

Abstract
Multi-site enzymes, defined as where multiple substrate molecules can bind simultaneously to the same enzyme molecule, play a key role in a number of biological networks, with the Escherichia coli protease ClpXP a well-studied example. These enzymes can form a low latency ‘waiting line’ of substrate to the enzyme's catalytic core, such that the enzyme molecule can continue to collect substrate even when the catalytic core is occupied. To understand multi-site enzyme kinetics, we study a discrete stochastic model that includes a single catalytic core fed by a fixed number of substrate binding sites. A natural queueing systems analogy is found to provide substantial insight into the dynamics of the model. From this, we derive exact results for the probability distribution of the enzyme configuration and for the distribution of substrate departure times in the case of identical but distinguishable classes of substrate molecules. Comments are also provided for the case when different classes of substrate molecules are not processed identically.
Keywords: queueing, multi-class, Michaelis–Menten, ClpXP, protease, enzyme
1. Introduction
The study of rate laws describing enzyme kinetics has a long history, with early representative works being that of Michaelis and Menten [1] and Monod. Analysis of these enzymatic bottlenecks is often assisted by coarse-graining the enzyme into a discrete set of states, e.g. a traditional two-state Michaelis–Menten approximation with distinct states corresponding to enzyme bound or unbound to substrate. However, even simple deterministic Michaelis–Menten models can be difficult to properly analyse without some form of approximation [2–4], and this difficulty is compounded when stochastic dynamics are also considered [5–12]. Given that the majority of cellular processes are governed by enzymatic reactions, progress in the development of intuitive but powerful mathematical approaches to enzymatic kinetics immediately impacts our understanding of many biological networks.
One recent approach to enzyme–substrate kinetics has been queueing theory. Queueing theory, which treats the routing of individual ‘customers’ between different ‘stations’ of ‘servers’, was historically developed for telecommunication networks and service centres [13]. A cursory glance over the queueing theory literature suggests that there are several notable strengths of a potential queueing theory approach to enzyme kinetics. First, queueing theory's fundamental assumption of an underlying discrete and stochastic model leads to a focus on results that are stochastic in origin, which meshes nicely with the realization that natural cellular networks are often noisy [14]. Second, queueing theory's assumption of finite bandwidth for the processing of customers by servers is entirely analogous to the finite bandwidth of reactions catalysed by enzyme. Finally, the heavy influence of queueing theory in a variety of disciplines, e.g. operations research, suggests that some biologically motivated questions tied to queueing may already have been answered in a different context. Pursuit of the queueing approach to biological problems has indeed been fruitful thus far, leading to progress in gene networks [15–19], metabolic networks [20,21], degradation pathways [22], signalling [23,24] and translational cross talk [25].
In this article, we extend a prior investigation [22] of ClpXP proteolytic kinetics. ClpXP is a well-studied protease in Escherichia coli that is responsible for degrading mistranslated proteins and (in healthy conditions) certain stress response factors [26–28]. ClpXP's function is greatly enhanced by the presence of multiple binding sites for substrate via interaction with a molecular chaperone, SspB [29–35]. Absence or dysfunction of the SspB molecule is strongly associated with reduced substrate affinity, since SspB bound to ClpX binding sites forms a ‘waiting line’ of substrate for the ClpXP catalytic core, thus preventing the catalytic core of ClpXP from being unoccupied by substrate for any appreciable duration of time. An appropriate queueing analogy for a single ClpXP molecule is then a server (catalytic core) that selects customers (protein substrate) at random from a finite capacity waiting line (SspB–substrate complex associated with ClpX binding sites) and processes (degrades) the customers (substrate). In the following, we analyse this model in some detail, and we argue that the multi-site nature of the enzyme drives the model into a regime well approximated by traditional queueing models. It is also worth mentioning that our investigation of multi-site enzyme kinetics is also multi-class, i.e. exactly treating the dynamics of multiple types of substrate competing for the same enzyme. This analysis is assisted by a new result, independent departure symmetry, which generalizes results originally derived for quasi-reversible queues. These multi-class results rely on the assumption that different classes of substrate are distinguishable but otherwise identical, but we will comment on why we anticipate that our results are not particularly sensitive to weakly breaking this assumption.
The coarse-grained models used to derive these results are sufficiently general that a number of other multi-site enzymes, e.g. AAA+ proteases other than ClpXP [36], may have similar waiting line behaviour. For example, the protease ClpAP uses the chaperone ClpS in much the same way ClpXP uses the chaperone SspB. Truly, given the many advantages of a multi-site motif, we anticipate our results will generalize to a wide array of enzymes in native and synthetic biological networks. Despite this, we will typically refer only to ClpXP when discussing results from our model, since it is one of the best understood multi-site enzymes.
In line with queueing theory [13], many of our results pertain to statistics of the ‘departure’ times of substrate from the enzyme, i.e. the times when substrate has been processed and expelled from the catalytic core. Understanding the statistics of departures is a natural way to characterize the behaviour of such discrete stochastic processes, e.g. the departures of a transcription process can be used to investigate burstiness of mRNA production [37]. We find that our multi-site enzyme models are far from bursty, with the departure process often being well represented by a Poisson process (independent exponential times between departures), which incidentally is the same result found in a variety of classical queueing networks [38].
The article is organized in the following way. In §2, a discrete stochastic model for multi-site enzyme kinetics is presented. Steady-state analysis in the case of a single class of substrate appears in §3, while the multi-class case is treated in §4. Section 5 investigates the influence of time-dependent substrate levels on single-class and multi-class settings. Concluding comments appear in §6.
2. Stochastic model definition
We wish to study a coarse-grained model for ClpXP (and similar enzymes), with a specific emphasis on multi-site dynamics, where multiple substrate molecules can bind and wait while another substrate molecule is being processed by the catalytic core. Illustrated in figure 1a, our queueing-inspired model for proteolytic degradation by ClpXP is chosen to be a system with N binding sites for substrate (the waiting line), a single catalytic core (the server) and R total species (classes) of substrate. When discussing ClpXP specifically, a value N = 6 would be reasonable based on the sixfold symmetry of ClpX, though the situation is made slightly more complex owing to SspB–substrate complexes typically binding to ClpXP as dimers [34] (we do not include this feature of ClpXP dynamics in the model). Unoccupied binding sites are occupied by substrate of class i with rate ηi(1 ≤ i ≤ R). An occupied binding site can deliver substrate of class i to be processed by an unoccupied catalytic core with rate ξi, and then the occupied catalytic core processes substrate of class i with rate μi. For reference, an analogous Michaelis–Menten system would only model the catalytic core.
Figure 1.
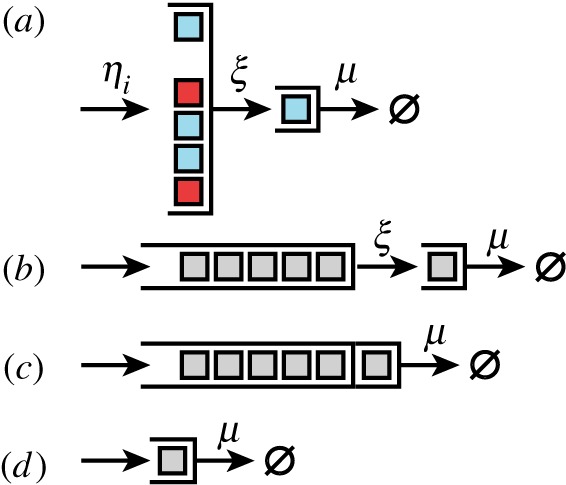
(a) A detailed model for multi-site enzymatic kinetics, e.g. for ClpXP degradation, with N binding sites for substrate (N = 6 in the figure). Free substrate of type i is recruited to each unoccupied site of the protease with rate ηi. Substrate bound to these sites can be passed along to an unoccupied catalytic core at rate ξ, which we typically assume is fast. Substrate bound to the catalytic core is processed, e.g. degraded, with rate μ. (b) Since the enzyme treats substrates of different types equally, by assumption, the enzyme's behaviour without regard to substrate class is that of a finite capacity queue feeding into another single unit queue. Then, the binding propensity of substrate becomes a function η · (N − M), where M is the number of occupied binding sites. (c) If bound substrate rapidly fills an empty catalytic core (ξ large), then the system reduces to a simple finite capacity queue with a single server. (d) For reference, a Michaelis–Menten enzyme (without substrate unbinding) can also be modelled as a queue with unit capacity. (Online version in colour.)
A set of reactions for the model are then
| 2.1 |
| 2.2 |
| 2.3 |
where i is a label for different kinds of substrate, Xn is the unoccupied nth binding site (1 ≤ n ≤ N), XnSi is the nth binding site occupied by substrate type i, Y is an unoccupied catalytic core and YSi is the catalytic core occupied by substrate type i. The rate constants ξi, and μi for passing and degrading substrate, respectively, can depend on substrate type i, but we set them to constants (μi = μ, ξi = ξ). This simplification in effect assumes that substrates are treated identically once bound, which will be important in our multi-class analysis. Furthermore, we suppose that ξ → ∞, i.e. that substrate is rapidly passed from occupied binding sites to an unoccupied catalytic site.
Free substrate is not treated explicitly in the analysis, but, instead, the rate constants ηi subsume the influence of free substrate concentration on the binding rate. Thus, we make a quasi-steady-state (Michaelis–Menten-like) approximation for the enzyme [10]. In the limit of large substrate count, where the concentration of substrate does not change appreciably on the time scale of enzyme kinetics, we predict that this approximation will be valid. It is worth noting that simpler models for multi-class enzyme kinetics have already been analysed in detail outside of the quasi-steady-state assumption [22], in which case free substrate can exhibit strongly correlated behaviour.
Finally, we neglect premature unbinding of substrate from the enzyme. This approximation is motivated by a large effective affinity of substrate to enzyme, e.g. owing to cooperative binding of the chaperone SspB. In the queueing interpretation, this is the same as supposing that customers do not renege, i.e. walk away from the service station owing to periods of inactivity. Refinement of the model to include the effects of substrate unbinding is straightforward, though it complicates analysis of the model.
Subsequent sections make use of the fact that, when analytic investigation is not easily achievable, the system represented by equations (2.1)–(2.3) maps precisely to a corresponding chemical master equation, which can be analysed numerically to find the probability for any system configuration [39]. For R substrate types, there are (R + 1)N+1 potential system configurations. Though the system size grows exponentially with N, the system with two classes of substrate and six binding sites has only 2187 probabilities to keep track of at any point in time, and if we also keep track of the last two departures (two additional virtual sites in the system occupied by the two substrates previously processed by the catalytic core), the number of probabilities increases to 8748. A modern computer can readily handle systems of these sizes, especially given the fact that the matrix governing evolution of this system is sparse. We use custom Python programs importing the SciPy package to perform these calculations [40].
3. Steady-state analysis for a single class of substrate
We first consider the dynamics of a multi-site enzyme in the case of a single class of substrate. This treatment reveals some of the most interesting properties of multi-site enzymes, including the ability for the binding sites of the enzyme to dramatically (cooperatively) increase the apparent affinity of the enzyme. Our analysis of a single class of substrate is applicable to several situations, including (i) when the enzyme is specific to a single class of substrate, (ii) when several very similar substrate classes are labelled as a single substrate class, and (iii) when only a single substrate class is sufficiently abundant to be relevant. The last two situations are relevant to ClpXP dynamics, since ClpXP generally degrades a variety of different substrates.
3.1. Steady-state distribution
At steady state, a multi-site enzyme is almost never fully depleted of substrate except at very low enzyme count, which implies that the enzyme is almost always busy processing bound substrate. We derive this property below by calculating the steady-state distribution of the enzyme configuration.
Define the occupation number Z, with 0 ≤ Z ≤ N + 1, such that the enzyme is devoid of substrate when Z = 0, while the enzyme possesses an occupied catalytic core and M = Z − 1 occupied binding sites for larger Z. Thus, the formula M = max(0, Z − 1) is a uniformly valid expression for the number of occupied binding sites. Also define the total rate of substrate binding η ≡ ∑iηi. The dynamics for Z can then be specified by the reactions
| 3.1 |
and
| 3.2 |
where we specify the propensities (i.e. excluding mass action terms), and where Θ(Z) is a Heaviside function (Θ(Z) = 1 for Z > 0, and Θ(Z) = 0 otherwise).
If Ps(z) is the steady-state distribution Pr(Z = z, t = ∞), then an elementary calculation for the one-dimensional master equation shows that the steady state is achieved by the zero net current (detailed balance) condition:
| 3.3 |
This recursion relationship is simpler when expressed in terms of the complementary variable 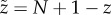 , i.e. the number of unoccupied sites. The recursion relationship in the new variable corresponds to the steady-state distribution for a complementary system with constant production rate μ and degradation rate (per substrate) η, excepting for a correction to the dynamics when
, i.e. the number of unoccupied sites. The recursion relationship in the new variable corresponds to the steady-state distribution for a complementary system with constant production rate μ and degradation rate (per substrate) η, excepting for a correction to the dynamics when 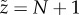 . It is well known that this complementary system should have a distribution similar to a Poisson distribution, and, with this inspiration, it is easy to find the steady-state distribution in the original variable
. It is well known that this complementary system should have a distribution similar to a Poisson distribution, and, with this inspiration, it is easy to find the steady-state distribution in the original variable
| 3.4 |
and
| 3.5 |
with λ ≡ μ/η, and  a normalization factor such that
a normalization factor such that 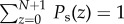 .
. 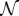 can be calculated numerically, but it also can be expressed in terms of incomplete gamma functions.
can be calculated numerically, but it also can be expressed in terms of incomplete gamma functions.
One limiting case is when 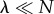 , i.e.
, i.e. 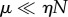 . This limit corresponds to the saturated regime, where the probability for the enzyme to be totally unoccupied (Ps(0)) is small. Since the distribution is then approximately a Poisson distribution (in the variable
. This limit corresponds to the saturated regime, where the probability for the enzyme to be totally unoccupied (Ps(0)) is small. Since the distribution is then approximately a Poisson distribution (in the variable 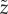 ), the normalization constant can be accurately approximated by the usual expression
), the normalization constant can be accurately approximated by the usual expression 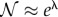 . The important quantity Ps(0) is then estimated to be
. The important quantity Ps(0) is then estimated to be
| 3.6 |
This value picks up a cooperativity of N + 1 with respect to the smallness parameter λ. Thus, multi-site enzymes can drastically increase their effective substrate affinity by leveraging a ‘waiting line’ for substrate. Figure 2a (large ηN) illustrates this effect. In its native context, ClpXP's multi-site nature allows it to efficiently degrade even small concentrations of potentially harmful prematurely terminated proteins.
Figure 2.
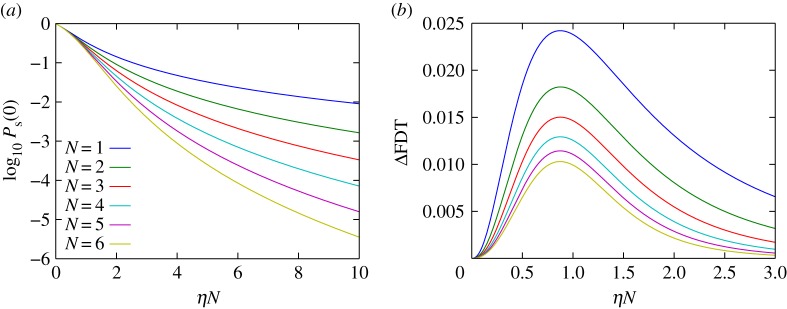
(a) For μ = 1 and ξ → ∞, the steady-state probability Ps(0) for the multi-site enzyme to be unoccupied as a function of ηN is presented (see equation (3.5)). A strong cooperative effect with increasing N is observed for large ηN, but this effect is lost for small ηN. (b) Integrated square deviation of the cumulative FDT distribution from a cumulative exponential distribution with the same mean departure time (see equations (3.14) and (3.15)). Maximal deviation from a perfect exponential is observed to occur near ηN = μ, which is where a broad distribution for Ps(z) is expected. Increasing the number of binding sites N decreases both typical and maximal deviation. Deviation from the ideal Poisson result tends to be small for all values of ηN (compare with a ‘large’ deviation of 1 when μ = 1).
Multi-site enzymes lose their effectiveness when 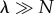 , i.e.
, i.e. 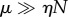 . In this case, the enzyme is mostly unoccupied, and the system tends to have at most one substrate bound, i.e.
. In this case, the enzyme is mostly unoccupied, and the system tends to have at most one substrate bound, i.e. 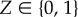 . The situation is then similar to the Michaelis–Menten case (figure 1d). We can then estimate the probability
. The situation is then similar to the Michaelis–Menten case (figure 1d). We can then estimate the probability
| 3.7 |
Thus, enzymes with different N parameter values but common μ parameter values act similarly for equal effective substrate binding rates ηN. Figure 2a (small ηN) illustrates this effect.
3.2. Enzymatic velocity
The steady-state velocity Vcat of the enzyme, i.e. the number of substrates processed per unit time, is μ times the probability that a substrate is occupying the catalytic core
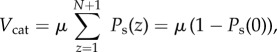 |
3.8 |
or approximately
| 3.9 |
and
| 3.10 |
leading as before to highly cooperative behaviour for 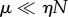 , and non-cooperative (hyperbolic) behaviour for
, and non-cooperative (hyperbolic) behaviour for 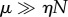 . It is worth reiterating that the strongly unsaturated case
. It is worth reiterating that the strongly unsaturated case 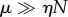 leads to kinetics of the usual Michaelis–Menten form.
leads to kinetics of the usual Michaelis–Menten form.
3.3. First departure time and next departure time
Knowledge of the functional form for the steady-state velocity Vcat would be largely sufficient for a deterministic approximation, but the stochastic nature of our model begs for an analysis that quantifies the probabilistic nature of enzymatic activity (this is a key feature of queueing models). Specifically, we desire to understand the stochastic process for the duration of time between successful enzymatic reactions, or, in queueing parlance, the departure process. A convenient measurement in this regard is that of the first departure time (FDT) T1, which is the duration of time to the next departure at steady state. A higher order statistic is the next departure time (NDT) ΔT = T2 − T1, which is the interval of time between the first and the second observed departures at steady state. When the steady-state departure process is a Poisson process, as occurs for a wide variety of queueing networks at steady state [41–43], then knowledge of the mean FDT or NDT is sufficient to define the departure process. For a renewal process, the NDT distribution is sufficient to define the dynamics of the system [44–46].
Consider first the cumulative probability function PFDT(τ1) ≡ Pr(T1 > τ1) for the FDT. Though this distribution can be difficult to obtain in general, the structure of our multi-site enzyme allows for a closed form solution in the limit ξ → ∞. If the enzyme is initially in a state Z ≥ 1, such that the catalytic core is occupied by substrate, then a single degradation reaction (rate μ) is sufficient to produce a departure. For initial state Z = 0, the FDT is instead the convolution of binding at least one substrate (rate ηN since Z = 0) followed by degradation (rate μ). A straightforward calculation reveals the conditional probabilities
| 3.11 |
and
| 3.12 |
The full FDT distribution is then the weighted sum
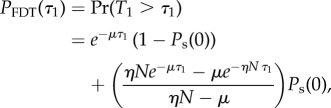 |
3.13 |
with Ps(0) obtained from equation (3.5).
As already mentioned, the departure process for a wide variety of queueing networks is Poisson, which has an exponential distribution,  , for some r > 0. Our multi-site enzyme model does not generally satisfy the conditions for this simple departure distribution; however, the cumulative distribution equation (3.13) can approximate an exponential function arbitrarily well depending on the value of λ = μ/η. This can be shown by calculating the integral squared deviation between the FDT cumulative distribution and a cumulative exponential distribution with the same mean departure time (r−1 = (1/μ) + (1/(ηN))Ps(0)), which after a straightforward calculation is
, for some r > 0. Our multi-site enzyme model does not generally satisfy the conditions for this simple departure distribution; however, the cumulative distribution equation (3.13) can approximate an exponential function arbitrarily well depending on the value of λ = μ/η. This can be shown by calculating the integral squared deviation between the FDT cumulative distribution and a cumulative exponential distribution with the same mean departure time (r−1 = (1/μ) + (1/(ηN))Ps(0)), which after a straightforward calculation is
| 3.14 |
| 3.15 |
Equation (3.15) approaches zero for both 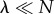 and
and 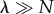 (figure 2b), suggesting these limits recover aspects of a Poisson departure process. The saturated regime, where Ps(0) is very small owing to cooperative effects, can lead to a particularly good approximation of the departure process by a Poisson process. An intuitive explanation for this is that if the catalytic core is always occupied, then departures occur with a constant rate μ, thus generating a Poisson process.
(figure 2b), suggesting these limits recover aspects of a Poisson departure process. The saturated regime, where Ps(0) is very small owing to cooperative effects, can lead to a particularly good approximation of the departure process by a Poisson process. An intuitive explanation for this is that if the catalytic core is always occupied, then departures occur with a constant rate μ, thus generating a Poisson process.
It is an interesting fact for our multi-site enzyme model that the NDT distribution Pr(ΔT > Δτ), describing the duration of time between the first and the second departures from a steady-state condition, is precisely the same functional form (swapping τ1 for Δτ) as the FDT distribution. A sketch for this calculation is relegated to appendix A. One can show in an entirely analogous manner that a queue with constant arrival rate and constant service rate has this same property [13], though this queue also satisfies this identity for higher order departures, while our multi-site model generally does not. This deviation from the queueing prediction is related to arrivals (substrate binding events) in our system not being Poisson, unlike the queueing case.
4. Steady-state analysis for multiple identical but distinguishable classes of substrate
Suppose that multiple classes of substrate are handled identically by the enzyme once bound. Then, as we will show, the analysis of the model at steady state in the case of multiple distinguishable but otherwise identical substrates follows almost immediately from the single-class case. In the biological context, this analysis is an idealization of the case where several similar classes of substrate compete for common processing, e.g. when fluorescent proteins YFP and CFP compete for degradation by ClpXP. We do not anticipate that our results are fragile to weakly breaking our underlying assumptions, as we briefly discuss in this section.
First note that since different classes of substrate are treated identically by the enzyme in our model, owing to ξi = ξ and μi = μ being class-independent constants (see equations (2.1)–(2.3)), the dynamics without regard to substrate class are the same as in §3. Recovery of the substrate class information from the single-class results is possible at steady state owing to a symmetry preserved by the system, where the class of each substrate within the system remains identically but independently distributed with respect to all other substrate molecules in the system. Because the statistics for a set of substrate molecules can be derived from a list of identical but independent random variables, we term this an ‘independent list symmetry’. Proof of this result can be done in a manner entirely analogous to the proof in Mather et al. [24]. We have included an intuitive explanation in figure 3, with further comments in appendix B.
Figure 3.
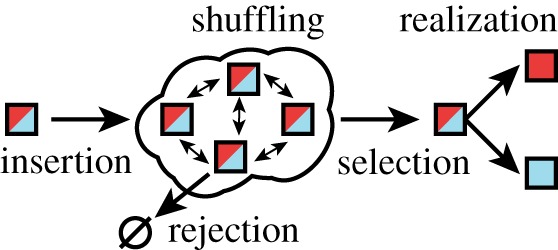
The independent departure symmetry can be intuitively understood. Suppose that we package boxes with either a red or blue ball according to fixed probabilities, e.g. a 40% chance of choosing a red ball (depicted as red/blue boxes). We place these boxes into a system (cloud shape) that can move the boxes around (shuffling), remove boxes from the system (rejection) or eject a box to be observed (selection). As long as all of these processes do not depend on the hidden information within the boxes, then since all boxes are independent but statistically identical, these actions do not entangle the probabilities of the boxes. Thus, upon peeking inside the selected box (realization), the colour of the ball inside will be independently distributed with the probabilities of inserted boxes. (Online version in colour.)
The probabilities for substrate class are entirely determined by the rate at which each class arrives into the system. Define the probabilities for arrivals of substrate class i
| 4.1 |
These probabilities are normalized, 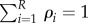 . Given that the enzyme treats different classes identically, it can be shown that independent list symmetry follows (perhaps after a short transient) if each ρi is constant in time. In principle, ηi values can then be time-dependent while keeping each ρi constant in time, but we only pursue steady-state results in this section.
. Given that the enzyme treats different classes identically, it can be shown that independent list symmetry follows (perhaps after a short transient) if each ρi is constant in time. In principle, ηi values can then be time-dependent while keeping each ρi constant in time, but we only pursue steady-state results in this section.
The steady-state distribution can be written in the following way. Define the random variable Q to be the state descriptor of the system. The component Qn (with 0 ≤ n ≤ N) describes the state of the catalytic site when n = 0 and the state of a particular binding site when n > 0. The value of Qn is an integer in the range 0 … R, with Qn = 0 indicating the site does not contain substrate, and with Qn > 0 indicating the class of substrate bound. Define a dependent random variable M describing the occupancy, with Mn = 0 if Qn = 0, and Mn = 1 if Qn > 0. Thus, Mn = Θ(Qn). For simplicity, define ρ0 = 1, which can be interpreted as that there is only one way to have an empty site. A system with independent list symmetry then has the probability distribution
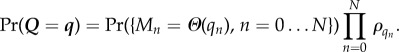 |
4.2 |
The term Pr(Mn = Θ(qn), n = 0 … N) is precisely the probability distribution for substrate to be bound to certain sites without regard to substrate class, which was explored previously in §3. The product Πnρqn then provides all the information pertaining to substrate class. In short, equation (4.2) corresponds to the class of each substrate for occupied sites being independently but identically distributed conditional on knowing which sites on the enzyme are occupied.
Another way to interpret equation (4.2) is that the independent list distribution maximizes entropy of the distribution given minimal constraints (appendix C). In short, the mean probabilities ρi provide all of the information relating to substrate class, and there exists no additional information on the substrate class for any particular site or sites in the queueing system. We anticipate but do not prove that this optimization principle leads to robustness, such that systems weakly breaking our independence assumption should have probability distributions close to maximal entropy distributions. Investigation of this hypothesis will be left for future work.
Given the distribution in equation (4.2), many results follow [22,24]. For example, it is easy to show that, for a definite number of substrate molecules, the frequency of substrate classes is multi-nomial. Now, we find that another significant consequence of independent list symmetry is independent departure symmetry. Intuitively, since the class of substrate located on the catalytic core is always identically but independently distributed relative to other substrate molecules, the probability distribution describing the class of each departure should also be independently distributed for each departure (appendix B). The full departure process can then be described by the probability distribution for the departures
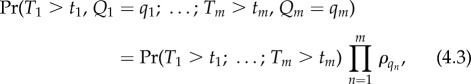 |
where Tn and Qn are the departure time and substrate class, respectively, at the nth departure. The function Pr(T1 > t1; … ; Tm > tm) encodes all of the information for the single substrate class system (e.g. from §3.3), and Πnρqn encodes the multi-class information.
We can compare equation (4.3) to classical results in queueing theory. For example, in quasi-reversible multi-class queueing networks [38,43,47,48], the departure process is equivalent to the superposition of independent Poisson processes for each substrate class. Quasi-reversibility then implies exponentially distributed times between each subsequent departure of a given class (or for all classes). The result in equation (4.3) includes the quasi-reversible result as a special case, but (4.3) can be much more general, allowing potentially complex statistical dependence between the times of different departures, but preserving independence for the class of different departures.
Given that we previously found that departures in our model are in a certain fashion well approximated by a Poisson process with appropriate mean rate (recall the discussion in §3.3), especially when  , and given that we now find independence of classes at each departure, then the departure process for multiple substrates strongly resembles independent Poisson processes for each class, as is found for many quasi-reversible queueing systems. Apparently, the multi-site nature of our model makes its departure process very much like a classical queueing process, which greatly simplifies our understanding of the model's behaviour. This tantalizing evidence begs for further enquiry but is left for future studies.
, and given that we now find independence of classes at each departure, then the departure process for multiple substrates strongly resembles independent Poisson processes for each class, as is found for many quasi-reversible queueing systems. Apparently, the multi-site nature of our model makes its departure process very much like a classical queueing process, which greatly simplifies our understanding of the model's behaviour. This tantalizing evidence begs for further enquiry but is left for future studies.
5. Time-dependent substrate binding rates
We can expect that the rates ηi vary as a function of time in a real system, e.g. owing to depletion of substrate through enzymatic activity. The probability distribution for the enzyme configuration in this case will not necessarily be well approximated by quasi-steady-state results. A reasonable question to ask therefore is how sensitive our multi-site enzymatic system is to fluctuations in ηi. We find numerically that multi-site enzymes tend to buffer fluctuations and thus remain close to certain quasi-steady-state assumptions, thus providing hope that the behaviour of multi-site enzymes in biological networks can be simplified using quasi-steady-state approaches.
Variation of the time-dependent rates ηi(t) can occur in at least two different ways. The simplest is in-phase variation, such that the substrate probabilities ρi from equation (4.1) are constant in time. In this case, the independent list symmetry can be maintained, and analysis of the single-class system with time-dependent η(t) = ∑iηi(t) is sufficient to analyse the full system. A central variable when studying the departure process is the probability P(0, t) ≡ Pr(Z = 0, t) for the enzyme to be unoccupied at a given time t (see §3). Analytical treatment of this situation is beyond the scope of this article, and so we numerically investigate how variation of the input rate leads to subsequent variation in the departure rate by measuring the variation in P(0, t) owing to periodic variation of η(t). We find that the multi-site structure leads to improved buffering of fluctuations in the saturated regime (figure 4).
Figure 4.
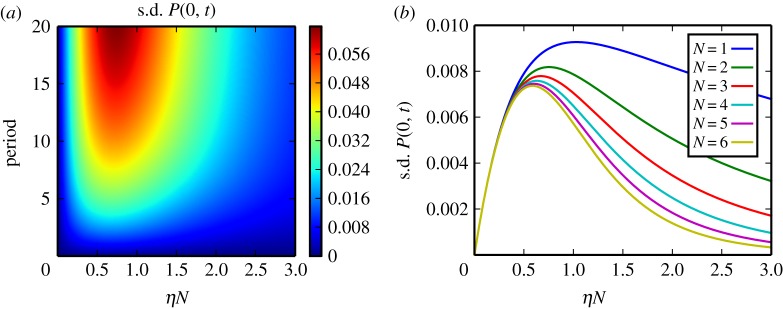
For a single-class system driven with periodic η(t), we measure the standard deviation of P(0, t), the probability for the enzyme to be devoid of substrate. The signal for η(t) is sinusoidal with given period and amplitude ±25% the mean value. (a) Results for the system with N = 3 for different drive periods and mean amplitude η. It is seen that the enzyme acts as a low-pass filter. (b) For the case with drive period = 1, we plot the oscillator response as a function of ηN. Increasing the number of binding sites N strongly suppresses the oscillatory response for large ηN.
The alternative variation in the rates ηi(t) is balanced variation, where η = ∑iηi(t) is constant, but each ηi(t) can be time dependent. The corresponding single-class version of the multi-class system has constant binding rate η and simply approaches steady state. Hence, all interesting non-stationary phenomena after a short transient are multi-class in origin. We attempt to distil such phenomena by measuring statistical dependence between the substrate classes of the first and the second most recent departures from the enzyme. Correlation between these departures is presented in figure 5. We find that, even in the worst case, deviations from the zero correlation limits are small. Similar results were found when we investigated the mutual information between the substrate classes for subsequent departures.
Figure 5.
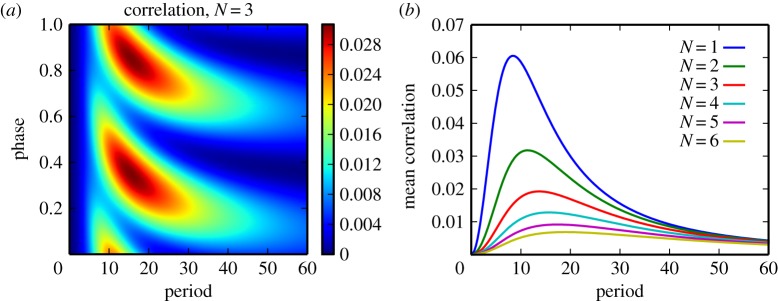
For a two-class system driven with periodic ηi(t) but constant η = ∑iηi(t), we measure the correlation between the index (either 1 or 2) of the last two departures. The signals for η1(t) and η2(t) are sinusoidal with given period and amplitude ±25% the mean value. The mean values over one period for the signals ηi(t) and η2(t) are taken to be identical. Consider this system in the limit that η → ∞, such that the system is fully saturated. (a) Correlation for the system with N = 3 for different drive periods. The correlation is plotted as a function of drive signal phase, which is normalized from 0 to 1. (b) Mean correlation averaged over phase for systems with different N and different drive periods. As in the single-class case, increasing N leads to suppressed fluctuations relative to the quasi-steady-state values. In this plot, we find the correlation is small relative to the maximum correlation of 1.
The present investigation suggests that the multi-site structure of the enzyme may effectively buffer both single-class and multi-class variation. Buffering in the single-class case is indeed reasonable, since an enzyme with many binding sites more rapidly becomes saturated (figure 2a). Buffering of fluctuations in the multi-class case is more interesting, since perturbation of the probability distribution can occur (in principle) even for saturated systems. A full analysis of this situation is beyond the scope of this article, but we attempt to provide an explanation in the limit where η → ∞ and N is very large. The system then requires a time greater than the natural time scale N/μ to significantly affect the abundance of each substrate class on the enzyme. Since this time scale is much larger than the time scale 2/μ to sample two departures, and since the class of departures is directly determined by the relative class abundance on the binding sites, then the system may remain in a quasi-steady state with respect to departures. Note that this result does not necessarily imply that the system is in quasi-steady state with respect to the instantaneous rates ηi(t).
6. Discussion
Development of a coherent biological queueing theory is ongoing, and many questions relating to its ultimate usefulness remain unanswered. Among these questions is just how often can a queueing formalism bring quantitative insight to a particular real biological system? In this article, we studied a model that was motivated by the known properties of the ‘waiting line’ structure of the E. coli protease ClpXP and related proteases.
We found that not only does a waiting line structure lead to a strong cooperative increase in the effective affinity of substrate to the enzyme, but the waiting line can also push the model deeply into a queueing regime, where the stochastic process describing the completion of enzymatic reactions at steady state is very similar to the departure process for traditional queueing models, i.e. Poisson processes. In the case of multiple classes of substrate, an apparently new queueing-inspired result, independent departure symmetry, was determined to hold for the departure processes of a wide range of enzyme models, even when traditional queueing theory does not apply. Finally, we numerically investigated the response of our enzyme to time-dependent perturbation, and we found preliminary evidence that the waiting line structure can effectively buffer the system from fluctuations.
Further investigation of multi-site enzyme kinetics is certainly warranted for a number of reasons. Especially, a fundamental assumption in our analysis was that different classes of substrate are treated identically by enzyme, i.e. ξi and μi did not depend on substrate class i. Breaking this assumption can be expected to break the independent list symmetry and independent departure symmetry discussed in this article, which greatly complicates the analysis of the model. The situation is not hopeless, however, as the theory for priority queues (preemptive and non-preemptive) itself has a long history and may assist in the analysis of enzymes with strong asymmetry in their processing rates. Preliminary investigation suggests that for these more general multi-site enzyme models in certain parameter regimes, such as saturated regimes (large ηi), the resulting departure process may be sufficiently simple to analyse.
A proper stochastic analysis of the accuracy of the Michaelis–Menten (quasi-steady-state) approximation for multi-site enzymes would also be welcomed. For substrate count sufficiently large, it is sensible that the quasi-steady-state approximation is valid. However, for small free substrate count, a number of details may have to be treated more carefully, e.g. competition for free substrate between enzymes, and the diffusion-limited dynamics of substrate binding to enzyme. Such details are outside the scope of this article, but we anticipate that queueing theory will also offer insight into this biophysical problem.
Acknowledgements
We would like to thank Ruth Williams and Nicholas Butzin for stimulating exchanges during the formation of this manuscript.
Appendix A. Formalism for next departure time calculation
Calculation of the NDT cumulative distribution Pr(ΔT > Δτ) can be done in the following way (see §3.3 for context). Define the joint probabilities Wn(τ1) that the FDT (labelled T1) exceeds τ1, and that Z has a certain value or range of values at τ1
| A 1 |
| A 2 |
| A 3 |
It is straightforward to show that these quantities satisfy the ODEs
| A 4 |
| A 5 |
| A 6 |
Equations (A 4)–(A 6) can be solved iteratively, starting from W0(τ1). With these solutions, define the infinitesimal joint probability dFn(τ1) that the FDT is precisely in a time dτ1 about τ1 and the state Z = 0 (n = 0) or Z ≥ 1 (n = 1) immediately after a departure (see, for example, [49] for a discussion of revising probabilities after a particular reaction). Then
| A 7 |
| A 8 |
Supposing these infinitesimal probabilities, the density (in τ1) for the cumulative probability for the next departure to be a time ΔT > Δτ can be derived in the same way as equation (3.13), leading to the equation
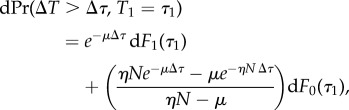 |
A 9 |
which can be integrated over τ1 to provide the full cumulative distribution
| A 10 |
Once this calculation has been done starting from the steady-state distribution for the multi-site model in equations (2.1)–(2.3) (with ξ → ∞), it is straightforward to show that the single-class FDT and NDT cumulative distributions have precisely the same functional form, i.e.
| A 11 |
though we do not provide the details of this calculation here.
Appendix B. Independent list symmetry and independent departure symmetry
We here provide a few additional details regarding the independent list symmetry and independent departure symmetry discussed in §4.
First, the preservation of independent list symmetry by a variety of queueing-associated operations can be demonstrated with minimal difficulty. Once this is done, it then is a trivial matter to extend the argument to time-continuous evolution [24]. We begin with an independent list probability distribution (see equation (4.2))
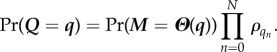 |
B 1 |
One of the most fundamental operations in queueing theory is the act of moving customers from location to location. Such operations can rather generally be represented by a permutation of the current state of the queueing network. Define an operator  and associated matrix
and associated matrix  that perform a permutation on the state vector,
that perform a permutation on the state vector,
| B 2 |
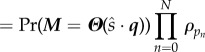 |
B 3 |
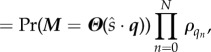 |
B 4 |
where 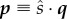 is a permutation of q. Equation (B 4) consists of two pieces, with
is a permutation of q. Equation (B 4) consists of two pieces, with 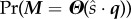 only relating to information on substrate occupancy in the permuted state, and
only relating to information on substrate occupancy in the permuted state, and 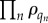 containing all information for the multi-class distribution. Equation (B 4) is precisely the same form as the independent list distribution in equation (B 1), and, thus, permutations of enzyme sites preserve the independent list symmetry.
containing all information for the multi-class distribution. Equation (B 4) is precisely the same form as the independent list distribution in equation (B 1), and, thus, permutations of enzyme sites preserve the independent list symmetry.
Another extremely important operation in queueing theory is the addition of a new customer to the network. Define an operation 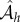 that adds a customer to site h, with an independently distributed random class determined according to the probabilities ρi in equation (4.1). Define an associated matrix
that adds a customer to site h, with an independently distributed random class determined according to the probabilities ρi in equation (4.1). Define an associated matrix 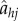 that converts the value at index h to value j. Then, operation of the arrival operator on an independent list distribution leads to the following equations (the notation Pr(Mh = z) is shorthand for the probability that M = Θ(q) for all indices except h, where Mh = z):
that converts the value at index h to value j. Then, operation of the arrival operator on an independent list distribution leads to the following equations (the notation Pr(Mh = z) is shorthand for the probability that M = Θ(q) for all indices except h, where Mh = z):
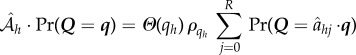 |
B 5 |
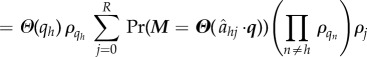 |
B 6 |
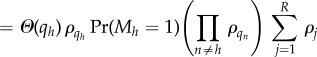 |
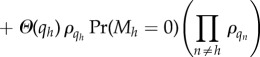 |
B 7 |
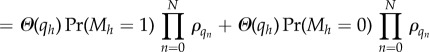 |
B 8 |
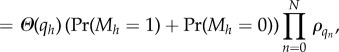 |
B 9 |
which is of the independent list form. Similarly for a deletion operator 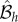 that replaces site h with an unoccupied site (δ(Z) = 1 if Z = 0, and δ(Z) = 0 otherwise)
that replaces site h with an unoccupied site (δ(Z) = 1 if Z = 0, and δ(Z) = 0 otherwise)
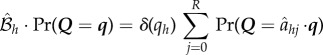 |
B 10 |
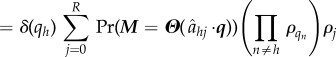 |
B 11 |
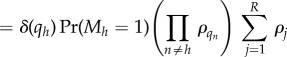 |
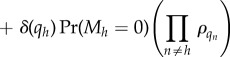 |
B 12 |
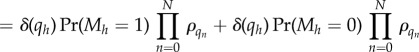 |
B 13 |
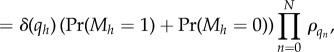 |
B 14 |
which is again of the independent list form. Note that the step between equations (B 12) and (B 13) used our convention that ρ0 = 1.
In this manner, it can be demonstrated that any composition of these shuffling, addition and removal operations leads to another independent list. An approach similar to that in Mather et al. [24] then extends the above results to the time-continuous case.
Once independent list symmetry has been shown to exist for a system, independent departures follow when the rule for selecting departures is independent of the class of customers in the system. To show this, we introduce an auxiliary queue that keeps track of the last several departures (figure 6). The method to prove independent list symmetry can then be applied to the joint system (departure queue plus the original system), since departures are simply customers moving from the original system to the departure queue. Thus, the departure queue maintains independent list symmetry, implying that adjacent departures are statistically independent from one another. We do not provide a full derivation here.
Figure 6.
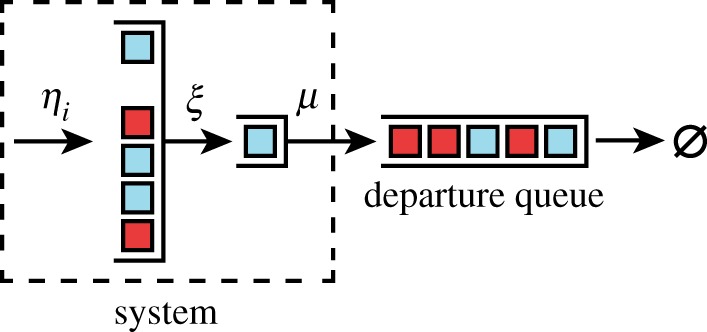
Independent departure symmetry follows from independent list symmetry. Consider a system with independent list symmetry (within the dashed box). Departures of single jobs from this system can be placed into an auxiliary departure queue that keeps track of the last several departures. Independent list symmetry is also preserved in the departure queue, owing to the arguments figure 3 and appendix B. Since this queue is an independent list, it is reasonable to believe that the probabilistic process for the substrate class of departures is a sequence of independently but identically distributed random variables. (Online version in colour.)
Appendix C. Independent list symmetry as a consequence of maximum entropy
The independent list distributions discussed in appendix B can be understood also through an entropy maximization principle. Intuitively, independent list distributions are the distributions of the greatest uncertainty conditional on known mean abundance for each substrate class in the queueing system. The fact that independent list distributions are in this way optimal suggests that small perturbations of the queueing system away from identical treatment of substrate will generate steady-state distributions near maximum entropy (deviation from maximum entropy is anticipated to be the second order in the perturbation strength, by optimality). We anticipate but do not prove then that maximum entropy distributions thus robustly predict steady-state distributions for a neighbourhood of systems approximated by identical but distinguishable substrate systems.
To demonstrate that independent list distributions satisfy maximum entropy, assume a queue (length N + 1) of a similar type as the above, but only allow non-empty queue sites. The more general case with both filled and empty sites is a straightforward generalization. Label for shorthand P(q) = Pr(Q = q), and define mk(q) to be the count of substrate class k in q, where k ≥ 1 for this discussion. We can define the entropy as the sum over all possible states of the queue
| C 1 |
We maximize the entropy subjected to several constraints. First, we constrain the distribution to be normalized
| C 2 |
We also constrain the mean count of each class k to be ρk,
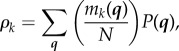 |
C 3 |
where mk(q)/N can be interpreted as the probability of finding a given substrate of class k in a particular queue q. It can be shown as a consequence of the above two constraints that
| C 4 |
but this is not an independent constraint. Using the method of Lagrange multipliers, we maximize a function
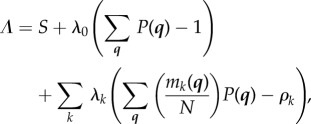 |
C 5 |
with respect to P(q) (for all possible q), λ0, and each λk. Maximization of Λ with respect to P(q) provides
| C 6 |
which implies
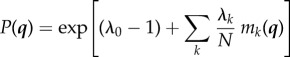 |
C 7 |
or
| C 8 |
We can adjust the parameters λ0 and each λk to fix this probability distribution in the form
| C 9 |
which can be shown to satisfy the two constraints of our maximization. Equation (C 9) can be rewritten as
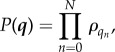 |
C 10 |
which is precisely the independent list distribution from before.
The more general case of queueing systems with unoccupied sites can be done by also constraining known probabilities Pr(M = Θ(q)) for occupancy (equation (B 1)). We do not perform these steps here.
Funding statement
This work was supported by the National Science Foundation (grant no. 1330180).
References
- 1.Briggs GE, Haldane JB. 1925. A note on the kinetics of enzyme action. Biochem. J. 19, 338–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Segel LA. 1988. On the validity of the steady-state assumption of enzyme-kinetics. Bull. Math. Biol. 50, 579–593. ( 10.1007/BF02460092) [DOI] [PubMed] [Google Scholar]
- 3.Stoleriu I, Davidson FA, Liu JL. 2004. Quasi-steady state assumptions for non-isolated enzyme-catalysed reactions. J. Math. Biol. 48, 82–104. ( 10.1007/s00285-003-0225-7) [DOI] [PubMed] [Google Scholar]
- 4.Bennett MR, Volfson D, Tsimring L, Hasty J. 2007. Transient dynamics of genetic regulatory networks. Biophys. J. 92, 3501–3512. ( 10.1529/biophysj.106.095638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aranyi P, Toth J. 1977. Full stochastic description of Michaelis–Menten reaction for small systems. Acta Biochim. Biophys. Hung. 12, 375–388. [PubMed] [Google Scholar]
- 6.Qian H, Elson E. 2002. Single-molecule enzymology: stochastic Michaelis–Menten kinetics. Biophys. Chem. 101, 565–576. ( 10.1016/S0301-4622(02)00145-X) [DOI] [PubMed] [Google Scholar]
- 7.Kou S, Cherayil B, Min W, English B, Xie X. 2005. Single-molecule Michaelis–Menten equations. J. Phys. Chem. B 109, 19 068–19 081. ( 10.1021/jp051490q) [DOI] [PubMed] [Google Scholar]
- 8.English BP, Min W, van Oijen AM, Lee KT, Luo GB, Sun HY, Cherayil BJ, Kou SC, Xie XS. 2006. Ever-fluctuating single enzyme molecules: Michaelis–Menten equation revisited. Nat. Chem. Biol. 2, 87–94. ( 10.1038/nchembio759) [DOI] [PubMed] [Google Scholar]
- 9.Yi M, Liu Q. 2010. Michaelis–Menten mechanism for single-enzyme and multi-enzyme system under stochastic noise and spatial diffusion. Phys. A Stat. Mech. Appl. 389, 3791–3803. ( 10.1016/j.physa.2010.05.041) [DOI] [Google Scholar]
- 10.Sanft KR, Gillespie DT, Petzold LR. 2011. Legitimacy of the stochastic Michaelis–Menten approximation. IET Syst. Biol. 5, 58–69. ( 10.1049/iet-syb.2009.0057) [DOI] [PubMed] [Google Scholar]
- 11.Agarwal A, Adams R, Castellani GC, Shouval HZ. 2012. On the precision of quasi steady state assumptions in stochastic dynamics. J. Chem. Phys. 137, 044105 ( 10.1063/1.4731754) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doka E, Lente G. 2012. Stochastic mapping of the Michaelis–Menten mechanism. J. Chem. Phys. 136, 054111 ( 10.1063/1.3681942) [DOI] [PubMed] [Google Scholar]
- 13.Gross D, Shortle JF, Thompson JM, Harris CM. 2013. Fundamentals of queueing theory. New York, NY: John Wiley & Sons. [Google Scholar]
- 14.Raser JM, O'Shea EK. 2004. Control of stochasticity in eukaryotic gene expression. Science 304, 1811–1814. ( 10.1126/science.1098641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peccoud J, Ycart B. 1995. Markovian modeling of gene-product synthesis. Theor. Popul. Biol. 48, 222–234. ( 10.1006/tpbi.1995.1027) [DOI] [Google Scholar]
- 16.Elgart V, Jia T, Kulkarni RV. 2010. Applications of Little's law to stochastic models of gene expression. Phys. Rev. E 82, 021901 ( 10.1103/PhysRevE.82.021901) [DOI] [PubMed] [Google Scholar]
- 17.Jia T, Kulkarni RV. 2011. Intrinsic noise in stochastic models of gene expression with molecular memory and bursting. Phys. Rev. Lett. 106, 058102 ( 10.1103/PhysRevLett.106.058102) [DOI] [PubMed] [Google Scholar]
- 18.Josić K, López JM, Ott W, Shiau L, Bennett MR. 2011. Stochastic delay accelerates signaling in gene networks. PLoS Comp. Biol. 7, e1002264 ( 10.1371/journal.pcbi.1002264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H, Gelenbe E. 2011. G-networks based two layer stochastic modeling of gene regulatory networks with post-translational processes. Interdiscip. Bio Central 3, 1–8. ( 10.4051/ibc.2011.3.2.0008) [DOI] [Google Scholar]
- 20.Kandemir-Cavas C, Cavas L, Yokes MB, Hlynka M, Schell R, Yurdakoc K. 2008. A novel application of queueing theory on the Caulerpenyne secreted by invasive Caulerpa taxifolia (Vahl) C. Agardh (Ulvophyceae, Caulerpales): a preliminary study. Mediterr. Mar. Sci. 9, 67–76. ( 10.12681/mms.144) [DOI] [Google Scholar]
- 21.Levine E, Hwa T. 2007. Stochastic fluctuations in metabolic pathways. Proc. Natl Acad. Sci. USA 104, 9224–9229. ( 10.1073/pnas.0610987104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mather WH, Cookson NA, Hasty J, Tsimring LS, Williams RJ. 2010. Correlation resonance generated by coupled enzymatic processing. Biophys. J. 99, 3172–3181. ( 10.1016/j.bpj.2010.09.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uygulanması İ. 2007. an application of queueing theory to the relationship between insulin level and number of insulin receptors. Türk Biyokimya Dergisi [Turk J. Biochem.] 32, 32–38. [Google Scholar]
- 24.Mather WH, Hasty J, Tsimring LS, Williams RJ. 2011. Factorized time-dependent distributions for certain multiclass queueing networks and an application to enzymatic processing networks. Queueing Syst. 69, 313–328. ( 10.1007/s11134-011-9216-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baumgartner BL, Bennett MR, Ferry M, Johnson TL, Tsimring LS, Hasty J. 2011. Antagonistic gene transcripts regulate adaptation to new growth environments. Proc. Natl Acad. Sci. USA 108, 21 087–21 092. ( 10.1073/pnas.1111408109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojtkowiak D, Georgopoulos C, Zylicz M. 1993. Isolation and characterization of ClpX, a new ATP-dependent specificity component of the Clp protease of Escherichia coli. J. Biol. Chem. 268, 22609. [PubMed] [Google Scholar]
- 27.Schweder T, Lee K, Lomovskaya O, Matin A. 1996. Regulation of Escherichia coli starvation sigma factor (sigma s) by ClpXP protease. J. Bacteriol. 178, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flynn J, Neher S, Kim Y, Sauer R, Baker T. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11, 671–683. ( 10.1016/S1097-2765(03)00060-1) [DOI] [PubMed] [Google Scholar]
- 29.Song HK, Eck MJ. 2003. Structural basis of degradation signal recognition by SspB, a specificity-enhancing factor for the ClpXP proteolytic machine. Mol. Cell 12, 75–86. ( 10.1016/S1097-2765(03)00271-5) [DOI] [PubMed] [Google Scholar]
- 30.Wah DA, Levchenko I, Rieckhof GE, Bolon DN, Baker TA, Sauer RT. 2003. Flexible linkers leash the substrate binding domain of SspB to a peptide module that stabilizes delivery complexes with the AAA+ ClpXP protease. Mol. Cell 12, 355–363. ( 10.1016/S1097-2765(03)00272-7) [DOI] [PubMed] [Google Scholar]
- 31.Dougan DA, Weber-Ban E, Bukau B. 2003. Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol. Cell 12, 373–380. ( 10.1016/j.molcel.2003.08.012) [DOI] [PubMed] [Google Scholar]
- 32.Bolon DN, Grant RA, Baker TA, Sauer RT. 2004. Nucleotide-dependent substrate handoff from the SspB adaptor to the AAA+ ClpXP protease. Mol. Cell 16, 343–50. ( 10.1016/j.molcel.2004.10.001) [DOI] [PubMed] [Google Scholar]
- 33.Hersch GL, Baker TA, Sauer RT. 2004. SspB delivery of substrates for ClpXP proteolysis probed by the design of improved degradation tags. Proc. Natl Acad. Sci. USA 101, 12 136–12 141. ( 10.1073/pnas.0404733101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGinness KE, Bolon DN, Kaganovich M, Baker TA, Sauer RT. 2007. Altered tethering of the SspB adaptor to the ClpXP protease causes changes in substrate delivery. J. Biol. Chem. 282, 11 465–11 473. ( 10.1074/jbc.M610671200) [DOI] [PubMed] [Google Scholar]
- 35.Chowdhury T, Chien P, Ebrahim S, Sauer RT, Baker TA. 2010. Versatile modes of peptide recognition by the ClpX N domain mediate alternative adaptor-binding specificities in different bacterial species. Protein Sci. 19, 242–254. ( 10.1002/pro.306) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 80, 587–612. ( 10.1146/annurev-biochem-060408-172623) [DOI] [PubMed] [Google Scholar]
- 37.Golding I, Paulsson J, Zawilski SM, Cox EC. 2005. Real-time kinetics of gene activity in individual bacteria. Cell 123, 1025–1036. ( 10.1016/j.cell.2005.09.031) [DOI] [PubMed] [Google Scholar]
- 38.Kelly FP. 1979. Reversibility and stochastic networks. New York, NY: John Wiley and Sons. [Google Scholar]
- 39.Gardiner CW, et al. 1985. Handbook of stochastic methods, vol. 3. Berlin, Germany: Springer. [Google Scholar]
- 40.Jones E, Oliphant T, Peterson P. 2001. SciPy: Open source scientific tools for Python See http://www.scipy.org/.
- 41.Burke PJ. 1956. The output of a queuing system. Oper. Res. 4, 699–704. ( 10.1287/opre.4.6.699) [DOI] [Google Scholar]
- 42.Jackson JR. 1963. Jobshop-like queueing systems. Manag. Sci. 10, 131–142. ( 10.1287/mnsc.10.1.131) [DOI] [Google Scholar]
- 43.Kelly FP. 1976. Networks of queues. Adv. Appl. Prob. 8, 416–432. ( 10.2307/1425912) [DOI] [Google Scholar]
- 44.Santos JE, Franosch T, Parmeggiani A, Frey E. 2005. Renewal processes and fluctuation analysis of molecular motor stepping. Phys. Biol. 2, 207–222. ( 10.1088/1478-3975/2/3/008) [DOI] [PubMed] [Google Scholar]
- 45.Qian H, Xie XS. 2006. Generalized Haldane equation and fluctuation theorem in the steady-state cycle kinetics of single enzymes. Phys. Rev. E 74, 010902 ( 10.1103/PhysRevE.74.010902) [DOI] [PubMed] [Google Scholar]
- 46.Saha S, Sinha A, Dua A. 2012. Single-molecule enzyme kinetics in the presence of inhibitors. J. Chem. Phys. 137, 045102 ( 10.1063/1.4737634) [DOI] [PubMed] [Google Scholar]
- 47.Muntz RR. 1972. Poisson departure processes and queueing networks. IBM Research Report RC 4145, IBM Thomas J. Watson Research Center, Yorktown Heights, NY, USA.
- 48.Kelly FP. 1975. Networks of queues with customers of different types. J. Appl. Prob. 12, 542–554. ( 10.2307/3212869) [DOI] [Google Scholar]
- 49.Mather WH, Hasty J, Tsimring LS. 2012. Fast stochastic algorithm for simulating evolutionary population dynamics. Bioinformatics 28, 1230–1238. ( 10.1093/bioinformatics/bts130) [DOI] [PMC free article] [PubMed] [Google Scholar]