

Abstract
Toxoplasma gondii infection has previously been described to cause dramatic changes in the host transcriptome by manipulating key regulators, including STATs, NF-κB, and microRNAs. Here, we report that Toxoplasma tachyzoites also mediate rapid and sustained induction of another pivotal regulator of host cell transcription, c-Myc. This induction is seen in cells infected with all three canonical types of Toxoplasma but not the closely related apicomplexan parasite Neospora caninum. Coinfection of cells with both Toxoplasma and Neospora still results in an increase in the level of host c-Myc, showing that c-Myc is actively upregulated by Toxoplasma infection (rather than repressed by Neospora). We further demonstrate that this upregulation may be mediated through c-Jun N-terminal protein kinase (JNK) and is unlikely to be a nonspecific host response, as heat-killed Toxoplasma parasites do not induce this increase and neither do nonviable parasites inside the host cell. Finally, we show that the induced c-Myc is active and that transcripts dependent on its function are upregulated, as predicted. Hence, c-Myc represents an additional way in which Toxoplasma tachyzoites have evolved to specifically alter host cell functions during intracellular growth.
INTRODUCTION
Toxoplasma gondii is a ubiquitous parasite that infects and reproduces in virtually any nucleated cell of warm-blooded animals. It is of great medical importance as infections in immunocompromised humans often lead to life-threatening disease. The ability of Toxoplasma to replicate in a wide host range is likely dependent on the capacity of the parasite to regulate conserved host cell pathways via its secreted factors. For example, the secreted protein ROP16 regulates immunologically important host transcription factors, STAT3 (1), STAT6 (2), and STAT5 (3), causing profound transcriptional changes shortly after invasion. Although many of the changes observed in infected host cells are strain specific and are important for the differences in virulence between the major strains of Toxoplasma (4), it is likely that there is also remodeling of host cells that occurs independently of strain type. Parasite effectors whose function is conserved across the three major types of Toxoplasma (e.g., Toxoplasma protein GRA16, which regulates the host p53 pathway [5]) have previously been identified. Such is likely an effective solution for those instances where the same need is faced by all strains, e.g., disarming defenses common to many hosts and/or meeting a survival challenge encountered in all cells.
We have previously reported that infection with Toxoplasma tachyzoites specifically upregulates microRNAs that belong to transcriptional loci miR-17∼92 and miR-106b∼25 (6). These microRNAs play an important role in the cell cycle and apoptosis of a host cell (7) and thereby likely affect the interplay between host and parasite. It has been reported that c-Myc is a direct transcriptional regulator of miR-17∼92 and miR-106b∼25 (8), and so we hypothesized that Toxoplasma induces this transcriptional regulator. The possibility of c-Myc involvement in this host-Toxoplasma interaction was supported by published results from microarray analysis of Toxoplasma-infected human foreskin fibroblasts (HFFs) that revealed 2- to 3-fold upregulation of c-Myc mRNA during Toxoplasma infection (9).
c-Myc is a tightly regulated transcription factor involved in vital cellular processes, including cell cycle progression, apoptosis, cell differentiation, and metabolism (10–12). To achieve its function, c-Myc forms a protein complex with its binding partner Max and regulates gene expression by binding to DNA of target genes (10, 12). In this way, c-Myc directly regulates a large network of host genes (it has been reported that nearly 15% of annotated mammalian genes are transcriptionally regulated by this factor [13]). The finding that c-Myc knockouts are embryonically lethal (14) further demonstrates the importance of this regulator in the biology of a cell and of the entire organism.
Here we present evidence that infection with Toxoplasma tachyzoites leads to rapid induction of host c-Myc. This is not the first example of such host-parasite interaction, as other transcription factors, e.g., STATs (1–3), NF-κB (15), and SRF (16), have previously been shown to be regulated upon infection. This phenotype is conserved across the three major types of Toxoplasma, suggesting that this interaction might have an important function in the intracellular biology of the parasite. Furthermore, we present evidence that c-Myc is actively regulated by the parasite and is not a result of a nonspecific immune response to infection. We also show that the induced c-Myc is active as a transcriptional regulator and that the genes that are known to be regulated by this key transcription factor are specifically upregulated in Toxoplasma-infected cells.
MATERIALS AND METHODS
Parasite culture and infections.
Toxoplasma and Neospora tachyzoites were propagated in human foreskin fibroblasts (HFFs) cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 2 mM l-glutamine, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin. Prior to infection, parasites were scraped and lysed using a 27-gauge needle, counted using a hemocytometer, and added to HFFs at a multiplicity of infection (MOI) of 1 unless otherwise stated. Time course infections were done under low-FBS conditions (i.e., in 0.5% FBS media) to reduce the background caused by FBS-mediated c-Myc induction. FBS stimulation of cells for use as a positive control was done by adding fresh 20% FBS to cells for 2 h (we found that the highest c-Myc induction due to FBS occurs around this time point). Heat-killed parasites were prepared by incubation of parasites at 50°C for 20 min. Supernatant treatment of HFFs was done by harvesting supernatant from a heavily infected HFF culture, filtering the supernatant through a 0.2-μm-pore-size filter, and adding it to HFF cultures. Treatment with 4-bromophenacyl bromide (4-BPB; Sigma-Aldrich) was done by incubating parasites for 15 min with 1 μM 4-BPB at room temperature and washing two times in phosphate-buffered saline (PBS), as previously described (17). Analysis of c-Jun N-terminal protein kinase (JNK) inhibition was done by incubating HFFs in DMEM containing 25 μM JNK inhibitor II (CAS 129-56-6; Calbiochem).
Parasite strains and generation of mCherry-expressing Neospora.
The following strains of Toxoplasma gondii were used: RH (18), Me49 (19), CTg (20), and RH Cre mCherry (21). The Neospora strain used was N. caninum NC-1 (22). The NC-1 mCherry strain was generated by transfection of NC-1Δhxgprt parasites (gift from Jon Boyle, University of Pittsburgh) with pTKO2c plasmid containing mCherry and HXGPRT cassettes (23). The linearized pTKO2c plasmid was used to transfect parasites by electroporation, and HXGPRT-positive (HXGPRT+) parasites were selected using complete DMEM supplemented with mycophenolic acid (MPA) and xanthine (XAN), as previously described (24). After successive passages in selective media, parasite clones were isolated by limiting dilution.
Western blotting.
Cell lysates were prepared at 20 h postinfection (hpi) (unless otherwise stated), and the total protein concentration of lysates was determined by a Bradford protein assay (Bio-Rad). Samples containing 20 μg of protein were boiled for 5 min, separated by SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. c-Myc was detected by incubation of membrane with rabbit anti-N-terminal c-Myc antibody (Abcam) followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG. The levels of HRP were detected using an ECL chemiluminescence kit (Pierce). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels served as a loading control. Membranes were stripped with stripping buffer (Thermo Scientific) and stained with mouse anti-GAPDH primary antibody and secondary horseradish peroxidase-conjugated goat anti-mouse IgG antibody. The levels of HRP were measured as stated above.
Immunofluorescence assay.
Infected cells grown on glass coverslips were fixed using 2.5% formaldehyde–PBS for 20 min. Samples were washed once with PBS and blocked using 3% BSA–PBS for 1 h at room temperature. Cells were permeabilized with 0.2% Triton X-100–3% BSA–PBS for 10 min at room temperature. c-Myc protein was detected with rabbit anti-N-terminal c-Myc (Abcam) and fluorochrome (488 nm)-conjugated anti-rabbit IgG. SAG1 protein was detected by mouse anti-SAG1 and fluorochrome (594 nm)-conjugated goat anti-mouse IgG. Vectashield with DAPI (4′,6-diamidino-2-phenylindole) stain (Vector Laboratories) was used to mount the coverslips on slides. Fluorescence was detected using a fluorescence microscope, and images were analyzed using ImageJ.
Generation and infection of reporter cells.
Two c-Myc reporters were used here. The first was a vector carrying green fluorescent protein (GFP) and luciferase (Luc) under the control of a c-Myc promoter (25), and the second was a vector carrying the c-Myc coding region fused to GFP and driven by a cytomegalovirus (CMV) promoter (26). These vectors were transfected into 10T1/2 fibroblasts (ATCC) as follows. The fibroblasts were washed with PBS and incubated in trypsin for 5 min at 37°C. After one wash with cytomix buffer, cells were resuspended in 400 μl of cytomix and transfected with 20 μg of reporter plasmid by electroporation. GFP+ cells were selected by fluorescence-activated cell sorting (FACS). After three rounds of FACS enrichment, cells were grown to confluence and infected with Toxoplasma or Neospora or mock infected under non-c-Myc-inducing conditions (0.5% FBS–cDMEM). The same methods were used for generating and infecting cells carrying a pCMV-GFP control vector lacking the c-Myc coding sequence.
Flow cytometry.
Cells were harvested by one wash with PBS and treatment with trypsin for 5 min at 37°C. After one PBS wash, cells were resuspended in 5% FBS–PBS and filtered using tubes with cell-strainer caps (BD Biosciences). Fluorescence was detected by the use of a DivaVan sorter and an Aria sorter (BD Biosciences) (Stanford FACS facility). Flow cytometry data were analyzed using FlowJo.
Luciferase assay.
Cell lysates were prepared using the reagents and protocol from a luciferase assay system (Promega). Briefly, cells were washed once with PBS and Promega lysis buffer was added to cover the cells. Cells were shaken at room temperature for 15 min, and subsequent freeze-thawing ensured complete lysis of the cells. Upon addition of the luciferase substrate, the luminescence of each sample was measured for 10 s using a Tecan Infinite M100 luminometer (Stanford High-Throughput Bioscience Center [HTBC] facility).
c-Myc binding ELISA.
Nuclear extracts of cells were prepared and analyzed using reagents and protocols provided with a TransAM c-Myc DNA-binding enzyme-linked immunosorbent assay (ELISA) (Active Motif). Binding of c-Myc to double-stranded oligonucleotides containing a c-Myc binding sequence was detected using a primary anti-c-Myc antibody and an HRP-conjugated secondary antibody. After addition of HRP substrate, colorimetric change in all wells was detected using an Epoch microplate spectrophotometer (BioTek). Competition experiments were done in the same way except for the addition of the appropriate oligonucleotides to the nuclear extracts prior to performing the ELISA.
Generation of microarray data.
RAW 264.7 (ATCC) cells were plated in 6-well plates. When confluent, cells were infected with type I Toxoplasma RH parasites at an MOI of 3. At 24 h postinfection (hpi), RNA was isolated using TRIzol reagent (Invitrogen) followed by ethanol precipitation according to the manufacturer's instructions. RNA was labeled using a 3′ IVT Express reaction, and labeled, fragmented cRNA was hybridized to a mouse Affymetrix array (Mouse 430 2.0) according to the manufacturer's protocol. Probe intensities were measured and processed into image analysis (.CEL) files by the PAN Facility at Stanford University. Intensity values were normalized using the RMA algorithm.
To identify enriched transcription factors, gene identifiers of genes with fold changes higher than 1.5 and lower than −1.5 between infected and mock-treated samples were entered into the ENCODE chromatin immunoprecipitation sequencing (ChIP-Seq) Significance Tool (27) (http://encodeqt.stanford.edu/hyper/). The expression of the most highly regulated c-Myc target genes was visualized using the TM4 Microarray Software Suite MultiExperiment Viewer (http://www.tm4.org). Ingenuity Pathway Analysis (IPA) was performed on a data set containing the Toxoplasma-regulated host genes known to be under the control of c-Myc based on prior ChIP-Seq analysis (ENCODE). Gene identifiers and corresponding fold-change values were uploaded, and each gene identifier was mapped to its corresponding gene in the Ingenuity Pathways Knowledge Base. Canonical Pathway Analysis, Network Analysis, and Downstream Effects Analysis were used to analyze the biological functions of the Toxoplasma-regulated c-Myc target genes.
Microarray data accession number.
The entire MIAME database-compliant data set from these experiments can be found in the NCBI's Gene Expression Omnibus (GEO) (accession number (GSE55298 ).
RESULTS
Toxoplasma infection results in specific upregulation of host c-Myc protein.
To determine if c-Myc is upregulated at the protein level during Toxoplasma infection, HFFs were infected with Toxoplasma RH (type I) tachyzoites or mock infected, and cell lysates were prepared 20 h postinfection (hpi). Western blot analysis of such lysates revealed a dramatic increase of c-Myc protein levels in Toxoplasma-infected cells relative to uninfected cells (Fig. 1A). The specificity of the anti-c-Myc antibody used in these studies was established by assaying cell lysates from mouse fibroblasts ectopically expressing c-Myc under the control of doxycycline (a gift from Dean Felsher [28]) with and without doxycycline (Fig. 1A). To determine if this effect was present in more biologically relevant cells, we repeated the experiments with bone marrow macrophages and 10T1/2 fibroblasts from mice; c-Myc was strongly upregulated in these cell lines, as well (data not shown).
FIG 1.
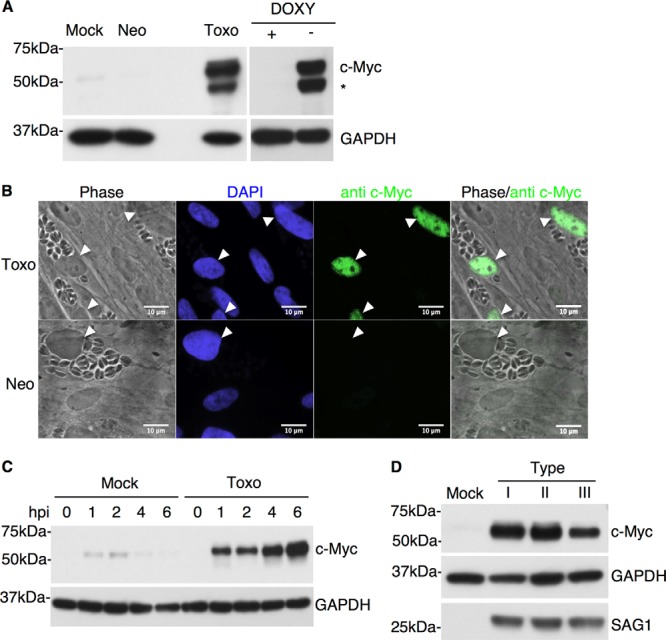
c-Myc levels are specifically and rapidly increased in Toxoplasma-infected cells. (A) Human foreskin fibroblasts (HFFs) were either mock infected (Mock) or infected at an MOI of 1 with Neospora (Neo) or Toxoplasma (Toxo) tachyzoites. Cell lysates were prepared 20 h postinfection (hpi), and Western blot analysis was performed with anti-c-Myc antibodies. Anti-GAPDH staining was used as a loading control. Cell lysates from mouse fibroblasts ectopically expressing human c-Myc under doxycycline (DOXY) control were used as a control for antibody specificity determinations. The asterisk (*) indicates a breakdown product of c-Myc that was variably detected in some lysates. Migration of size markers is shown to the left (sizes in kDa). (B) HFFs were infected at an MOI of 1 with either Toxoplasma (Toxo) or Neospora (Neo) tachyzoites. Cells were examined 20 hpi for c-Myc levels by IFA using anti-c-Myc antibodies. DAPI was used to identify the host cell nuclei. White arrowheads indicate the nuclei of parasite-infected cells. (C) HFFs were infected with Toxoplasma or mock infected. Cell lysates were prepared at the following time points: 0, 1, 2, 4, and 6 hpi. c-Myc levels were analyzed by Western blotting as described for panel A. (D) HFFs were infected with type I (RH), type II (Me49), or type III (CTg) Toxoplasma parasites, and cell lysates were prepared at 48 hpi and analyzed by Western blotting as described for panel A. Anti-SAG1 was used to ensure that similar levels of infection were achieved in all samples.
To determine where in the cell the induced c-Myc resides and whether the effect is restricted to infected cells only, immunofluorescence analysis (IFA) was performed on Toxoplasma RH-infected HFFs at 20 hpi. The results showed that c-Myc accumulates in the host nucleus and that this occurred only in infected cells and not in the neighboring, uninfected cells (Fig. 1B). Therefore, c-Myc upregulation is not due to a soluble or trans-acting factor in the supernatant but is specifically induced in parasite-containing cells.
Previous work by Zeiner et al. revealed that the upregulation of a subset of microRNAs is specific to Toxoplasma infection, as infection with a closely related apicomplexan parasite, Neospora caninum, does not lead to this change (6). To determine whether induction of c-Myc is also specific to Toxoplasma, we infected HFFs with either Toxoplasma (RH) or Neospora (NC-1) tachyzoites. Visual comparison using light microscopy was used to ensure that the infected cell cultures exhibited similar levels of infection, and immunoblotting was used to analyze the c-Myc levels 20 hpi. The results showed that the effect is specific to Toxoplasma: Neospora did not elicit an increase in c-Myc levels at any time tested from 0 to 48 hpi (Fig. 1A and data not shown). This result was confirmed by IFA: c-Myc was not observed in the nucleus of cells heavily infected with Neospora (Fig. 1B). Thus, c-Myc induction is a specific trait of Toxoplasma and not a nonspecific host cell response to parasitic infection.
To determine the timing of the c-Myc upregulation, c-Myc protein levels were assayed at 0, 1, 2, 4, and 6 hpi in Toxoplasma RH- or mock-infected lysates. To avoid high background levels of c-Myc, the infection was performed under low (0.5%)-serum conditions (growth factors within serum are known stimulators of c-Myc [29]). Mock-infected cells showed a low level of c-Myc upregulation at 1 and 2 hpi (likely because of the low levels of serum present in the media transferred during infection); this background disappeared, however, by 4 hpi (Fig. 1C). c-Myc levels in Toxoplasma-infected cells relative to mock-infected cells were markedly increased by as early as 1 hpi (Fig. 1C). Remarkably, this upregulation of c-Myc was sustained throughout the time of infection, as lysates harvested at late time points (20 and 48 hpi) continued to exhibit high levels of c-Myc (Fig. 1A and D). Thus, Toxoplasma infection leads to a rapid and sustained upregulation of host c-Myc.
The three major types of Toxoplasma, referred to as types I, II, and III, often exhibit type-specific differences in their impact on a host cell (4, 30). These differences have been a useful tool in identifying novel, strain-specific virulence genes in Toxoplasma (31). To determine if the c-Myc induction phenotypes differ between the three major types, c-Myc protein levels were assessed in HFFs infected with type I (RH), type II (Me49), or type III (CTg) parasites. The results (Fig. 1D) showed that c-Myc levels were increased in infected host cells relative to mock-treated cells in all three Toxoplasma strains; the slightly smaller amount of c-Myc signal seen upon infection with the type III strain was not a reproducible difference relative to infections with the other strains. Thus, c-Myc upregulation is a phenotype common to all three of the dominant Toxoplasma types.
Three Toxoplasma effectors have been identified that alter host signaling pathways that could, potentially, drive the c-Myc upregulation: ROP16 subverts the STAT pathway (1–3), GRA15 regulates host NF-κB (15), and GRA24 activates p38 mitogen-activated protein kinase (MAPK) (32). To exclude the possibility that c-Myc induction is a downstream effect of any of these previously identified effectors, strains deficient in ROP16 (2), GRA15 (15), and GRA24 (32) were assayed for the ability to induce c-Myc. Lysates of cells infected with strains deficient in each of these loci exhibited levels of c-Myc upregulation similar to those seen in cells infected with the wild-type Toxoplasma strain (data not shown). Although the known activities of ROP5 (33) do not include any that are known to induce c-Myc, we also tested knockout strains for this effector and again saw no difference from wild-type results in the ability to induce c-Myc (data not shown). Thus, none of these effectors are likely responsible for the observed induction of c-Myc. Further evidence for this conclusion comes from the fact that three of these effectors (GRA15, ROP5, and ROP16) are highly polymorphic and their effects on the host cell are dramatically different between the three Toxoplasma types (1, 15, 33, 34) and yet no type-specific difference in the induction of c-Myc was observed, as noted above.
c-Myc is actively induced by Toxoplasma.
Two possible models could explain c-Myc upregulation in host cells harboring Toxoplasma and its absence in host cells harboring Neospora: (i) Toxoplasma but not Neospora actively upregulates host c-Myc, or (ii) the mere presence of a parasite in the host cell triggers this alteration in c-Myc levels, and Neospora, but not Toxoplasma, actively suppresses this response. To discriminate between these two possibilities, HFFs were coinfected with Toxoplasma and Neospora. Toxoplasma and Neospora are morphologically similar, so the two parasites were distinguished using antibodies to the SAG1 Toxoplasma-specific surface marker. IFA of infected cells revealed that c-Myc induction by Toxoplasma is a dominant phenotype, as this protein was induced in all doubly infected cells (Fig. 2). These data indicate that Toxoplasma actively induces host c-Myc rather than c-Myc induction being a general host response to parasite invasion that Neospora suppresses.
FIG 2.
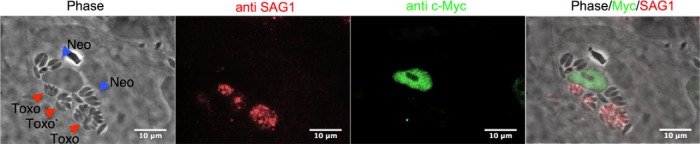
Mixed infection by Toxoplasma and Neospora reveals that c-Myc is actively induced by Toxoplasma. HFFs were infected at an MOI of 1 with a mixture of Toxoplasma and Neospora tachyzoites. c-Myc levels were examined by IFA 20 hpi using anti-c-Myc antibodies. Toxoplasma and Neospora parasites were differentiated using antibodies to the Toxoplasma surface marker SAG1. Toxo, Toxoplasma gondii (red triangles); Neo, Neospora caninum (blue triangles).
Active invasion is necessary for c-Myc induction.
To determine if active parasite invasion is necessary for c-Myc upregulation, HFFs were treated with heat-killed or live Toxoplasma RH parasites. Immunoblots of cell lysates revealed that exposure to heat-killed parasites did not result in an induction of c-Myc (Fig. 3A). This result indicates that c-Myc upregulation is a result of active parasite invasion or processes (e.g., secreted proteins) associated with active invasion. In addition, cells treated with supernatant from Toxoplasma-infected cultures did not exhibit c-Myc upregulation (Fig. 3A). Thus, c-Myc induction is not a result of extracellularly secreted host or parasite factors (e.g., Toxoplasma proteins secreted from dense granules prior to invasion).
FIG 3.
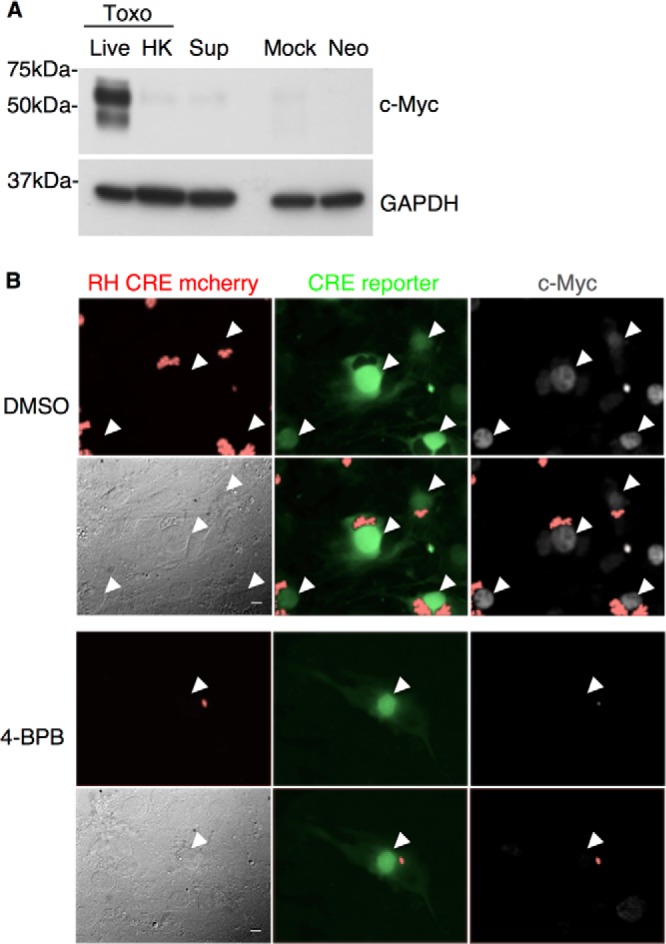
Active invasion is necessary but not sufficient for c-Myc induction. (A) From left to right, HFFs were infected at an MOI of 1 with untreated (Live) or heat-killed (HK) Toxoplasma RH parasites (Toxo), treated with supernatant from Toxoplasma-infected culture (Sup), mock infected (Mock), or infected with Neospora (Neo). Cell lysates were prepared at 20 hpi and subjected to Western blot analysis of c-Myc and GAPDH levels as described for Fig. 1A. (B) Toxoplasma RH Cre mCherry parasites were treated with DMSO (upper set of 6 panels) or 4-bromophenacyl bromide (4-BPB; lower set of 6 panels) prior to infection. Cre reporter cells, which turn from red to green upon Cre-mediated recombination (Koshy et al. 21), were infected at an MOI of 1. c-Myc levels were determined by IFA 20 hpi. White arrows indicate nuclei of infected cells. All six panels in each group show the same field, with filters to detect mCherry (red; upper left), GFP (green; upper middle), c-Myc (white; upper right), phase (lower left), mCherry and GFP (lower middle), or mCherry and c-Myc (lower right). Scale bars in phase (lower left) panels indicate the distance of 10 μm.
The rapid c-Myc upregulation in infected host cells suggests that c-Myc might be induced by a Toxoplasma effector secreted into host cells early during invasion/infection; therefore, the role of parasite rhoptry secretion in c-Myc induction was tested. To assess this, the inhibitor 4-bromophenacyl bromide (4-BPB) was used to partially inhibit rhoptry secretion. Treatment with this chemical inhibitor results in a far longer time being needed for invasion (no invasion is seen for about 4 to 6 h after treated parasites are added to HFF monlayer) and a block in replication of the parasites that do eventually enter (17). Cre reporter 10T1/2 fibroblasts, which turn on enhanced green fluorescent protein (eGFP) upon Cre-mediated recombination (21), were infected with a strain of Toxoplasma RH that injects Cre recombinase fused to a rhoptry protein (toxofilin) upon invasion (21). Prior to infection, parasites were pretreated with 4-BPB or dimethyl sulfoxide (DMSO) as a negative control. IFA of the cultures 20 hpi revealed that c-Myc was not induced in the cells infected with 4-BPB-treated parasites (Fig. 3B) even though sufficient rhoptry protein had been injected to turn on the eGFP reporter. Thus, either rhoptry proteins are not involved in the induction of c-Myc or insufficient quantities are injected by 4-BPB-treated parasites within the necessary time frame to elicit the effect. These data further show that invasion itself is not sufficient for the induction.
Toxoplasma induces c-Myc by transcriptional upregulation.
c-Myc is a tightly regulated factor, and there are many pathways by which a cell controls its levels and activity (35, 36). Data from a previous microarray analysis showed that c-Myc mRNA exhibits an ∼2-fold increase in Toxoplasma-infected HFFs (9). To confirm that this effect is also occurring in other cell types, we repeated the microarray analyses with a mouse macrophage cell line (RAW 264.7) infected for 24 h with Toxoplasma RH parasites at an MOI of 3 and saw an ∼5.5-fold upregulation of c-Myc mRNA in infected cells relative to mock-treated cells (Fig. 4A).
FIG 4.
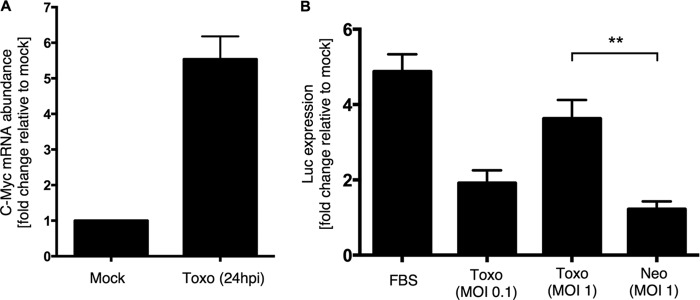
c-Myc is transcriptionally regulated in Toxoplasma-infected cells. (A) RAW 264.7 macrophages were infected with Toxoplasma RH parasites at an MOI of 3 [Toxo (24hpi)], or treated with syringe-lysed uninfected HFF cultures (Mock), in duplicate. At 24 hpi, RNA was prepared from each culture and analyzed by hybridization to microarrays. The graph depicts the fold increase in c-Myc mRNA abundance relative to mock-treated cells. (B) Cells expressing a reporter containing the fusion of the c-Myc promoter with a luciferase coding region (Luc) were infected with Toxoplasma (Toxo) at an MOI of 0.1 or 1 or Neospora (Neo) at an MOI of 1 or were treated with 20% FBS (FBS) as a positive control for c-Myc induction. Infected cells were lysed 20 hpi, and lysates were analyzed for luminescence upon addition of luciferin substrate. All values are relative to mock-infected samples. Statistical analysis was done by unpaired t test; a double asterisk (**) indicates P < 0.005.
To discriminate between transcriptional activation and RNA stabilization as a cause of the c-Myc mRNA increase, host cells transfected with a reporter plasmid carrying GFP and luciferase (Luc) driven by the promoter from the c-Myc gene were used (25). This reporter has no other c-Myc gene elements, and so any upregulation should be a specific indication of an increase in transcription. These reporter cells were infected with Toxoplasma RH or with Neospora or were mock treated. Serum-stimulated cells served as a positive control for the induction of c-Myc transcription (29). Luciferase activity was measured in cell lysates at 20 hpi. Toxoplasma-infected cells exhibited markedly increased luciferase activity relative to mock-treated cells (nearly 2-fold and 4-fold induction were observed for cells infected at MOIs of 0.1 and 1, respectively), while Neospora-infected cells exhibited little if any increase (Fig. 4B). These data suggest that transcriptional activation is at least in part responsible for the induction of c-Myc during Toxoplasma infection.
c-Myc protein levels are stabilized during Toxoplasma infection.
The demonstration of an effect on c-Myc transcription did not preclude a posttranscriptional effect, as there is no a priori reason why the induction could not be mediated at more than one level. Indeed, the levels of c-Myc protein itself are known to be regulated at several posttranscriptional steps (35, 36) and so we examined whether c-Myc is also being regulated in this manner during Toxoplasma infection. To do this, we used a reporter containing only the coding region of c-Myc fused to GFP and driven by a CMV promoter (26). Since this reporter does not include the c-Myc promoter or any DNA elements known to be inducible by c-Myc, any changes in expression detected upon Toxoplasma infection should be due to processes other than transcription (i.e., mRNA stability, translation, and/or protein stability). A cell population stably expressing this reporter was infected with Toxoplasma RH or Neospora or was mock treated. Parasites expressing mCherry were used here to distinguish between infected and uninfected cells. At 20 hpi, cells were harvested and fluorescence was analyzed by flow cytometry. The data revealed that Toxoplasma-infected reporter cells exhibited a substantial (∼8-fold) increase in green fluorescence relative to mock-treated cells, while Neospora had a far less pronounced effect (∼2-fold; Fig. 5A). As expected, we did not observe an increase in green fluorescence in Toxoplasma-infected cells carrying a pCMV-GFP reporter without any c-Myc coding or promoter elements (Fig. 5A). These results indicate that, in addition to transcriptional activation of c-Myc, Toxoplasma also appears to upregulate the expression of this gene on a posttranscriptional level.
FIG 5.

Toxoplasma infection leads to stabilization of c-Myc protein. (A) Reporter cells expressing either the c-Myc coding region fused to GFP and driven by a CMV promoter (pCMV-c-Myc-GFP) (top panels) or the pCMV-GFP control (bottom panels) were infected at an MOI of 1 with Toxoplasma (Toxo, gray) or Neospora (Neo, gray) or were mock infected (Mock, black). Cells were harvested 20 hpi and analyzed by flow cytometry. Highly infected cells were distinguished by the presence of mCherry-expressing parasites, and the results for these cells only are shown. (B) HFFs were infected with Toxoplasma at an MOI of 1 for 20 h or treated with medium containing 20% FBS for 2 h. Cell lysates were prepared at 0, 30, 60, 90, and 120 min after the addition of 30 μM cycloheximide (CHX). Western blotting was done as described for Fig. 1A. GAPDH staining served as a loading control. The graph represents band intensities normalized to GAPDH and relative to time zero. One phase decay curve was fitted to the data points.
Regulation of protein half-life is an efficient strategy employed by mammalian cells to control the levels of important factors, and c-Myc has been shown to be regulated at this level (via phosphorylation, acetylation, and ubiquitination) (36). Therefore, we next tested whether the increase in c-Myc protein might be due to decreased turnover in Toxoplasma-infected cells. Toxoplasma-infected cells and FBS-stimulated cells were treated with cycloheximide (CHX) to block protein translation and, thus, production of newly synthesized c-Myc. Cell lysates prepared at 0, 30, 60, 90, and 120 min after CHX treatment were analyzed by immunoblotting. The half-life of c-Myc protein in quiescent cells has been reported to be 20 to 30 min (37), and a similar result was observed for the cells briefly stimulated by FBS, with a half-life of ∼30 min (Fig. 5B). Toxoplasma infection, however, resulted in a dramatic decrease in c-Myc protein turnover, with a half-life of greater than 2 h. These data indicate that at least one way that Toxoplasma infection controls c-Myc levels posttranscriptionally is through protein stabilization.
Inhibition of JNK leads to loss of c-Myc upregulation in infected cells.
JNK is known to be one of the regulators of c-Myc transcription and protein stability (36, 38–40). To test the involvement of JNK in Toxoplasma-mediated c-Myc induction, HFFs were pretreated with a specific inhibitor of JNK 30 min prior to Toxoplasma infection or FBS treatment. Lysates of infected cells were prepared at 5 hpi (this early time point was chosen to reduce the possibility of off-target effects that could result in decreased parasite growth and thereby decreased c-Myc abundance). Visual analysis of the cells in addition to anti-SAG1 staining was used to ensure that similar levels of infection were achieved across the samples. Western blot analysis revealed dramatically decreased levels of c-Myc in infected cells treated with JNK inhibitor compared to the untreated infected cells (Fig. 6). As expected, FBS-stimulated cells also exhibited a decrease of c-Myc in the presence of the JNK inhibitor (Fig. 6). These data suggest that the JNK signaling pathway plays a role in the induction of c-Myc by Toxoplasma, although we cannot exclude the possibility that the inhibitor had an off-target effect on the host or parasite that caused the observed decreases in c-Myc.
FIG 6.
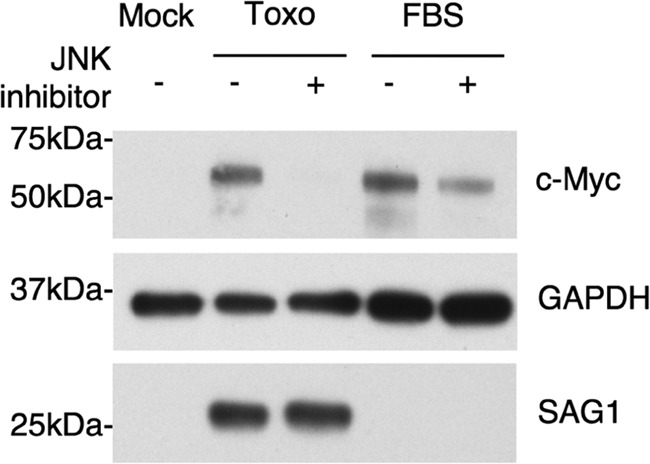
JNK inhibitor treatment abolishes induction of c-Myc in Toxoplasma-infected cells. HFFs were treated with 25 μM JNK inhibitor or with DMSO for 30 min prior to infection. Cells were infected at an MOI of 1 with Toxoplasma RH parasites or mock infected, and cell lysates were harvested 5 hpi. FBS-stimulated cells pretreated with either JNK inhibitor or DMSO served as a control for JNK inhibition. Whole-cell lysates were subjected to Western blot analysis using anti-c-Myc and anti-GAPDH antibodies. Anti-SAG1 was used to ensure that similar levels of infection were achieved in all samples.
Upregulation of c-Myc correlates with increased binding of c-Myc target DNA.
To determine whether the upregulated c-Myc is active as a transcription factor, we examined its ability to bind the appropriate DNA elements, using an ELISA that measures the binding of c-Myc to double-stranded oligonucleotides containing a c-Myc consensus sequence. HFFs were either mock treated or infected with Toxoplasma RH or Neospora, and FBS induction was used as a positive control. Nuclear extracts of cells were prepared at 20 hpi, and the DNA binding activity of c-Myc was assessed by ELISA, where the levels of c-Myc bound to the target DNA were detected by a primary c-Myc antibody and a secondary HRP-conjugated antibody. The results indicated that lysates of Toxoplasma-infected cells exhibited a substantial level of DNA binding by c-Myc relative to mock-treated cells whereas the Neospora-infected cells did not (Fig. 7A). To establish that this increase in DNA binding was sequence specific, we employed a competition assay with oligonucleotides containing either wild-type or mutated c-Myc binding sequences. These oligonucleotides were added to nuclear extracts of Toxoplasma RH-infected HFFs, and c-Myc binding to target DNA sequence was then assessed by ELISA, as described above. The results showed that wild-type but not mutant c-Myc binding sequences effectively blocked binding by c-Myc in nuclear extracts of Toxoplasma-infected cells, reducing it to the levels seen for the mock-treated or Neospora-infected cells (Fig. 7B). Hence, the c-Myc protein induced by Toxoplasma infection exhibits increased target DNA binding and is, therefore, likely active as a transcription factor in such cells.
FIG 7.
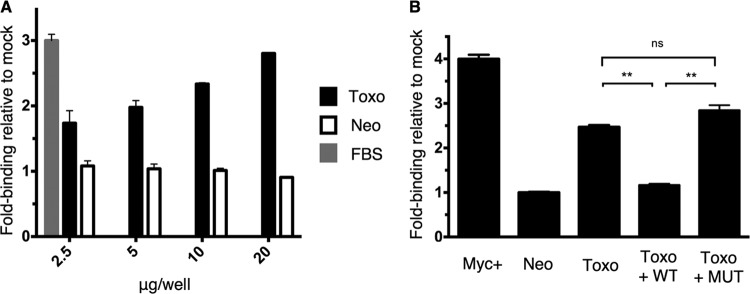
c-Myc target DNA binding is increased in Toxoplasma-infected cells. (A) HFFs were infected at an MOI of 1 with Toxoplasma (Toxo, black) or Neospora (Neo, white), mock treated, or FBS treated (FBS, gray). Nuclear extracts from cells were prepared at 20 hpi and added in increasing amounts (μg/well) to wells containing double-stranded oligonucleotides representing the c-Myc consensus binding sequence. Binding of c-Myc was detected by a primary antibody specific to c-Myc and an HRP-conjugated secondary antibody. After addition of HRP substrate, colorimetric change in all wells was detected by a plate reader. FBS induction served as a positive control for c-Myc induction using the 2.5 μg/well amount. All values are relative to the results obtained with mock-treated cells at the relevant concentration. (B) HFFs were infected at an MOI of 1 with Toxoplasma (Toxo) or Neospora (Neo), and nuclear extracts were prepared at 20 hpi. A 20-pmol volume of oligonucleotides containing the wild-type or mutated c-Myc binding sequence was added to 10 μg of nuclear extracts. The binding of c-Myc to wells coated with c-Myc target DNA was detected as described for panel A. Nuclear extract of Jurkat cells served as a positive control. All values are relative to mock-treated cells. Statistical analysis was done by unpaired t test; “ns” indicates that the results were not significant (P > 0.05), and a double asterisk (**) indicates P < 0.005.
c-Myc-regulated target genes are induced in Toxoplasma-infected cells.
Infection with Toxoplasma tachyzoites is well known to substantially regulate a large number of host genes (9). To assess whether c-Myc upregulation contributes to this effect, we asked whether there is enrichment for c-Myc-regulated genes among the genes whose expression is modulated in infected cells. This was done by microarray analysis of RNA generated from RAW macrophages infected at an MOI of 3 with Toxoplasma RH tachyzoites for 24 h. From this data set, significantly regulated genes (those that exhibited at least a 1.5-fold change relative to the levels in mock-infected cells) were selected and inputted into the ENCODE ChIP-Seq Significance tool analysis (27) (http://encodeqt.stanford.edu/hyper/). The results of this analysis indicated that c-Myc target genes are highly enriched among the genes that are regulated during Toxoplasma infection (q-value = 1.26e-4; q-value is defined as the minimum false discovery rate at which a transcription factor can still be considered significant (27). Among the other transcription factors whose target genes were regulated in infected cells were NF-κB (q-value = 2.64e-6), previously shown to control transcription in Toxoplasma-infected cells (15), and the c-Myc binding partner, Max (q-value = 3.22e-4). The 50 c-Myc target genes that showed the greatest change upon Toxoplasma infection were all increased in expression relative to the levels in uninfected cells and are represented in a heat map (Fig. 8). The finding that all of these most highly regulated c-Myc target genes were upregulated (as opposed to repressed) could be due to the fact that the host cells used for the infections were in stationary phase when infected and so genes that c-Myc downregulates might have already been at a low expression level. In addition, downregulation is typically more difficult to detect when studying cell-autonomous effects such as the c-Myc activation we report here because uninfected cells are not affected and thus dampen the average apparent effect. Lastly, studies on c-Myc in other settings have noted that it more often functions as a gene inducer than as a repressor (41, 42).
FIG 8.

Toxoplasma-infected cells show altered RNA levels for genes known to be regulated by c-Myc. Infection of RAW 264.7 cells and subsequent microarray analysis were performed as described for Fig. 4A. The ENCODE ChIP-Seq Significance Tool (http://encodeqt.stanford.edu/hyper/) was used to identify the enriched ENCODE transcription factors that regulate host genes whose expression was significantly different in the Toxoplasma-infected cells relative to the mock-infected cells. Of the host genes found by ENCODE ChIP-Seq Significance analysis to be c-Myc targets, the 50 most highly regulated are depicted in the heat map. These 50 genes are shown in descending order, with the first, MARCKS, showing the highest fold difference between the average results determined for two biological duplicates of Toxoplasma-infected samples and the average results determined for two mock-infected samples as determined by microarray analysis. The scale bar indicates the colors corresponding to log2 values for the two mock-infected (mk1 and mk 2) and the two RH-infected (WT1 and WT2) samples.
IPA of the c-Myc target genes regulated during Toxoplasma infection revealed that there is a significant enrichment of genes known to be involved in host functions such as cellular development (P < 0.001), cellular growth and proliferation (P < 0.001), immune function (P < 0.001), cell death and survival (P < 0.001), and metabolism (P < 0.001). Interestingly, many of the Toxoplasma-induced c-Myc targets in our data set have been shown to be positive regulators of cell survival and viability and negative regulators of apoptosis and cell cycling (specifically, genes regulating the G2/M checkpoint were enriched [P = 0.00132]), which is consistent with previous reports on the regulation of apoptosis and cell cycle by this parasite (43–45). Altogether, these data suggest that the upregulation of c-Myc in Toxoplasma-infected cells has a substantial impact on the host transcriptome and, likely, the overall phenotype of the infected cell.
DISCUSSION
Here we described a novel interaction between Toxoplasma gondii and the infected host cell involving c-Myc, a key host transcriptional regulator. The observation that c-Myc induction occurs only in Toxoplasma-infected cells (and not in the neighboring uninfected cells), together with the result that heat-killed parasites lose the c-Myc phenotype, strongly suggests that a Toxoplasma effector exists that is secreted into the host cell and co-opts host c-Myc. Our results with 4-BPB suggest that the protein involved does not originate in the rhoptries, as parasites that were able to invade, a process that requires rhoptry protein injection, did not exhibit c-Myc induction. The injection of rhoptry proteins in the presence of 4-BPB into these cells was further indicated by the fact that the host cells turned green, a process dependent on injection of the rhoptry-originating Cre recombinase. Although we cannot exclude the possibility that 4-BPB treatment prevented the secretion of the required amount of rhoptry proteins within the time frame necessary for c-Myc induction, overall our data are most consistent with the c-Myc-inducing effector originating from the dense granules. These secretory organelles are thought to release their contents after invasion and throughout the process of intracellular growth (46), and it is, therefore, likely that a halt in parasite growth, as occurs with 4-BPB treatment, would substantially decrease the amount of dense granule proteins introduced into the infected host cell.
A growing number of dense granule proteins (GRAs) have recently been described that interface with the host cell in important ways, including effects on gene expression; e.g., Toxoplasma dense granule protein GRA15 has been shown to regulate host NF-κB (15), Toxoplasma factor GRA16 has been shown to subvert the host p53 pathway (5), and GRA24 has been found to regulate p38 MAP kinase of the host (32). The hypothetical effector involved in c-Myc upregulation could act either directly or indirectly via manipulation of the many pathways Toxoplasma infection is known to impact, such as STAT-3 (1, 47), NF-κB (15, 48), PI3 kinase (49, 50), and MAP kinases (36, 39, 51). Our results with the ROP16, GRA15, and GRA24 knockouts indicate that these effectors are not responsible for the Toxoplasma-dependent c-Myc induction, suggesting that STAT-3, NF-κB, and p38 MAP kinase are not involved in c-Myc induction in infected cells. Our JNK inhibition data suggest that the unidentified Toxoplasma effector could act through the JNK pathway to induce c-Myc. We cannot, however, exclude the possible involvement of other MAP and PI3 kinase pathways yet to be investigated.
As mentioned above, Toxoplasma is known to regulate conserved host pathways, and it is believed that its broad host range is due to its ability to co-opt almost any host cell it infects. It is, therefore, not surprising that c-Myc, a highly conserved regulator (human and mouse c-Myc homologs have 90.3% amino acid identity, and c-Myc homologs in human and chicken contain 72.5% amino acid identity), is targeted during Toxoplasma infection. The finding that c-Myc is induced by all three major types of Toxoplasma further suggests that this interaction plays an important role in the biology of this parasite. The finding that a closely related apicomplexan parasite, Neospora caninum, lacks the c-Myc phenotype uncovers yet another example of differences in how these two parasites evolved to interact with their host cells and suggests that c-Myc induction has a Toxoplasma-specific function. This interaction, in addition to the other pathways differentially regulated by the two parasites (e.g., p38 MAPK regulation by the parasite factor GRA24 [32]), could potentially explain the great difference in the host ranges of the two apicomplexan organisms. Importantly, our experiments involving heat-killed and 4-BPB-treated parasites showed that this induction is actively elicited by the parasite, suggesting that it is not an immune host response mediated by sensing of the parasite's surface.
c-Myc is upregulated during infection by another apicomplexan parasite, Theileria (52). It is not yet clear whether the induction of c-Myc in Theileria- and Toxoplasma-infected cells occurs by related mechanisms; however, our data indicate that, similarly to Theileria infection, host c-Myc is upregulated in Toxoplasma-infected cells by both transcriptional activation and protein stabilization. It has been reported that c-Myc has antiapoptotic effects in the context of Theileria infection that result in transformation of lymphocytes (52). Although transformation has not been reported in Toxoplasma-infected cells, possibly due to the fact that lysis of the infected host cell precedes its transformation, the ability of Toxoplasma to regulate apoptosis in infected host cells has been previously described (45), and this effect could facilitate Toxoplasma proliferation. Consistent with these findings are the IPA results showing that many of the induced c-Myc targets are known to be negative regulators of apoptosis.
We have attempted to address the role of c-Myc upregulation in Toxoplasma growth using RNA interference (RNAi) to knock down its expression (c-Myc knockouts are embryonically lethal [14] and so cannot be used). These experiments were uninformative, however, because the knockdown of c-Myc rapidly caused the host cells to become nonviable, independently of any infection (data not shown). In addition, we tested the effects of a chemical inhibitor of c-Myc, 10058-F4 (53), and found that the growth of Toxoplasma tachyzoites was inhibited in cells treated with this inhibitor. While consistent with the possibility that c-Myc upregulation plays a role in Toxoplasma growth, these results are inconclusive as the growth of Neospora was also halted, suggesting that 10058-F4 has nonspecific effects on the parasites themselves or on some other host factor that both require (data not shown).
Among the Toxoplasma-induced c-Myc targets are genes that belong to networks responsible for regulating pathways critical for cell function such as cell proliferation, apoptosis, metabolism, and immune function. As an obligate intracellular parasite, Toxoplasma depends on its host to supply small molecules such as glucose or amino acids (54). Therefore, a possible role of c-Myc regulation could be to manipulate host metabolic pathways to ensure that Toxoplasma tachyzoites have enough nutrients to scavenge from their host. Another possibility is that Toxoplasma co-opts c-Myc to regulate the host cell cycle. Our findings using IPA of gene expression changes in Toxoplasma-infected cells are consistent with previous reports that have shown that Toxoplasma can induce host cell cycle arrest at a G2/M checkpoint (43, 44). Restricting the proliferation ability of the host cell could be a strategy to ensure that enough nutrients are available for the parasite instead of being utilized by the host cell. Like Theileria, Toxoplasma may be manipulating c-Myc to prevent host apoptosis and prolong survival, thereby securing its intracellular niche. Similarly, c-Myc-mediated regulation of host immune function could serve as protection from host immune recognition, enabling Toxoplasma to survive and proliferate inside the host. Determining the precise role of c-Myc upregulation during Toxoplasma infection will require the identification and subsequent deletion of the Toxoplasma factor responsible for this effect so that the impact of such a deletion can be assessed during in vitro and in vivo growth.
ACKNOWLEDGMENTS
This work was supported by NIH RO1-AI21423 (J.C.B.), a Microbiology and Immunology departmental training grant (5 T32 AI007328) (M.F.), and a National Science Foundation Graduate Research Fellowship and a Stanford University DARE Doctoral Fellowship (A.J.S.).
We thank Xiaolin Wu from NIH/NCI for providing the plasmid carrying GFP and Luc driven by a c-Myc promoter, Hiroyoshi Ariga from Hokkaido University for providing the vector carrying the c-Myc coding region fused to GFP and driven by a CMV promoter, and the laboratory of Christina Smolke for providing the pCMV-GFP vector. We thank Dean Felsher, Peter Choi, and Yulin Li from Stanford University for providing lysates of cells expressing doxycycline-repressible c-Myc and c-Myc RNAi knockdown reagents and for their expertise in the area of c-Myc biology. We thank the Stanford FACS facility personnel Bianca Gomez and Tim Knaak for providing technical support during flow cytometry experiments. We also thank the members of the Boothroyd laboratory and a former member, Gusti Zeiner, for their critical input and useful discussions. Kerry Buchholz, Lena Pernas, and Moritz Treeck provided helpful input on the manuscript.
Footnotes
Published ahead of print 14 February 2014
REFERENCES
- 1.Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T, Soldati-Favre D, Takeda K. 2009. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 206:2747–2760. 10.1084/jem.20091703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ong YC, Reese ML, Boothroyd JC. 2010. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 285:28731–28740. 10.1074/jbc.M110.112359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen KD, Hu K, Whitmarsh RJ, Hassan MA, Julien L, Lu D, Chen L, Hunter CA, Saeij JP. 2013. Toxoplasma gondii rhoptry 16 kinase promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect. Immun. 81:2156–2167. 10.1128/IAI.01185-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saeij JPJ, Boyle JP, Boothroyd JC. 2005. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 21:476–481. 10.1016/j.pt.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 5.Bougdour A, Durandau E, Brenier-Pinchart MP, Ortet P, Barakat M, Kieffer S, Curt-Varesano A, Curt-Bertini RL, Bastien O, Coute Y, Pelloux H, Hakimi MA. 2013. Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13:489–500. 10.1016/j.chom.2013.03.002 [DOI] [PubMed] [Google Scholar]
- 6.Zeiner GM, Norman KL, Thomson JM, Hammond SM, Boothroyd JC. 2010. Toxoplasma gondii infection specifically increases the levels of key host microRNAs. PLoS One 5:e8742. 10.1371/journal.pone.0008742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendell JT. 2008. miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222. 10.1016/j.cell.2008.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435:839–843. 10.1038/nature03677 [DOI] [PubMed] [Google Scholar]
- 9.Blader IJ, Manger ID, Boothroyd JC. 2001. Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J. Biol. Chem. 276:24223–24231. 10.1074/jbc.M100951200 [DOI] [PubMed] [Google Scholar]
- 10.Gardner L, Lee L, Dang C. 2002. c-myc Protooncogene, p 555–561 In Bertino J. (ed), The encyclopedia of cancer, 2nd ed. Academic Press, New York, NY [Google Scholar]
- 11.Dang CV, Lewis BC. 1997. Role of oncogenic transcription factor c-Myc in cell cycle regulation, apoptosis and metabolism. J. Biomed. Sci. 4:269–278. 10.1007/BF02258350 [DOI] [PubMed] [Google Scholar]
- 12.Dang CV, Resar LM, Emison E, Kim S, Li Q, Prescott JE, Wonsey D, Zeller K. 1999. Function of the c-Myc oncogenic transcription factor. Exp. Cell Res. 253:63–77. 10.1006/excr.1999.4686 [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Lee JH, Iyer VR. 2008. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One 3:e1798. 10.1371/journal.pone.0001798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. 1993. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7:671–682. 10.1101/gad.7.4.671 [DOI] [PubMed] [Google Scholar]
- 15.Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, Saeij JP. 2011. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208:195–212. 10.1084/jem.20100717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiley M, Teygong C, Phelps E, Radke J, Blader IJ. 2011. Serum response factor regulates immediate early host gene expression in Toxoplasma gondii-infected host cells. PLoS One 6:e18335. 10.1371/journal.pone.0018335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ravindran S, Lodoen MB, Verhelst SH, Bogyo M, Boothroyd JC. 2009. 4-Bromophenacyl bromide specifically inhibits rhoptry secretion during Toxoplasma invasion. PLoS One 4:e8143. 10.1371/journal.pone.0008143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfefferkorn ER, Pfefferkorn LC. 1976. Toxoplasma gondii: isolation and preliminary characterization of temperature-sensitive mutants. Exp. Parasitol. 39:365–376. 10.1016/0014-4894(76)90040-0 [DOI] [PubMed] [Google Scholar]
- 19.Kasper LH, Ware PL. 1985. Recognition and characterization of stage-specific oocyst/sporozoite antigens of Toxoplasma gondii by human antisera. J. Clin. Invest. 75:1570–1577. 10.1172/JCI111862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfefferkorn ER, Pfefferkorn LC, Colby ED. 1977. Development of gametes and oocysts in cats fed cysts derived from cloned trophozoites of Toxoplasma gondii. J. Parasitol. 63:158–159. 10.2307/3280129 [DOI] [PubMed] [Google Scholar]
- 21.Koshy AA, Fouts AE, Lodoen MB, Alkan O, Blau HM, Boothroyd JC. 2010. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods 7:307–309. 10.1038/nmeth.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubey JP, Hattel AL, Lindsay DS, Topper MJ. 1988. Neonatal Neospora caninum infection in dogs: isolation of the causative agent and experimental transmission. J. Am. Vet. Med. Assoc. 193:1259–1263 [PubMed] [Google Scholar]
- 23.Caffaro CE, Koshy AA, Liu L, Zeiner GM, Hirschberg CB, Boothroyd JC. 2013. A nucleotide sugar transporter involved in glycosylation of the Toxoplasma tissue cyst wall is required for efficient persistence of bradyzoites. PLoS Pathog. 9:e1003331. 10.1371/journal.ppat.1003331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soldati D, Boothroyd JC. 1993. Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260:349–352. 10.1126/science.8469986 [DOI] [PubMed] [Google Scholar]
- 25.Sotelo J, Esposito D, Duhagon MA, Banfield K, Mehalko J, Liao HL, Stephens RM, Harris TJR, Munroe DJ, Wu XL. 2010. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. U. S. A. 107:3001–3005. 10.1073/pnas.0906067107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arabi A, Rustum C, Hallberg E, Wright AP. 2003. Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J. Cell Sci. 116:1707–1717. 10.1242/jcs.00370 [DOI] [PubMed] [Google Scholar]
- 27.Auerbach RK, Chen B, Butte AJ. 2013. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics 29:1922–1924. 10.1093/bioinformatics/btt316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsher DW, Bishop JM. 1999. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 4:199–207. 10.1016/S1097-2765(00)80367-6 [DOI] [PubMed] [Google Scholar]
- 29.Cheng M, Wang D, Roussel MF. 1999. Expression of c-Myc in response to colony-stimulating factor-1 requires mitogen-activated protein kinase kinase-1. J. Biol. Chem. 274:6553–6558. 10.1074/jbc.274.10.6553 [DOI] [PubMed] [Google Scholar]
- 30.Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. 2005. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect. Immun. 73:695–702. 10.1128/IAI.73.2.695-702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boothroyd JC. 25 April 2013. Have it your way: how polymorphic, injected kinases and pseudokinases enable toxoplasma to subvert host defenses. PLoS Pathog. 10.1371/journal.ppat.1003296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braun L, Brenier-Pinchart MP, Yogavel M, Curt-Varesano A, Curt-Bertini RL, Hussain T, Kieffer-Jaquinod S, Coute Y, Pelloux H, Tardieux I, Sharma A, Belrhali H, Bougdour A, Hakimi MA. 2013. A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J. Exp. Med. 210:2071–2086. 10.1084/jem.20130103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reese ML, Zeiner GM, Saeij JP, Boothroyd JC, Boyle JP. 2011. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. U. S. A. 108:9625–9630. 10.1073/pnas.1015980108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327. 10.1038/nature05395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Levens D. 2006. Making myc. Curr. Top. Microbiol. Immunol. 302:1–32 [DOI] [PubMed] [Google Scholar]
- 36.Vervoorts J, Luscher-Firzlaff J, Luscher B. 2006. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 281:34725–34729. 10.1074/jbc.R600017200 [DOI] [PubMed] [Google Scholar]
- 37.Bartholomeusz G, Talpaz M, Bornmann W, Kong LY, Donato NJ. 2007. Degrasyn activates proteasomal-dependent degradation of c-Myc. Cancer Res. 67:3912–3918. 10.1158/0008-5472.CAN-06-4464 [DOI] [PubMed] [Google Scholar]
- 38.Noguchi K, Kitanaka C, Yamana H, Kokubu A, Mochizuki T, Kuchino Y. 1999. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J. Biol. Chem. 274:32580–32587. 10.1074/jbc.274.46.32580 [DOI] [PubMed] [Google Scholar]
- 39.Alarcon-Vargas D, Ronai Z. 2004. c-Jun-NH2 kinase (JNK) contributes to the regulation of c-Myc protein stability. J. Biol. Chem. 279:5008–5016. 10.1074/jbc.M312054200 [DOI] [PubMed] [Google Scholar]
- 40.Iavarone C, Catania A, Marinissen MJ, Visconti R, Acunzo M, Tarantino C, Carlomagno MS, Bruni CB, Gutkind JS, Chiariello M. 2003. The platelet-derived growth factor controls c-myc expression through a JNK- and AP-1-dependent signaling pathway. J. Biol. Chem. 278:50024–50030. 10.1074/jbc.M308617200 [DOI] [PubMed] [Google Scholar]
- 41.Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. 2012. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151:56–67. 10.1016/j.cell.2012.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D. 2012. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151:68–79. 10.1016/j.cell.2012.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunet J, Pfaff AW, Abidi A, Unoki M, Nakamura Y, Guinard M, Klein JP, Candolfi E, Mousli M. 2008. Toxoplasma gondii exploits UHRF1 and induces host cell cycle arrest at G2 to enable its proliferation. Cell. Microbiol. 10:908–920. 10.1111/j.1462-5822.2007.01093.x [DOI] [PubMed] [Google Scholar]
- 44.Molestina RE, El-Guendy N, Sinai AP. 2008. Infection with Toxoplasma gondii results in dysregulation of the host cell cycle. Cell. Microbiol. 10:1153–1165. 10.1111/j.1462-5822.2008.01117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lüder CG, Gross U. 2005. Apoptosis and its modulation during infection with Toxoplasma gondii: molecular mechanisms and role in pathogenesis. Curr. Top. Microbiol. Immunol. 289:219–237 [DOI] [PubMed] [Google Scholar]
- 46.Laliberté J, Carruthers VB. 2008. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell. Mol. Life Sci. 65:1900–1915. 10.1007/s00018-008-7556-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiuchi N, Nakajima K, Ichiba M, Fukada T, Narimatsu M, Mizuno K, Hibi M, Hirano T. 1999. STAT3 is required for the gp130-mediated full activation of the c-myc gene. J. Exp. Med. 189:63–73. 10.1084/jem.189.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.La Rosa FA, Pierce JW, Sonenshein GE. 1994. Differential regulation of the c-myc oncogene promoter by the NF-kappa B rel family of transcription factors. Mol. Cell. Biol. 14:1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim L, Denkers EY. 2006. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J. Cell Sci. 119:2119–2126. 10.1242/jcs.02934 [DOI] [PubMed] [Google Scholar]
- 50.Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, Kuo MT. 2012. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 72:2622–2633. 10.1158/0008-5472.CAN-11-3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valère A, Garnotel R, Villena I, Guenounou M, Pinon JM, Aubert D. 2003. Activation of the cellular mitogen-activated protein kinase pathways ERK, P38 and JNK during Toxoplasma gondii invasion. Parasite 10:59–64 [DOI] [PubMed] [Google Scholar]
- 52.Dessauge F, Hilaly S, Baumgartner M, Blumen B, Werling D, Langsley G. 2005. c-Myc activation by Theileria parasites promotes survival of infected B-lymphocytes. Oncogene 24:1075–1083. 10.1038/sj.onc.1208314 [DOI] [PubMed] [Google Scholar]
- 53.Huang MJ, Cheng YC, Liu CR, Lin S, Liu HE. 2006. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 34:1480–1489. 10.1016/j.exphem.2006.06.019 [DOI] [PubMed] [Google Scholar]
- 54.Blader IJ, Saeij JP. 2009. Communication between Toxoplasma gondii and its host: impact on parasite growth, development, immune evasion, and virulence. APMIS 117:458–476. 10.1111/j.1600-0463.2009.02453.x [DOI] [PMC free article] [PubMed] [Google Scholar]