

Abstract
A persistent and nonresolving inflammatory response to accumulating Aβ peptide species is a cardinal feature in the development of Alzheimer's disease (AD). In response to accumulating Aβ peptide species, microglia, the innate immune cells of the brain, generate a toxic inflammatory response that accelerates synaptic and neuronal injury. Many proinflammatory signaling pathways are linked to progression of neurodegeneration. However, endogenous anti-inflammatory pathways capable of suppressing Aβ-induced inflammation represent a relatively unexplored area. Here we report that signaling through the prostaglandin-E2 (PGE2) EP4 receptor potently suppresses microglial inflammatory responses to Aβ42 peptides. In cultured microglial cells, EP4 stimulation attenuated levels of Aβ42-induced inflammatory factors and potentiated phagocytosis of Aβ42. Microarray analysis demonstrated that EP4 stimulation broadly opposed Aβ42-driven gene expression changes in microglia, with enrichment for targets of IRF1, IRF7, and NF-κB transcription factors. In vivo, conditional deletion of microglial EP4 in APPSwe-PS1ΔE9 (APP-PS1) mice conversely increased inflammatory gene expression, oxidative protein modification, and Aβ deposition in brain at early stages of pathology, but not at later stages, suggesting an early anti-inflammatory function of microglial EP4 signaling in the APP-PS1 model. Finally, EP4 receptor levels decreased significantly in human cortex with progression from normal to AD states, suggesting that early loss of this beneficial signaling system in preclinical AD development may contribute to subsequent progression of pathology.
Keywords: Aβ peptide, Alzheimer's disease, microglia, neuroinflammation, PGE2, receptor
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease, with an expected tripling by 2050 as a result of an expanding aging population (Hebert et al., 2003). The limited efficacy of current AD treatment strategies underscores the need for a more complete understanding of AD pathogenesis to identify novel therapeutic targets. The inflammatory response is one component of AD pathogenesis wherein microglia, the innate immune cells of the brain, become highly activated in response to accumulating Aβ peptide species and produce toxic cytokines and reactive oxygen species (Akiyama et al., 2000; Heneka and O'Banion, 2007). The ultimate role of this chronic inflammatory response remains controversial: activated microglia may be toxic to neurons, but they may also exert beneficial effects, including clearance of toxic molecules, such as Aβ peptide species or generation of trophic and reparative factors (Wyss-Coray, 2006). Strategies that promote the beneficial phagocytic role of microglia while preventing the transition to a toxic inflammatory response could therefore represent attractive targets for AD prevention or therapy.
In AD model mice, Aβ42 signaling through Toll-like receptors (TLRs) (Landreth and Reed-Geaghan, 2009) drives canonical cytokine and chemokine pathways, such as TNF-α (He et al., 2007), IL12b (Vom Berg et al., 2012), and CCL3 (Passos et al., 2009) that contribute to Aβ42-induced neuronal damage and cognitive decline. Although many studies have identified proinflammatory pathways in AD, fewer anti-inflammatory pathways have been identified. Of these, signaling through the fractalkine receptor, CX3CR1, has been most extensively studied (Cardona et al., 2006; Lee et al., 2010). However, the identification of additional endogenous anti-inflammatory pathways is highly relevant to AD and other neurodegenerative diseases characterized by nonresolving and toxic inflammatory responses.
To this end, we have sought to clarify the role of prostaglandin-E2 (PGE2), a pivotal immune signaling molecule and a primary target of NSAIDs, in models of Aβ42 toxicity. We and others have found so far that three of the four G-protein coupled receptors for PGE2 (the EP1, EP2, and EP3 receptors) exert proinflammatory and/or proamyloidogenic effects in AD mouse models (Liang et al., 2005; Shi et al., 2012; Zhen et al., 2012). In contrast, we recently identified a striking anti-inflammatory role for the microglial EP4 receptor in a model of lipopolysaccharide (LPS)-induced innate immunity (Shi et al., 2010). Given this finding, we asked whether the EP4 receptor could be a protective target in models of AD.
Here we report anti-inflammatory effects of microglial EP4 receptor signaling in cultured microglia and in the APP-PS1 mouse model of AD. EP4 signaling broadly suppresses the activation of target genes for NF-κB and interferon regulatory factors (IRFs), transcription factors that are central regulators of the microglial response to Aβ42. Moreover, we find that EP4 signaling potentiates phagocytosis of Aβ42 by microglia. In vivo, in APP-PS1 mice lacking microglial EP4, we find a converse upregulation of inflammatory gene expression, oxidative stress, and amyloid accumulation at early stages of pathology. Our findings identify EP4 receptor signaling as a novel anti-inflammatory pathway in models of AD neuroinflammation.
Materials and Methods
Materials.
Aβ42 was obtained from rPeptide and prepared in oligomeric form as described previously (Yang et al., 2008). Briefly, HFIP-prepared Aβ42 was resuspended in DMSO (0.1 mg in 10 μl) followed by 1:10 dilution in Ham's F12 culture medium (Mediatech) at 4C for 24 h before use. This stock solution of 222 μm (molarity based on original Aβ42 monomer concentration) was then diluted for cell treatment experiments. The EP4 agonist AE1–329 and the EP4 antagonist AE3–208 were generous gifts from ONO Pharmaceuticals. Their specificity for the EP4 receptor has been established previously (Suzawa et al., 2000; Kabashima et al., 2002).
Human brain tissue.
Temporal and parietal cortex from control, mild cognitive impairment (MCI), and AD patients (Alzheimer's Disease Research Center, University of Washington, Seattle) was derived from subjects 79–88 years of age with a postmortem delay of 2.5–8 h.
Animals.
This study was conducted in accordance with National Institutes of Health guidelines, and protocols were approved by the Institutional Animal Care and Use Committee at Stanford University. C57BL/6J EP4lox/lox mice (Schneider et al., 2004) were kindly provided by Drs. Richard and Matthew Breyer (Vanderbilt University School of Medicine, Nashville). C57BL/6J Cd11b-Cre mice (Boillée et al., 2006) were kindly provided by Dr. G. Kollias (Alexander Fleming Biomedical Sciences Research Center, Vari, Greece) and Dr. Donald Cleveland (University of California San Diego). APPSwe-PS1ΔE9 mice (APP-PS1) (Jankowsky et al., 2001) were kindly provided by Dr. David Borchelt and backcrossed to a C57BL/6J background for n > 12 generations. APP-PS1 or CD11bCre mice were serially crossed to EP4lox/lox mice to produce APP-PS1;EP4lox/lox and CD11bCre;EP4lox/lox mice. These mice were interbred, as were APP-PS1;EP4+/+ and CD11bCre;EP4+/+ mice, to produce the APP-PS1;EP4-WT and APP-PS1;EP4-cKO mice used in this study. The female mice used for this study were aged to 5 months before being transcardially perfused with cold saline. One brain hemisphere was postfixed in 4% PFA for 24 h for use in immunohistochemistry; the other hemisphere was dissected and frozen for qPCR and levuglandin analysis.
Primary microglia isolation.
Primary microglia were isolated from the brains of postnatal day 7 C57BL/6J mouse pups obtained from Charles River Laboratories. Primary microglia were isolated using the Neural Tissue Dissociation Kit (P), MACS Separation Columns (LS), and magnetic CD11b Microbeads from Miltenyi Biotec. Microglia were grown in culture for 3–5 days before being treated in each experiment.
Cell culture.
Primary microglia and murine immortalized microglial BV-2 cells were grown in DMEM supplemented with 10% heat-inactivated FBS (HyClone) and 100 U/ml each penicillin and streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Immunocytochemistry.
Primary mouse microglia were plated on poly-l-lysine-coated coverslips and fixed with 4% PFA after 5 d in culture. Immunocytochemistry for mouse EP4 was performed using a chicken antibody directed against the mouse EP4 receptor (1:500), described and validated in (Liang et al., 2011), and a rat monoclonal antibody directed against mouse CD11b (1:500, AbD Serotec). Fluorescently labeled secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Chicken serum (Jackson ImmunoResearch Laboratories) was used as a negative control in place of the primary EP4 receptor antibody. Images were acquired using a Leica DM5500 Q confocal microscope (Leica Microsystems).
qPCR.
RNA isolation, cDNA production, and SYBR-Green based qPCR (QuantiTect SYBR Green Kit, QIAGEN) were performed as described in detail previously (Shi et al., 2010) using the standard curve method and normalizing to 18S and GAPDH. Melting curve analysis confirmed the specificity of each reaction. Samples without reverse transcription served as negative controls. Primers were designed by PrimerQuest (Integrated DNA Technologies) or PrimerBank (Spandidos et al., 2010) and synthesized by Integrated DNA Technologies. Primer sequences were as follows: 18S: CGGCTACCACATCCAAGGAA and GCTGGAATTACCGCGGCT; CCL3: ATACAAGCAGCAGCGAGTACCAGT and AATCTTCCGGCTGTAGGAGAAGCA; COX-2: TGCAAGATCCACAGCCTACC and GCTCAGTTGAACGCCTTTTG; GAPDH:TGCACCACCAACTGCTTAG and GATGCAGGGATGATGTTC; Il12b: TGGTTTGCCATCGTTTTGCTG and ACAGGTGAGGTTCACTGTTTCT; iNOS: TGACGGCAAACATGACTTCAG and GCCATCGGGCATCTGGTA; IRF1: GGCCGATACAAAGCAGGAGAA and GGAGTTCATGGCACAACGGA; IRF7: CCCCAGCCGGTGATCTTTC and CACAGTGACGGTCCTCGAAG; Nur77: TGCACAGCTTGGGTGTTGATGTTC and TGTGCTCCTTCAGACAGCTAGCAA; Nurr1: TCTGCGCTTAGCATACAGGTCCAA and CAGCAATGCAGGAGAAGGCAGAAA; and TNF-α: TCATTCCTGCTTGTGGCAGGGG and GTGGTTTGCTACGACGTGGGCT.
Cell viability quantification.
Primary microglia were plated and treated with oligomeric Aβ42 or vehicle for 24 h before addition of 200 μg/ml Trypan Blue (Invitrogen). The ratio of trypan blue-negative cells to the total number of cells counted (>300 cells counted per condition) was calculated as a measure of cell viability.
Cytokine and chemokine ELISA.
ELISA assays for mouse TNF-α and CCL3 (R&D Systems) were performed as detailed in the manufacturer's protocol and quantified using a SpectraMax M5 plate reader (Molecular Devices).
Phagocytosis of FITC-Aβ.
Cells were pretreated for 3 h with the indicated concentrations of EP4 agonist or Cytochalasin D (Cell Biolabs) before addition of fluorescent Aβ. FITC-labeled Aβ42 (rPeptide) was prepared as described previously (Shie et al., 2005a) before being added to cells at a final concentration of 1 μm. After 1, 6, or 24 h of incubation, cells were washed with PBS followed by addition of 200 μg/ml Trypan Blue (Invitrogen) to quench extracellular fluorescence. Intracellular fluorescence was then assayed using a SpectraMax M5 plate reader (Molecular Devices). Background signal from wells with no plated cells was subtracted from all experimental values.
Microarray analysis.
RNA from primary microglia was isolated using Trizol (Invitrogen) followed by the RNeasy Mini Kit (QIAGEN). RNA quality was assessed using a BioAnalyzer (Agilent) and determined to be sufficient for microarray analysis (RNA Integrity Number > 9.9 for all samples). cDNA synthesis, labeling, hybridization, and scanning were performed by the Stanford Protein and Nucleic Acid (PAN) Facility using GeneChip Mouse Gene 1.0 ST arrays (Affymetrix). Microarray data were statistically analyzed using Partek software (Partek) to identify differentially expressed genes and for unsupervised clustering to create the heat map of EP4/Aβ-responsive genes. Data have been deposited in the Gene Expression Omnibus, accession number GSE55627. DAVID functional annotation software (National Institute of Allergy and Infectious Diseases, National Institutes of Health) (Huang da et al., 2009) was used to identify KEGG molecular pathways significantly over-represented among the lists of differentially expressed genes. Ingenuity Pathway Analysis (Ingenuity Systems) was used to identify transcription factor pathways over-represented among the genes differentially expressed by Aβ42 and EP4 agonist treatment. Gene Set Enrichment Analysis (GSEA) software from the Broad Institute (Subramanian et al., 2005) was used to identify Transcription Factor Target (TFT) gene sets differentially enriched by Aβ42 and EP4 agonist treatment. In the GSEA analysis, microarray data from Aβ42-alone versus Vehicle-alone comparison (Aβ42 effect) were first assessed for TFT enrichment. The top 20 Aβ42-responsive TFT sets (all with normalized enrichment score > 1.60, nominal p < 0.0001, FDR p < 0.05) were then assayed in the Aβ42+EP4 Agonist versus Aβ42-Veh comparison to calculate normalized enrichment scores.
γ-Ketoaldehyde adduct quantification.
Cortex samples were processed and analyzed by liquid-chromatography electrospray-ionization multistage mass spectrometry (LC/ESI/MS/MS) as previously described (Zagol-Ikapitte et al., 2005).
Aβ42 and Aβ40 ELISA.
Levels of total guanidine-extracted Aβ40 and Aβ42 peptides were measured by ELISA as previously described (Liang et al., 2005) using mouse monoclonal antibody 6E10 as a capture antibody and biotinylated mouse monoclonal antibodies 12F4 (Aβ42) and B10 (Aβ40) as detection antibodies (antibodies from Covance).
Quantification of Aβ plaque density.
Immunostaining for Aβ plaques using the 6E10 antibody (Covance) and staining for dense-core plaques using Congo Red (Sigma) were performed as previously described (Liang et al., 2005). Briefly, every sixth section (40 μm) through the hippocampus was stained and imaged (n = 10 sections per mouse). Images were quantified for the area above threshold in the region of interest (hippocampus) using Volocity 5.1 software (PerkinElmer).
Immunohistochemistry in human tissue.
Sections from parietal cortex (Alzheimer's Disease Research Center, University of Washington, Seattle) were first treated with citrate buffer (10 mm sodium citrate, 0.05% Tween 20, pH 6.0) to retrieve antigens for staining. Sections were then sequentially immunostained, first for EP4 with a rabbit antibody directed against human EP4 (1:200, Cayman Chemical) and developed with DAB (Polysciences), then for Aβ using the mouse monoclonal antibody 6E10 (1:1000, Covance) and developed with VIP solution (Vector Laboratories). Biotinylated secondary antibodies (Vector Laboratories) were used at a dilution of 1:250. The M.O.M. kit (Vector Laboratories) was used to reduce background staining with the mouse antibody. Rabbit and mouse sera (Jackson ImmunoResearch Laboratories) were used as negative controls in place of primary antibodies on adjacent sections.
Western blot.
Western blot was performed and quantified as previously described (Shi et al., 2010, 2012). Briefly, BV2 cell lysates were made using cell lysis buffer (Cell Signaling Technology) supplemented with protease inhibitors (Roche) and phosphatase inhibitor mixture (Sigma). Lysates were run on NuPAGE 4–12% polyacrylamide gels (Invitrogen) and transferred to PVDF membranes. In vitro studies used a rabbit monoclonal antibody for phospho-STAT1 (Tyr701, Clone D4A7, 1:1000; Cell Signaling Technology) and a rabbit polyclonal antibody for total-STAT1 (1:1000, #9172; Cell Signaling Technology). Quantitative Western blots of synaptic proteins were performed as previously described (Shi et al., 2012) with normalization to either tubulin or actin. Western blot from human temporal cortex lysates was performed using a rabbit polyclonal antibody directed against human EP4 (1:500; Cayman Chemical). A mouse monoclonal antibody directed against β actin (1:10,000; Sigma) served as a loading control.
Statistical analysis.
Data are expressed as mean ± SEM. Comparisons were made using Student's t test (for two groups), one-way ANOVA with Dunnett's post test (for more than two groups across one variable, with post test comparisons to the control group), or two-way ANOVA with Bonferroni multiple comparisons (for groups across two variables, with post test comparisons between individual groups). All comparison tests were two-tailed. Results with p < 0.05 were considered significant.
Results
EP4 receptor signaling attenuates the microglial inflammatory response to Aβ42
We first confirmed that primary mouse microglia express the EP4 receptor using an antibody specific for the mouse peptide sequence (Liang et al., 2011) (Fig. 1A), consistent with our previous studies identifying microglial expression of the EP4 receptor in mouse brain (Shi et al., 2010). To assess the effect of EP4 receptor signaling on Aβ42-induced inflammation, we established an in vitro model of primary mouse microglia stimulated with oligomeric Aβ42; oligomeric Aβ42 constitutes a highly pathologic Aβ peptide species that induces synaptic and neuronal injury and cognitive decline in models of AD (Lesné et al., 2006). We prepared oligomeric Aβ42 according to methods previously validated by atomic force microscopy (Yang et al., 2008) and found that this preparation at a dose of 10 μm induced a robust inflammatory transcriptional response in microglia (Fig. 1B–E). This inflammatory response was not associated with cell death, as we detected no increase in cell death among primary microglia treated with oligomeric Aβ42 for 24 h (94.3 ± 0.7% live cells for vehicle, 95.4 ± 0.9% live cells for 10 μm Aβ42, n = 9 samples per group, p = 0.38). Notably, similar concentrations of Aβ42 (10–12 μm) have previously been established for acute induction of inflammatory and neurotoxic responses in primary murine microglia (Shie et al., 2005a; Halle et al., 2008).
Figure 1.
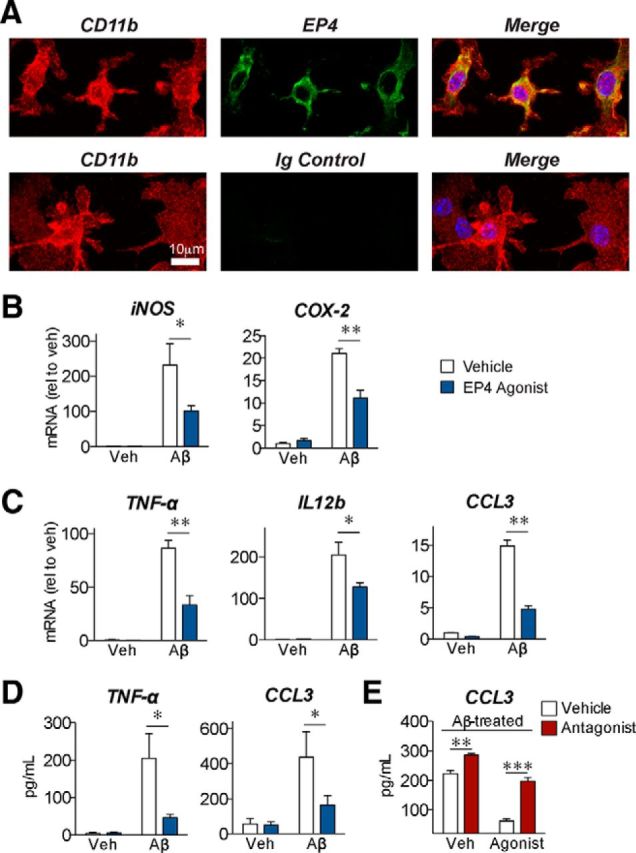
EP4 receptor signaling reverses the inflammatory response to Aβ42 in primary microglia. A, Immunostaining in primary cultured mouse microglia demonstrates expression of EP4 receptor (green) in CD11b-positive (red) cells. Nuclei were stained with Hoechst (blue). No signal was observed with immunoglobulin control for the EP4 receptor antibody (bottom). B–E, Primary microglia were cotreated for 6 h with oligomeric Aβ42 (10 μm), the EP4 agonist AE1–329 (100 nm), or the EP4 antagonist AE3–208 (100 nm). B, qRT-PCR demonstrates that EP4 agonist (blue) attenuates expression of the oxidative enzymes iNOS and COX-2 in response to Aβ42. C, qRT-PCR demonstrates that EP4 stimulation attenuates expression of the cytokines TNF-α and IL12b, and the chemokine CCL3, in response to Aβ42. D, Supernatant ELISA demonstrates that EP4 stimulation attenuates levels of secreted TNF-α and CCL3 in response to Aβ42. E, Supernatant ELISA demonstrates that EP4 agonist treatment attenuates, whereas EP4 antagonist treatment (red bars) potentiates, the levels of secreted CCL3. For each experiment, n = 3 microglia isolations from independent mouse cohorts. *p < 0.05 (Bonferroni multiple comparison test). **p < 0.01 (Bonferroni multiple comparison test). ***p < 0.001 by (Bonferroni multiple comparison test).
To test the effect of EP4 receptor signaling on Aβ42-mediated inflammatory responses, we cotreated primary microglia with oligomeric Aβ42 and the EP4-receptor-specific agonist AE1–329 (Suzawa et al., 2000). We found that EP4 receptor stimulation significantly attenuated mRNA levels for inflammatory genes, including the oxidative enzymes iNOS and COX-2 (Fig. 1B), the cytokines TNF-α and IL12b, and the chemokine CCL3 (Fig. 1C). ELISA quantification demonstrated that EP4 receptor signaling reduced the Aβ42-induced secretion of TNF-α and CCL3 proteins from microglia (Fig. 1D). We next asked whether EP4 receptor signaling is not only sufficient but also necessary in suppressing the inflammatory response to Aβ42. Although we were unable to culture and assess EP4 knock-out microglia because of the perinatal lethality of the EP4−/− genotype in the C57B6 background (Nguyen et al., 1997), we used a pharmacological approach to inhibit the EP4 receptor. We found that the selective EP4 receptor antagonist AE3–208 (Kabashima et al., 2002) increased CCL3 secretion in Aβ42-treated microglia and reversed the effect of the agonist (Fig. 1E). In parallel, we tested the effect of EP4 receptor signaling in BV-2 immortalized murine microglial cells; here we found a similar attenuation of Aβ42-induced inflammatory responses (Fig. 2A). Together, these results identify a potent action of EP4 receptor signaling to suppress Aβ42-induced production of inflammatory factors by microglia.
Figure 2.

EP4 receptor signaling reduces inflammatory responses and promotes phagocytosis of Aβ42 by BV2 microglial cells. A, BV2 microglial cells were cotreated with oligomeric Aβ42 (5 μm) or the EP4 agonist AE1–329 (100 nm) for 6 h. qRT-PCR demonstrates that EP4 receptor stimulation attenuates the expression of the cytokine TNF-α and the chemokine CCL3 in response to Aβ42. n = 5 or 6 independent samples per group. **p < 0.01 (Bonferroni multiple comparison test). B–E, BV2 microglial cells were pretreated for 3 h with the EP4 agonist AE1–329, PGE2, or the phagocytosis inhibitor cytochalasin-D before addition of FITC-labeled Aβ (1 μm) for the indicated times. B, Images of BV2 cells immunostained for CD11b (white) and nuclei (Hoechst, blue) demonstrate that pretreatment with EP4 agonist (3 h, 100 nm) increases uptake of FITC-Aβ42 (6 h, internalized Aβ indicated with arrows). C, EP4 agonist pretreatment dose-dependently increases intracellular FITC-Aβ42 levels after either 1 or 6 h of FITC-Aβ42 incubation. n = 12 (1 h) or 7 (6 h) samples per group. p (one-way ANOVA). *p < 0.05, versus vehicle treatment (Dunnett's post test). **p < 0.01, versus vehicle treatment (Dunnett's post test). ***p < 0.001, versus vehicle treatment (Dunnett's post test). D, EP4 agonist (100 nm) treatment increases, whereas PGE2 (100 nm) treatment decreases, intracellular FITC-Aβ42 levels after 6 h. E, Cytochalasin-D (CyD, 2 μm) decreases intracellular FITC-Aβ42 levels after 6 h. D, E, n = 7 or 8 samples per group. *p < 0.05, versus vehicle treatment (unpaired t test). ***p < 0.001, versus vehicle treatment (unpaired t test).
EP4 receptor signaling potentiates phagocytosis of Aβ
Previous studies have found that inflammatory cytokines such as TNF-α reduce the ability of microglia to effectively phagocytose and clear Aβ (El Khoury et al., 2003; Hickman et al., 2008). To determine whether the anti-inflammatory role we observed for EP4 receptor signaling was conversely associated with increased phagocytosis, we assessed the ability of BV2 microglial cells to phagocytose FITC-labeled Aβ42 after stimulation with EP4 agonist (Fig. 2B). We observed a dose-dependent increase in intracellular FITC-Aβ42 levels with EP4 agonist treatment (Fig. 2C). Notably, this observed increase in phagocytosis with EP4 receptor stimulation is in contrast to the decreased phagocytosis previously demonstrated for PGE2 signaling through the EP2 receptor in macrophages and microglia (Aronoff et al., 2004; Shie et al., 2005a). To determine the overall effect of PGE2 on FITC-Aβ42 phagocytosis, we treated BV2 cells with PGE2 and observed a significant decrease in intracellular FITC-Aβ42 signal (Fig. 2D), consistent with previous studies. This finding suggests that, while the overall effect of PGE2 is to decrease phagocytosis, differential signaling through EP4 or EP2 receptors may modulate this effect. As an important control for our assays, we found that Cytochalasin-D, an established inhibitor of phagocytosis, also significantly reduced the intracellular FITC-Aβ42 signal (Fig. 2E). We next attempted to confirm these findings in primary microglia; however, even after 24 h of incubation with FITC-Aβ42, intracellular FITC-Aβ42 levels remained undetectably low in primary microglia (7.62 ± 0.93 arbitrary fluorescence units in primary microglia after 24 h, compared with 30.43 ± 1.48 in control BV2 cells after 6 h and 11.55 ± 0.82 in Cytochalasin-D-treated BV2 cells after 6 h). This finding, while precluding us from assessing phagocytosis in primary microglia, is consistent with previously reported differences in Aβ phagocytosis between BV2 cells and primary microglia (Jiang et al., 2008). As a whole, however, these data suggest that EP4 receptor signaling potentiates phagocytosis of Aβ42 in contexts where cells exhibit sufficient basal levels of phagocytosis.
EP4 signaling opposes genome-wide Aβ42-induced transcriptional changes in microglia through NF-κB, IRF1, and IRF7 transcription factors
To determine whether EP4 receptor signaling broadly opposed the transcriptional changes brought about by Aβ42, we turned to an unbiased approach by performing microarray analysis on RNA from primary microglia treated with oligomeric Aβ42 and/or the EP4 agonist AE1–329 for 6 h. We identified 597 Aβ42-responsive transcripts with significant expression changes (fold change > 2.0; FDR p < 0.05) in Aβ42-only compared with vehicle-only treatment groups, 92 genes differentially regulated in Aβ42+EP4 agonist versus Aβ42+Veh, and 78 genes differentially regulated between EP4 agonist versus vehicle (Fig. 3A). Unsupervised hierarchical clustering based on the expression of the 92 genes regulated in Aβ42+EP4 agonist versus Aβ42+Veh revealed a striking distinction among treatment groups: many Aβ42-upregulated transcripts were decreased in expression with EP4 agonist cotreatment, and many Aβ42-downregulated transcripts were conversely increased in expression with EP4 agonist cotreatment (Fig. 3B).
Figure 3.

Microarray analysis identifies transcriptional targets of EP4 receptor signaling in suppressing the microglial inflammatory response to oligomeric Aβ42. A, Genome-wide microarray analysis was performed on primary microglia cotreated for 6 h with oligomeric Aβ42 (10 μm) and/or the EP4 agonist AE1–329 (100 nm). The Venn diagram displays the numbers of transcripts significantly regulated (fold change > 2.0, FDR p < 0.05) in each comparison. B, Unsupervised hierarchical clustering based on the expression of the 92 EP4/Aβ42-responsive transcripts (fold change > 2.0, FDR p < 0.05 in Aβ42+EP4 agonist vs Aβ42+Veh comparison) demonstrates that EP4 receptor stimulation opposes many gene expression changes induced by Aβ42. C, DAVID functional annotation analysis reveals several KEGG pathways significantly over-represented among the 597 transcripts regulated by Aβ42 and among the 116 Aβ42-responsive transcripts for which EP4 receptor stimulation opposed the Aβ42 effect by >1.5-fold (Table 1). Pathways with FDR (p < 0.05) in this comparison are in bold. D, Shown are the differentially expressed transcripts from the KEGG pathways with FDR (p < 0.05) (green: Aβ42-up, EP4-down; red: Aβ42-down, EP4-up).
To better understand the nature of the genes regulated in opposite directions by Aβ42 and EP4 agonist, we narrowed our list of 597 Aβ42-responsive transcripts to those in which the EP4 agonist reversed the Aβ42 effect by >1.5-fold (116 transcripts, Table 1). Although the stringent cutoffs for this list excluded several of the candidate inflammatory genes we had previously examined (all of which showed the same direction of change between our qPCR and microarray studies), we aimed here to use an unbiased approach to uncover mechanisms underlying the microglial response to Aβ42. DAVID functional annotation analysis (Huang da et al., 2009) of these 116 transcripts, as well as the original list of 597 Aβ42 responsive transcripts, demonstrated several significantly over-represented KEGG pathways, all of which corresponded to inflammatory signaling networks (Fig. 3C). These included microglial pathways previously associated with Aβ42, including TLR, cytokine, and chemokine signaling; interestingly, this analysis also identified several transcriptional pathways that have been less well characterized in the response to Aβ42, including nod-like receptor, RIG-1-like receptor, and cytosolic DNA-sensing pathways characterized primarily for their role in the interferon-mediated antiviral immune response. The transcripts included in the most significantly over-represented KEGG pathways encoded cytokines, chemokines, growth factors, membrane receptors, and transcription factors (Fig. 3D), suggesting that EP4 receptor signaling antagonizes the inflammatory response to Aβ42 at multiple levels.
Table 1.
Aβ-responsive genes in microglia for which EP4 signaling opposed the effect of Aβ by >1.5-fold
| RefSeq//gene symbol//gene assignment | Gene | Fold change | p | Fold change | p |
|---|---|---|---|---|---|
| Genes upregulated by Aß42 oligomers | Aß42o-veh versus Con-Veh | Aß42o + EP4 ag versus Aß42o-veh | |||
| NM_001033122 // Cd69 // CD69 antigen | Cd69 | 127.22 | 3.2E-06 | −1.92 | 1.6E-01 |
| NM_021274 // Cxcl10 // chemokine (C-X-C motif) ligand 10 | Cxcl10 | 24.75 | 5.5E-04 | −1.75 | 3.6E-01 |
| NM_138648 // Olr1 // oxidized low density lipoprotein (lectin-like) receptor 1 | Olr1 | 21.60 | 2.9E-09 | −1.89 | 4.0E-04 |
| NM_054055 // Slc13a3 // solute carrier family 13 | Slc13a3 | 19.03 | 2.1E-09 | −1.71 | 7.3E-04 |
| NR_027852 // Cd40 // CD40 antigen | Cd40 | 16.96 | 2.1E-11 | −2.69 | 8.8E-08 |
| NM_010260 // Gbp2 // guanylate binding protein 2 | Gbp2 | 16.91 | 2.3E-05 | −1.76 | 1.2E-01 |
| NM_153564 // Gbp5 // guanylate binding protein 5 | Gbp5 | 16.53 | 1.2E-06 | −1.54 | 8.1E-02 |
| NM_008204 // H2-M2 // histocompatibility 2, M region locus 2 | H2-M2 | 16.21 | 1.3E-10 | −1.73 | 3.8E-05 |
| NM_009890 // Ch25h // cholesterol 25-hydroxylase | Ch25h | 16.07 | 1.7E-08 | −2.52 | 7.2E-05 |
| NM_030701 // Niacr1 // niacin receptor 1 | Niacr1 | 15.24 | 8.4E-11 | −1.78 | 1.6E-05 |
| NM_172845 // Adamts4 // a disintegrin-like and metallopeptidase | Adamts4 | 11.24 | 3.3E-08 | −2.88 | 1.9E-05 |
| NM_008331 // Ifit1 // interferon-induced protein with tetratricopeptide repeats | Ifit1 | 10.35 | 2.9E-04 | −2.16 | 7.9E-02 |
| NM_172648 // Ifi205 // interferon activated gene 205 | Ifi205 | 10.23 | 3.2E-05 | −2.05 | 3.2E-02 |
| NM_008599 // Cxcl9 // chemokine (C-X-C motif) ligand 9 | Cxcl9 | 6.89 | 6.0E-07 | −3.97 | 7.8E-06 |
| NM_021384 // Rsad2 // radical S-adenosyl methionine domain containing 2 | Rsad2 | 6.84 | 2.7E-03 | −1.98 | 1.7E-01 |
| NM_008332 // Ifit2 // interferon-induced protein with tetratricopeptide repeats | Ifit2 | 6.46 | 3.5E-03 | −3.13 | 3.7E-02 |
| NM_010846 // Mx1 // myxovirus (influenza virus) resistance 1 | Mx1 | 6.43 | 4.1E-04 | −3.21 | 6.8E-03 |
| NR_029565 // Mir155 // microRNA 155 | Mir155 | 6.19 | 5.3E-09 | −1.59 | 1.8E-04 |
| NM_011909 // Usp18 // ubiquitin specific peptidase 18 | Usp18 | 5.89 | 4.0E-04 | −2.75 | 1.1E-02 |
| NR_003508 // Mx2 // myxovirus (influenza virus) resistance 2 | Mx2 | 5.77 | 1.1E-05 | −3.30 | 1.8E-04 |
| NM_029758 // Fam49a // family with sequence similarity 49, member A | Fam49a | 5.48 | 6.8E-07 | −1.70 | 2.5E-03 |
| NM_001045481 // Ifi203 // interferon activated gene 203 | Ifi203 | 5.46 | 9.2E-04 | −1.79 | 1.2E-01 |
| NM_020557 // Cmpk2 // cytidine monophosphate (UMP-CMP) kinase 2, mitochondrial | Cmpk2 | 5.40 | 2.8E-03 | −2.16 | 8.8E-02 |
| NM_018734 // Gbp3 // guanylate binding protein 3 | Gbp3 | 5.26 | 1.7E-05 | −2.22 | 2.4E-03 |
| NM_013652 // Ccl4 // chemokine (C-C motif) ligand 4 | Ccl4 | 5.00 | 2.7E-08 | −1.64 | 1.9E-04 |
| NM_029084 // Slamf8 // SLAM family member 8 | Slamf8 | 4.96 | 6.1E-07 | −2.02 | 2.6E-04 |
| NR_029806 // Mir221 // microRNA 221 | Mir221 | 4.86 | 8.5E-08 | −1.85 | 1.0E-04 |
| NM_001114665 // Fnbp1l // formin binding protein 1-like | Fnbp1l | 4.55 | 2.8E-09 | −1.87 | 2.8E-06 |
| NM_145545 // Gbp6 // guanylate binding protein 6 | Gbp6 | 4.25 | 3.1E-05 | −1.52 | 4.1E-02 |
| NM_145066 // Gpr85 // G protein-coupled receptor 85 | Gpr85 | 4.19 | 6.0E-09 | −1.57 | 4.1E-05 |
| NM_010657 // Hivep3 // human immunodeficiency virus type I enhancer binding pr | Hivep3 | 4.14 | 2.8E-08 | −1.77 | 3.1E-05 |
| NM_145968 // Tagap // T-cell activation Rho GTPase-activating protein | Tagap | 4.05 | 1.5E-09 | −1.57 | 1.0E-05 |
| NM_010104 // Edn1 // endothelin 1 | Edn1 | 3.91 | 1.6E-06 | −2.49 | 3.2E-05 |
| NM_008607 // Mmp13 // matrix metallopeptidase 13 | Mmp13 | 3.87 | 6.2E-07 | −1.82 | 2.5E-04 |
| NM_022331 // Herpud1 // homocysteine-inducible, endoplasmic reticulum stress-ind | Herpud1 | 3.68 | 1.6E-09 | −1.83 | 6.9E-07 |
| NM_001033415 // Shisa3 // shisa homolog 3 (Xenopus laevis) | Shisa3 | 3.63 | 2.9E-08 | −2.34 | 7.5E-07 |
| NM_010755 // Maff // v-maf musculoaponeurotic fibrosarcoma oncogene family | Maff | 3.54 | 1.0E-06 | −1.64 | 8.9E-04 |
| NM_001039530 // Parp14 // poly (ADP-ribose) polymerase family, member 14 | Parp14 | 3.38 | 2.4E-06 | −1.61 | 1.8E-03 |
| NR_029807 // Mir222 // microRNA 222 | Mir222 | 3.33 | 6.2E-07 | −1.74 | 1.9E-04 |
| NM_008390 // Irf1 // interferon regulatory factor 1 | Irf1 | 3.32 | 3.8E-07 | −1.63 | 2.7E-04 |
| NM_016850 // Irf7 // interferon regulatory factor 7 | Irf7 | 3.24 | 2.7E-03 | −1.56 | 1.4E-01 |
| NM_001042611 // Cp // ceruloplasmin | Cp | 3.21 | 5.0E-05 | −1.92 | 2.3E-03 |
| NM_013654 // Ccl7 // chemokine (C-C motif) ligand 7 | Ccl7 | 3.09 | 1.1E-04 | −1.95 | 3.0E-03 |
| NM_183177 // Zfp811 // zinc finger protein 811 | Zfp811 | 3.05 | 2.1E-07 | −1.70 | 5.6E-05 |
| NM_009425 // Tnfsf10 // tumor necrosis factor (ligand) superfamily, member 10 | Tnfsf10 | 2.98 | 2.0E-03 | −2.35 | 7.7E-03 |
| NM_008330 // Ifi47 // interferon gamma inducible protein 47 | Ifi47 | 2.96 | 6.8E-04 | −2.08 | 6.8E-03 |
| NM_181545 // Slfn8 // schlafen 8 | Slfn8 | 2.89 | 1.5E-05 | −1.73 | 1.4E-03 |
| NM_028968 // Ifitm7 // interferon induced transmembrane protein 7 | Ifitm7 | 2.88 | 4.1E-07 | −1.74 | 5.1E-05 |
| NM_001134457 // Fam55c // family with sequence similarity 55, member C | Fam55c | 2.87 | 2.9E-06 | −1.71 | 3.9E-04 |
| NM_027835 // Ifih1 // interferon induced with helicase C domain 1 | Ifih1 | 2.87 | 2.5E-06 | −1.82 | 1.5E-04 |
| NM_178607 // Rnf24 // ring finger protein 24 | Rnf24 | 2.84 | 2.5E-06 | −1.52 | 1.6E-03 |
| ENSMUST00000131035 // Rnf213 // ring finger protein 213 | Rnf213 | 2.82 | 1.9E-04 | −1.86 | 4.7E-03 |
| NM_011331 // Ccl12 // chemokine (C-C motif) ligand 12 | Ccl12 | 2.80 | 1.3E-06 | −2.76 | 1.5E-06 |
| NM_001037713 // Xaf1 // XIAP associated factor 1 | Xaf1 | 2.79 | 6.0E-05 | −1.88 | 1.5E-03 |
| NM_011333 // Ccl2 // chemokine (C-C motif) ligand 2 | Ccl2 | 2.78 | 5.9E-05 | −1.76 | 2.8E-03 |
| NM_023141 // Tor3a // torsin family 3, member A | Tor3a | 2.74 | 1.8E-05 | −1.76 | 9.6E-04 |
| NM_019440 // Irgm2 // immunity-related GTPase family M member 2 | Irgm2 | 2.66 | 2.7E-05 | −2.10 | 1.9E-04 |
| NM_009873 // Cdk6 // cyclin-dependent kinase 6 | Cdk6 | 2.61 | 5.7E-10 | −2.41 | 1.2E-09 |
| NM_194336 // Mpa2l // macrophage activation 2 like | Mpa2l | 2.60 | 3.2E-05 | −1.71 | 1.6E-03 |
| NM_183201 // Slfn5 // schlafen 5 | Slfn5 | 2.56 | 4.4E-05 | −1.80 | 1.1E-03 |
| NM_011579 // Tgtp1 // T-cell specific GTPase 1 | Tgtp1 | 2.56 | 1.3E-05 | −2.31 | 3.0E-05 |
| NM_023386 // Rtp4 // receptor transporter protein 4 | Rtp4 | 2.52 | 2.1E-05 | −1.62 | 1.7E-03 |
| NM_011579 // Tgtp1 // T-cell specific GTPase 1 | Tgtp1 | 2.50 | 1.1E-05 | −2.19 | 3.5E-05 |
| NM_009277 // Trim21 // tripartite motif-containing 21 | Trim21 | 2.48 | 2.5E-06 | −1.69 | 1.4E-04 |
| NM_183029 // Igf2bp2 // insulin-like growth factor 2 mRNA binding protein 2 | Igf2bp2 | 2.47 | 1.6E-07 | −1.62 | 1.9E-05 |
| NM_008326 // Irgm1 // immunity-related GTPase family M member 1 | Irgm1 | 2.42 | 3.9E-04 | −2.23 | 7.3E-04 |
| NM_021788 // Sap30 // sin3 associated polypeptide | Sap30 | 2.38 | 4.5E-06 | −1.75 | 1.1E-04 |
| NM_011057 // Pdgfb // platelet derived growth factor, B polypeptide | Pdgfb | 2.36 | 1.2E-05 | −2.65 | 4.6E-06 |
| NR_003507 // Oas1b // 2′-5′ oligoadenylate synthetase 1B | Oas1b | 2.35 | 5.7E-06 | −1.63 | 3.1E-04 |
| NM_010828 // Cited2 // Cbp/p300-interacting transactivator | Cited2 | 2.32 | 3.1E-06 | −1.75 | 6.3E-05 |
| NM_025992 // Herc6 // hect domain and RLD 6 | Herc6 | 2.31 | 2.4E-05 | −1.64 | 8.5E-04 |
| NM_008356 // Il13ra2 // interleukin 13 receptor, alpha 2 | Il13ra2 | 2.29 | 2.0E-07 | −1.72 | 4.9E-06 |
| NM_145636 // Il27 // interleukin 27 | Il27 | 2.25 | 4.4E-08 | −1.86 | 3.6E-07 |
| NM_010215 // Il4i1 // interleukin 4 induced 1 | Il4i1 | 2.21 | 3.0E-07 | −2.33 | 1.8E-07 |
| NR_029728 // Mirlet7c-1 // microRNA let7c-1 | Mirlet7c-1 | 2.15 | 5.6E-06 | −2.03 | 1.0E-05 |
| NM_013730 // Slamf1 // signaling lymphocytic activation molecule family member 1 | Slamf1 | 2.14 | 1.0E-06 | −1.59 | 4.3E-05 |
| NM_027320 // Ifi35 // interferon-induced protein 35 | Ifi35 | 2.14 | 2.6E-04 | −1.57 | 6.5E-03 |
| NM_172689 // Ddx58 // DEAD (Asp-Glu-Ala-Asp) box polypeptide 58 | Ddx58 | 2.08 | 1.1E-04 | −1.66 | 1.3E-03 |
| NM_007829 // Daxx // Fas death domain-associated protein | Daxx | 2.07 | 1.3E-04 | −1.54 | 3.6E-03 |
| NM_008360 // Il18 // interleukin 18 | Il18 | 2.06 | 7.1E-05 | −2.07 | 6.7E-05 |
| NM_020583 // Isg20 // interferon-stimulated protein | Isg20 | 2.06 | 6.5E-04 | −1.53 | 1.3E-02 |
| NM_016960 // Ccl20 // chemokine (C-C motif) ligand 20 | Ccl20 | 2.05 | 7.1E-04 | −1.58 | 9.5E-03 |
| NR_029535 // Mir99a // microRNA 99a | Mir99a | 2.03 | 6.3E-06 | −1.87 | 1.6E-05 |
| Genes downregulated by Aß42 oligomers | |||||
| NM_008676 // Nbr1 // neighbor of Brca1 gene 1 | Nbr1 | −2.02 | 7.0E-09 | 1.51 | 4.7E-07 |
| NM_001037957 // Dyrk1b // dual-specificity tyrosine-(Y)-phosphorylation | Dyrk1b | −2.02 | 5.4E-08 | 1.90 | 1.2E-07 |
| NM_026875 // Ypel3 // yippee-like 3 (Drosophila) | Ypel3 | −2.04 | 2.3E-08 | 1.62 | 4.8E-07 |
| NM_011505 // Stxbp4 // syntaxin binding protein 4 | Stxbp4 | −2.05 | 6.9E-04 | 1.59 | 8.5E-03 |
| NM_021356 // Gab1 // growth factor receptor bound protein 2-associated protein 1 | Gab1 | −2.06 | 5.5E-07 | 2.82 | 3.4E-08 |
| NM_010591 // Jun // Jun oncogene | Jun | −2.08 | 6.2E-06 | 1.82 | 2.8E-05 |
| NM_007797 // Ctla2b // cytotoxic T lymphocyte-associated protein 2 beta | Ctla2b | −2.12 | 7.1E-07 | 2.16 | 5.9E-07 |
| NM_010847 // Mxi1 // Max interacting protein 1 | Mxi1 | −2.13 | 2.0E-08 | 3.92 | 1.8E-10 |
| ENSMUST00000162022 // Glis3 // GLIS family zinc finger 3 | Glis3 | −2.23 | 2.0E-05 | 1.65 | 5.4E-04 |
| NM_028149 // Fbxl20 // F-box and leucine-rich repeat protein 20 | Fbxl20 | −2.25 | 3.8E-08 | 1.82 | 3.9E-07 |
| NM_175445 // Rassf2 // Ras association (RalGDS/AF-6) domain family member 2 | Rassf2 | −2.26 | 2.0E-09 | 2.46 | 9.1E-10 |
| NM_025979 // Mastl // microtubule associated serine/threonine kinase-like | Mastl | −2.27 | 5.6E-04 | 1.60 | 1.4E-02 |
| NM_172589 // Lhfpl2 // lipoma HMGIC fusion partner-like 2 | Lhfpl2 | −2.32 | 1.1E-06 | 1.80 | 1.7E-05 |
| NM_133667 // Pdk2 // pyruvate dehydrogenase kinase, isoenzyme 2 | Pdk2 | −2.35 | 2.3E-06 | 2.09 | 7.0E-06 |
| NM_011454 // Serpinb6b // serine (or cysteine) peptidase inhibitor | Serpinb6b | −2.52 | 6.5E-07 | 1.70 | 4.0E-05 |
| NM_134250 // Havcr2 // hepatitis A virus cellular receptor 2 | Havcr2 | −2.61 | 4.6E-07 | 1.60 | 9.6E-05 |
| NM_021897 // Trp53inp1 // transformation related protein 53 inducible | Trp53inp1 | −2.74 | 7.8E-10 | 1.96 | 1.9E-08 |
| NM_007635 // Ccng2 // cyclin G2 | Ccng2 | −2.83 | 2.0E-06 | 1.89 | 7.7E-05 |
| NM_001081278 // Tbc1d4 // TBC1 domain family, member 4 | Tbc1d4 | −3.01 | 5.2E-08 | 1.63 | 2.7E-05 |
| NM_007901 // S1pr1 // sphingosine-1-phosphate receptor 1 | S1pr1 | −3.02 | 2.4E-07 | 2.60 | 7.6E-07 |
| NM_011050 // Pdcd4 // programmed cell death 4 | Pdcd4 | −3.28 | 2.9E-06 | 1.60 | 1.8E-03 |
| NM_133212 // Tlr8 // toll-like receptor 8 | Tlr8 | −3.55 | 3.5E-08 | 1.88 | 7.5E-06 |
| NM_145933 // St6gal1 // beta galactoside alpha 2,6 sialyltransferase 1 | St6gal1 | −3.59 | 7.4E-11 | 2.00 | 9.7E-09 |
| NM_146073 // Zdhhc14 // zinc finger, DHHC domain containing 14 | Zdhhc14 | −3.71 | 7.1E-09 | 2.39 | 1.8E-07 |
| NM_010658 // Mafb // v-maf musculoaponeurotic fibrosarcoma oncogene family | Mafb | −3.99 | 7.1E-10 | 2.09 | 1.0E-07 |
| NM_011882 // Rnasel // ribonuclease L | Rnasel | −4.29 | 1.0E-07 | 2.51 | 3.5E-06 |
| NM_175116 // Lpar6 // lysophosphatidic acid receptor 6 | Lpar6 | −5.30 | 1.6E-09 | 1.54 | 5.4E-05 |
| NM_009183 // St8sia4 // ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransf | St8sia4 | −5.53 | 1.9E-10 | 2.53 | 2.4E-08 |
| NM_009911 // Cxcr4 // chemokine (C-X-C motif) receptor 4 | Cxcr4 | −5.65 | 2.4E-12 | 1.74 | 2.0E-08 |
| NM_010427 // Hgf // hepatocyte growth factor | Hgf | −5.66 | 1.1E-09 | 1.72 | 9.7E-06 |
| NM_001042591 // Arrdc3 // arrestin domain containing 3 | Arrdc3 | −6.74 | 1.2E-08 | 1.95 | 3.8E-05 |
| NM_007564 // Zfp36l1 // zinc finger protein 36, C3H type-like 1 | Zfp36l1 | −8.01 | 7.3E-08 | 1.64 | 2.3E-03 |
| NM_146042 // Rnf144b // ring finger protein 144B | Rnf144b | −8.34 | 2.0E-11 | 1.70 | 1.1E-06 |
We next asked which transcription factors could mediate the antagonistic effects of Aβ42 and EP4 receptor signaling on the microglial inflammatory response. To answer this, we performed Ingenuity Pathway Analysis on the set of 116 differentially expressed transcripts. This analysis identified two transcription factor pathways most significantly over-represented among these transcripts, centering on NF-κB (Fig. 4A) and IRF 1 and 7 (Fig. 4B). A number of studies have previously identified NF-κB as a downstream effector of Aβ42-mediated inflammatory effects in microglia, through Aβ42 binding to TLR2, TLR4, and the RAGE (for review, see Landreth and Reed-Geaghan, 2009; Glass et al., 2010). The IRF family of transcription factors has been most well characterized in the antiviral immune response, where tight regulation of IRF expression and activity control the transcription of Type I interferons (for review, see Honda et al., 2006). IRF transcriptional activity, however, has been less well characterized in the inflammatory response to Aβ42. To confirm this finding by an independent analysis, we performed GSEA (Subramanian et al., 2005) to compare our array samples with gene sets enriched for different transcription factor binding sites in their promoters. Microglia treated with Aβ42 were highly enriched for gene expression from several transcription factor sets, including sets representing NF-κB and IRF-1 binding sites (normalized enrichment score > 1.60, nominal p < 0.0001, FDR q < 0.05). Moreover, the samples cotreated with EP4 agonist and Aβ42 were negatively enriched, compared with treatment with Aβ42 only, for all of these NF-κB and IRF-1 target gene sets (Fig. 4C). Together, these data indicate that positive regulation of IRF and NF-κB transcriptional activity by Aβ42, and negative regulation by EP4 receptor signaling, may underlie the anti-inflammatory effect of EP4 in microglia.
Figure 4.
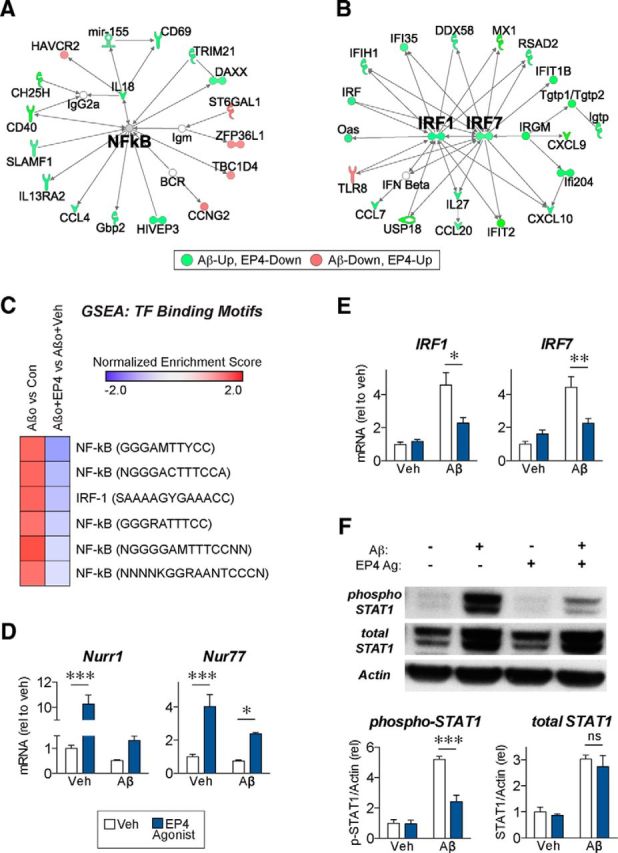
NF-κB and IRF pathways mediate the antagonistic effects of the EP4 receptor in microglial inflammatory responses to oligomeric Aβ42. A, B, Ingenuity Pathway Analysis was performed on the 116 Aβ42-responsive genes for which EP4 agonist treatment opposed the Aβ42 effect (fold change > 1.5 in Aβ42+EP4 agonist vs Aβ42+Veh comparison) in primary microglia cotreated for 6 h with oligomeric Aβ42 (10 μm) and/or the EP4 agonist AE1–329 (100 nm). Simplified diagrams of the two most significantly enriched pathways from these genes, centering on NF-κB and IRF1/IRF7, are displayed. C, GSEA for transcription factor (TF) binding motifs shows positive enrichment for NF-κB and IRF1 binding motifs in the Aβ42 vs Con comparison, and negative enrichment for the same sites in the Aβ42+EP4 agonist versus Aβ42+Veh comparison. D, qRT-PCR demonstrates that EP4 receptor stimulation increases expression of Nurr1 and Nur77, two nuclear receptors that repress NF-κB activity. E, qRT-PCR confirms that EP4 receptor stimulation attenuates expression of IRF1 and IRF7 in response to Aβ42. For each experiment, n = 3 microglia isolations from independent mouse cohorts. *p < 0.05 (Bonferroni multiple comparison test). **p < 0.01 (Bonferroni multiple comparison test). ***p < 0.001 (Bonferroni multiple comparison test). F, Western blot from BV2 microglial cells cotreated for 8 h with Aβ42- (10 μm) and/or the EP4 agonist AE1–329 (100 nm) demonstrates that Aβ42 increases both total and Tyr701-phosphorylated STAT1 levels, whereas EP4 agonist treatment attenuates phospho-STAT1 levels without altering total STAT1 protein level. n = 6 independent samples per group. ***p < 0.001 (Bonferroni multiple comparison test). ns, p > 0.05 (Bonferroni multiple comparison test).
Although our microarray analysis did not identify any changes in NF-κB subunit expression, we identified several EP4-regulated genes whose expression may contribute to the suppression of NF-κB activity. In particular, the anti-inflammatory nuclear receptors Nurr1 (gene name Nr4a2) and Nur77 (gene name Nr4a1) were significantly increased by EP4 agonist treatment (Fig. 4D). Notably, Aβ42 significantly suppressed overall expression for both Nurr1 and Nur77 (F(1,8) = 159.2, p < 0.0001 for Nurr1 and F(1,8) = 6.48, p = 0.0344 for Nur77 effects of Aβ42 treatment). Previous studies have demonstrated that Nurr1 directly binds to NF-κB on inflammatory gene promoters and, by recruitment of an inhibitory CoREST complex, clears NF-κB from the promoters and thereby represses target gene expression (Saijo et al., 2009). Similarly, Nur77 overexpression reduces, whereas Nur77 deletion enhances, the expression of NF-κB targets in inflammatory macrophages (Saijo et al., 2009; Hanna et al., 2012). These results suggest that EP4 receptor signaling can regulate NF-κB activity through the expression of anti-inflammatory nuclear receptors, adding to previously published mechanisms in which EP4 suppresses NF-κB activity through unique EP4 binding partners that retain NF-κB in the cytosol (Minami et al., 2008) or through inhibition of the Akt/IKK/I-κB pathway that mediates NF-κB nuclear translocation (Shi et al., 2010).
Unlike our results for NF-κB, the microarray results for IRF1 and IRF7 suggested that these transcription factors were themselves transcriptionally modulated by Aβ42 and EP4 receptor signaling. qPCR from primary microglia confirmed that Aβ42 treatment increased IRF1 and IRF7 mRNA levels with significant attenuation by EP4 agonist treatment (Fig. 4E). Previous findings have established that IRF7 expression can be maintained by a positive feedback loop: Type I interferons signal through their receptor to phosphorylate STAT1, which then translocates to the nucleus to promote the expression of the Irf7 gene and the resulting expression of Type I interferons (Marié et al., 1998; Honda et al., 2006). To test whether this pathway is active in the microglial responses to Aβ42 and EP4, we performed Western blots from BV2 microglial cells and found that levels of phosphorylated STAT1 were highly increased by Aβ42 and significantly attenuated with EP4 agonist cotreatment (Fig. 4F). These data suggest that EP4 receptor signaling antagonizes not only the expression of IRF1 and IRF7 but also exerts an anti-inflammatory function by suppressing the positive feedback loop controlling further IRF transcription. Together, these data suggest a mechanism in which Aβ42 stimulates IRF1/7 and NF-κB transcription of inflammatory genes, whereas EP4 receptor signaling, through downregulation of IRF1/7 and upregulation of Nurr1/Nur77, represses the transcription of proinflammatory IRF and NF-κB target genes in microglia.
Deletion of microglial EP4 receptor enhances inflammation and amyloid burden in APP-PS1 mice
To test the effect of microglial EP4 receptor signaling in vivo in a model of AD, we turned to a conditional knock-out approach to delete EP4 in microglia. We used mice carrying the EP4lox allele (Schneider et al., 2004) to generate CD11bCre;EP4+/+ and CD11bCre;EP4lox/lox mice in which the EP4 receptor is selectively deleted in cells of the monocytic lineage, including microglia. By crossing these mice to APPSwe-PS1ΔE9 (APP-PS1) AD model mice, we generated APP-PS1mice of four genotypes: APP-PS1;EP4+/+, APP-PS1;EP4lox/lox, APP-PS1;CD11bCre;EP4+/+, and APP-PS1;CD11bCre;EP4lox/lox. The first three of these APP-PS1 genotypes have functional EP4 receptor signaling in microglia, and demonstrated no significant differences among them for all assays tested. Therefore, we considered these genotypes as APP-PS1;EP4 wild-type (APPS;EP4-WT) and compared mice from this group to APP-PS1;CD11bCre;EP4lox/lox mice (APPS;EP4cKO) and to nontransgenic controls (Fig. 5).
Figure 5.

EP4 receptor conditional knock-out in microglia enhances inflammation and amyloid deposition early at 5 months in APPSwe-PS1ΔE9 (APPS) mice. Microglial EP4 receptor conditional knock-out (EP4-cKO) 5 month female and 9 month male APPS or nontransgenic (Non-tg) mice were generated using the CD11b-Cre; EP4lox/lox strategy. A, qRT-PCR from hippocampal RNA demonstrates elevated CCL3 expression in APPS EP4-cKO mice at 5 months but not 9 months, and a trend toward increased IL-1β in 5 month APPS-EP4cKO mice. B, LC/ESI/MS/MS analysis of cortical lysates demonstrates elevated γ-ketoaldehyde adduct levels in young APPS EP4-cKO mice at 5 months. Aβ40 (C) and (D) Aβ42 ELISAs from guanidine-extracted cerebral cortex demonstrate increased Aβ levels in EP4-cKO mice in 5 month but not 9 month APPS-EP4cKO mice. E, Immunostaining for 6E10-positive Aβ plaques (arrows) and quantification of Aβ plaque area in the hippocampus (F) demonstrates increased Aβ plaque area and density in APPS EP4-cKO mice at 5 months of age. G, Congo Red staining for plaque cores (white arrows) and quantification of Congo Red-positive area in the hippocampus and cerebral cortex (H) demonstrates increased dense-core plaque percentage area in APPS EP4-cKO mice (A–H; n = 4–11 mice per group). *p < 0.05 (unpaired t test). **p < 0.01 (unpaired t test). ***p < 0.0001 (unpaired t test). I, J, Quantitative Western analysis of PSD-95 demonstrates significant decrease in 5 month (n = 4/genotype; p < 0.05) and in 9 month cohorts (n = 3–5 mice per genotype; ANOVA p = 0.032, post hoc Bonferroni multiple comparison. *p < 0.05, APP-PS1;Cd11bCre; EP4+/+ versus APP-PS1;Cd11Cre;EP4lox/lox, with conditional deletion of EP4 in APP-PS1 mice.
Because EP4 receptor activation was prominently anti-inflammatory in cultured microglia in response to acute stimulation with Aβ42 oligomers, we hypothesized that the inflammatory and oxidative responses might be exacerbated in aging APP-PS1 mice with conditional deletion of EP4 in microglia. The temporal course and the magnitude of the inflammatory response are quite different between acute Aβ42 stimulation of microglial cells in vitro (Figs. 123-4) and the more chronic evolving inflammatory response to Aβ42 generation in transgenic APP-PS1 mice. However, qPCR of hippocampal mRNA demonstrated modest increases in expression of selected inflammatory proteins, including CCL3 and IL1β in 5 month female APPS:EP4-cKO mice, an effect that disappeared in older 9 month male APPS:EP4-cKO cohorts (Fig. 5A,B). Our previous studies indicate no differences in levels of inflammation and Aβ peptide levels between male and female genders at 5 months in this model, which coincides with the onset of Aβ plaque deposition (data not shown). The disappearance at 9 months of differences between the inflammatory profiles of APPS-WT and APPS-EP4cKO may occur because of a ceiling effect in older APP-PS1 mice where the inflammatory responses are much greater, and where further increases in inflammation from EP4 microglial deletion may be difficult to detect.
Inflammatory genes that were highly regulated in vitro, including iNOS, TNF-α, COX-2, IRF1, and IRF7, were not differentially regulated in whole hippocampus at either age between nontransgenic and APP-PS1 mice, or between APPS-WT and APPS;EP4cKO mice (data not shown). However, levels of oxidative protein modification were significantly increased early at 5 months in APPS:EP4-cKO mice compared with both nontransgenic and APPS-WT mice, but this effect was not seen at 9 months of age. Here, we examined the generation of a class of reactive aldehydes, the γ-ketoaldehydes, that are formed through nonenzymatic lipid peroxidation by reactive oxygen species and through enzymatic COX-2 activity, two processes active during inflammation. The aldehyde moiety of γ-ketoaldehydes readily reacts with the ε-amine of lysines, resulting in the covalent addition of a hydrophobic aldehyde to proteins that can be measured as lysyl-lactams by LC/ESI/MS/MS. Lactam levels, a highly sensitive readout of inflammatory oxidative protein injury, are elevated in the hippocampus of AD patients (Zagol-Ikapitte et al., 2005) and increase the toxicity of Aβ42 in cultured neurons (Boutaud et al., 2006). We found that young but not older EP4-cKO mice had elevated lactam adduct levels in cerebral cortex (Fig. 5B), indicative of heightened protein damage by reactive aldehydes.
Because our previous data showed that EP4 receptor signaling increased phagocytosis of Aβ42, we next tested whether deletion of the microglial EP4 receptor would lead to enhanced Aβ peptide deposition in APP-PS1 mice. ELISA of cortical extracts demonstrated that APPS;EP4-cKO mice had elevated levels of both Aβ42 and Aβ40 at 5 months, but this effect disappeared by 9 months of age (Fig. 5C,D), a pattern similar to that observed for inflammatory gene expression and lipid peroxidation. Additional confirmation of increased Aβ accumulation in 5 month APPS-WT and APPS-EP4cKO mice was performed (Fig. 5E–H). Here, amyloid plaque quantification by both 6E10 immunostaining for total Aβ (Fig. 5E,F) and Congo Red staining for the β-pleated sheets of dense-core amyloid plaques (Fig. 5G,H) demonstrated a significant increase in amyloid deposition in hippocampus of 5 month EP4-cKO mice (1.76-fold for 6E10; 2.14-fold for Congo Red; Fig. 5G). Similar results were obtained in cerebral cortex (Fig. 5H).
Because inflammatory and oxidative stress can impact on synaptic viability, we assayed candidate presynaptic and postsynaptic markers in 5 month and 9 month cerebral cortex and tested whether deletion of anti-inflammatory EP4 signaling may impact on levels of synaptic proteins. Quantitative Western analysis of presynaptic proteins synaptophysin, synapsin 1, and SNAP-25 did not show differences between genotypes at either age, but the postsynaptic marker PSD-95 was significantly reduced at both ages in APPS-EP4cKO compared with APPS-WT cohorts (Fig. 5I,J). Together, these findings demonstrate in vivo that microglial EP4 signaling reduces oxidative inflammation and limits Aβ deposition early at 5 months in the APP-PS1 model of AD, but this effect is lost at the later age of 9 months; loss of the postsynaptic marker PSD-95, however, appears sustained with increasing age.
EP4 receptor protein levels are reduced in the brains of AD patients
To assess the potential clinical relevance of EP4 receptor signaling in the progression of AD, the cellular expression pattern of the EP4 receptor was examined by immunohistochemistry in cortical sections from AD, MCI, and age-matched control patients (Fig. 6A–D). Control cortex demonstrated high levels of EP4 receptor staining in cells with both neuronal and glial morphology, including small cells resembling microglia (Fig. 6A). The overall level of EP4 receptor staining was reduced in MCI and AD patients, most strikingly in neurons (Fig. 6B,C). This is especially interesting in light of recent in vivo studies identifying neuronal EP4 receptor signaling as a protective pathway in models of cerebral ischemia (Liang et al., 2011). In both MCI and AD patients, we observed EP4-receptor-positive cells with microglial morphology adjacent to Aβ plaques (Fig. 6B,C). To quantify the levels of EP4 receptor in human brain, we performed Western blot on temporal cortex lysates from AD, MCI, and age-matched control patients and found a significant reduction in EP4 receptor levels in both the MCI and AD patient samples compared with controls (Fig. 6E,F), reflecting potentially a combined loss of neuronal and glial EP4 receptor. Notably, this decrease in EP4 expression is in the opposite direction from the increase in the proinflammatory EP3 receptor expression observed in both human AD cortex and in APP-PS1 mouse hippocampus (Shi et al., 2012). To determine whether EP4 receptor expression is modulated similarly in APP-PS1 mice as it is in human AD brain, we assessed EP4 mRNA expression levels by qPCR from the hippocampus of 12 month APP-PS1 mice. Here, we found no difference in EP4 expression between WT and APP-PS1 mice (1.00 ± 0.20, n = 6; vs 1.06 ± 0.14, n = 5; p = 0.82 by unpaired t test). This divergence between human AD and the APP-PS1 mouse model could be reflective of several ways in which mouse models of AD fail to recapitulate the full human disease. For instance, mouse AD models generally exhibit little or no neuronal loss, whereas human AD is characterized by progressive and extensive neuronal loss. In light of this, the observed decrease of EP4 receptor levels in MCI and AD brain could be reflective of either loss of EP4 protein from dying neurons or a shift in EP receptor expression patterns among multiple cell types away from anti-inflammatory EP4 receptor expression and toward proinflammatory EP2 and EP3 receptor expression. Although the precise mechanisms remain unclear for the differential EP receptor expression profiles in MCI and AD, future clinical studies assessing expression patterns among different cell types in AD brain may shed light on these pathways in disease pathogenesis.
Figure 6.

EP4 receptor protein levels are depleted in the cortex of AD patients. Sections from parietal cortex of (A) age-matched control, (B) MCI, and (C) AD patients were immunostained for EP4 receptor (brown) and Aβ (purple). Images from cortical layer V demonstrate EP4 receptor expression in the cell bodies and dendrites of neurons (N) and in smaller cells with glial morphology (G). Overall EP4 receptor staining levels were reduced in MCI and AD brain, particularly in neurons. Small EP4-receptor-positive cells with microglial morphology were identified near amyloid plaques in MCI and AD brain. D, Adjacent sections stained with control immunoglobulin demonstrate low background staining. Scale bars, 20 μm. E, Western blot. F, Quantification from control, MCI, and AD temporal cortex lysates demonstrates decreased EP4 receptor expression in MCI and AD brain. n = 3 samples per group. p (one-way ANOVA). *p < 0.05 (Dunnett's post test).
Discussion
Our findings identify EP4 receptor signaling in microglia as a potent suppressor of the inflammatory response to immunogenic Aβ42 oligomers. Using both in vitro and in vivo approaches, we demonstrate that EP4 receptor stimulation attenuates, whereas microglial EP4 receptor deletion enhances, the oxidative and cytokine/chemokine responses to Aβ42. Our studies suggest a model by which EP4 receptor signaling and Aβ42 exert opposing effects on microglia. Whereas Aβ42 induces expression of transcripts associated with NF-κB, IRF1, and IRF7 networks, EP4 receptor signaling suppresses these effects and may stimulate phagocytosis of Aβ42. When EP4 receptor signaling is inhibited or absent, Aβ42-induced inflammatory responses are enhanced, with additional amplification through the previously reported positive feedback cycle of COX-2 expression, PGE2 production, and proinflammatory signaling through the EP2 and EP3 receptors (Shie et al., 2005a, b, 2012). Our results suggest that EP4 receptor signaling suppresses and interrupts this feedforward inflammatory loop, and they establish a mechanism by which one ligand, PGE2, may exert opposing effects on microglia depending on the EP receptor subtype(s) expressed and activated (Fig. 7).
Figure 7.

Model of early and late effects of EP4 signaling in Aβ42-associated AD neuroinflammation. A, Aβ42 oligomers initiate an inflammatory response in microglia through IRF and NF-κB transcriptional pathways, resulting in expression of many proinflammatory genes, including COX-2, cytokines such as TNF-α, and reactive oxygen species (ROS) generating enzymes such as iNOS. COX-2 is the rate-limiting step in the production of PGE2, which can signal through its four EP receptors, EP1-EP4. EP4 receptor signaling suppresses expression of proinflammatory IRF and genes regulated by NF-κB and significantly blunts the inflammatory response to Aβ while promoting phagocytosis and amyloid clearance. B, In the absence of EP4 receptor signaling, Aβ-driven inflammation proceeds unchecked. Increased COX-2 activity and PGE2 production though the proinflammatory EP2 and EP3 PGE2 receptors (Liang et al., 2005; Shie et al., 2005a; Shi et al., 2012) further amplify the inflammatory gene response leading to a feedforward cycle of persistent and unresolved inflammation.
Although our findings point to a broad immunosuppressive function of EP4 signaling in Aβ42 models of neuroinflammation, it is important to note several limitations inherent in the models we tested. In purified primary microglia, EP4 signaling elicited a profound anti-inflammatory effect in response to acute stimulation with Aβ42 oligomers; in vivo, we observed a more muted effect on inflammatory gene expression in whole hippocampi of 5 month cohorts that disappeared at later stages of pathology in 9 month cohorts. This contrast could be the result of several factors, most importantly differences between an acute in vitro response to Aβ42 treatment in a purified microglial population versus a chronic in vivo response in whole hippocampus to low-level transgenic overexpression of Aβ for several months. A potentially more sensitive in vivo readout, namely, measurement of lysyl-lactam adducts formed from the reaction of γ-ketoaldehydes with proteins, supports the anti-inflammatory role for the EP4 receptor in APP-PS1 mice. Lactam adducts are permanent and covalent modifications of proteins; thus, lactam levels represent a cumulative index of protein oxidative injury. Similarly, the sustained loss of the postsynaptic marker PSD-95 in both young and older APPS-EP4-cKO cohorts may be similarly indicative of cumulative synaptic injury. In vivo, the parallel increases in inflammatory gene expression, lysine adducts, and Aβ peptides in APPS-EP4cKO mice early at 5 months, but not at later stages at 9 months, suggests an early and beneficial effect of EP4 signaling at the onset of Aβ peptide accumulation that targets the nascent inflammatory and oxidative responses. The sustained loss of PSD-95 in 9 month APPS-EP4-cKO mice further suggests that this early beneficial effect of EP4 signaling has ramifications at later stages of pathology.
Loss of microglial EP4 early in the progression of pathology in 5 month APPS-PS1 mice increased inflammatory and amyloid pathology. At this stage, there is minimal inflammation and Aβ peptide deposition. However, later at 9 months of age, when amyloid deposition, secondary inflammatory responses, and synaptic injury are well underway in APP-PS1 mice, the effect of cKO of microglia EP4 was less evident. This age dependence in EP4 effect may represent a ceiling effect, whereby eliminating anti-inflammatory EP4 signaling may not significantly alter the already robust inflammatory response; alternatively, it may suggest that microglia, which can change significantly in their inflammatory and phagocytic phenotype with progressive Aβ deposition in mutant APP models (Krabbe et al., 2013), may not successfully engage the anti-inflammatory EP4 signaling pathway.
The in vitro microarray results are helpful in identifying mechanisms by which the EP4 receptor suppresses Aβ42-mediated inflammation, as well as the overall microglial response to oligomeric Aβ42. We confirm the well-established role of NF-κB in microglia exposed to Aβ42 (Glass et al., 2010) and identify Nurr1 and Nur77 as novel anti-inflammatory targets of EP4 receptor signaling that may suppress this Aβ42-driven NF-κB response. While innate immune stimulation with LPS has been found to increase Nurr1 expression by microglia (Saijo et al., 2009), we found that Aβ42 indeed reduced expression of both Nurr1 and Nur77 by microglia. This suggests that Nurr1/Nur77 depletion may contribute to the proinflammatory effect of Aβ42 and that EP4 signaling may oppose Aβ42-induced inflammation by restoring the expression of these nuclear receptors. Notably, this mechanism adds to previously reported mechanisms for EP4 receptor antagonism of NF-κB transcriptional activity in models of LPS-induced innate immunity (Minami et al., 2008; Shi et al., 2010).
The second Aβ42-associated transcription factor network identified in our microarray centered on IRF1 and/or IRF7 transcription factors and the Type I interferon response. Here, EP4 receptor signaling suppressed Aβ42-induced IRF1 and IRF7 transcript levels and the phosphorylation of the relevant transcription factor STAT1. The regulation of IRF7 is particularly interesting, given that this transcription factor has been described as the master regulator of Type I interferon responses to viral infection (Honda et al., 2005). Although this transcriptional network has not been widely studied in AD, a growing literature suggests that Type I interferons and IRFs are elevated in microglia and astrocytes in diverse models of neurodegeneration including amyotrophic lateral sclerosis (Wang et al., 2011), prion disease (Field et al., 2010), and axonal injury (Wang et al., 2011). Although our studies did not detect elevated IRF transcript levels in APP-PS1 mice, future studies of more severe AD models could clarify this finding and more directly connect AD models to the extensive literature on IRF signaling in the innate immune response to viruses and other pathogens.
Quantitative Western analysis and immunocytochemistry demonstrated that levels of EP4 receptor decline significantly with progression from control to MCI and AD states. This suggests that loss of this beneficial anti-inflammatory signaling pathway may contribute to disease acceleration in preclinical development of AD. It is interesting to note that MCI and AD patients show increased expression of the proinflammatory EP3 (Shi et al., 2012) and EP2 receptors in cerebral cortex (J. Johansson, unpublished results). This contrasting expression pattern of inflammatory and anti-inflammatory PGE2 EP receptors is consistent with a shift away from anti-inflammatory and neuroprotective EP4 receptor expression and toward toxic EP2/3 receptor expression in the brain, even at the earliest stages of cognitive impairment (Fig. 7). These expression changes likely occur across multiple cell types, as we observed visible decreases in neuronal EP4 receptor staining in MCI and AD brain. Consistent with this, protective effects of the EP4 receptor are exerted through multiple cell types, including neurons and endothelial cells in models cerebral ischemia and excitotoxicity (Liang et al., 2011). Importantly, EP4 receptor signaling may also exert detrimental effects in other cell types: for example, internalization of the EP4 receptor has been shown to increase the production of Aβ, potentially through its interaction with presenilin-1 (Hoshino et al., 2009). These results underscore the need to investigate signaling in multiple cell types (e.g., in neurons that produce Aβ and in microglia that mount an inflammatory response to Aβ) to gain a more complete understanding of how molecular pathways contribute to complex neurodegenerative diseases. As of yet, mechanisms that regulate the differential expression of EP receptor subtypes among different cell types remain largely unexplored, but our studies suggest that identifying these mechanisms could yield insight into the shifting function of PGE2 signaling over the development of neurodegenerative disease.
In addition, our results may help clarify the role of NSAIDs, which reduce the production of PGE2 through inhibition of COX-1 and/or COX-2. Epidemiological evidence indicates that NSAID use is associated with reduced risk of AD in cognitively normal aging populations (in t' Veld et al., 2001; McGeer and McGeer, 2007; Vlad et al., 2008); however, NSAIDs are ineffective once cognitive change begins (Breitner et al., 2011). In light of recent studies, preventive effects of NSAIDs may be the result, at least in part, of reduced PGE2 signaling through inflammatory EP2 and EP3 receptors: deletion of either EP2 or EP3 reduces inflammatory damage and amyloid deposition in mouse models of AD (Liang et al., 2005; Shi et al., 2012), and microglial EP2 receptor signaling is potently proinflammatory in models of neuroinflammation and neurodegeneration (Johansson et al., 2013). On the other hand, the harmful effects of NSAIDs in AD may occur in part from reduced PGE2 signaling through the anti-inflammatory EP4 receptor: our results suggest that EP4 signaling attentuates the inflammatory response to Aβ42 at early stages of APP-PS1 pathology. Together, PGE2 signaling through its EP receptors suggest that PGE2 can exert both toxic and beneficial effects in models of AD.
In conclusion, our studies identify EP4 receptor signaling as a potent mechanism through which microglia suppress toxic inflammatory responses to Aβ42 and potentiate phagocytosis of Aβ42 using in vitro culture and in vivo conditional knock-out strategies. Moreover, these results identify NF-κB, IRF1, and IRF7 as nodal transcription factors in the microglial response to Aβ42 and demonstrate that they are suppressed by EP4 receptor signaling. These findings support future approaches targeting the EP4 receptor to suppress toxic microglial inflammatory responses in AD and other neurodegenerative diseases.
Footnotes
This work was supported by the American Federation for Aging Research RO1AG030209 and R21AG033914 to K.I.A., R21AG042194 to K.I.A. and O.B., and UL1TR000445 to O.B., Alzheimer's Association to K.I.A., National Science Foundation Graduate Research Fellowship to N.S.W., and National Institutes of Health NRSA F31AG039195 to N.S.W. We thank the Stanford PAN facility for assistance with microarray experiments and Dr. Yoon-Jae Cho for helpful advice with GSEA analysis.
The authors declare no competing financial interests.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Boutaud O, Montine TJ, Chang L, Klein WL, Oates JA. PGH2-derived levuglandin adducts increase the neurotoxicity of amyloid beta1–42. J Neurochem. 2006;96:917–923. doi: 10.1111/j.1471-4159.2005.03586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN. Extended results of the Alzheimer's disease anti-inflammatory prevention trial. Alzheimers Dement. 2011;7:402–411. doi: 10.1016/j.jalz.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field R, Campion S, Warren C, Murray C, Cunningham C. Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun. 2010;24:996–1007. doi: 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, Hedrick CC. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ Res. 2012;110:416–427. doi: 10.1161/CIRCRESAHA.111.253377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He PP, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Heneka MT, O'Banion MK. Inflammatory processes in Alzheimer's disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Namba T, Takehara M, Nakaya T, Sugimoto Y, Araki W, Narumiya S, Suzuki T, Mizushima T. Prostaglandin E2 stimulates the production of amyloid-beta peptides through internalization of the EP4 receptor. J Biol Chem. 2009;284:18493–18502. doi: 10.1074/jbc.M109.003269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/S1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson JU, Pradhan S, Lokteva LA, Woodling NS, Ko N, Brown HD, Wang Q, Loh C, Cekanaviciute E, Buckwalter M, Manning-Bog AB, Andreasson KI. Suppression of inflammation with conditional deletion of the prostaglandin E2 EP2 receptor in macrophages and brain microglia. J Neurosci. 2013;33:16016–16032. doi: 10.1523/JNEUROSCI.2203-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, Tsuboi K, Sugimoto Y, Kobayashi T, Miyachi Y, Ichikawa A, Narumiya S. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, Miller KR, Prokop S, Kettenmann H, Heppner FL. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. doi: 10.1371/journal.pone.0060921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreth GE, Reed-Geaghan EG. Current topics in microbiology and immunology. Berlin: Springer; 2009. Toll-like receptors in Alzheimer's disease; pp. 137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Liang X, Wang Q, Hand T, Wu L, Breyer RM, Montine TJ, Andreasson K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer's disease. J Neurosci. 2005;25:10180–10187. doi: 10.1523/JNEUROSCI.3591-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Lin L, Woodling NS, Wang Q, Anacker C, Pan T, Merchant M, Andreasson K. Signaling via the prostaglandin E(2) receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. J Clin Invest. 2011;121:4362–4371. doi: 10.1172/JCI46279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging. 2007;28:639–647. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Minami M, Shimizu K, Okamoto Y, Folco E, Ilasaca ML, Feinberg MW, Aikawa M, Libby P. Prostaglandin E receptor type 4-associated protein interacts directly with NF-kappaB1 and attenuates macrophage activation. J Biol Chem. 2008;283:9692–9703. doi: 10.1074/jbc.M709663200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Camenisch T, Snouwaert JN, Hicks E, Coffman TM, Anderson PA, Malouf NN, Koller BH. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- Passos GF, Figueiredo CP, Prediger RDS, Pandolfo P, Duarte FS, Medeiros R, Calixto JB. Role of the macrophage inflammatory protein-1alpha/CC chemokine receptor 5 signaling pathway in the neuroinflammatory response and cognitive deficits induced by beta-amyloid peptide. J Pathol. 2009;175:1586–1597. doi: 10.2353/ajpath.2009.081113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM, Breyer MD. Generation of a conditional allele of the mouse prostaglandin EP4 receptor. Genesis. 2004;40:7–14. doi: 10.1002/gene.20048. [DOI] [PubMed] [Google Scholar]
- Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010;184:7207–7218. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang Q, Johansson JU, Liang X, Woodling NS, Priyam P, Loui TM, Merchant M, Breyer RM, Montine TJ, Andreasson K. Inflammatory prostaglandin E(2) signaling in a mouse model of Alzheimer disease. Ann Neurol. 2012;72:788–798. doi: 10.1002/ana.23677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Breyer RM, Montine TJ. Microglia lacking E prostanoid receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. J Pathol. 2005a;166:1163–1172. doi: 10.1016/S0002-9440(10)62336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shie FS, Montine KS, Breyer RM, Montine TJ. Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia. 2005b;52:70–77. doi: 10.1002/glia.20220. [DOI] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141:1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- Vlad SC, Miller DR, Kowall NW, Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL. Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med. 2012;18:1812–1819. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- Wang R, Yang B, Zhang D. Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia. 2011;59:946–958. doi: 10.1002/glia.21167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yang T, Knowles JK, Lu Q, Zhang H, Arancio O, Moore LA, Chang T, Wang Q, Andreasson K, Rajadas J, Fuller GG, Xie Y, Massa SM, Longo FM. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS One. 2008;3:e3604. doi: 10.1371/journal.pone.0003604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagol-Ikapitte I, Masterson TS, Amarnath V, Montine TJ, Andreasson KI, Boutaud O, Oates JA. Prostaglandin H(2)-derived adducts of proteins correlate with Alzheimer's disease severity. J Neurochem. 2005;94:1140–1145. doi: 10.1111/j.1471-4159.2005.03264.x. [DOI] [PubMed] [Google Scholar]
- Zhen G, Kim YT, Li RC, Yocum J, Kapoor N, Langer J, Dobrowolski P, Maruyama T, Narumiya S, Doré S. PGE2 EP1 receptor exacerbated neurotoxicity in a mouse model of cerebral ischemia and Alzheimer's disease. Neurobiol Aging. 2012;33:2215–2219. doi: 10.1016/j.neurobiolaging.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]