromosomes with no telomeres, the triple mutant has no recombination intermediates at telomeres (29). We sought to determine whether deletion of rad51+ suppressed the prometaphase arrest of the pot1Δ rqh1-hd double mutant. Indeed, deletion of rad51+ did suppress prometaphase arrest (Fig. 4D). This further supports a link between recombination intermediates at telomeres and SAC activation in the pot1Δ rqh1-hd double mutant.
Accumulation of replication intermediates at rDNA loci also activates the SAC.
Our results suggest that the accumulation of recombination intermediates at telomeres in the pot1Δ rqh1-hd double mutant activates the SAC. We next sought to determine whether the accumulation of recombination or replication intermediates at other loci, such as rDNA, also activated the SAC. The rqh1-hd single mutant accumulates recombination and replication intermediates at chromosomes, including rDNA loci, and enters mitosis with those intermediates when the cell cycle is released from hydroxyurea (HU)-mediated DNA replication block (24, 27, 42). We analyzed M phase progression in the rqh1-hd mutant after release from HU-mediated DNA replication block by monitoring the elongation of the mitotic spindle. The mitotic spindle elongated normally in most rqh1-hd single mutants (Fig. 5A). However, arrest of spindle elongation was detected in ca. 40% of the rqh1-hd cells released from HU arrest. Moreover, the arrest was Bub1 dependent (Fig. 5A). These results suggest that the accumulation of recombination and replication intermediates at internal chromosomes, including rDNA loci, also activates the SAC.
Given that release from HU-mediated DNA replication block may generate DNA damage in the rqh1-hd single mutant in addition to the accumulation of recombination and replication intermediates, DNA damage itself might underlie SAC activation. Deletion of rad51+ suppresses aberrant mitosis in rqh1 mutant cells released from HU arrest, suggesting that Rad51 generates aberrant recombination and replication intermediates in the rqh1 mutant (43). Thus, deletion of rad51+ in rqh1-hd would reduce the accumulation of recombination and replication intermediates but not DNA damage itself. To determine that the accumulation of recombination and replication intermediates, but not DNA damage itself, was the reason for SAC activation in rqh1-hd cells released from HU arrest, we used the rqh1-hd rad51Δ double mutant. Cell cycle progression in the rqh1-hd rad51Δ double mutant was monitored after cells were released from HU-mediated arrest. Unlike the rqh1-hd single mutant, the rqh1-hd rad51Δ double mutant did not arrest in prometaphase, suggesting that recombination and replication intermediates, but not DNA damage itself, are required for SAC activation (Fig. 5A).
The rqh1 single mutant has defects in rDNA segregation (42). Relieving replication fork arrest at the replication fork barriers by deletion of reb1+ suppresses the defect in rDNA segregation, suggesting that the aberrant replication intermediates generated at rDNA loci underlie the rDNA segregation defect in the rqh1 mutant (42, 44, 45). We sought to determine whether relieving replication fork arrest at the replication fork barriers suppressed prometaphase arrest in rqh1-hd cells released from HU-mediated arrest. Interestingly, prometaphase arrest in rqh1-hd cells was suppressed by the deletion of reb1+ (Fig. 5A). This suggests that the aberrant replication intermediates generated at rDNA loci underlie prometaphase arrest in rqh1-hd cells released from HU-mediated arrest.
We also confirmed SAC activation by monitoring Cut2-GFP signals after releasing cells from HU-mediated arrest. In the rqh1-hd mutant, the Cut2-GFP signal disappeared in cells in which spindle microtubules elongated normally, whereas the Cut2-GFP signal did not disappear in cells in which spindle microtubule elongation was arrested (Fig. 5B). This further suggests the SAC is activated when replication intermediates accumulate at rDNA loci. Mad2 foci were present for more than 8 min in the prometaphase-arrested rqh1-hd mutants (n = 9, 100%), suggesting a defect in kinetochore-microtubule attachment (Fig. 5C). The Mad2 foci detected colocalized with Mis6, a kinetochore protein, demonstrating that Mad2 localized to kinetochores (Fig. 5D).
Bub1 contributes to the viability of the rqh1-hd mutant in the presence of HU.
Our results suggest that the accumulation of replication intermediates at rDNA results in SAC activation. We next sought to determine whether SAC activation contributed to the viability of rqh1-hd mutants exposed to HU, which accumulated recombination and replication intermediates. The rqh1-hd single mutant, but not the bub1 single mutant, was sensitive to HU (46) (Fig. 6). Interestingly, the rqh1-hd bub1Δ double mutant was more sensitive to HU than either single mutant, demonstrating that SAC activation by Bub1 contributes to the viability of the rqh1-hd mutant in the presence of HU.
FIG 6.

Bub1 contributes to the viability of the rqh1-hd mutant in the presence of HU. A spotting assay using 10-fold serial dilutions of cells was performed. Sensitivity to HU was assessed by spotting wild-type, bub1Δ, and rqh1-hd bub1Δ cells onto YEA plates in the absence or presence of the indicated concentrations of HU at 30°C.
DISCUSSION
Recombination intermediates accumulate at telomeres during M phase in the pot1Δ rqh1-hd double mutant (29). In the present study, we found that the pot1Δ rqh1-hd double mutant arrested at prometaphase in a manner dependent on Mad2 and Bub1, suggesting that the arrest is SAC-dependent (Fig. 1A and B). Moreover, Cut2 was not degraded in the prometaphase-arrested pot1Δ rqh1-hd double mutant (Fig. 1C), further supporting SAC activation. The SAC detects defects in kinetochore-microtubule attachment. Bub1 and Mad2 localize to the kinetochore when proper kinetochore-microtubule attachment has not been achieved. Indeed, we found that Bub1 and Mad2 foci persisted for long periods of time in the prometaphase-arrested pot1Δ rqh1-hd double mutant (Fig. 2). Moreover, Mis12-GFP signals corresponding to kinetochores continued to move between the two SPBs in the prometaphase-arrested pot1Δ rqh1-hd double mutant (Fig. 3A). These facts suggest that proper kinetochore-microtubule attachment cannot be achieved in the prometaphase-arrested pot1Δ rqh1-hd double mutant.
We found that deletion of chk1+ or mutation of the kinase domain in chk1+ suppressed TBZ sensitivity and the accumulation of recombination intermediates at telomeres in the pot1Δ rqh1-hd double mutant (Fig. 4A and B). These results support our model in which the accumulation of recombination intermediates underlies the TBZ sensitivity of the pot1Δ rqh1-hd double mutant. It remains unclear how Chk1 contributes to the accumulation of the intermediates. Given that Chk1 is required for cell cycle arrest at the G2/M transition, deletion of chk1+ may reduce the time in which recombination intermediates can accumulate. Another possibility is that Chk1 contributes directly to the recombination-dependent telomere maintenance pathway by controlling the proteins involved in this pathway. Importantly, the prometaphase arrest of the pot1Δ rqh1-hd double mutant was suppressed by the deletion of chk1+ or rad51+ (Fig. 4D), suggesting a link between the accumulation of recombination intermediates and SAC activation.
We also found that rqh1-hd cells arrested at prometaphase when the cells were released from HU arrest, which causes recombination and replication intermediates to accumulate at chromosome regions, including rDNA loci (Fig. 5A). The arrest was Bub1 dependent (Fig. 5A), and Cut2 was not degraded in prometaphase-arrested rqh1-hd cells, suggesting that the SAC is activated under these conditions (Fig. 5B). Neither the rqh1-hd rad51Δ double mutant nor the rqh1-hd reb1Δ double mutant arrested at prometaphase, suggesting that Rad51-dependent replication intermediates generated at replication fork sites at rDNA loci cause SAC activation (Fig. 5A). The structure of the Rad51-dependent aberrant replication intermediates generated at the rDNA loci remains unclear. Rad51-dependent template exchange occurs during the restart of stalled replication forks in the rqh1 mutant (47). Therefore, we assume that template exchange between sister chromatids generates the aberrant replication intermediates. Mad2 foci persisted for long periods of time in prometaphase-arrested rqh1-hd cells that were released from HU arrest (Fig. 5C). These data suggest that the accumulation of replication intermediates at rDNA in the rqh1-hd single mutant also causes a defect in kinetochore-microtubule attachment. The rqh1-hd bub1Δ double mutant was more sensitive to HU than either single mutant, suggesting that Bub1 contributes to the viability of the rqh1-hd mutant by arresting cells in prometaphase when recombination and/or replication intermediates accumulate (Fig. 6). This emphasizes the importance of SAC activation when aberrant replication intermediates have accumulated. Unlike in the rqh1-hd bub1Δ double mutant, the HU sensitivity of the rqh1 mad2 double mutant is similar to that of each single mutant (42). Thus, Bub1 may be more important than Mad2 for the survival of rqh1-hd mutants on HU plates.
Drosophila and vertebrate Chk1 are involved in SAC activation (16, 48, 49). The S. pombe crb2 mutant arrests in prometaphase in a Chk1-dependent fashion in response to replication stress induced by a topoisomerase I inhibitor (11). We also found that the prometaphase arrest of the pot1Δ rqh1-hd double mutant was Chk1 dependent. However, the involvement of S. pombe Chk1 in the prometaphase arrest of the pot1Δ rqh1-hd double mutant may be indirect because deletion of chk1+ in the pot1Δ rqh1-hd double mutant suppressed the accumulation of recombination intermediates. Unlike the pot1Δ rqh1-hd double mutant, prometaphase arrest in rqh1-hd cells that were released from HU arrest was not suppressed by the deletion of chk1+ or the concomitant deletion of chk1+ and cds1+ (unpublished data). These results suggest that neither the DNA damage checkpoint nor the replication checkpoint is involved in the SAC-dependent prometaphase arrest of rqh1-hd cells released from HU arrest.
Uncapped Drosophila melanogaster telomeres activate the SAC (30). In this case, BubR1, a homologue of S. pombe Mad3, localizes to uncapped telomeres. However, we detected Mad3 foci only on mitotic spindles in the prometaphase-arrested pot1Δ rqh1-hd double mutant, suggesting that Mad3 does not localize to telomeres (data not shown). Moreover, we found that the accumulation of replication intermediates at rDNA loci also activated the SAC. Therefore, the mechanism of SAC activation in response to uncapped D. melanogaster telomeres may differ from the mechanism of SAC activation in response to the accumulation of recombination or replication intermediates during M phase in S. pombe.
In conclusion, our results suggest that recombination intermediates at telomeres or replication intermediates at rDNA activate the SAC, possibly by affecting proper kinetochore-microtubule attachment.
ACKNOWLEDGMENTS
We thank P. Baumann, J. Murray, M. Yanagida, N. Nakazawa, Y. Watanabe, T. Sakuno, T. Toda, D. Hirata, S. Hauf, S. Saitoh, R. Tesin, P. Hernandez, and N. Walworth and the National Bio Resource Project of Japan for providing plasmids and strains. The time-lapse imaging was performed at the Research Center for the Mathematics of Chromatin Live Dynamics of Hiroshima University.
This study was supported by Grants-in-Aid for Scientific Research in Priority Areas from the Ministry of Education, Culture, Sports, Science, and Technology of Japan to M.U.
Footnotes
Published ahead of print 27 January 2014
REFERENCES
- 1.Elowe S. 2011. Bub1 and BubR1: at the interface between chromosome attachment and the spindle checkpoint. Mol. Cell. Biol. 31:3085–3093. 10.1128/MCB.05326-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kadura S, Sazer S. 2005. SAC-ing mitotic errors: how the spindle assembly checkpoint (SAC) plays defense against chromosome mis-segregation. Cell Motil. Cytoskeleton 61:145–160. 10.1002/cm.20072 [DOI] [PubMed] [Google Scholar]
- 3.Skoufias DA, Andreassen PR, Lacroix FB, Wilson L, Margolis RL. 2001. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc. Natl. Acad. Sci. U. S. A. 98:4492–4497. 10.1073/pnas.081076898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waters JC, Chen RH, Murray AW, Salmon ED. 1998. Localization of Mad2 to kinetochores depends on microtubule attachment, not tension. J. Cell Biol. 141:1181–1191. 10.1083/jcb.141.5.1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carr AM. 2002. DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair (Amst.) 1:983–994. 10.1016/S1568-7864(02)00165-9 [DOI] [PubMed] [Google Scholar]
- 6.Humphrey T. 2000. DNA damage and cell cycle control in Schizosaccharomyces pombe. Mutat. Res. 451:211–226. 10.1016/S0027-5107(00)00051-8 [DOI] [PubMed] [Google Scholar]
- 7.Finn K, Lowndes NF, Grenon M. 2012. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci. 69:1447–1473. 10.1007/s00018-011-0875-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langerak P, Russell P. 2011. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366:3562–3571. 10.1098/rstb.2011.0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raleigh JM, O'Connell MJ. 2000. The G2 DNA damage checkpoint targets both Wee1 and Cdc25. J. Cell Sci. 113(Pt 10):1727–1736 [DOI] [PubMed] [Google Scholar]
- 10.Rhind N, Furnari B, Russell P. 1997. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev. 11:504–511. 10.1101/gad.11.4.504 [DOI] [PubMed] [Google Scholar]
- 11.Collura A, Blaisonneau J, Baldacci G, Francesconi S. 2005. The fission yeast Crb2/Chk1 pathway coordinates the DNA damage and spindle checkpoint in response to replication stress induced by topoisomerase I inhibitor. Mol. Cell. Biol. 25:7889–7899. 10.1128/MCB.25.17.7889-7899.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. 2010. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 20:328–332. 10.1016/j.cub.2009.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim EM, Burke DJ. 2008. DNA damage activates the SAC in an ATM/ATR-dependent manner, independently of the kinetochore. PLoS Genet. 4:e1000015. 10.1371/journal.pgen.1000015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikhailov A, Cole RW, Rieder CL. 2002. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 12:1797–1806. 10.1016/S0960-9822(02)01226-5 [DOI] [PubMed] [Google Scholar]
- 15.Royou A, Macias H, Sullivan W. 2005. The Drosophila Grp/Chk1 DNA damage checkpoint controls entry into anaphase. Curr. Biol. 15:334–339. 10.1016/j.cub.2005.02.026 [DOI] [PubMed] [Google Scholar]
- 16.Su TT. 2011. Safeguarding genetic information in Drosophila. Chromosoma 120:547–555. 10.1007/s00412-011-0342-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugimoto I, Murakami H, Tonami Y, Moriyama A, Nakanishi M. 2004. DNA replication checkpoint control mediated by the spindle checkpoint protein Mad2p in fission yeast. J. Biol. Chem. 279:47372–47378. 10.1074/jbc.M403231200 [DOI] [PubMed] [Google Scholar]
- 18.Yang C, Wang H, Xu Y, Brinkman KL, Ishiyama H, Wong ST, Xu B. 2012. The kinetochore protein Bub1 participates in the DNA damage response. DNA Repair (Amst.) 11:185–191. 10.1016/j.dnarep.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain D, Cooper JP. 2010. Telomeric strategies: means to an end. Annu. Rev. Genet. 44:243–269. 10.1146/annurev-genet-102108-134841 [DOI] [PubMed] [Google Scholar]
- 20.Cooper JP, Nimmo ER, Allshire RC, Cech TR. 1997. Regulation of telomere length and function by a Myb-domain protein in fission yeast. Nature 385:744–747. 10.1038/385744a0 [DOI] [PubMed] [Google Scholar]
- 21.Miller KM, Cooper JP. 2003. The telomere protein Taz1 is required to prevent and repair genomic DNA breaks. Mol. Cell 11:303–313. 10.1016/S1097-2765(03)00041-8 [DOI] [PubMed] [Google Scholar]
- 22.Baumann P, Cech TR. 2001. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292:1171–1175. 10.1126/science.1060036 [DOI] [PubMed] [Google Scholar]
- 23.Caspari T, Murray JM, Carr AM. 2002. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 16:1195–1208. 10.1101/gad.221402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doe CL, Dixon J, Osman F, Whitby MC. 2000. Partial suppression of the fission yeast rqh1− phenotype by expression of a bacterial Holliday junction resolvase. EMBO J. 19:2751–2762. 10.1093/emboj/19.11.2751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray JM, Lindsay HD, Munday CA, Carr AM. 1997. Role of Schizosaccharomyces pombe RecQ homolog, recombination, and checkpoint genes in UV damage tolerance. Mol. Cell. Biol. 17:6868–6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Onoda F, Seki M, Miyajima A, Enomoto T. 2001. Involvement of SGS1 in DNA damage-induced heteroallelic recombination that requires RAD52 in Saccharomyces cerevisiae. Mol. Gen. Genet. 264:702–708. 10.1007/s004380000358 [DOI] [PubMed] [Google Scholar]
- 27.Stewart E, Chapman CR, Al-Khodairy F, Carr AM, Enoch T. 1997. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J. 16:2682–2692. 10.1093/emboj/16.10.2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watt PM, Hickson ID, Borts RH, Louis EJ. 1996. SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144:935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takahashi K, Imano R, Kibe T, Seimiya H, Muramatsu Y, Kawabata N, Tanaka G, Matsumoto Y, Hiromoto T, Koizumi Y, Nakazawa N, Yanagida M, Yukawa M, Tsuchiya E, Ueno M. 2011. Fission yeast Pot1 and RecQ helicase are required for efficient chromosome segregation. Mol. Cell. Biol. 31:495–506. 10.1128/MCB.00613-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musaro M, Ciapponi L, Fasulo B, Gatti M, Cenci G. 2008. Unprotected Drosophila melanogaster telomeres activate the spindle assembly checkpoint. Nat. Genet. 40:362–366. 10.1038/ng.2007.64 [DOI] [PubMed] [Google Scholar]
- 31.Nanbu T, Takahashi K, Murray JM, Hirata N, Ukimori S, Kanke M, Masukata H, Yukawa M, Tsuchiya E, Ueno M. 2013. Fission yeast RecQ helicase Rqh1 is required for the maintenance of circular chromosomes. Mol. Cell. Biol. 33:1175–1187. 10.1128/MCB.01713-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakuno T, Tada K, Watanabe Y. 2009. Kinetochore geometry defined by cohesion within the centromere. Nature 458:852–858. 10.1038/nature07876 [DOI] [PubMed] [Google Scholar]
- 33.Moreno S, Klar A, Nurse P. 1991. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194:795–823. 10.1016/0076-6879(91)94059-L [DOI] [PubMed] [Google Scholar]
- 34.Sugawara N. 1988. DNA sequences at the telomeres of the fission yeast Schizosaccharomyces pombe. Ph.D. thesis Harvard University, Cambridge, MA [Google Scholar]
- 35.Kumada K, Nakamura T, Nagao K, Funabiki H, Nakagawa T, Yanagida M. 1998. Cut1 is loaded onto the spindle by binding to Cut2 and promotes anaphase spindle movement upon Cut2 proteolysis. Curr. Biol. 8:633–641. 10.1016/S0960-9822(98)70250-7 [DOI] [PubMed] [Google Scholar]
- 36.Nasmyth K, Peters JM, Uhlmann F. 2000. Splitting the chromosome: cutting the ties that bind sister chromatids. Science 288:1379–1385. 10.1126/science.288.5470.1379 [DOI] [PubMed] [Google Scholar]
- 37.Toyoda Y, Furuya K, Goshima G, Nagao K, Takahashi K, Yanagida M. 2002. Requirement of chromatid cohesion proteins Rad21/Scc1 and Mis4/Scc2 for normal spindle-kinetochore interaction in fission yeast. Curr. Biol. 12:347–358. 10.1016/S0960-9822(02)00692-9 [DOI] [PubMed] [Google Scholar]
- 38.Saitoh S, Takahashi K, Yanagida M. 1997. Mis6, a fission yeast inner centromere protein, acts during G1/S and forms specialized chromatin required for equal segregation. Cell 90:131–143. 10.1016/S0092-8674(00)80320-7 [DOI] [PubMed] [Google Scholar]
- 39.Tang NH, Takada H, Hsu KS, Toda T. 2013. The internal loop of fission yeast Ndc80 binds Alp7/TACC-Alp14/TOG and ensures proper chromosome attachment. Mol. Biol. Cell 24:1122–1133. 10.1091/mbc.E12-11-0817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goshima G, Saitoh S, Yanagida M. 1999. Proper metaphase spindle length is determined by centromere proteins Mis12 and Mis6 required for faithful chromosome segregation. Genes Dev. 13:1664–1677. 10.1101/gad.13.13.1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Capasso H, Palermo C, Wan S, Rao H, John UP, O'Connell MJ, Walworth NC. 2002. Phosphorylation activates Chk1 and is required for checkpoint-mediated cell cycle arrest. J. Cell Sci. 115:4555–4564. 10.1242/jcs.00133 [DOI] [PubMed] [Google Scholar]
- 42.Win TZ, Mankouri HW, Hickson ID, Wang SW. 2005. A role for the fission yeast Rqh1 helicase in chromosome segregation. J. Cell Sci. 118:5777–5784. 10.1242/jcs.02694 [DOI] [PubMed] [Google Scholar]
- 43.Miyabe I, Morishita T, Hishida T, Yonei S, Shinagawa H. 2006. Rhp51-dependent recombination intermediates that do not generate checkpoint signal are accumulated in Schizosaccharomyces pombe rad60 and smc5/6 mutants after release from replication arrest. Mol. Cell. Biol. 26:343–353. 10.1128/MCB.26.1.343-353.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krings G, Bastia D. 2004. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc. Natl. Acad. Sci. U. S. A. 101:14085–14090. 10.1073/pnas.0406037101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez-Gorostiaga A, Lopez-Estrano C, Krimer DB, Schvartzman JB, Hernandez P. 2004. Transcription termination factor reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol. Cell. Biol. 24:398–406. 10.1128/MCB.24.1.398-406.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laursen LV, Ampatzidou E, Andersen AH, Murray JM. 2003. Role for the fission yeast RecQ helicase in DNA repair in G2. Mol. Cell. Biol. 23:3692–3705. 10.1128/MCB.23.10.3692-3705.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Freon K, Murray JM, Carr AM, Baldacci G. 2010. Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol. Cell 39:346–359. 10.1016/j.molcel.2010.07.015 [DOI] [PubMed] [Google Scholar]
- 48.Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. 2009. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc. Natl. Acad. Sci. U. S. A. 106:5159–5164. 10.1073/pnas.0806671106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zachos G, Gillespie DA. 2007. Exercising restraints: role of Chk1 in regulating the onset and progression of unperturbed mitosis in vertebrate cells. Cell Cycle 6:810–813. 10.4161/cc.6.7.4048 [DOI] [PubMed] [Google Scholar]