

Abstract
Background and Purpose
Fostamatinib is an inhibitor of spleen tyrosine kinase (TK). In patients, fostamatinib treatment was associated with increased BP. Some TK inhibitors cause BP elevation, by inhibiting the VEGF receptor 2 (VEGFR2). Here, we have assessed the mechanistic link between fostamatinib-induced BP elevation and inhibition of VEGF signalling.
Experimental Approach
We used conscious rats with automated blood sampling and radio telemetry and anaesthetized rats to measure cardiovascular changes. Rat isolated aorta and isolated hearts, and human resistance vessels in vitro were also used. NO production by human microvascular endothelial cells was measured with the NO-dependent probe, DAF-FM and VEGFR2 phosphorylation was determined in mouse lung, ex vivo.
Key Results
In conscious rats, fostamatinib dose-dependently increased BP. The time course of the BP effect correlated closely with the plasma concentrations of R406 (the active metabolite of fostamatinib). In anaesthetized rats, infusion of R406 increased BP and decreased femoral arterial conductance. Endothelial function was unaffected, as infusion of R406 did not inhibit hyperaemia-or ACh-induced vasodilatation in rats. R406 did not affect contraction of isolated blood vessels. R406 inhibited VEGF-stimulated NO production from human endothelial cells in vitro, and treatment with R406 inhibited VEGFR2 phosphorylation in vivo. R406 inhibited VEGF-induced hypotension in anaesthetized rats.
Conclusions and Implications
Increased vascular resistance, secondary to reduced VEGF-induced NO release from endothelium, may contribute to BP increases observed with fostamatanib. This is consistent with the elevated BP induced by other drugs inhibiting VEGF signalling, although the contribution of other mechanisms cannot be excluded.
Keywords: drug-induced, BP, TK, VEGF, endothelial function, R406, fostamatinib
Introduction
Drug-induced BP elevation is a recognized effect of several drugs that inhibit VEGF receptor (VEGFR) signalling (Robinson et al., 2010; Thanigaimani et al., 2011; receptor nomenclature follows Alexander et al., 2013). For example, sunitinib, sorafenib and pazopanib are drugs approved for the treatment of a variety of cancers. These drugs inhibit VEGFR2 at clinically therapeutic exposures and are associated with a 15–60% incidence of hypertension in the clinic (Keefe et al., 2011; McTigue et al., 2012). The mechanism by which these and other VEGF receptor tyrosine kinase inhibitors (RTKi) elevate arterial BP has been the focus of several investigations, which together suggest that inhibition of VEGF signalling in the vasculature is involved. VEGF is a key regulator of vascular homeostasis (Robinson et al., 2010). In particular, ligand binding to VEGFR2 on endothelial cells leads to stimulation of NOS, and the resulting release of NO causes smooth muscle relaxation and vasodilatation (Facemire et al., 2009). Inhibition of VEGF itself (e.g. by the anti-VEGF bevacizumab) or direct inhibition of VEGFR2 therefore impairs this vasodilatory response, leading to BP elevation.
Fostamatinib (the pro-drug of the active metabolite R406) is a small-molecule kinase inhibitor with activity on spleen TK (SYK) and has completed phase III clinical studies for the treatment of rheumatoid arthritis (Braselmann et al., 2006; McInnes and Schett, 2007; Riccaboni et al., 2010; Weinblatt et al., 2010). Fostamatinib inhibits SYK-mediated immune signalling in many cell types involved in inflammation and tissue damage in rheumatoid arthritis and so may inhibit key steps in the progression of this disease (Wong et al., 2004). As is common for RTKi that bind to the ATP binding pocket of the protein, fostamatinib inhibits several kinases (including VEGFR2) other than the intended primary target, when assessed in isolated enzyme assays (Davis et al., 2011; Metz et al., 2011). This off-target kinase activity is, however, lower than SYK inhibition, in assays measuring cellular function (Braselmann et al., 2006).
In phase II clinical studies in patients with rheumatoid arthritis, fostamatinib has been associated with a mean increase in systolic BP of approximately 3 mmHg between baseline and 1 month after treatment initiation, compared with a decrease of 2 mmHg with placebo (Weinblatt et al., 2010; Genovese et al., 2011). In all cases, BP elevation responded to antihypertensive treatment or a reduction in the dose of fostamatinib. The purpose of this study is to understand the physiological and molecular mechanisms for fostamatinib-induced BP elevation. The data presented here suggest that fostamatinib-induced BP elevation in rats is the consequence of increased vascular resistance. Data suggest that this may be in part mediated by impaired vasorelaxation due to inhibition of VEGF-induced endothelial NO release. These findings suggest that the BP elevation mechanism for fostamatinib may be related to that seen in other members of the RTKi class. However, given that fostamatinib inhibits a number of other kinases, we cannot exclude the possibility that targets other than VEGFR2 may also contribute to BP elevation.
Methods
Animal care and use
Animal care and experimental procedures at the AstraZeneca facility in the UK were performed under the authority of a valid Home Office Project Licence and conformed to the UK Animals (Scientific Procedures) Act, 1986 (UK Animals, Scientific Procedures Act, 1986). Telemetry procedures were performed under the authority of the AALAS approved Institutional Animal Care and Use Committee (IACUC) at the AstraZeneca facility in Waltham, Massachusetts USA. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 113 animals were used in the experiments described here. Rats were supplied by Harlan Laboratories, Bicester, UK or Charles River Laboratories, Raleigh, NC USA and transferred to the AstraZeneca facility where they were maintained in a 12-hour light:dark cycle and were given full access to a standard rat chow and drinking water at all times. Animals were acclimatized for at least 7 days before being used for experiments
Cardiovascular recording in conscious telemetered rats
Arterial BP was measured using radiotelemetry combined with automated blood sampling as previously described (Kamendi et al., 2010). Briefly, twenty-four male Sprague-Dawley rats (300-350g), were implanted with TL11M2-C50-PXT transmitters (Data Sciences International, St. Paul, MN USA) and a femoral vein catheter under isoflurane anesthesia at Charles River Laboratories (Raleigh, NC USA). Details of the preparation of the telemetered rats are given in the Supporting Information. Animals were transferred to the AstraZeneca facility and, after the acclimatisation period, the femoral catheters were connected to a Culex® automated blood sampler (BASi, West Lafayette, IN USA). BP was recorded before and up to 24-hours following oral dosing with 10 mL·kg−1 vehicle, 10, 30, or 100 mg·kg−1 of fostamatinib. Blood samples (150 μL) were automatically collected to determine the plasma concentration of R406.
General haemodynamics, VEGF-induced vasodilatation and endothelial function in anaesthetised rats
For all studies in anaesthetized animals, male Sprague-Dawley rats were surgically prepared for the measurement of arterial BP, ECG, and femoral arterial blood flow (FBF)(1mm flow probe, Transonic Systems Inc., Ithaca, NY USA) under isofluorane anaesthesia (0.5 to 5%) with mechanical ventilation. Femoral vascular conductance (FVC) was calculated as FBF/MABP (mean arterial BP). Arterial blood gases and rectal temperature were monitored and blood samples were taken via the arterial catheter to determine plasma R406 concentrations.
For general haemodynamics, left ventricular pressure (LVP) was also measured from a catheter inserted into the left ventricle (Scisense Inc., London, ON Canada). Animals (n = 8 per group) received a single 30 min i.v. infusion of vehicle, 3 or 5 mg·kg−1 R406 at a dose volume of 1.0 mL·kg−1 (rate 0.03 mL·kg−1min−1).
For VEGF-induced vasodilatation experiments, rats (n = 6 per group) received a single i.v infusion of vehicle or 4.5 mg·kg−1 R406 administered over 20 min at a dose volume of 1.5 mL·kg−1. At 15 min into the infusion, a bolus dose of VEGF (55 μg·kg−1) was administered i.v using a dose volume of 0.8 mL·kg−1. Animals were monitored for a further 15 min following the cessation of the infusion.
Endothelial function was tested by examining the effects of R406 on reactive hyperaemia (RH) and acetylcholine (ACh)-induced vasodilatation. Animals (n = 5 to 7 per group) received two consecutive 20 minutes i.v. infusions of vehicle, or vehicle followed by 4.5 mg/kg R406 using a dose volume of 1.5 mL·kg−1. Separate groups of animals (n = 6 per group) were given vehicle (water for injection) or 10 mg·kg−1 L-NAME, as an i.v. infusion over 1 minute in a dose volume of 1 mL·kg−1. For RH, hindlimb ischaemia was induced at 10 min into each infusion (for R406 and vehicle), or before and after dosing (for L-NAME and vehicle), by occluding the femoral artery, proximal to the flow probe, with a cotton ligature for a 5 min period, after which time the occluder was released to permit reperfusion and reactive hyperaemia. For pharmacologically-induced vasodilatation, 0.1 μg ACh was administered as an intra-arterial bolus in a 20 μL volume at approximately 15 min into each infusion (for R406 and vehicle), or before and after dosing (for L-NAME and vehicle). The arterial catheter was positioned so that the tip lay in the aorta proximal to the iliac bifurcation.
Cardiovascular data was recorded using ART Gold (DataSciences international) for telemetry experiments and HEM version 4.2 (Notocord, Croissy Sur Seine, FRANCE) for anaesthetised studies.
Determination of R406 plasma concentrations from in vivo studies
Blood samples were taken using K2-EDTA as anticoagulant, centrifuged, and stored at −70°C until analysed. The plasma concentration of R406 was determined by liquid-liquid extraction and liquid chromatography followed by detection by mass spectrometry. Where free plasma concentrations are quoted, this is based on plasma protein binding in the rat of 97.9%. Details of the subsequent pharmacokinetic-pharmacodynamic (PKPD) modelling are given in the Supporting Information (Table S3).
Measurement of phosphorylated VEGFR2 (pVEGFR2) from murine studies
To assess the effects of fostamatinib on VEGFR-2 phosphorylation in mouse lung in vivo, fostamatinib (100 mg kg−1) or vehicle (1% polysorbate) was given to male nude (nu/nu genotype) Alderley Park (Swiss-derived) mice. In order to increase the basal VEGFR2 phosphorylation level, a bolus dose of VEGF-A (30 μg per mouse) was given intravenously 5 min before humanely killing the animals at 3 h after the oral dose of fostamatinib. Lungs were removed and immediately snap frozen in liquid nitrogen. The amount of VEGFR2 phosphorylation was determined as previously described (Smith et al., 2007).
Endothelial cell assays
VEGF-stimulated NO production by human microvascular endothelial cells (HMECs; from AstraZeneca Tissue Culture Collection, UK) was determined using the NO-dependent probe, DAF-FM (4-amino-5-methylamino-2′,7′-difluorofluorescein. HMECs were cultured in MCDB131 medium supplemented with MVGS (Invitrogen) and 2mM L-alanyl-L-glutamine in 0.1% gelatin coated flasks. For each experiment, cells were seeded into black, clear bottom 96 well plates (Costar) at 6 × 104 cm2 for 24 h, then serum-starved overnight. R406, pazopanib and anisomycin (a reference NOS inhibitor) were dissolved in DMSO then diluted 1:200 into DAF reaction buffer (DAF-FM diacetate 0.2 μmol·L−1; L-arginine 1mmol·L−1; Reaction Buffer) to achieve final working concentrations. These were then applied to cells and incubated at 37°C for 20 min. VEGF and Hoechst nuclear stain (Invitrogen H3570) were added to give final concentrations of 50ng·mL−1 and 2μg·mL−1, respectively. Plates were incubated for a further 30 minutes at 37°C. Cells were washed with PBS and read using an ImageXpress Micro (Molecular Devices). Images were analysed using the Metaexpress® multi-wavelength cell scoring algorithm measuring the cytoplasmic integrated intensity per cell.
Rat isolated heart
Isolated hearts from rats were prepared according to the Langendorff method and used as described by Stucker et al., (1985). Briefly, male Wistar rats (250-310g) were anaesthetised (Imalgen1000, 1.5 mL·kg−1, i.p.) and 100 units of heparin also injected. The heart was removed and rinsed in Krebs-Henseleit buffer at 4°C. The aorta was cannulated for retrograde perfusion of the coronary arteries at a constant pressure of 70 mmHg with a Krebs-Henseleit buffer containing 25 mmol L−1 NaHC03, 1.8 mmol L−1 CaCl2 and 1 mmol·L−1 Na lactate (pH 7.4) at 36.5 to 37°C and gassed continuously with 95%O2-5%CO2. The heart was placed in thermostatically regulated chamber and was constantly rinsed with the liquid ejected from the pulmonary artery. Coronary flow was measured by the perfusion pump. A metal cannula with a latex balloon was inserted into the left ventricle and connected to a pressure transducer. Left ventricular dP/dt+ and heart rate were calculated from the left ventricular pressure signal. Each heart was either exposed to ascending concentrations of R406 (up to 300 nmol·L−1) or 0.1% DMSO as a time-matched vehicle control (n = 6 hearts per group).
Rat isolated aorta
The effects of R406 in rat isolated aorta were determined using methods similar to those previously described (University of Edinburgh. Dept. of Pharmacology, 1970). Male Wistar rats (250–310 g), were killed and the thoracic aortae removed. The rings were mounted between metal hooks and suspended in a 25 ml organ bath containing Krebs buffer warmed to 36-37oC, oxygenated with 95% O2, 5% CO2. The endothelium was not removed. Tissues were placed under a 1g resting tension and equilibrated in the Krebs solution using tissue bath wash outs at 10 min intervals for 1 h, resting tension being restored manually after each wash. Following this, tissues were sensitised to phenylephrine at a final bath concentration of 10 μmol·L−1, tissues washed after each response, until reproducible contractions were obtained (typically after the 4th sensitisation).
Once reproducible contractions to phenylephrine were obtained, tissues were washed a final time and then constricted with 10 μmol·L−1 phenylephrine (final bath concentration). Time was allowed for this contraction to stabilise, typically between 10 and 20 min, to provide a constant baseline. R406 was added to the bath in increasing concentrations up to 1 μmol·L−1. A high concentration of 100 μmol·L−1 R406 was formulated in 20% N-methyl-2-pyrrolidone (NMP) (Fisher Scientific, Leicestershire, UK) and 80% distilled water and diluted further into buffer to give the range of lower concentrations used in the assay. The same vehicle without test compound was also diluted in the same way to give a matched vehicle sample for each active compound concentration added, thus both compound and vehicle increased in concentration in the assay.
In a second set of studies, rat aortae were prepared as above and sensitised to phenylephrine in the same manner, and then washed with Krebs buffer. At this point a dose-response to phenylephrine was generated over a range of concentrations (10 nmol·L−1–100 μmol·L−1). After this first dose-response, tissues were washed until tension had returned to the original 1g baseline. R406, at final bath concentrations of 0.7 or 2 μmol·L−1, or 100 μmol·L−1 L-NAME, or vehicle control were then added to individual tissues and allowed to equilibrate for 90 minutes. At that point, still in the presence of test compound or vehicle, a second phenylephrine dose-response was conducted.
Human isolated blood vessels
Ethical approval was received in advance of the research and informed consent was obtained from all patients in accordance with the Declaration of Helsinki. Skin samples were obtained from residual tissue of patients undergoing elective cosmetic surgery. Excised tissue was dissected into vessel segments approximately 2 mm in length, and the endothelium was left intact. Contractile responses were measured using wire myography as previously described (Lynch et al. 2013). Briefly, tissue were exposed to high extracellular K+ (62.5 mmol·L−1), ACh (10 μmol·L−1) and U46619 (100 nmol·L−1) to ensure contractile responses were intact, and then washed with physiological saline solution prior to testing R406. Tissues which did not respond were rejected. Following this procedure, contractile responses were measured in response to ascending concentrations of R406, up to 1 μmol L−1.
Data analysis
Data from the conscious telemetered rat and general haemodynamic studies were modelled using a repeated measures (mixed effects) model. Dose, time points and their interaction were fitted as fixed effects; animal was fitted as a random effect. The average of two baseline values was included as a covariate in the general haemodynamics experiment, but not deemed necessary in the telemetry study. Differences between each dose group and the vehicle group at each post-dose time point were evaluated using a two-sided test, with P < 0.05 considered significant. Student's t distribution (general haemodynamics experiment) or Tukey's post hoc test (telemetry) were used for comparisons.
In the studies with anaesthetised rats, data were obtained on endothelial function and VEGF-induced vasodilatation. For the endothelial function testing, since both R406 and the positive control, L-NAME, induced changes in baseline haemodynamic parameters, the maximum increase in femoral blood flow (FBF) and femoral vascular conductance (FVC) from pre-occlusion baseline were calculated for the first (control) hyperaemia and for the hyperaemia after administration of R406, L-NAME or their respective vehicles. The increase in FVC induced during the hyperaemia was quantified by calculating the area under the curve (AUC) of the first 2 or 5 min of the response. The increase in FVC evoked by intra-arterial administration of ACh was quantified in a similar manner; however, due to its more transient effects, the AUC0-30s was calculated. Changes in the AUC parameters, induced by the test substances or vehicles, were calculated by subtracting the control response from that obtained after test substance administration. Effects of treatments on endothelial function and on VEGF-induced hypotension were compared to their respective time-matched, vehicle controls using a two-sided unpaired Student's t-test assuming equal variance between groups.
Data from the isolated heart preparations were analysed with a two-way ANOVA with repeated measures followed, if P<0.05, by a Bonferroni post hoc test, comparing the treated and control groups. Data from the assays using isolated blood vessels (rat or human) were analysed by two-tailed unpaired Student's t-test. A P value of <0.05 was considered significant.
Materials
Fostamatinib, R406, pazopanib and anisomycin were obtained from the AstraZeneca compound collection. For oral dosing of fostamatinib, the vehicle used was an aqueous solution containing 0.1% sodium carboxymethylcellulose, 0.1% methyl paraben and 0.02% propyl paraben and for intravenous dosing of R406, the vehicle was 40% DMA/40% PEG 400/20% of 75% HP-β-CD. Human VEGF-A (165 isoform) was expressed in Sf21 insect cells using the baculovirus expression system and purified from cell lysates using heparin-Sepharose affinity chromatography followed by C2-C18 mixed bed reversed-phase chromatography. Other compounds used were purchased from Sigma unless stated otherwise.
Results
Effects of fostamatinib on BP in conscious rats
In conscious telemetered rats, single oral doses of 10, 30 and 100 mg·kg−1 of fostamatinib caused a dose-dependent elevation in BP that returned to baseline within approximately 24 h of dosing (Figure 1A–C). Significant peak increases in mean arterial BP (MABP) varied from approximately 10% following administration of 10 mg·kg−1 of fostamatinib to 15% following 100 mg·kg−1. The time course of the BP effect correlated closely with changes in R406 plasma concentrations, suggesting a pharmacologically mediated effect. The relationship between R406 plasma concentration and increase in MABP was well described by the sigmoid Emax model (Figure 1D). From these data, the EC50 estimate for MABP was 790 and 17 nmol·L−1, based on total and free plasma concentrations respectively. Emax was estimated to 11% change from baseline and γ was estimated to 1.6. Similar EC50 values were also returned for changes in diastolic and systolic arterial BP (DABP and SABP; see Supporting Information). The 100 mg·kg−1 dose was also associated with a decreased heart rate that did not reach significance (data not shown). The increase in BP in conscious rats with fostamatinib treatment is consistent with our previous data with a VEGF receptor inhibitor (Curwen et al., 2008).
Figure 1.
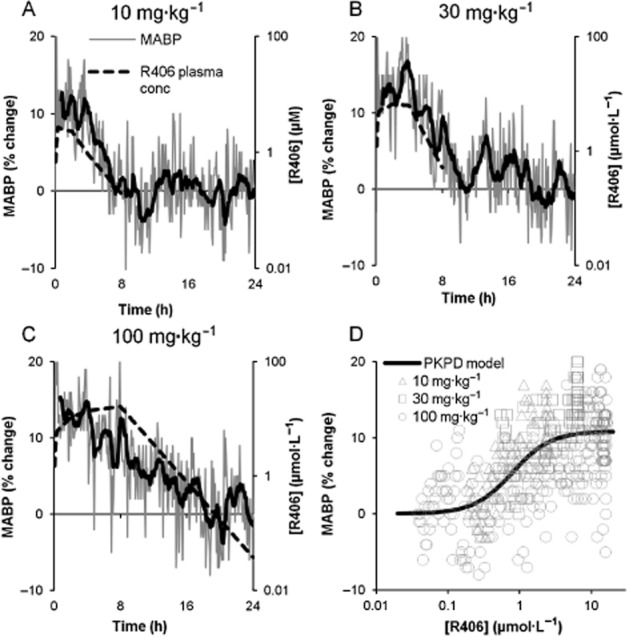
Pharmacokinetic : pharmacodynamic (PK/PD) relationship for R406 and BP. (A–C) Changes in MABP in conscious telemetered rats following treatment with single oral doses of 10, 30 and 100 mg·kg−1 fostamatinib. Each graph shows vehicle-adjusted % change from baseline MABP versus time (fine grey line, left Y-axes) and total R406 plasma concentration, including interpolated data (dashed line, right Y-axes) versus time. Moving average for MABP was calculated over 10 consecutive observations and plotted at the time point of the last observation (bold black line). D shows the fit of the PKPD model (black line) along with the observed data (individual symbols), where the observed data are composed of the measured BP at each time point plotted against an observed or interpolated total R406 plasma concentration at the same time point.
Haemodynamic effects of R406 in anaesthetized rats
In order to fully characterize the relationship between BP and other haemodynamic effects, an anaesthetized rat model was used to determine the effects of R406 on a number of cardiovascular parameters. Figure 2 and Table 1 illustrate the general haemodynamic actions of 3 and 5 mg·kg−1 R406 when administered intravenously over 30 min. R406 caused a maximum increase of 10% in MABP (83 ± 5 compared with 75 ± 3 mmHg in the vehicle group, P < 0.05). R406 caused a significant reduction in femoral arterial blood flow (FBF) and femoral vascular conductance (FVC) of similar magnitude in both dose groups (26–30% reduction for FBF and 36% reduction for FVC; P < 0.01; Figure 2C and E, Table 1). Decreases in heart rate (5%) and LV dP/dt+ (an index of contractility; 13%) were observed in the 5 mg·kg−1 group only (P < 0.05) possibly reflecting a greater degree of reflex compensation at the higher dose (Figure 2B and D, Table 1). Maximum R406 free plasma concentrations were 82 ± 5 and 129 ± 17 nmol·L−1 for the 3 and 5 mg·kg−1 groups respectively.
Figure 2.
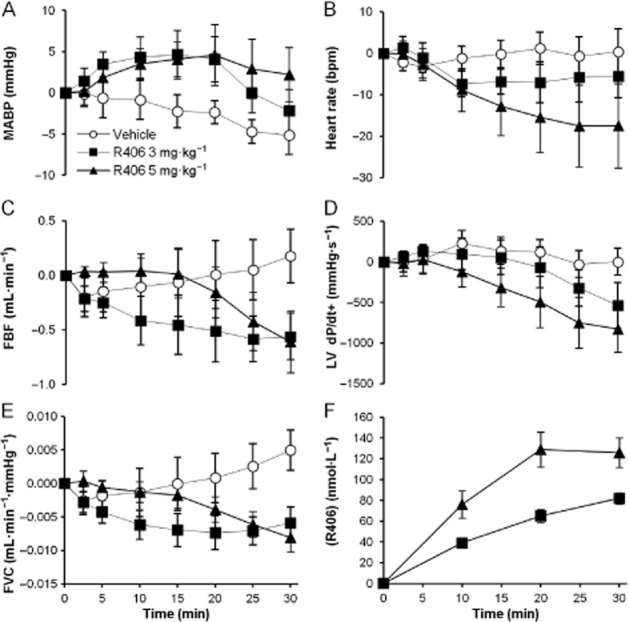
Effect of R406 on haemodynamic parameters in anaesthetized rats. The effects of i.v. infusion of 3 or 5 mg·kg−1 R406 on (A) MABP, (B) heart rate (bpm), (C) FBF, (D) left ventricular contractility index (LV dP/dt+) and (E) FVC over a 30 min period. Panel F contains the corresponding unbound plasma concentrations in nmol·L−1. Data are shown as mean ± SEM change from baseline, n = 8 per group. Peak effects are given in Table 1.
Table 1.
Effects of R406 on haemodynamic parameters in anaesthetized rats
| Parameter | R406 3 mg·kg−1 | R406 5 mg·kg−1 |
|---|---|---|
| SABP | +10% | +7%* |
| DABP | +9% | +13%* |
| MABP | +10% | +10%* |
| Heart rate | −2% | −5%* |
| LV systolic BP | −3% | +1% |
| LVEDP | −32% | −13% |
| dP/dt+ | −7% | −13%* |
| dP/dt− | +8% | −8% |
| FBF | −30%* | −26%* |
| FVC | −36%** | −36%** |
Peak effects of 3 or 5 mg·kg−1 R406 on haemodynamic parameters (SABP, DABP and MABP, LV parameters (LVEDP, LV end diastolic pressure), indexes of cardiac contractility and relaxation (dP/dt+ and dP/dt-) and FBF and its derivative FVC over a 30 min period. Data expressed as percent change from time-matched vehicle data following baseline adjustment for vehicle and treated groups. n = 8 per group.
P < 0.05,
P < 0.01; significantly different from time-matched vehicle data; analysis of covariance. Further details are given in the Supporting Information.
Effects of R406 on VEGF signalling and NO production
R406 has a Kd of 20–40 nmol·L−1 for VEGFR2 in isolated enzyme assays (Davis et al., 2011; Metz et al., 2011). R406 also inhibits auto-phosphorylation of VEGFR2 in human umbilical venous endothelial cells with an IC50 of 333 nmol·L−1 (T. Gururaja, unpubl. obs.). VEGF exerts its vasodilatory effect by stimulating the production of endothelial NO. Therefore, we determined the effects of R406 on VEGF-stimulated NO production in HMEC cultures. Figure 3A shows the results from typical experiments where pretreatment with R406, or the VEGFR2 inhibitor pazopanib, caused a concentration-dependent inhibition of VEGF-stimulated NO production, as determined by cytosolic DAF fluorescence. Based on five independent experiments, R406 and pazopanib inhibited NO production with IC50 values of 339 (136; 838) nmol·L−1 and 25 (10; 63) nmol·L−1 respectively (means with 95% confidence limits).
Figure 3.
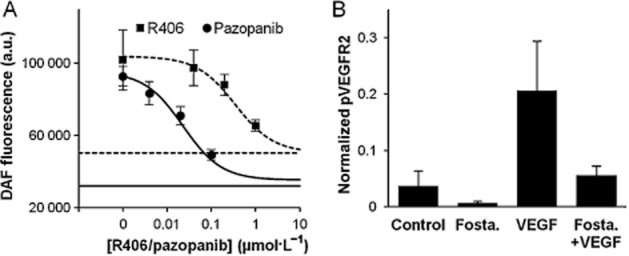
Effects of R406 and fostamatinib on VEGF signalling in vitro and in vivo. (A) R406 inhibits VEGF-stimulated release of NO from HMECs. DAF-FM-loaded cells were pre-incubated with R406 or pazopanib for 20 min in independent experiments, and stimulated for 30 min with 50 ng·mL−1 VEGF. Cytoplasm-specific fluorescence was captured using an ImageXpress plate imager. Basal fluorescence in each experiment was established by incubating cells with 5 μM anisomycin, a potent NOS inhibitor (horizontal and dotted lines). The bottom of each concentration-response curve was fitted to these minima. Results are expressed as non-cumulative concentration-effects curves expressed as mean (±SEM; n = 5) for a representative experiment. a.u., arbitrary units. (B) Lung p-VEGFR2 levels in mice after dosing with 100 mg·kg−1 fostamatinib (Fosta) and/or 20 μg VEGF. pVEGFR2 levels are normalized to total VEGFR2 and GAPDH.
Inhibition of the VEGFR2 kinase activity was also determined in vivo.
Normal lung tissue endothelial cells in mice express high levels of total and phosphorylated VEGFR2 and provide a convenient ex vivo assay (Smith et al., 2007). In our experiments, treatment of mice with fostamatinib decreased the levels of pVEGFR2 in lung tissue (Figure 3B).
Effects of R406 on VEGF-induced hypotension in anaesthetized rats
Consistent with previous studies (Yang et al., 1996; Horowitz et al., 1997), i.v. bolus administration of VEGF had an acute hypotensive effect: a dose of 55 μg·kg−1 VEGF administered to vehicle-treated animals caused a maximum 19% (20 ± 3 mmHg) reduction in MABP at 2 min after administration (Figure 4A). VEGF also caused an increase in FBF and FVC. Administration of 4.5 mg·kg−1 R406 inhibited this effect so that only a 1% (1 ± 1 mmHg) fall in MABP was seen with the same dose of VEGF (P < 0.001). VEGF-induced increases in FBF and FVC were also inhibited by R406 (P < 0.01; Figure 4B and C). These data are consistent with our previous findings demonstrating that VEGF receptor inhibitors can inhibit VEGF-induced hypotension in anaesthetized rats (Wedge et al., 2002; Curwen et al., 2008).
Figure 4.
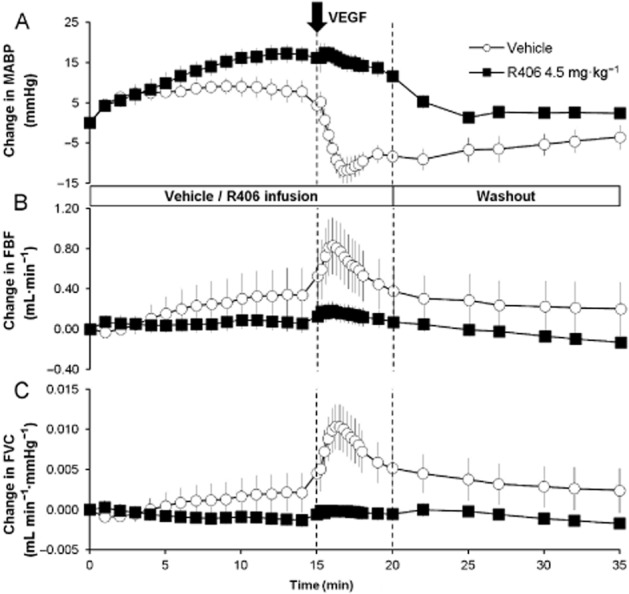
Effect of R406 on VEGF-induced vascular responses in anaesthetized rats. The effects of i.v. infusion of vehicle or 4.5 mg·kg−1 R406 on (A) MABP, (B) FBF and (C) FVC when given in conjunction with VEGF. Vehicle or R406 were administered for 20 min as an i.v. infusion. At 15 min into the infusion, a bolus 55 μg·kg−1 dose of VEGF was administered. Parameters were monitored for a further 15 min after the 20 min infusion period. Data are shown as mean ± SEM change from baseline, n = 6 per group. The vertical line at 15 min represents the time for the VEGF injection, and the line at 20 min represents the end of the R406/vehicle infusion and start of the washout.
Effects of R406 on vascular and cardiac contraction in vitro
Data from the anaesthetized rat study demonstrated that infusion of R406 resulted in decreases in femoral conductance, dP/dt+ and heart rate. In order to determine if these were direct cardiac and vascular responses, we examined the effects of R406 on vascular tone of isolated blood vessels and cardiac function of rat isolated hearts. R406 (up to 1 μmol·L−1) produced no contractile or relaxant responses in rat aorta that were pre-constricted with 10 μmol·L−1 phenylephrine (n = 4, P > 0.05, Figure 5A). R406 (up to 1 μmol·L−1) did not directly constrict human isolated resistance vessels (n = 6, P > 0.05, Figure 5A). The effects of pre-incubation with R406 (0.7 or 2 μmol·L−1) on the phenylephrine concentration-response curve were determined in rat aorta. During the 60–90 min pre-incubation period, R406 had no contractile effect (data not shown), and had no effect on the phenylephrine response (n = 4, P > 0.05, Figure 5B). Pretreatment with L-NAME enhanced the peak response to phenylephrine by 28% (n = 4, P < 0.05, Figure 5B), consistent with inhibition of basal NO activity (see Al-Zobaidy et al., 2011). Finally, R406 was tested for its effects in a Langendorff-perfused rat isolated heart model. In this model, R406 (up to 0.3 μmol·L−1) had no effect on coronary flow, heart rate or dP/dt+, (n = 6 hearts, P > 0.05, Figure 5C).
Figure 5.
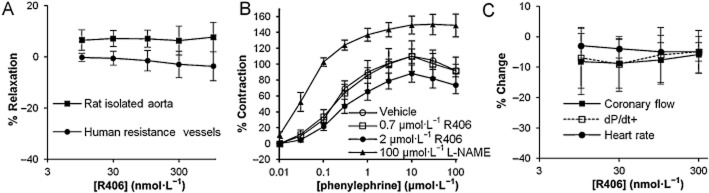
Effects of R406 on rat isolated aorta, human isolated resistance vessels and rat isolated heart. (A) Contraction of rat isolated aorta (n = 4) and human s.c. resistance arteries (n = 6) in the presence of R406. Data are presented as % relaxation relative to baseline and vehicle time-matched control. (B) Rat aorta concentration-response curves to phenylephrine following pre-incubation with vehicle, R406 (0.7 and 2 μmol·L−1) and L-NAME (100 μmol·L−1). Data are presented as % contraction relative to the maximal response seen with the first phenylephrine concentration-response curve. (C) Effects of R406 on coronary flow, dP/dt+ and heart rate in Langendorff-perfused rat isolated hearts, n = 6 hearts. Data are presented as vehicle adjusted mean (±SEM) % change relative to baseline.
Effects of R406 on endothelial function in anaesthetized rats
Given that R406 caused a decrease in femoral arterial conductance in vivo, an investigation into a possible effect on endothelial function was conducted. The effects of R406 were assessed in anaesthetized rats using two different endothelial function tests: reactive hyperaemia (RH) and ACh-induced dilatation (see Figure 6).
Figure 6.
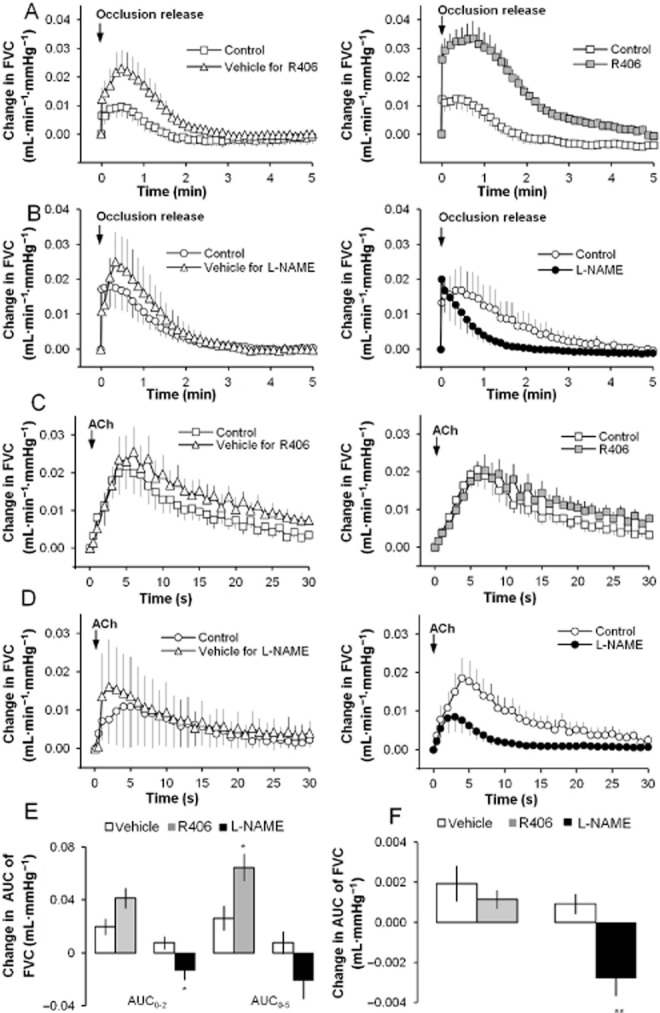
Effect of R406 on endothelial function in anaesthetized rats. (A) The effects of vehicle (for R406) and R406 (4.5 mg·kg−1) on the increase in FVC induced during reactive hyperaemia (RH). (B) The effects of vehicle (for L-NAME) and L-NAME (10 mg·kg−1) on the increase in FVC induced during RH. (C) The effects of vehicle (for R406) and R406 (4.5 mg·kg−1) on the increase in FVC induced by intra-arterial injection of ACh (0.1 μg). (D) The effects of vehicle (for L-NAME) and L-NAME (10 mg·kg−1) on the increase in FVC induced by intra-arterial injection of ACh (0.1 μg). (E) The effects of R406, L-NAME and corresponding vehicles on RH. The RH was quantified as the AUC of the of the FVC response (first 2 or 5 min following occlusion release). Histograms show changes from the control RH. (F) The effects of R406, L-NAME and corresponding vehicles on the ACh-induced vasodilatation. The vasodilatation was quantified as the AUC of the FVC response (first 30 s following injection). Time-effect plots show means ± SEM (n = 5–7 rats) of change from baseline data. Histograms show changes from the control ACh response. * P < 0.05, **P < 0.01; significantly different from vehicle; unpaired t-test.
Effects on RH
Occlusion of the femoral artery for 5 min caused a reproducible increase in FBF and FVC upon release, with peak increases occurring within 1 min. In the vehicle and R406 group, a control RH was produced during an infusion of vehicle and followed, 20 min later, by another RH during either a second infusion of vehicle or R406, respectively. The vehicle itself had an effect on the RH increasing the response versus the control response and a slightly larger increase versus control was observed after administration of 4.5 mg·kg−1 R406 (Figure 6A). The RH was quantified by calculating the AUC of the FVC response and while R406 had no statistically significant effect on the initial phase of the RH (AUC0–2 min), the AUC of the entire RH (AUC0–5 min) was increased when compared with the vehicle data (increase of 0.064 ± 0.010 mL·mmHg−1 after 4.5 mg·kg−1 compared with increase of 0.026 ± 0.009 mL·mmHg−1 in the vehicle group, P < 0.05; Figure 6E).
Inhibition of NOS with 10 mg·kg−1 L-NAME caused a reduction of the RH while the vehicle for L-NAME had no marked effect on the RH response (Figure 6B). L-NAME reduced both the AUC0–2 min and the AUC0–5 min of the RH; however, only the reduction of the initial phase was statistically significantly (decrease of 0.013 ± 0.008 mL·mmHg−1 after 10 mg·kg−1 compared with an increase of 0.008 ± 0.005 mL·mmHg−1 in the vehicle group, P < 0.05; Figure 6E). The results indicate that the hyperaemic response is mediated, in part, by mechanisms involving NOS, which, to the best of our knowledge, is a novel finding in the rat.
Effect on ACh-induced response
Intra-arterial bolus injection of ACh (0.1 μg) caused a vasodilatation, as shown by a transient increase in FBF and FVC, with the peak effects occurring within 5–10 s.
The femoral vascular response to ACh was not markedly affected by the administration of R406 or its vehicle when compared with the control response performed 20 min earlier (Figure 6C). The vascular effect of ACh was quantified by calculating an AUC of the FVC response and no significant difference between the effects of R406 or its vehicle was observed (Figure 6F). In contrast, the femoral vascular response induced by ACh was reduced after administration of L-NAME (decrease of 0.003 ± 0.001 mL·mmHg−1 compared with an increase of 0.001 ± 0.001 mL·mmHg−1 in the vehicle group, P < 0.01; Figure 6D and F) confirming previous findings that the vasodilatation induced by ACh is mediated in part by NO (Furchgott and Zawadzki, 1980).
Discussion and conclusions
BP elevation is a recognized side effect of treatments that inhibit VEGF signalling (Robinson et al., 2010; Keefe et al., 2011; Thanigaimani et al., 2011). A number of mechanisms have been proposed for the hypertensive effect of these drugs, and it is likely that the contribution of VEGF in maintaining basal vascular tone is involved (Bhargava, 2009; Lazarus and Keshet, 2011). Based on a systematic investigation in non-clinical models, the data presented here clearly demonstrated that fostamatinib evoked a dose-dependent elevation in BP that was indicative of a direct pharmacological effect. Additional data indicated that the BP elevation in rats was driven, at least in part, by an increase in vascular resistance. Both in vitro and in vivo data also demonstrated VEGF signalling was inhibited by R406. Importantly, treatment with R406 could inhibit VEGF-induced vasodilatation and hypotension in anaesthetized rats. These data are in agreement with similar observations made with other RTKIs (Curwen et al., 2008; Franklin et al., 2009; Kappers et al., 2010; 2012,; Brave et al., 2011; Blasi et al., 2012; Nagasawa et al., 2012). Taken together, the current study suggests that impaired vasorelaxation, resulting from reduced VEGF-mediated endothelial NO release, may contribute to fostamatinib-induced BP elevation. This hypothesis is consistent with the BP-elevating mechanism of other kinase inhibitors whose pharmacological profiles overlap to some extent with that of R406 (for instance, inhibition of VEGFR2; Robinson et al., 2010; Keefe et al., 2011; Thanigaimani et al., 2011).
Clinical investigations have shown that TK inhibitors can have complex effects on vascular function (Steeghs et al., 2008; Mayer et al., 2011). The data presented here show that R406 had a very specific inhibitory effect on NO-dependent aspects of endothelial function in the rat model. Firstly, VEGF-induced vasodilatation, a response known to be NO-dependent, was almost completely inhibited by R406. Secondly, R406 had no effect on the femoral arterial vasodilatation induced by ACh, an effect that has been shown previously, both clinically and non-clinically, to be due in part to endothelial NO release (Furchgott and Zawadzki, 1980; Vallance et al., 1989; Rees et al., 1990). Thirdly, R406 also had no inhibitory effect on the RH evoked in the rat femoral vascular bed. While the initial phase of the RH in response to transient periods of ischaemia involves vasodilator metabolites and myogenic responses, endothelial NO release has been shown to contribute significantly to the later phase in animals and humans (Yamabe et al., 1992; O'Leary et al., 1994; Meredith et al., 1996; Joannides et al., 2006). Finally, R406 did not inhibit the basal NO release present in rat aorta. Overall, these data show that it is possible to specifically inhibit one of the many pathways that converge on endothelial NOS activation while leaving other pathways unaffected. It is also convincing evidence that the fostamatinib-induced BP elevation seen in the rat is not due to general depression of endothelial function or decreased smooth muscle function, but rather due to a specific inhibition of tonic VEGF-induced NO release.
It is of interest that R406 enhanced the RH response (Figure 6A). It is not clear at this time what is the mechanism for this enhanced response might be, but one possibility is that it reflects activities of R406 at additional secondary targets. However, further investigative studies will be required to understand this phenomenon.
One apparent inconsistency between fostamatinib and other TK inhibitors is the amount of VEGFR2 inhibition expected at therapeutic doses, and the effect on BP. For most drugs that inhibit VEGFR2 as the primary target, therapeutic plasma levels of free drug exceed the VEGFR2 IC50 by 6-to 300-fold (based on values derived from HUVECs; McTigue et al., 2012). Of note in clinical studies of rheumatoid arthritis with fostamatinib, predicted therapeutic levels of R406 are sevenfold below the equivalent VEGFR2 IC50. However, this apparent difference can be reconciled by the magnitude of BP elevation observed with these TK inhibitors, and their relative potencies at VEGFR2. In phase II clinical trials, the mean increase in systolic BP seen with fostamatinib is approximately 5 mmHg in patients with rheumatoid arthritis (Weinblatt et al., 2010; Genovese et al., 2011). However, other VEGFR2 inhibitors show much greater increases in BP in the clinic (value in brackets refer to the mean mmHg increase in systolic BP): cediranib (19, Robinson et al., 2010), pazopanib (18, Heath et al., 2013), sorafenib (21, Veronese et al., 2006), sunitinib (25, Eechoute et al., 2012) and vandetanib (12, Mayer et al., 2011). Therefore, the comparatively lower increase in BP seen with fostamatinib may reflect the lower inhibition of VEGFR2 at therapeutic levels. This is consistent with a large amount of clinical data showing that treatment-induced hypertension for these approved drugs is dose-dependent and correlates with the potency of VEGFR2 inhibition (Chen and Cleck, 2009).
One important aspect that we have not directly addressed is whether the proposed mechanism for BP elevation in the rat translates to that in humans. A number of TK inhibitors that are known to elevate BP in humans also elevate BP in several non-clinical species, including rats (Curwen et al., 2008; Franklin et al., 2009; Kappers et al., 2010; 2012,; Brave et al., 2011; Blasi et al., 2012; Nagasawa et al., 2012). This suggests that the mechanisms for BP elevation by RTKIs may be common across species. Importantly, the effects we have shown on BP in rats occur at similar R406 plasma concentrations to those that occur at predicted therapeutic doses in humans (the mean unbound maximum R406 plasma concentration was ∼49 nmol·L−1 with 100 mg fostamatinib b.i.d.; M. Braddock, unpubl. obs.). In addition, we have shown that R406 inhibits VEGF-induced NO production from human endothelial cells, providing some limited evidence that the mechanism for BP elevation identified in our models may translate to humans.
This study has focused on the role that inhibition of VEGF signalling plays in acute changes in vascular tone that occur in response to fostamatinib. For this reason, alternative mechanisms for BP elevation have not been examined in detail. Numerous theories have been postulated to explain the effects of VEGFR2 inhibition on BP (Bhargava, 2009). These include microvascular rarefaction, reduced NO production and increased circulating levels of vasoactive hormones such as endothelin-1 and angiotensin II (Veronese et al., 2006; Steeghs et al., 2008; Mayer et al., 2011; Belcik et al., 2012; Blasi et al., 2012; Lankhorst et al., 2012; 2012,; Nagasawa et al., 2012). The acute changes seen in BP in this study probably exclude a structural or pathological change as a mechanism for BP elevation (i.e. microvascular rarefaction or kidney pathology). We have also shown that fostamatinib increases BP after angiotensin signalling is blocked by treatment with an ACE inhibitor (Lengel et al., in preparation). This suggests that the pressor effect of fostamatinib is not mediated by the angiotensin signalling system. However, as we have not measured levels of blood-borne factors that may cause BP elevation (e.g. endothelin-1 and catecholamines), we cannot exclude this as a possible mechanism for fostamatinib-induced BP elevation. Another possibility is that fostamatinib increases BP via an increased sympathetic vasomotor tone and this may explain the lack of vascular effects in vitro. We have not addressed this in the current studies but we believe it is unlikely since levels of tissue radioactivity were either near or below quantifiable levels in the brain and spinal cord in a quantitative whole-body autoradiography study in rats given radiolabelled fostamatinib (A. Lordi, unpubl. data). An effect at the level of the sympathetic ganglia cannot, however, be excluded.
In our studies, we have measured the effects of R406 only on the femoral vascular bed, much of which perfuses hindlimb skeletal muscle. However, a significant proportion of the cardiac output is directed to the skeletal muscle vasculature, and small changes in the radius of resistance vessels in skeletal muscle have been shown to greatly influence total peripheral resistance (Thomas et al., 1990). This is exemplified by the effects of L-NAME, which increases BP and also concomitantly increases total peripheral, regional and femoral vascular resistances in rats (Gardiner et al., 1990). Therefore, it is reasonable to assume that the R406-induced decrease in femoral vascular conductance (the inverse of resistance) can be taken as an index of increased total peripheral resistance.
BP elevation has been observed in a proportion of patients treated with fostamatinib in clinical trials. Based on the non-clinical data presented here, a potential mechanism underlying clinical responses may be increased vascular resistance as a consequence of impaired vasorelaxation, resulting from reduced VEGF-induced NO release from the endothelium. This hypothesis is consistent with the hypertensive mechanism of other TK inhibitors whose pharmacological profile share overlap with R406 (e.g. inhibition of VEGFR2). However, given the complexity of BP regulation, it is possible that inhibition of VEGFR2 is not the sole mechanism for fostamatinib-induced BP elevation. These data suggest that, in the clinic, established anti-hypertensive treatments used to manage BP elevation and hypertension observed with marketed anti-VEGF drugs may also be appropriate for fostamatinib-induced BP elevation.
Acknowledgments
The authors would like to thank Grace Jefferies for assistance in running the rat aorta assays, Dr Jean-Pierre Valentin, Dr Pierre Lainee and Dr Mike Rolf for helpful discussions and Dr Chris Pollard for critically reviewing the manuscript. The rat isolated heart study was performed on behalf of AstraZeneca at CEROM, Paris, France. The human isolated resistance vessel study was performed on behalf of AstraZeneca at Biopta, Glasgow, UK.
Glossary
- DABP
diastolic arterial BP
- dP/dt +/−
the rate of left ventricle pressure rise and decline
- FBF
femoral arterial blood flow
- FVC
femoral vascular conductance
- HMEC
human microvascular endothelial cells
- LV
left ventricular
- MABP
mean arterial BP
- RH
reactive hyperaemia
- RTKi
receptor TK inhibitors
- SABP
systolic arterial BP
- TK
tyrosine kinase
Funding
This work was funded by AstraZeneca.
Conflicts of interest
All authors were employees of AstraZeneca at the time this study was conducted.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12559
Methods
Preparation of telemetered rats PKPD modelling
Results
Table S1. Effects of R406 on haemodynamic parameters in anaesthetized rats.
Table S2. Effects of R406 and L-NAME on hyperaemia parameters in anaesthetized rats.
Table S3. Parameter estimates (CV%) for PKPD models.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. British Journal of Pharmacology. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zobaidy MJ, Craig J, Brown K, Pettifor G, Martin W. Stimulus-specific blockade of nitric oxide-mediated dilatation by asymmetric dimethylarginine (ADMA) and monomethylarginine (L-NMMA) in rat aorta and carotid artery. Eur J Pharmacol. 2011;673:78–84. doi: 10.1016/j.ejphar.2011.10.024. [DOI] [PubMed] [Google Scholar]
- Belcik JT, Qi Y, Kaufmann BA, Xie A, Bullens S, Morgan TK, et al. Cardiovascular and systemic microvascular effects of anti-vascular endothelial growth factor therapy for cancer. J Am Coll Cardiol. 2012;60:618–625. doi: 10.1016/j.jacc.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava P. VEGF kinase inhibitors: how do they cause hypertension? Am J Physiol Regul Integr Comp Physiol. 2009;297:R1–R5. doi: 10.1152/ajpregu.90502.2008. [DOI] [PubMed] [Google Scholar]
- Blasi E, Heyen J, Patyna S, Hemkens M, Ramirez D, John-Baptiste A, et al. Sunitinib, a receptor tyrosine kinase inhibitor, increases blood pressure in rats without associated changes in cardiac structure and function. Cardiovasc Ther. 2012;30:287–294. doi: 10.1111/j.1755-5922.2011.00278.x. [DOI] [PubMed] [Google Scholar]
- Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- Brave SR, Odedra R, James NH, Smith NR, Marshall GB, Acheson KL, et al. Vandetanib inhibits both VEGFR-2 and EGFR signalling at clinically relevant drug levels in preclinical models of human cancer. Int J Oncol. 2011;39:271–278. doi: 10.3892/ijo.2011.1022. [DOI] [PubMed] [Google Scholar]
- Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- Curwen JO, Musgrove HL, Kendrew J, Richmond GH, Ogilvie DJ, Wedge SR. Inhibition of vascular endothelial growth factor-a signaling induces hypertension: examining the effect of cediranib (recentin; AZD2171) treatment on blood pressure in rat and the use of concomitant antihypertensive therapy. Clin Cancer Res. 2008;14:3124–3131. doi: 10.1158/1078-0432.CCR-07-4783. [DOI] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Eechoute K, Veldt van der AA, Oosting S, Kappers MH, Wessels JA, Gelderblom H, et al. Polymorphisms in endothelial nitric oxide synthase (eNOS) and vascular endothelial growth factor (VEGF) predict sunitinib-induced hypertension. Clin Pharmacol Ther. 2012;92:503–510. doi: 10.1038/clpt.2012.136. [DOI] [PubMed] [Google Scholar]
- Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54:652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin PH, Banfor PN, Tapang P, Segreti JA, Widomski DL, Larson KJ, et al. Effect of the multitargeted receptor tyrosine kinase inhibitor, ABT-869 [N-(4-(3-amino-1H-indazol-4-yl)phenyl)-N′-(2-fluoro-5-methylphenyl)urea], on blood pressure in conscious rats and mice: reversal with antihypertensive agents and effect on tumor growth inhibition. J Pharmacol Exp Ther. 2009;329:928–937. doi: 10.1124/jpet.108.144816. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Gardiner SM, Compton AM, Kemp PA, Bennett T. Regional and cardiac haemodynamic effects of NG-nitro-L-arginine methyl ester in conscious, Long Evans rats. Br J Pharmacol. 1990;101:625–631. doi: 10.1111/j.1476-5381.1990.tb14131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese MC, Kavanaugh A, Weinblatt ME, Peterfy C, DiCarlo J, White ML, et al. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: a three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011;63:337–345. doi: 10.1002/art.30114. [DOI] [PubMed] [Google Scholar]
- Heath EI, Infante J, Lewis LD, Luu T, Stephenson J, Tan AR, et al. A randomized, double-blind, placebo-controlled study to evaluate the effect of repeated oral doses of pazopanib on cardiac conduction in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71:565–573. doi: 10.1007/s00280-012-2030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier C, Berry C, Petrie MC, O'Dwyer PJ, Hamilton C, Brown A, et al. Effects of urotensin II in human arteries and veins of varying caliber. Circulation. 2001;103:1378–1381. doi: 10.1161/01.cir.103.10.1378. [DOI] [PubMed] [Google Scholar]
- Horowitz JR, Rivard A, Zee van der R, Hariawala M, Sheriff DD, Esakof DD, et al. Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol. 1997;17:2793–2800. doi: 10.1161/01.atv.17.11.2793. [DOI] [PubMed] [Google Scholar]
- Joannides R, Bellien J, Thuillez C. Clinical methods for the evaluation of endothelial function-a focus on resistance arteries. Fundam Clin Pharmacol. 2006;20:311–320. doi: 10.1111/j.1472-8206.2006.00406.x. [DOI] [PubMed] [Google Scholar]
- Kamendi HW, Brott DA, Chen Y, Litwin DC, Lengel DJ, Fonck C, et al. Combining radio telemetry and automated blood sampling: a novel approach for integrative pharmacology and toxicology studies. J Pharmacol Toxicol Methods. 2010;62:30–39. doi: 10.1016/j.vascn.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Kappers MH, Esch van JH, Sluiter W, Sleijfer S, Danser AH, Meiracker van den AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56:675–681. doi: 10.1161/HYPERTENSIONAHA.109.149690. [DOI] [PubMed] [Google Scholar]
- Kappers MH, Beer de VJ, Zhou Z, Danser AH, Sleijfer S, Duncker DJ, et al. Sunitinib-induced systemic vasoconstriction in swine is endothelin mediated and does not involve nitric oxide or oxidative stress. Hypertension. 2012;59:151–157. doi: 10.1161/HYPERTENSIONAHA.111.182220. [DOI] [PubMed] [Google Scholar]
- Keefe D, Bowen J, Gibson R, Tan T, Okera M, Stringer A. Noncardiac vascular toxicities of vascular endothelial growth factor inhibitors in advanced cancer: a review. Oncologist. 2011;16:432–444. doi: 10.1634/theoncologist.2010-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankhorst S, Kappers MH, Esch van JH, Danser AH, Meiracker van den AH. Mechanism of hypertension and proteinuria during angiogenesis inhibition: evolving role of endothelin-1. J Hypertens. 2012;31:444–454. doi: 10.1097/HJH.0b013e32835c1d1b. [DOI] [PubMed] [Google Scholar]
- Lazarus A, Keshet E. Vascular endothelial growth factor and vascular homeostasis. Proc Am Thorac Soc. 2011;8:508–511. doi: 10.1513/pats.201102-021MW. [DOI] [PubMed] [Google Scholar]
- Lynch J, Regan C, Beatch G, Gleim G, Morabito C. Comparison of the intrinsic vasorelaxant and inotropic effects of the antiarrhythmic agents vernakalant and flecainide in human isolated vascular and cardiac tissues. J Cardiovasc Pharmacol. 2013;61:226–232. doi: 10.1097/FJC.0b013e31827afd29. [DOI] [PubMed] [Google Scholar]
- McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue M, Murray BW, Chen JH, Deng YL, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci U S A. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer EL, Dallabrida SM, Rupnick MA, Redline WM, Hannagan K, Ismail NS, et al. Contrary effects of the receptor tyrosine kinase inhibitor vandetanib on constitutive and flow-stimulated nitric oxide elaboration in humans. Hypertension. 2011;58:85–92. doi: 10.1161/HYPERTENSIONAHA.110.168120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith IT, Currie KE, Anderson TJ, Roddy MA, Ganz P, Creager MA. Postischemic vasodilation in human forearm is dependent on endothelium-derived nitric oxide. Am J Physiol. 1996;270:H1435–H1440. doi: 10.1152/ajpheart.1996.270.4.H1435. [DOI] [PubMed] [Google Scholar]
- Metz JT, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nat Chem Biol. 2011;7:200–202. doi: 10.1038/nchembio.530. [DOI] [PubMed] [Google Scholar]
- Nagasawa T, Hye Khan MA, Imig JD. Captopril attenuates hypertension and renal injury induced by the vascular endothelial growth factor inhibitor sorafenib. Clin Exp Pharmacol Physiol. 2012;39:454–461. doi: 10.1111/j.1440-1681.2012.05699.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary DS, Dunlap RC, Glover KW. Role of endothelium-derived relaxing factor in hindlimb reactive and active hyperemia in conscious dogs. Am J Physiol. 1994;266:R1213–R1219. doi: 10.1152/ajpregu.1994.266.4.R1213. [DOI] [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol. 1990;101:746–752. doi: 10.1111/j.1476-5381.1990.tb14151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccaboni M, Bianchi I, Petrillo P. Spleen tyrosine kinases: biology, therapeutic targets and drugs. Drug Discov Today. 2010;15:517–530. doi: 10.1016/j.drudis.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD. Hypertension induced by vascular endothelial growth factor signaling pathway inhibition: mechanisms and potential use as a biomarker. Semin Nephrol. 2010;30:591–601. doi: 10.1016/j.semnephrol.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NR, James NH, Oakley I, Wainwright A, Copley C, Kendrew J, et al. Acute pharmacodynamic and antivascular effects of the vascular endothelial growth factor signaling inhibitor AZD2171 in Calu-6 human lung tumor xenografts. Mol Cancer Ther. 2007;6:2198–2208. doi: 10.1158/1535-7163.MCT-07-0142. [DOI] [PubMed] [Google Scholar]
- Steeghs N, Gelderblom H, Roodt JO, Christensen O, Rajagopalan P, Hovens M, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. 2008;14:3470–3476. doi: 10.1158/1078-0432.CCR-07-5050. [DOI] [PubMed] [Google Scholar]
- Stucker O, Vicaut E, Villereal MC, Ropars C, Teisseire BP, Duvelleroy MA. Coronary response to large decreases of hemoglobin-O2 affinity in isolated rat heart. Am J Physiol. 1985;249:H1224–H1227. doi: 10.1152/ajpheart.1985.249.6.H1224. [DOI] [PubMed] [Google Scholar]
- Thanigaimani S, Kichenadasse G, Mangoni AA. The emerging role of vascular endothelial growth factor (VEGF) in vascular homeostasis: lessons from recent trials with anti-VEGF drugs. Curr Vasc Pharmacol. 2011;9:358–380. doi: 10.2174/157016111795495503. [DOI] [PubMed] [Google Scholar]
- Thomas GR, Walder CE, Thiemermann C, Vane JR. Regional vascular resistance and haemodynamics in the spontaneously hypertensive rat: the effects of bradykinin. J Cardiovasc Pharmacol. 1990;15:211–217. doi: 10.1097/00005344-199002000-00006. [DOI] [PubMed] [Google Scholar]
- University of Edinburgh, Department of Pharmacology. Pharmacological Experiments on Isolated Preparations. Edinburgh: E. and S. Livingstone; 1970. [Google Scholar]
- Vallance P, Collier J, Moncada S. Effects of endothelium-derived nitric oxide on peripheral arteriolar tone in man. Lancet. 1989;2:997–1000. doi: 10.1016/s0140-6736(89)91013-1. [DOI] [PubMed] [Google Scholar]
- Veronese ML, Mosenkis A, Flaherty KT, Gallagher M, Stevenson JP, Townsend RR, et al. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24:1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645–4655. [PubMed] [Google Scholar]
- Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–1312. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- Wong BR, Grossbard EB, Payan DG, Masuda ES. Targeting Syk as a treatment for allergic and autoimmune disorders. Expert Opin Investig Drugs. 2004;13:743–762. doi: 10.1517/13543784.13.7.743. [DOI] [PubMed] [Google Scholar]
- Yamabe H, Okumura K, Ishizaka H, Tsuchiya T, Yasue H. Role of endothelium-derived nitric oxide in myocardial reactive hyperemia. Am J Physiol. 1992;263:H8–14. doi: 10.1152/ajpheart.1992.263.1.H8. [DOI] [PubMed] [Google Scholar]
- Yang R, Thomas GR, Bunting S, Ko A, Ferrara N, Keyt B, et al. Effects of vascular endothelial growth factor on hemodynamics and cardiac performance. J Cardiovasc Pharmacol. 1996;27:838–844. doi: 10.1097/00005344-199606000-00011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Effects of R406 on haemodynamic parameters in anaesthetized rats.
Table S2. Effects of R406 and L-NAME on hyperaemia parameters in anaesthetized rats.
Table S3. Parameter estimates (CV%) for PKPD models.